

Abstract
Accumulation of pathological tau protein is a major hallmark of Alzheimer’s disease. Tau protein spreads from the entorhinal cortex to the hippocampal region early in the disease. Microglia, the primary phagocytes in the brain, are positively correlated with tau pathology, but their involvement in tau propagation is unknown. We developed an adeno-associated virus–based model exhibiting rapid tau propagation from the entorhinal cortex to the dentate gyrus in 4 weeks. We found that depleting microglia dramatically suppressed the propagation of tau and reduced excitability in the dentate gyrus in this mouse model. Moreover, we demonstrate that microglia spread tau via exosome secretion, and inhibiting exosome synthesis significantly reduced tau propagation in vitro and in vivo. These data suggest that microglia and exosomes contribute to the progression of tauopathy and that the exosome secretion pathway may be a therapeutic target.
The development of tau pathology in Alzheimer’s disease is correlated with progressive cognitive dysfunction and neuronal loss. Neurofibrillary tangles (NFTs), composed of abnormally phosphorylated tau protein, first appear in the entorhinal cortex (EC) before any sign of symptoms and then progress in a hierarchical pattern, distributing to the hippocampus and neocortex1. A growing body of evidence suggests that pathological tau protein spreads between cells and recruits native tau to be transformed into fibrillar aggregates in which tau pathology develops2–8. Remarkably, recent studies have shown that the inoculation of preformed fibrillar tau protein into tau transgenic mice can cause Alzheimer’s disease–like NFT pathology in connected brain regions in fairly rapid manner2–5. Transgenic mouse models expressing a human tau (hTau) mutation specifically in the EC show that misfolded tau spreads through anatomically connected neurons, suggesting trans-synaptic transmission of tau aggregates6–8. Moreover, preformed tau aggregates can also spread beyond synaptic connections, suggesting the existence of nonsynaptic propagation pathways7,9. It has been a matter of debate as to how tau protein is released from neurons and then transduced to recipient neurons to initiate tau propagation. Tau protein can be secreted from neurons by synaptic excitation, such as glutamatergic neuro-transmission in vivo10. Tau fibrils can be transferred from cell to cell in vitro and in vivo11. Extracellular vesicles are emerging as a player in cellular communication and transport of pathogenic proteins related to Alzheimer’s disease12. Notably, tau protein is identified in specific extracellular vesicles called exosomes in the cerebrospinal fluid (CSF) of patients with Alzheimer’s disease13. Furthermore, the exosomal tau protein level in blood is elevated in prodromal Alzheimer’s disease14. These previous findings suggest possible mechanisms of tau transmission, with an observable dependence on tau’s structural nature. Exact mechanisms involving secretory pathways have yet to be elucidated, which motivates our further study of microglia, resident phagocytes with secretory properties.
Microglia, resident mononuclear phagocytes in the CNS, constantly survey the brain parenchyma using their branched projections to detect tissue damage or microbial insults. Engaged in tissue defense as primary innate immune cells in the brain, they phagocytose not only dying cells, debris and protein aggregates, but also living neurons or synapses15,16. Recent attention has focused on the function of microglia in exocytosing microvesicles, such as exosomes, as vehicles for antigen presentation, cytokines and microRNAs17. Although evidence suggests the involvement of microglia in tau pathology of Alzheimer’s disease18,19, the role of microglia in tau propagation has never been explicitly tested. Microglia could aid tau dissemination because they are highly endocytic (via phagocytosis) and exocytic (via exosomes). Thus we hypothesized that microglia may facilitate tau protein propagation between neurons by phagocytosing and exocytosing tau protein. To test this hypothesis, we developed a rapid tau propagation mouse model that shows tau transmission between the EC and the dentate gyrus (DG) within 28 d after the injection of adeno-associated virus (AAV) expressing a tau transgene in a neuron-specific manner.
RESULTS
Development of rapid tau propagation model
Previously reported mouse models take ~2 years for tau propagation from the EC to DG6–8. To accelerate the study of tau propagation, we developed a model exhibiting rapid tau propagation using an AAV vector that expresses the transgene under the control of the neuron-specific synapsin-1 promoter (AAV2/6-SYN1)20. AAV2/6-SYN1 expressing the human P301L tau 1–441 mutant (AAV-tau hereafter) and AAV2/6-SYN1-green fluorescent protein (AAV-GFP hereafter) or AAV-GFP alone as control were stereotactically injected into the medial entorhinal cortex (MEC) layer II/III region of 4-month-old C57BL/6 male mice (Supplementary Fig. 1a,b). GFP expression was identified in MEC layer II/III at 7 days post-injection (dpi) (Supplementary Fig. 1c). Immunofluorescence against NeuN (a neuronal marker), Iba1 (a microglial and myeloid cell marker), glial fibrillary acidic protein (GFAP, an astrocytic marker) and GFP in the MEC revealed expression of GFP in neurons but not in microglia or astrocytes, confirming neuron-specific gene expression in this system (Supplementary Fig. 1d–f). The expression of tau protein was detected by monoclonal antibodies AT8 (specific to tau phosphorylated on Ser202 and Thr205 (pTau)) and HT7 (specific to hTau). AT8+ pTau colocalized with GFP in the MEC at both 7 and 28 dpi in AAV-GFP/tau mice (Fig. 1a). Layer II EC neurons project axon terminals to the outer molecular layer (OML) of the DG of the hippocampus via the perforant path. GFP+ axon terminals of EC neurons were detected in the OML of both AAV-GFP and AAV-GFP/tau-injected mice at 7 dpi. They were also immunoreactive to AT8 in AAV-GFP/tau mice (Fig. 1a), demonstrating the distribution of tau protein from the MEC to the OML. The dendrites of dentate granule cells are connected to axon terminals of EC neurons in the OML, where trans-synaptic transmission of tau protein is believed to take place. Dentate granule cells became AT8+ by 28 dpi (Fig. 1a), suggesting the accumulation of pTau. No GFP signal was detected in the granule cell layer (GCL) in AAV-GFP or AAV-GFP/tau mice, ruling out the possibility that this was due to the infection of granule neurons by AAV-tau. There was no pTau or hTau signal in AAV-GFP mice at either 7 or 28 dpi (Fig. 1a). In addition, we performed a combination of in situ hybridization and immunofluorescence to detect the expression of the recombinant gene and endogenous murine Mapt (mTau) mRNA in vivo. cRNA probes were designed against the 3′-UTR WPRE (woodchuck hepatitis virus post-transcriptional regulatory element) sequence for AAV-derived genes and the 3′-UTR sequence of mTau to minimize cross-reactivity. We detected mTau mRNA signal in all neurons in the MEC or DG (Fig. 1b,c) and WPRE mRNA signal in the GFP+ cells in the MEC (Fig. 1b), but not in the GCL (Fig. 1c). Notably, HT7+ signals were detected in the GCL by 7 dpi with AAV-GFP/tau mice and increased by 28 dpi (Fig. 1d), showing hTau transmission from the OML to the GCL. These data confirm the transmission of hTau but not AAV from MEC neurons to GCL neurons.
Figure 1.
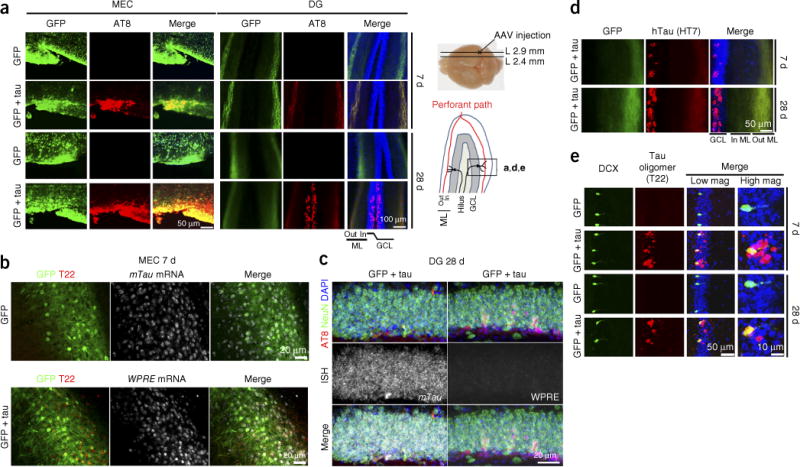
Tau propagates to the DG after the injection of AAV-GFP/tau in the MEC of the mouse brain. (a) C57BL/6 mice at 4 months of age were injected in the MEC with AAV-GFP or AAV-GFP/tau, then sacrificed at 7 or 28 dpi and subjected to immunofluorescence for AT8 (pTau at pSer202 and Ser205, red), GFP (green) and DAPI (blue) in the MEC (left panels) and the dentate gyrus (DG) regions (right panels). The scheme of the right side depicts the anatomical orientations of the images; L, lateral; Out ML, outer molecular layer; In ML, inner molecular layer; GCL, granule cell layer. (b) Combined immunofluorescence at 7 dpi of GFP (green) and T22 (tau oligomer, red) and in situ hybridization of the 3′ UTR of mTau mRNA (top, white) or the AAV-derived 3′-UTR WPRE (bottom, white) in the MEC of AAV-GFP (top) or AAV-GFP/tau-injected mice (bottom). (c) Combined immunofluorescence for GFP (green), AT8 (red) and DAPI (blue) and in situ hybridization (ISH) of mTau (left, white) or WPRE mRNA (right, white) in the DG GCL of AAV-GFP/tau-injected mice at 28 dpi. (d) HT7 (hTau, red), GFP (green) and DAPI (blue) in the DG of AAV-GFP/tau injected mice at 7 and 28 dpi. (e) DCX (doublecortin: immature neuronal marker, green), T22 (red) and DAPI (blue) in the GCL of AAV-GFP or AAV-GFP/tau-injected mice at 7 and 28 dpi; scale bars: 50 μm for low magnifications and 10 μm for high magnifications. All immunofluorescence images are representative of 3 independent experiments.
Next we examined tau pathology development by immunofluorescence to T22 (a tau oligomer–specific marker), CP13 (specific to pSer202 pTau), HT7, doublecortin (DCX; an immature neuronal cell marker), NeuN (a mature neuronal marker), P2ry12 (a microglia-specific marker), Iba1 (a mononuclear phagocyte marker), GFAP (an astrocytic marker) and activated caspase-3 (an apoptotic cell marker). We detected T22+ cells in the GCL, and some of them colocalized with DCX+ in AAV-GFP/tau mice at both 7 and 28 dpi (Figs. 1e and 2a). Co-immunostaining with tau markers and several cellular markers revealed that T22+ cells were 56% NeuN+ mature neurons, 35% DCX+ immature neurons and 0.8% GFAP+ astrocytes at 7 dpi, and 38% DCX+ immature neurons at 28 dpi (Fig. 2b–g). There was no T22 staining in AAV-GFP mice at either 7 or 28 dpi. CP13+ cells were 81% DCX+ immature neurons at 7 dpi, which was reduced to 29% at 28 dpi (Fig. 2e–g), suggesting the maturation of tau-bearing neurons from CP13+DCX+ to AT8+NeuN+ cells. Notably, 11% of HT7+ cells colocalized with P2ry12+ microglia (Fig. 2e–g) at 7 dpi, showing the phagocytosis of hTau by microglia. At 28 dpi, AT8+ cells were 66% NeuN+ mature neurons, 34% DCX+ immature neurons, 18% P2RY12+ microglia and 5% GFAP+ astrocytes. Microglia showed accumulation around AT8+mTau+ cells in the DG (Fig. 2c,d). HT7+ cells were 34% and 43% DCX+ immature neurons at 7 and 28 dpi, respectively. No cells were stained by MC1 (detecting an immature tau aggregation form preceding oligomerization) or thioflavin-S (filamentous tau) in the GCL at either time point. Moreover, 80% of the AT8+ cells in the GCL were positive for activated caspase-3 in AAV-GFP/tau mice at 28 dpi (Fig. 2g and Supplementary Fig. 2a), exhibiting cytopathic changes in tau-bearing neurons in the DG. The number of Iba1+ microglia was unchanged between AAV-GFP and AAV-GFP/tau mice in the DG at 7 and 28 dpi (Supplementary Fig. 3a,b). These data corroborate with previously reported studies that microglia phagocytose apoptotic tau-bearing neurons15,16,18,21,22.
Figure 2.
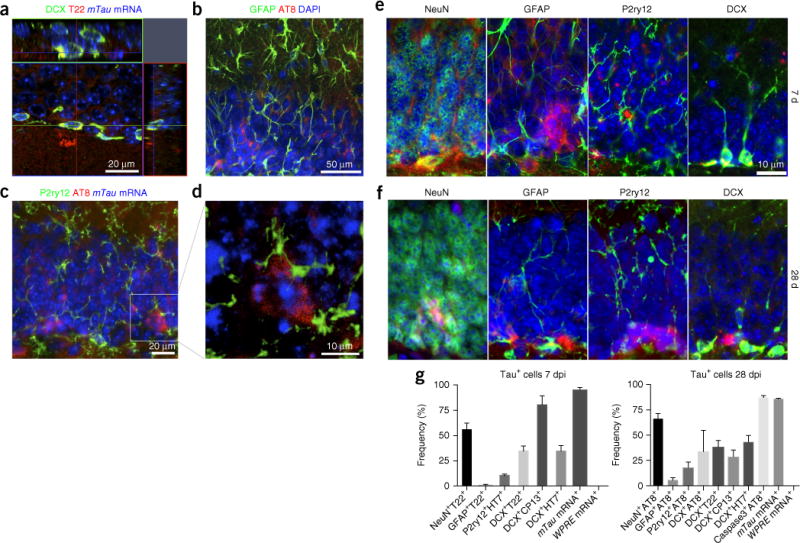
Immunofluorescence for tau with cellular markers. (a) Stacked sequential confocal microscopy imaging at 7 dpi of the GCL in the C57BL/6 mouse brain injected with AAV-GFP/tau into the MEC. DCX, green; T22, red; DAPI, blue. Scale bar, 20 μm. (b,c) Immunofluorescence at 28 dpi of the GCL in the C57BL/6 mice brain injected with AAV- GFP/tau in the MEC: GFAP (astrocytes, green), AT8 (red) and DAPI (blue) or P2ry12 (microglia, green), AT8 (red) and mTau mRNA (in situ hybridization, blue). (d) Stacked sequential confocal microscopy imaging of sections in c. AT8+ and mTau mRNA+ tau-bearing neurons are surrounded by P2ry12+ microglia (green). Scale bar, 10 μm. (e,f) Immunofluorescence of the GCL of a AAV-GFP/tau-injected mouse brain for cellular markers (green) and tau markers (red) at 7 (e) or 28 dpi (f). Scale bar, 10 μm. (e) From left to right, NeuN (mature neurons) and T22, GFAP and T22, P2ry12 and HT7 (hTau), and DCX and T22. (f) NeuN and AT8, GFAP and AT8, P2ry12 and AT8, and DCX and AT8. (g) Quantification of tau+ cells in AAV-GFP/tau-injected mice at 7 (left) or 28 dpi (right). The values represent double-positive cells with the cellular marker and the tau marker as a percentage of total tau marker–positive cells in the DG (mean ± s.e.m.). For mTau and WPRE mRNA, in situ hybridization was performed before the immunostaining for tau markers (T22 for 7 dpi and AT8 for 28 dpi).
Depletion of microglia suppresses tau propagation
To determine whether microglia actually phagocytose tau aggregates, we performed confocal and electron microscopic imaging of PS19 mice expressing P301S human tau (isoform 1N4R, containing four microtubule-binding repeats and lacking exon 3). PS19 mice develop tau aggregation by 3 months of age, neuroinflammation by 4 months and hippocampal neurodegeneration by 12 months23. Confocal microscopy imaging revealed colocalization of AT8+ pTau with Iba1+ microglia in the hippocampal region (Fig. 3a and Supplementary Fig. 4a). Consistent with this observation, double-immunogold labeling and electron microscopy identified PHF1 (pTau at pSer396 and Ser404)-immunoreactive electron-dense materials within Iba1+ microglial processes (Fig. 3a and Supplementary Fig. 4b). Some microglia engulfing tau aggregates were closely apposed to PHF1+ neuronal cells. These data demonstrate that microglia phagocytose tau aggregates in vivo, which provides further support for the hypothesis that microglia facilitate tau transmission from neuron to neuron via efficient phagocytosis and exocytosis of tau protein.
Figure 3.
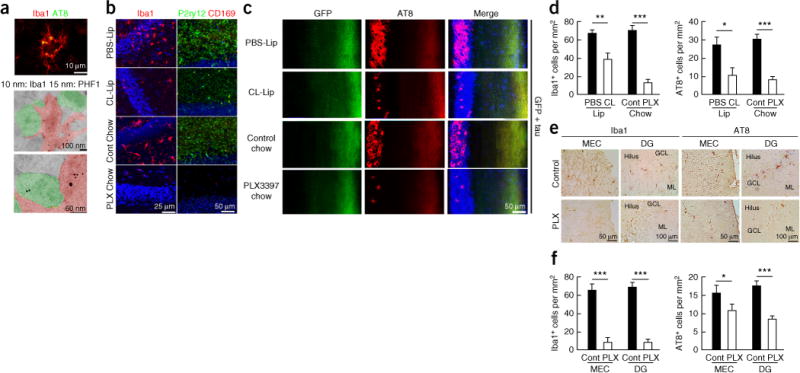
Microglial depletion suppresses tau propagation in the DG of AAV-GFP/tau-injected mice and PS19 tau mice. (a) Colocalization of tau with microglia in the hippocampal region of PS19 (P301S tau transgenic) mouse brain by laser-scanning confocal microscopy (top) and immunogold electron microscopy with two different gold particle sizes (middle and bottom). (b) Microglia depletion of AAV-GFP/tau mice by ICV injection of clodronate liposome (CL-Lip) or feeding with PLX3397 chow (290 p.p.m.), followed by immunofluorescence for Iba1 (mononuclear phagocyte marker; left, red), or P2ry12 (right, green) and CD169 (infiltrating monocyte marker, red) in the DG. (c) Immunofluorescence for AT8 (red), GFP (green) and DAPI (blue) in the DG at 28 dpi. (d) Quantification of the number of Iba1+ and AT8+ cells in the GCL of AAV-GFP/tau mice in c. Iba1+ cells in the PBS group (n = 3 mice, 18 sections) and CL group (n = 3 mice, 18 sections): P = 0.0013, t(34) = 3.506; Iba1+ cells in control (n = 4 mice, 16 sections) and PLX33973397 chow groups (n = 4 mice, 11 sections), P < 0.0001, t(25) = 7.185; AT8+ cells in PBS (n = 3 mice, 16 sections) and CL groups (n = 3 mice, 14 sections): P = 0.017, t(28) = 3.470; AT8+ cells in control (n = 4 mice, 18 sections) and PLX3397 chow groups (n = 4 mice, 16 sections): P < 0.0001, t(32) = 4.605. All unpaired t-tests. (e) Microglial depletion in the PS19 mice at 4.5 months of age after feeding PLX3397 chow for 4 weeks, followed by immunohistochemistry for Iba1 (red) and AT8 (red) and quantification of the number of Iba1+ and AT8+ cells in the MEC and DG. (f) Quantification of the number of Iba1+ and AT8+ cells in the GCL of PS19 mice in e. Iba1+ cells in the MEC in control (n = 4 mice, 23 sections) and PLX3397 chow groups (n = 3 mice, 12 sections): P < 0.0001, t(33) = 7.647; Iba1+ cells in the DG in control (n = 4 mice, 20 sections) and PLX3397 chow groups (n = 3 mice, 11 sections), P < 0.0001, t(32) = 10.35; AT8+ cells in control (n = 4 mice, 19 sections) and PLX3397 chow groups (n = 4 mice, 14 sections): P = 0.033, t(28) = 2.243, AT8+ cells in control (n = 4 mice, 18 sections) and PLX3397 chow groups (n = 4 mice, 14 sections): P < 0.0001, t(30) = 4.791. All unpaired t-tests. Error bars represent s.e.m. *P < 0.05, **P < 0.01 and ***P < 0.001 as determined by unpaired Student’s t-test.
To test this hypothesis, we depleted microglia in AAV-GFP/tau injected mice by two different methods: intracerebroventricular (ICV) infusion of clodronate liposome (CL-Lip) or feeding of chow containing PLX3397, an inhibitor of colony stimulating factor 1 receptor (CSF1R), which specifically depletes microglia24. For the ICV infusion, we implanted a guide cannula into the lateral ventricle of the mouse brain. AAV-GFP/tau was injected into the MEC and CL- or PBS-Lip was ICV injected through the implanted cannula for 1 month (Supplementary Fig. 5a,b). For the PLX3397 experiment, mice were fed PLX3397-containing or control chow for 1 month to deplete microglia before AAV-GFP/tau injection and were maintained on the same chow for 1 month after AAV injection. Both CL-lip and PLX3397 treatment of AAV-GFP/tau mice reduced Iba1+ and P2ry12+ microglia in the DG, and CD169+ infiltrating monocytes were lacking (Fig. 3b). Remarkably, CL-Lip or PLX3397 treatment caused, respectively, 72% and 86% reductions in AT8+ pTau in the GCL of AAV-GFP/tau mice as compared to the control groups (Fig. 3c,d). We also observed 70% and 86% reductions of Iba1+ microglia as compared to PBS-lip or control chow–treated group, respectively (Fig. 3d). PLX3397 treatment did not affect the expression of hTau in the injection sites as determined by human tau–specific ELISA (Supplementary Fig. 6a). There was no change in the gene expression of either anti- or proinflammatory molecules in the hippocampus of control chow–treated AAV-GFP/tau mice as compared to untreated C57BL/6 mice (Supplementary Fig. 6b). PLX3397 treatment, however, significantly reduced all tested cytokine genes as compared to control chow treatment in the AAV-GFP/tau group and C57BL/6 mice (P < 0.05 or lower; Supplementary Fig. 6b). This indicates that AAV-GFP/tau injection to the MEC does not induce hippocampal inflammation and that microglial depletion reduces the basal expression of pro- and anti-inflammatory cytokines.
We cross-examined the effect of microglial depletion on tau propagation using PS19 mice at 3.5 months of age. PS19 mice were treated with PLX3397 chow for 4 weeks. PLX3397-treated PS19 mice showed more than 90% reduction of Iba1+ microglia in both the EC and DG as compared to the control chow-treated group (Fig. 3e,f). Moreover, PLX3397-treated PS19 tau mice showed 47% and 70% reductions of AT8+ pTau in the EC and DG, respectively, as compared to the control group (Fig. 3f). These data from two independent tauopathy models with two independent microglia depletion approaches show that microglia play a major role in tau propagation from the EC to DG.
Tau propagation reduces excitability in the DG
To examine the physiological effect of rapid tau accumulation in the GCL and the depletion of microglia in the AAV-injection mouse model, we performed field recordings in the DG after tetanic stimulation of the perforant path in AAV-GFP and AAV-GFP/tau mice with control or PLX3397 chow feeding. Recordings were obtained from a total of 61 DG fields in 61 slices. The number of subjects was 3 per group and the number of fields per group was 16 AAV-GFP, 10 AAV-GFP/tau, 16 AAV-GFP with PLX3397 and 19 AAV-GFP/tau mice with PLX3397. We analyzed only fields with evoked population spikes of at least 1 mV in amplitude, which represented >90% of all fields (for example, Fig. 4a). There was no significant difference in the slope of the field excitatory postsynaptic potential between groups, indicating that the DG synaptic response per se was not affected by tau expression in the MEC either with or without microglial depletion (Fig. 4b). However, the population spike amplitude was significantly lower at every stimulus intensity beyond 60 μA in the AAV-GFP/tau group as compared to both AAV-GFP and AAV-GFP/tau with microglia-depletion groups (P < 0.01 as determined by two-way ANOVA; Fig. 4c), while those of the latter two groups were not significantly different. These findings indicate that AAV-GFP/tau injection into the MEC reduces the excitability (as measured by the population spike) of dentate granule cells as compared to that in the AAV-GFP group and that normal responses can be evoked in AAV-GFP/tau mice in which microglia are depleted, consistent with our hypothesis that microglia are critical to the modification of neurophysiological function by tau in tau-bearing dentate granule cell synapses. Tetanizing stimuli elicited an increase in amplitude of the postsynaptic response in ~60% of the fields, with the amplitude of the increase being variable, ranging from ~16% to >100% potentiation across groups. There was no difference in the percentage of fields that potentiated or in the magnitude of potentiation between groups (data not shown).
Figure 4.
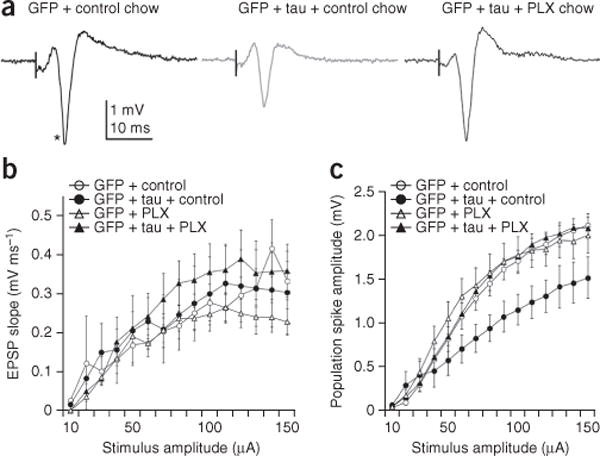
Reduced population spike responses in AAV-GFP/tau mice are rescued by depletion of microglia. (a) Field potential responses in the dentate granule cell layer to a 100-μA stimulus in the middle molecular layer in hippocampal slices prepared from the following: left, AAV-GFP (control); middle, AAV-GFP/tau; right PLX3397-treated (microglia depleted) AAV-GFP/tau mice. Vertical black line represents time of stimulus and asterisk denotes the population spike. (b) Mean input-output relationships for the excitatory postsynaptic potential (EPSP) slope for all fields in slices prepared from AAV-GFP mice fed control chow (open circles; n = 3 mice, 16 slices), AAV-GFP/tau mice fed control chow (filled circles, n = 3 mice, 10 slices), AAV-GFP mice fed PLX3397 chow (open triangles; n = 3 mice, 16 slices) and AAV-GFP/tau mice fed PLX3397 chow (filled triangles, n = 3 mice, 19 slices). (c) Mean input-output relationships for the population spike for all fields in the same groups as b. Error bars in b and c represent s.e.m. P = 0.0003, F(14,364) = 2.932 for AAV-GFP + control versus AAV-GFP/tau + control; P = 0.0605, F(14,350) = 1.668 for AAV-GFP/tau + control versus AAV-GFP + PLX3397; P = 0.0057, F(14,392) = 2.263 for AAV-GFP/tau + control versus AAV-GFP/tau + PLX3397, as determined by two-way repeated-measurement ANOVA.
Microglia phagocytose and secrete tau in exosomes
Microglia efficiently secrete microvesicles, particularly exosomes, as a part of their antigen presentation and cytokine release mechanisms17. Tau protein is found in the exosomal fractions in the cerebrospinal fluid of patients with early-stage Alzheimer’s disease13. Thus, we investigated whether microglia and other brain cells phagocytose and secrete tau protein in exosomes. Primary cultured murine microglia, astrocytes and neurons were incubated with pre-aggregated human tau 1–441 protein (hTau, pre-aggregated to oligomer forms; Supplementary Fig. 7a,b), and the secreted exosome fractions were examined for the presence of tau (Fig. 5a). Microglia efficiently phagocytosed hTau, whereas neurons or astrocytes exhibited less uptake of hTau than microglia (Fig. 5b). The uptake of tau by microglia was significantly inhibited by cytochalasin D, a specific inhibitor of phagocytosis (P < 0.05, Supplementary Fig. 7c,d). Tau in NFTs is often ubiquiti-nated, which is indicative of failed proteasomal or lysosomal clearance. Since ubiquitination of some proteins is a cue for their insertion into multivesicular bodies before their incorporation into exosomes, we tested whether lipopolysaccharide (LPS; Toll-like receptor 4 ligand) would induce tau ubiquitination25. LPS treatment of microglia induced ubiquitination of hTau in the cell lysates as a ladder of molecular weights upon gel electrophoresis (Fig. 5b), which was confirmed by immunoprecipitation of the cell lysate by anti-ubiquitin antibody and immunoblotting using the Tau46 monoclonal antibody specific to the amino acid sequence 404–441 of hTau (Fig. 5b). Ubiquitinated tau was barely detected in the cell lysates of neurons or astrocytes (Fig. 5b).
Figure 5.
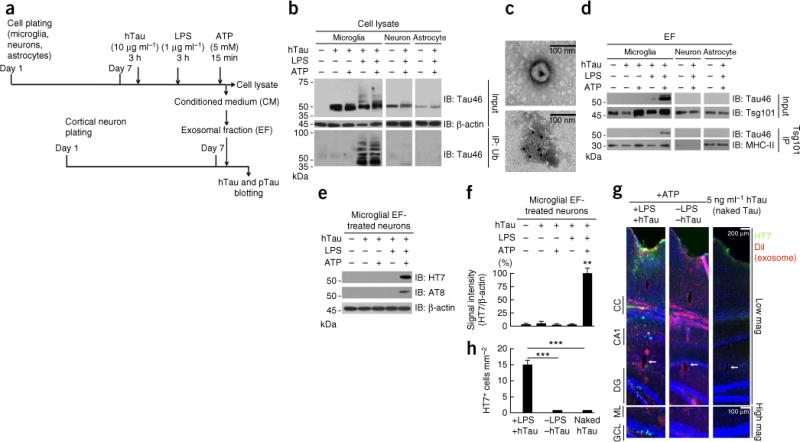
Microglia phagocytose and secrete human tau in exosomes, which facilitates tau propagation to neurons in vitro and in vivo. (a) Experimental timeline for in vitro tau transmission from primary cultured murine microglia, neurons or astrocytes to neurons. (b) Cell lysates (Input) were immunoblotted (IB) for total tau (Tau46) and β-actin (upper panels), or subjected to immunoprecipitation (IP) with anti-ubiquitin (Ub) antibody and immunoblotting with Tau46 (lower panels). (c) Electron micrograph of purified exosomes from microglia (top) and immunoelectron micrograph of exosomes with Tsg101 immunogold labeling (10-nm gold particles, bottom). (d) The exosomal fraction (EF) separately collected from conditioned media was immunoblotted for Tau46 or Tsg101 (upper panels). The EF was also subjected to immunoaffinity purification with Tsg101 antibody and immunoblotted for Tau46 or MHC-II (another exosomal marker, lower panels). (e) The EF was applied to primary cultured murine cortical neurons for 4 h, and after washing, the neuron cell lysates were tested for the uptake of hTau with HT7 or AT8. Results are representative of 3 independent experiments. (f) Quantification of band intensity in c. **P < 0.0001, F(4,10) = 253.3, n = 3 per group as determined by one-way ANOVA and Tukey post hoc test. Error bars in b and c represent s.e.m. (g) The DiI-labeled EF containing tau (+LPS +tau) or no tau (−LPS −tau), or 5 ng μl−1 of naked human tau aggregates, were injected into the OML of DG in C57BL/6 mice and sacrificed 21 dpi for immunofluorescence for HT7 (green), exosome (DiI, red) and DAPI (blue) labeling along the injection tract (top) and in the DG regions (bottom). CC, corpus callosum; arrows, injection site in the OML. (h) Quantification of HT7+ cells. Error bars represent s.e.m. ***P < 0.0001, F(2,27) = 36.08 as determined by one-way ANOVA and Tukey post hoc test for +LPS +tau (n = 4 mice, 12 sections), −LPS −tau (n = 3 mice, 9 sections) and naked tau (n = 3 mice, 9 sections) groups. Full-length blots are presented in Supplementary Figure 10.
ATP, released from damaged cells as an alarmin, stimulates purinergic receptors and induces exocytosis of exosomes25. Our exosome preparation from ATP-stimulated microglia-conditioned medium was enriched in microvesicles 50–100 nm in diameter, which were immunoreactive to Tsg101, an exosomal marker, as determined by electron microscopy (Fig. 5c). ATP stimulation of microglia, but not neurons or astrocytes, led to secretion of tau in the exosome fraction in an LPS treatment–dependent manner (Fig. 5d). Since it is possible that tau protein detected by the immunoblotting was not in exosomes but co-enriched in the exosomal fraction, we further immunoaffinity-purified exosomes from the enriched fraction using an anti-Tsg101 antibody as described26. Tau protein was detected in purified exosomes from microglia treated with LPS and ATP but not in purified exosomes from neurons or astrocytes similarly treated (Fig. 5d), confirming the presence of tau protein in exosomes from microglia. No AT8+ pTau was detected in microglial exosomes by immunoblotting (data not shown). To determine whether exosomes transduce hTau to neurons, we applied the microglial exosomal fraction to primary cultured murine cortical neurons. We detected both hTau and pTau in neurons by immunoblotting using HT7 and AT8 antibodies, respectively (Fig. 5e,f), demonstrating that hTau is transmitted from exosomes to neurons and is phosphorylated.
Efficient transduction of tau via exosomes in vivo
To demonstrate the transduction of tau protein from exosomes into neurons in vivo, we labeled the purified tau-containing exosomes from hTau-phagocytosed microglia with DiI (dioctadecyl-3,3,3′,3′- tetramethylindocarbocyanine) (Supplementary Fig. 8b) and stere-otactically injected them into the OML of the DG in C57BL/6 mice. We examined the brain tissue 3 weeks after injection using HT7 human tau antibody by immunofluorescence microscopy. Both DiI-labeled exosomes and hTau were transduced in the GCL of the mice injected with tau-containing exosomes, but there was no hTau signal in the GCL of mice injected with exosomes without tau or an equal amount of naked pre-aggregated hTau (5 ng μl−1; Fig. 5g,h and Supplementary Fig. 8c,d). These data demonstrate that microglia play a major role in phagocytosis and secretion of tau protein in exosomes and that microglia-derived exosomes transduce tau to neurons more efficiently than the naked form in vivo.
Characterization of tau-containing exosomes from PS19 mice
To test the existence of tau in exosomes in vivo, we isolated brain exosomes from PS19 mice by sequential centrifugation and filtration as described27. We detected HT7+ hTau, PHF1+ pTau and Tsg101+ immunoreactivity by immunoelectron microscopy (Fig. 6a), demonstrating the existence of tau in exosomes in tau mouse brain. We measured the size of extracellular vesicles immunogold-labeled for tau. Tau+ extracellular vesicles in the exosome range (50–100 nm in diameter) constituted 68% of total extracellular vesicles (Fig. 6b). The average sizes of extracellular vesicles were 106.4 ± 29.6 nm for tau− vesicles (n = 261) and 99.1 ± 31.5 nm for tau+ (n = 107, Fig. 6c). These data demonstrate that exosomes are a major subgroup of tau-bearing extracellular vesicles in PS19 mouse brain.
Figure 6.
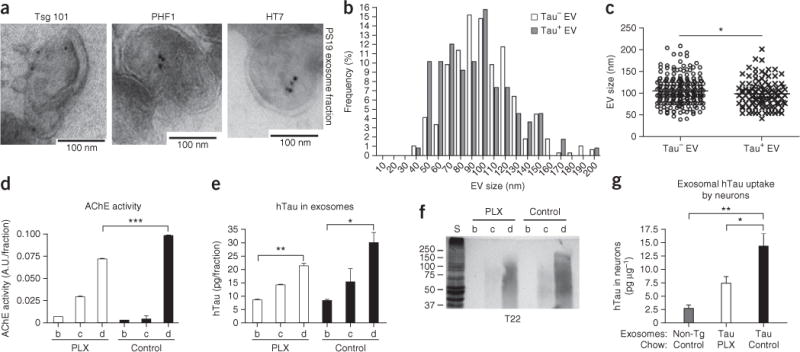
Characterization of tau-containing extracellular vesicles from microglia depleted PS19 mouse brain. (a) Electron micrograph from the exosomal fraction of PS19 tau mouse brains. Immunogold labeling of Tsg101 (exosome), PHF1 (pTau) and HT7 (hTau). (b,c) Histogram of the size of tau+ (n = 107) or tau− extracellular vesicles (EVs, n = 261) (b) and their quantification (c). *P = 0.0371, t(366) = 2.092 as determined by unpaired Student’s t-test. (d) AChE activity of sucrose gradient ultracentrifugation fractions. A.U., arbitrary units. ***P < 0.0001, F(5,6) = 539.3 as determined by one-way ANOVA and Tukey post hoc test (n = 3 per group). (e) Quantification of hTau in each fraction by hTau-specific ELISA. P = 0.0078, F(5,6) = 9.632 as determined by one-way ANOVA and Tukey post hoc test (n = 3 per group). (f) Immunoblotting of tau oligomers using T22 antibody. (g) Ten micrograms of fraction d (exosome fraction) from non-transgenic (non-Tg), PS19 tau mice with a 4-week treatment with PLX3397 chow (tau PLX) or PS19 mice with control chow treatment (tau control) were applied to primary cultured murine cortical neurons as described in Figure 5a, and exosome-transferred hTau in the neurons was quantified by hTau ELISA. P = 0.0026, F(2,9) = 12.34 as determined by one-way ANOVA and Tukey post hoc test (n = 3 per group). Error bars represent s.e.m. *P < 0.05 and **P < 0.01. Results are representative of 3 independent experiments.
We next investigated whether exosomes isolated from PS19 mice could transduce tau into primary cultured neurons and whether depletion of microglia would affect this transduction. PS19 mice were fed either control or PLX3397 chow for 4 weeks, and brain exosomes were isolated by sucrose gradient ultracentrifugation27. Exosomal fractions were conventionally determined by acetylcholine esterase (AChE) activity28, which was enriched in fractions c and d after sucrose step-gradient ultracentrifugation of brain homogenates (Fig. 6d). AChE activity in fraction d was significantly reduced in the PLX3397 group as compared to the control group (P < 0.0001, Fig. 6d). PXL3397 treatment led to a trend toward reduced total hTau in fraction d (Fig. 6e). Moreover, T22+ oligomeric hTau was mostly enriched in fraction d and was reduced in the PLX3397-treated PS19 group (Fig. 6f). Finally, we applied 10 μg of exosomal fraction d from each group to primary cultured murine cortical neurons. The exosomes isolated from PS19 transferred hTau to neurons ex vivo, and this was significantly reduced by PLX3397 treatment of PS19 mice (P < 0.05, Fig. 6g). These data demonstrate that (i) exosomes isolated from PS19 mouse brains contain hTau oligomers, (ii) microglial depletion partially reduces the hTau contents in exosomes, (iii) exosomal tau is readily transmissible to neurons and (iv) microglial depletion suppresses exosomal transmission of tau ex vivo.
Inhibition of sphingomyelinase-2 suppresses tau propagation
Next we examined whether pharmacologic inhibition of exosome synthesis could reduce tau secretion from microglia. Ceramide is an essential lipid for a subtype of exosome biogenesis in multivesicular bodies, and its synthesis is regulated by neutral sphingomyelinase-2 (nSMase2, also known as sphingomyelin phosphodiesterase 3, Smpd3). Therefore, inhibition of nSMase2 reduces the synthesis of exosomes29. We targeted nSMase2 in microglia by two different approaches: silencing nSMase2 expression by short interfering RNA or pharmacological inhibition of nSMase2 activity by GW4869. Treatment of microglia with siRNA against nSMase2 for 24 h suppressed Smpd3 (nSMase2) gene expression by 55% as compared to scrambled siRNA-treated microglia (Supplementary Fig. 8a). Microglia were also separately treated with either GW4869 or control vehicle, 5% dimethylsulfoxide (DMSO) in PBS, for 24 h. Both groups of microglia were incubated with hTau and stimulated with LPS and ATP, and the collected exosomal fraction from microglial conditioned medium was subjected to immunoblotting using Tau46 and Tsg101 antibodies. Strikingly, either silencing nSMase2 expression or inhibiting nSMase2 activity in microglia reduced secretion of hTau in exosomes (Fig. 7a,b). Subsequent treatment of primary cultured neurons with tau-containing exosomal fractions from microglia showed significantly reduced transduction of hTau into neurons by nSMase2 siRNA treatment (36.3 ± 7.13% of scrambled siRNA–treated group, P = 0.0017, F(2,2) = 5.022, n = 3 per group; Fig. 7c) and by GW4869 treatment (36.0 ± 10.97% of DMSO-treated group, P = 0.0060, F(2,2) = 4.439, n = 3 per group; Fig. 7d). These data reveal that nSMase2 is a potential therapeutic target for inhibiting tau propagation.
Figure 7.
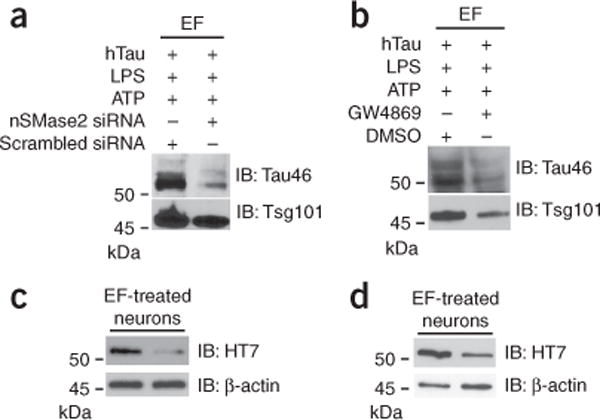
nSMase2 silencing or inhibition reduces exosomal secretion and exosome-mediated tau transmission from microglia to neurons. (a,b) Primary cultured microglia were either transfected with siRNA against murine nSMase2 gene or scrambled siRNA (a) or treated with 3 μM GW4869 in 0.5% DMSO in DMEM for 24 h (b), followed by treatment with oligomeric hTau, LPS and ATP as described for Figure 5a. The exosomal fraction (EF) from microglia (10 μg per lane) was immunoblotted with Tau46 and Tsg101. (c,d) Microglia-derived exosome-treated neuronal cell lysates were immunoblotted for hTau (HT7) or β-actin. Results are representative of 3 independent experiments. Full-length blots are presented in Supplementary Figure 10.
Finally, we examined the therapeutic effect of the CNS-penetrant GW4869 on an AAV-GFP/tau mouse model in vivo. GW4869 or control vehicle was intraperitoneally administered once a day for 1 month after AAV-GFP/tau injection into the MEC of C57BL/6 mice. GW4869 reduced the number of AT8+ cells in the GCL by 75% in AAV-GFP/tau mice as compared to the DMSO-treated groups (37.89 ± 3.57 AT8+ cells per mm2 in the DMSO-treated group and 10.90 ± 2.14 AT8+ cells per mm2 in the GW4869-treated group, P = 0.0026, F(2,2) = 3.018, n = 3 per group; Fig. 8a). In addition, GW4869 treatment of PS19 tau mice from 3.5 months of age reduced the number of AT8+ cells in the GCL by 52% (P = 0.0013) but had no effect on AT8+ cells in the MEC (Fig. 8b,c). Consistent with these data, dot blot analysis using T22 antibody revealed a reduction in tau oligomer accumulation of 44% in hippocampal (P = 0.0136) but not MEC regions in the GW4869-treated group as compared to the DMSO-treated group (Fig. 8d). GW4869 treatment did not alter the expression of tau at the injection sites as determined by human tau–specific ELISA (Supplementary Fig. 9a). There was no difference in cytokine gene expression in the hippocampus of DMSO-injected AAV-GFP/tau mice or GW4869-treated AAV-GFP/tau group as compared to untreated C57BL/6 mice, except for a significant increase in the anti-inflammatory cytokine mRNA Il10 by GW4869 treatment (P < 0.001, Supplementary Fig. 9b). These results from two different tau mouse models demonstrate that inhibition of nSMase2 activity by a CNS-penetrant drug, GW4869, is highly effective in slowing tau propagation in vivo. The reduction of tau spread in the GCL by PLX3397 or GW4849 in two different tau mouse models is accompanied by a reduction in activated caspase-3+ cells in the same region (Supplementary Fig. 2b,c), indicating the pathobiological significance of this tau spreading mechanism.
Figure 8.
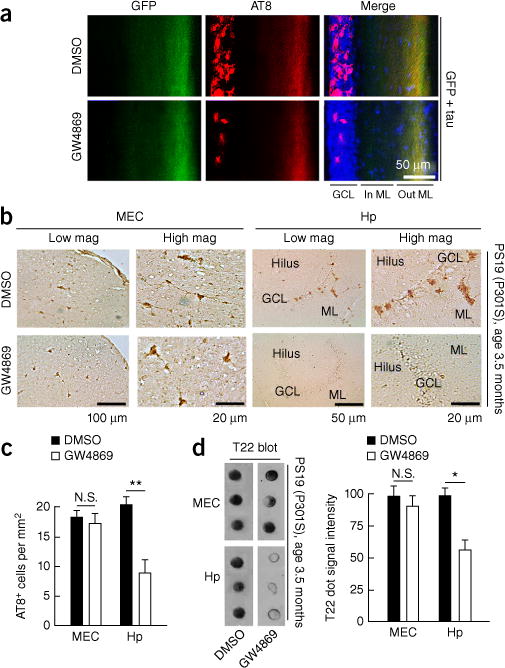
Inhibition of nSMase2 suppresses tau propagation in the DG of AAV-GFP/tau-injected and PS19 tau mice. (a) C57BL/6 mice at 4 months of age were injected with AAV-GFP/tau into the MEC and injected daily intraperitoneally (i.p.) with GW4869 or control vehicle for 4 weeks after AAV injection. Immunofluorescence image shows GFP (green), AT8 (red) and DAPI (blue) and quantification of AT8+ cells in the GCL of the DG in AAV-GFP/tau injected mice. GCL, granule cell layer; In ML, inner molecular layer; Out ML, outer molecular layer. (b,c) PS19 mice at 3.5 months of age were treated with daily i.p. injection of GW4869 (1.25 mg per kg per day in 200 μl of 5% DMSO) or control vehicle for 4 weeks and sacrificed for immunofluorescence with AT8 and quantification of AT8+ cells in the EC and the GCL of the DG in PS19 mice. N.S., no significance (P = 0.8067), t(19) = 0.2481 between DMSO (n = 3 mice, 11 sections) and GW4869-treated groups (n = 3 mice, 10 sections) in the MEC; **P = 0.0013, t(22) = 3.698 between DMSO (n = 3 mice, 12 sections) and GW4869-treated groups (n = 3 mice, 12 sections), as determined by unpaired Student’s t-test. Hp, hippocampus. Error bars represent s.e.m. (d) T22 dot blot of brain tissue lysate of the EC and DG from PS19 mice. N.S., no significance (P = 0.7512), t(4) = 0.3397; *P = 0.0136, t(4) = 4.228, as determined by unpaired Student’s t-test for each brain region (n = 3 mice per group). Error bars in d represent s.e.m.
DISCUSSION
In this study, we confirmed with two different tau mouse models that tau propagation and accompanying neurotoxicity is sensitive to microglial depletion and inhibition of nSMase2 activity. We conclude that microglia play a previously unrecognized and significant role in the propagation of tau protein and neurotoxicity in the brain. The multiple independent lines of our investigation show that microglia promote tau propagation. The importance of microglia in tau propagation is emphasized by the marked reduction of tau propagation by depletion of microglia in two independent models of tauopathy: in the DG and EC regions of PS19 mice, as well as in the DG of the rapid AAV model. The role of microglia in tauopathy indicates that tau propagation can occur through both trans-synaptic and non-trans-synaptic pathways. Microglia-mediated tau propagation could provide an explanation for the evolution of Braak staging, which shows lateral propagation of tau pathology in neurons that are not connected by synapses, such as those from the transentorhinal cortex to higher order sensory association areas of the occipitotemporal gyrus in Braak III stage Alzheimer’s disease. Lateral (nonsynaptic) propagation of tau has also been documented in rTgECtau mice, which reversibly express human variant tau P301L primarily in layer II EC7. The mechanism of this spread was previously unknown. Our discovery of tau propagation through microglial mechanisms provides one highly plausible explanation for the nonsynaptic spread of tau pathology in human brains.
The study of tau propagation has been delayed by the long time courses required to observe the propagation in rodent brains. We describe here a mouse model that is based on AAV-tau injection. This model has several advantages over the rTgECtau mice used in previous studies. The rTgECtau model takes ~2 years to develop tau accumulation in the DG, and tau expression is not specific to the EC in the rTgECtau mice; hTau is also expressed in the mossy fibers and DG granule cells6. In contrast, the AAV-tau injection model exhibits tau pathology in 28 d, is anatomically specific to the EC and has the additional benefit that it is done in C57BL/6 mice. The benefit of the AAV-tau injection model is highlighted by observations that the evolution of tau pathology in this model is similar to that observed in the rTgECtau model, thus providing both speed and pathological fidelity.
The cell biology of microglia in the propagation of tau is still unknown. Our study suggests that microglia phagocytose tau-containing cytopathic neurons or synapses and secrete tau protein in exosomes, which efficiently transmit tau to neurons. Our study demonstrates that the secretion of tau-containing exosomes from microglia is sensitive to nSMase2 inhibition, suggesting that ceramide synthesis is critical for the exosome biogenesis. Our study demonstrates that microglia phagocytose tau, as determined by laser-scanning confocal microcopy and double immunogold labeling of PS19 mouse brain tissue sections with Iba1 and tau markers (HT7 and PHF1). We do not know how microglia phagocytose tau protein from neurons. Microglial phagocytosis of cytopathic but live neurons is known as phagoptosis30. Since we found that tau-bearing neurons were also apoptotic (positive for activated caspase-3), their axonal terminals may express phosphatidylserine for efficient recognition and phagocytosis by microglia. In addition, tau protein shifts in distribution from the axonal to the dendritic compartment upon aggregation and impairs postsynaptic activity31. In accord with this, AAV-GFP/tau-injected mice also showed impairment in excitability in our study. Microglia may efficiently phagocytose tau-containing inactive synapses, which is known as synaptic pruning16. Thus, cytopathic changes of neurons and inactivated dendritic spines may attract microglial phagocytosis and enhance transport of tau from neurons to microglia.
Immunoelectron microscopy of ultrathin-sections of purified exosomes from PS19 mouse brains showed the tau inside the inner leaflet of exosomes. This is also the first time, to our knowledge, that rapid tau propagation has been observed in non-transgenic mice; previous reports have shown tau accumulation in C57BL/6 mice, but not until 11 months after injection32,33. We show evidence that depletion of microglia reduces the content of tau in the brain exosomal fraction purified from PS19 mice. Furthermore, the microglial depletion significantly suppressed exosomal transmission of tau to neurons ex vivo. These data shows that microglia-derived exosomes substantially transmit tau to neurons. In support of this interpretation, higher levels of myeloid cell–derived microvesicles, including exosomes, in the CSF are strongly associated with higher levels of tau in the CSF in Alzheimer’s disease34. Furthermore, there are high levels of pTau (pThr181 and pSer396) in neural exosomes isolated from the blood from patients with prodromal Alzheimer’s disease14. These clinical studies strongly support our findings that exosomes are an important component of the pathobiology of Alzheimer’s disease.
Our study shows that systemic inhibition of nSMase2 by GW4869 in PS19 mice is effective in suppressing tau propagation in the DG but not in the EC region. This suggests that the EC region is either less dependent on exosome-mediated tau propagation or less efficient in the distribution of GW4869. Since the EC is one of the first regions of tau accumulation in PS19 mice, the former possibility would suggest that initial tau accumulation is independent of exosome synthesis and that tau accumulation in the DG is exosome-dependent and is secondary to tau accumulation in the EC region.
Previous studies demonstrated that intracranial injection of preformed fibrillar tau protein into tau transgenic mice accelerates propagation of NFT-like mature tau aggregates, which more closely resemble authentic Alzheimer’s disease–associated human tangles4,5. The spread of NFT pathology as disease progresses, however, does not necessarily mean that NFTs are the only tau species capable of spreading from neuron to neuron. In human Alzheimer’s disease, AT8+ NFT-containing neurons appear in late-stage NFTs, whereas T22 antibody can stain thioflavin-S–negative early tau accumulations. In this study, we first observed accumulation of T22+ tau oligomers and CP13+ pTau, with no mature AT8+ pTau accumulation in the GCL. This suggests that tau oligomers are efficiently transduced from EC to DG neurons in the AAV-GFP/tau injected mouse model, which is followed by development of AT8+ pTau in the somatodendritic compartment. This is consistent with the sequence of conformational changes from normal to pathogenic tau aggregation35. We also found that DCX+ immature neurons were transduced with HT7+ hTau. DCX+ neurons might take up exosomes efficiently because exosomes are enriched in phosphatidylserine36. DCX+ immature neurons are known to efficiently detect phosphatidylserine and phagocytose apoptotic cells by means of the intracellular engulfment protein ELMO1 (ref. 37). Thus, the DCX+ immature neurons might recognize the phosphatidylserine of exosomes and efficiently phagocytose them.
Whether fibrillar or oligomeric tau is toxic to neurons has been a matter of debate38. Indeed, passive immunization of tau mice expressing P301L tau mice with tau oligomer-specific monoclonal antibody reduces tauopathy development and restores spatial learning, demonstrating the potent toxicity of tau oligomers in tau mice39. In accord with this observation, we also found a reduction in neuronal excitability in the DG of AAV-GFP/tau injected mice after stimulation of the perforant path. This reduction was rescued in AAV-GFP/tau mice depleted of microglia, demonstrating that accumulation of non-fibrillar tau in dentate granule cells is toxic to neurons and that microglia affect their physiological function.
In summary, we demonstrate that microglia and exosomes help propagate tau pathology in the mammalian brain. This work adds tau to the growing list of other pathological proteins transmitted through the exosomal pathway, including α-synuclein and prions40–42. Exosomal protein transfer might be a generally applicable mechanism of spreading pathogenic proteins in neurodegenerative disorders. The putative role of microglial and exosomal spread of tau in Alzheimer’s disease is important because it enables new approaches for delaying disease progression. Research in this area should be further accelerated by the development of this rapid tau propagation model.
METHODS
Methods and any associated references are available in the online version of the paper.
ONLINE METHODS
AAV and animal surgery
All experimental procedures using animals were approved by the Institutional Animal Care and Use Committee at Boston University School of Medicine. Animals were housed individually for postoperative monitoring and in a standard light/dark cycle. Recombinant AAV serotype 6 (AAV6) vectors were generated by transiently transfecting the respective genome plasmid (consisting of AAV-2 ITRs, the human synapsin-1 gene promoter driving transgene expression of either EGFP or human P301L tau 1–441, the woodchuck hepatitis virus post-transcriptional control element (WPRE) and a bovine growth hormone polyadenylation site) with the DP6 helper plasmid into human embryonic kidney (HEK293) cells. Viral particles were purified by iodixanol step gradient ultracentrifugation, affinity chromatography on a heparin FPLC column and overnight dialysis against PBS. Titers were determined by Q-PCR and purity > 95% by SDS-PAGE and Coomassie brilliant blue staining. AAV viral particles (VPs) were unilaterally injected into the medial entorhinal cortex (MEC) of male C57BL/6 mouse brain at 4 months of age as descried43. Briefly, mice were anesthetized by 1–2% isoflurane in a 95% O2 and 5% CO2 mixture. After mice were immobilized in a stereotactic apparatus (model 963, David Kopf Instruments, Tujunga, CA), a midline skin incision was made over the bregma. AAV was stereotaxically injected using a motorized microinjector (cat. no. 53311, Stoelting, Wood Dale, IL) and a 10-μl gas-tight microsyringe (cat. no. 80301, Hamilton, Reno, NV) into layer II/III of MEC unilaterally via the following coordinates: anteroposterior, −4.75 mm; lateral, 2.9; dorsoventral, −4.6. For AAV-GFP/tau injection, AAV-tau (9 × 107 VP) and AAV-GFP (1 × 107 VP) was injected in a total of 1 μl into the MEC. For AAV-GFP alone, 1 × 107 VP were injected in a total of 1 μl. To monitor the effect of AAV injection on microgliosis, the same titer of AAV-GFP or AAV-GFP/tau (1 × 108 VP in 1 μl) was injected into the MEC. The animals were sacrificed at 7 or 28 d post infection (dpi).
P301S tau transgenic (PS19) mouse model
The PS19 transgenic mouse line, which expresses human tau (isoform 1N4R, containing four microtubule- binding repeats and lacking exon 3) carrying the FTDP-17-linked P301S mutation, was purchased from Jackson Laboratory (cat. no. 008169, Bar Harbor, ME)23. Non-transgenic control strain B6C3F1 (cat. no. 100010, Jackson Laboratory) was used to maintain PS19 transgene hemizygotes. Adult male PS19 mice at 3.5 months of age were tested for the study. This mouse model does no develop appreciable neuronal loss in the hippocampal region until 6 months of age4.
Intracerebroventricular cannula implantation and clodronate liposome infusion
The intracerebroventricular (ICV) cannula implantation into mouse brains was performed as described44. A stainless steel guide cannula (22G, 2.2 mm below, cat. no. C312GS-5/SPC, Plastics One, Roanoke, VA) with a plastic insert was stereotaxically implanted into the left lateral ventricle (anteroposterior, −0.22 mm; lateral, −1.0 mm) and fixed using Loctite 454 cyanoacrylate adhesive (cat. no. 0008670, Alzet, Cupertino, CA). This was followed by the stereotaxic injection of AAV into the MEC as described above as a part of the single animal surgery. One day after the cannula implantation, 5 μl of clodronate liposome (CL-Lip, 5 mg ml−1, ClodronateLiposome) or PBS-liposome (PBS-Lip, Encapsula NanoSciences) were injected through the injector tubing (24 G, 2.7 mm, cat. no. C312IS-5/SPC, Plastics One), which was attached to a 10-μl gas-tight microsyringe. Liposome injection was repeated once a week, at 7, 14 and 21 dpi (see Supplementary Fig. 5b for the experimental design). No obvious behavioral or health problems were observed after the cannula implantation and during the CL-Lip treatment. The effect of microglial depletion on tau propagation was analyzed by the number of AT8+ cells in the granule cell layer (GCL) of the dentate gyrus (DG) normalized by the GFP+ outer molecular layer (OML) area for AAV-GFP/tau mice, and the number of AT8+ cells in the EC and GCL normalized by the size of EC layer II/III or the GCL area for PS19 mice.
CSF1R inhibitor treatment
The CSF1R inhibitor PLX3397 incorporated into rodent chow at 290 p.p.m. (delivering daily doses of approximately 45 mg kg−1) was provided along with control chow from Plexxikon Inc. (Berkeley, CA)24. C57BL/6 mice at 3.5 months of age were fed PLX3397 or control chow for 4 weeks before and after the AAV injection and then sacrificed (n = 4 per group). PS19 mice at 3.5 months of age were fed PLX3397 or control chow for 4 weeks and sacrificed for biochemical and neuropathological analyses. No obvious behavioral or health problems were observed during the PLX3397 treatment except patchy whitening of the fur. The effect of microglial depletion on tau propagation was analyzed by the number of AT8+ cells in the GCL area normalized by the size of the GFP+ OML area for AAV-GFP/tau mice, and the number of AT8+ cells in the EC and GCL normalized by the size of EC layer II/III or the GCL area for PS19 mice.
Immunohistochemistry
Fixed tissue sections were subjected to immunofluorescence using the following antibodies: AT8 mouse monoclonal antibody (mAb) (phospho-tau, Ser202 and Thr205, 1:500, cat. no. MN1020, Thermo Fisher Scientific Inc.; cat. no. MN1020, Waltham, MA); CP13 mAb (phospho-tau, Ser202, 1:500, kindly provided by P. Davies (Albert Einstein College of Medicine)45, MC1 mAb (conformation-specific tau, 1:500, P. Davies)46, PHF1 mAb (phospho-tau Ser396 and Ser404, 1:500, P. Davies)47; HT7 mAb (1:500, cat. no. MN1000, Thermo Fisher Scientific); T22 rabbit polyclonal antibody (pAb) (1:250, kindly provided by R. Kayed (University of Texas Medical Branch)35, ionized calcium-binding adaptor molecule 1 (Iba1) rabbit pAb (1:1,000, cat. no. 019-19741, Wako, Richmond, VA); P2ry12 rabbit pAb (1:2,000, generated in the laboratory of O.B.)48, CD169 rat mAb (1:500, cat. no. MCA884GA, AbD Serotec, Raleigh, NC), cleaved caspase-3 rabbit pAb (1:500, cat. no. Asp175, Cell Signaling Technology, Danvers, MA); GFP rabbit pAb (1:1,000, cat. no. SC-8334, Santa Cruz Biotechnology, Dallas, TX); doublecortin (DCX) goat pAb (1:100, cat. no. SC-8066, Santa Cruz Biotechnology); DCX rabbit pAb (1:300, cat. no. 4604, Cell Signaling, Danvers, MA). This was followed by incubation with species-specific Alexa Fluor (488, 546, 568 and 647)-conjugated secondary antibodies (1:1,000, cat. no. A-11011, A-11029, A-11031, A-11034, A-20206, A-11003, A-10040, A-21245 and A-21447, Molecular Probes/Invitrogen, Grand Island, NY). Immunostained images were captured using an inverted fluorescence microscope attached to a monochromatic CCD camera (TE2000-U, Nikon Instruments) and the cell number or fluorescence intensity was quantified using ImageJ software (NIH). For immunohistochemistry of PS19 mouse brains, an anti-mouse or anti-rabbit EnVision+ kit (cat. no. K4001, K4003, Dako, Carpinteria, CA) and conventional 3,3′-diaminobenzidine (DAB, cat. no. SK-4100, Vector Laboratories, Inc., Burlingame, CA) were used as a chromogen and cell numbers were analyzed by counting cell bodies using a Nikon Eclipse E600 microscope and color CCD camera (Nikon Instruments). All of the commercially available antibodies were validated by the manufacturer. T22 rabbit polyclonal antibody was a product of the Kayed laboratory and was validated in ref. 35. P2ry12 rabbit polyclonal was a product of the laboratory of O.B. and was validated in refs. 48,49.
Image capture, analysis and quantification
NIS-Elements (Nikon Instruments) and ImageJ software (version 1.43; NIH, Bethesda, MD) were used to capture and crop images and merge channels into composite RGB images. Photoshop (Adobe) was used to adjust individual channels. In all cases, identical adjustments were applied across all images used in an experiment for each channel. Three-dimensional deconvolution software was used for the development of z-stack images using Zen software (Zeiss). Cell number or fluorescence intensity was quantified with ImageJ. For immunofluorescence image quantification, the number of immunoreactive cells in the granule layer or the DG was manually counted using the Cell Counter plug-in on the gridded images and normalized to the specified area (mm2) using 20× original magnification images (4 sections per animal, 3 or 4 animals per group). Area of the granule layer and DG was measured using the ImageJ Measure tool. For DAB IHC, immunoreactive cells in the granule layer and in the DG were counted and normalized to the size of corresponding area (mm2) using 20× original magnification images (4 sections per animal, 3 or 4 animals per group). Microglia density was determined by counting the number of Iba1+ cells in the DG (hilus, subgranular zone and granule cell layer) from at least fifteen 20× objective original magnification images per group.
In situ hybridization
In vitro transcription of cRNA probes against the murine Mapt 3′ UTR (mTau) and AAV 3′ UTR transgene WPRE were constructed in the pBluescript II KS(−) vector. The cRNA sequence for mTau is the 627-bp reverse complement of positions 1606–2236 of murine Mapt mRNA (NM_001038609.2), which has minimal overlap with the human MAPT mRNA sequence. Since there is considerable overlap between the inserted human tau 1–441 gene sequence and the mTau mRNA sequence, the 645-bp reverse complement of the 3′-UTR post-translational regulatory element (WPRE) sequence in the AAV vector was used for the cRNA probe of AAV transgenes. After linearization of the vector, digoxigenin (DIG)-labeled cRNA riboprobe was synthesized using DIG labeling mix (cat. no. 1 277 073, Roche Diagnostics GmbH, Mannheim, Germany) and Riboprobe In Vitro Transcription Systems (cat. no. P1420, Promega, Madison, WI) and purified using ProbeQuant G-50 micro columns (cat. no. GE28-9034, GE Healthcare, Boston, MA). Enhanced fluorescence in situ hybridization was conducted for the detection of mTau and WPRE mRNA on brain tissue sections as described50 using anti-DIG-POD Fab fragments (cat. no. 11 207 733 910, Roche, Branford, CT), the TSA Plus Cy5 fluorescence system (cat. no. NEL745, Perkin Elmer, Waltham, MA) and the HybriWell sealing system (cat. no. HBW20, Grace Bio-Labs, Bend, OR). After the in situ hybridization, the tissue sections were processed for immunofluorescence in darkness as described above.
Laser-scanning confocal microscopy
For Scale imaging, the fixed mouse brains were horizontally sectioned by vibratome with 0.5 mm thickness, then treated with Scale2A solution (4 M urea, 0.1% Triton-X, 10% glycerol) for 2 weeks at 4 °C in the darkness51. The Scale-treated tissue sections were placed in 35-mm dishes on the stage of a LSM 710 microscope (Carl Zeiss Microscopy GmbH, Jena, Germany)52. Cleared brains were imaged while in Scale solution using a 20×, 1.0 NA Plan-Apochromat lens and 488 nm excitation on an LSM710 NLO confocal microscope. Other studies included confocal scanning 1-μm optical sections of fixed brain sections with appropriate excitation wavelengths using 20×, 0.8 NA or 40×, 1.40 NA objectives. Multicolor stacked images were created using Zen software (Zeiss).
Electrophysiology
Preparation of slices
Mice were anesthetized with isoflurane and then sacrificed by decapitation, and their brains were extracted and placed in oxygenated (95% O2 and 5% CO2) ice-cold Ringer’s solution containing (in mM) 25 NaHCO3, 124 NaCl, 1 KCl, 2 KH2PO4, 10 glucose, 2.5 CaCl2, 1.3 MgCl2 and 0.01 picrotoxin (pH 7.4; Sigma-Aldrich, St. Louis, MO). After blocking, cortical hemispheres were sectioned with a vibrating microtome into 8 to 10 coronal sections containing the hippocampus (300 μm thickness). Slices were maintained in oxygenated room-temperature Ringer’s solution for equilibration for at least 1 h before field potential recordings. Following equilibration, individual slices were situated in submersion-type recording chambers (Harvard Apparatus, Holliston, MA) affixed to the stages of Nikon E600 infrared-differential interference contrast (IR-DIC) microscopes (Micro Video Instruments, Avon, MA). During recordings slices were superfused with oxygenated room-temperature Ringer’s solution at a rate of 2.5 ml/min.
Field potential recordings
Concentric bipolar tungsten stimulating electrodes were positioned in the middle molecular layer of the DG while recording electrodes were positioned in the dentate granule cell body layer. Inter-electrode distance was typically <200 μm. Field potential recordings were made using recording pipettes fabricated from non-heparinized microhematocrit capillary tubes (Thermo Fisher Scientific, Pittsburg, PA) on a Flaming and Brown horizontal pipette puller (Model P-87, Sutter Instrument, Novato, CA) and filled with the external solution. EPC-9 and EPC-10 patch-clamp amplifiers and PatchMaster software were used for data acquisition and FitMaster analysis software was used for data analyses (HEKA Elektronik, Lambrecht, Germany). Stimuli (100 μs in duration) were applied at 0.05 Hz at amplitudes increasing in increments of 10 μA from 0 to 150 μA, to generate input (stimulus intensity)–output (response amplitude) curves for each field. Following establishment of the input-output relationship, a baseline was established by stimulating at the stimulus intensity that elicited a ~50% response amplitude at 0.01 Hz for a period of 10 min. To elicit long-term potentiation, a tetanizing stimulus consisting of five 1-s pulses of 100 Hz were applied at intervals of 20 s. Following application of the tetanizing stimulus, the baseline was run for an additional 10- to 30-min period and the input-output relationship again assessed.
Primary tissue culture of murine microglia, astrocytes and cortical neurons
Primary cultures of mouse microglia and astrocytes were prepared from CD-1 mouse P0 pups as described53–55. Cells were maintained in complete DMEM (Dulbecco’s modified essential medium, 10% FBS and penicillin/streptomycin) (cat. no.11965-118, 10082-147 and 15140-122, Life Technologies/Invitrogen, Grand Island, NY). Murine monocyte colony stimulating factor (M-CSF, 10 ng/ml, cat. no. 4238, BioVision, Milpitas, CA) was added to the complete DMEM for microglial culture during the first 7 d after plating. Murine primary cortical neurons were prepared from E16 embryonic brains as described54 and maintained in serum-free Neurobasal medium with 2% B27 supplement, 2 mM L-glutamine and penicillin/streptomycin for 7 d (cat. no. 21103-049, 17504-001, 25030-081, Life Technologies/Invitrogen). Neurons express endogenous tau protein by 7 d in vitro (DIV7)56.
Tau phagocytosis assay
Purified recombinant 2N4R human tau 1–441 (hTau, 500 μg/ml, cat. no. T-1001-1, rPeptide, Bogart, GA) was preincubated with 30 μM heparin (cat. no. 07980, Stemcell Technologies, Vancouver, Canada) to be aggregated for 3 h (0–72 h for Supplementary Fig. 7)57. Primary-cultured murine microglia, astrocytes and neurons were incubated with 10 μg/ml of recombinant human tau for 3 h at 37 °C with 5% CO2. The cells were washed twice with DMEM to remove serum factors in the complete DMEM and stimulated with or without lipopolysaccharide (from E. coli 0111:B4, 1 μg/ml, cat. no. L3024, Sigma-Aldrich, St. Louis, MO) for 4 h. After the LPS stimulation, media were removed and the cells were treated with or without 5 mM ATP (Sigma-Aldrich) for 15 min. The media were collected as conditioned media (CM) and the cells were washed twice with ice-cold PBS and stored at −80 °C. Collected CM were centrifuged, filtered through 0.22-μm pore-size membrane (EMD Millipore) and stored at −80 °C. Independently cultured recipient murine cortical neurons were treated with 5 μl of exosomal fraction in a total volume of 250 μl for 4 h at DIV7. As for the quantification of phagocytosis in microglia, after the two washes with PBS, cells were incubated with 0.25% trypsin/EDTA for 10 min to digest extracellular tau, followed by addition of 1 mM phenylmethylsulfonyl fluoride (PMSF) to block trypsin activity and then cell lysis in ice-cold RIPA buffer. The cell lysates were incubated at 95 °C for 3 min to heat-inactivate proteases and total proteins were quantified by BCA assay (cat. no. PI-23221, Pierce/ThermoFisher Scientific Inc.,) and hTau by ELISA (human tau ELISA kit, cat. no. KHB0041, Life Technologies). The same experiments were also performed using a murine microglial cell line (BV-2, cat. no. ICLC ATL03001, CABRI)58, which was cultured in complete DMEM and under the same conditions.
Preparation of exosomal fraction
The exosomal fraction (EF) was prepared from CM from tissue-cultured murine neuronal cells as described13. Briefly, 2 ml of microglial CM was centrifuged at 2,000g for 10 min, and the supernatant was centrifuged at 10,000g for 30 min at 4 °C. The supernatant was then centrifuged at 100,000g for 90 min at 4 °C. The pellet fraction was resuspended in the same volume of ice-cold PBS and centrifuged at 100,000g for 90 min at 4 °C to remove any contaminating non-exosomal proteins. The pellet was resuspended with 20 μl of PBS for immunoelectron microscopy or biochemistry. The EF from PS19 mouse brain tissues was prepared as previously described with a minor modification: 0.2-μm-mesh filtration was replaced by serial centrifugation27. After the ultracentrifugation, the washed pellet was resuspended with either the same volume of PBS for the immunoelectron microscopy study or 2 ml of 0.95 M sucrose for sucrose step gradient ultracentrifugation at 200,000g for 16 h at 4 °C as described. The fractions are designated a (0.25 M sucrose), b (0.25–0.8 M sucrose), c (0.8–0.95 M sucrose), d (0.95–1.3 M sucrose), e (1.3–1.65 M sucrose), f (1.65–2.0 M sucrose) and g (2.0 M sucrose) from the lightest to heaviest sucrose density. Fractioned samples were subjected to acetylcholine esterase assay using Ellman’s reagent (5,5′-dithiobis-(2-nitrobenzoic acid) to identify brain exosomal fractions (fractions c and d in this study) as described59. Exosome fractions were subjected to immunoelectron microscopy or biochemical studies.
Immunoblotting
Protein lysates from whole-cell or EF samples were prepared in RIPA lysis buffer containing 1% NP-40, 0.5% sodium deoxycholate and 0.1% SDS in the presence of 1 mM PMSF, 1 mM NaF and 1 mM Na3VO4. The protein samples (20 μg) were subjected to 4–15% gradient gel SDS-PAGE and immunoblotted with AT8 mouse mAb (1:500), Tau46 mouse mAb (total tau, 1:500, cat. no. 4019, Thermo Scientific Pierce, cat. no. 4019), HT7 mouse mAb (specific to hTau, 1:500), T22 rabbit pAb (specific to tau oligomer, 1:500 for dot blot) (5), Tsg101 rabbit pAb (exosomal marker, 1:200, cat. no. ab30871, Abcam, Cambridge, MA), MHC-II rat mAb (I-A/I-E, exosomal marker, cat. no. 556999, 1:500, BD Bioscience/Pharmingen, San Jose, CA) and β-actin mouse mAb (1:1,000, cat. no. A5441, Sigma-Aldrich). hTau protein was also quantified by a human tau ELISA kit (cat. no. KHB0041, Life Technologies). All of the commercially available antibodies were validated by the manufacturers.
Tau ubiquitination assay
Cell lysates were boiled for 5 min to completely denature proteins and to disrupt noncovalent protein-protein interactions, then incubated with anti-ubiquitin (Ub) rabbit pAb (1 μg per 100 μg total protein, cat. no. SC-9133, Santa Cruz Biotechnology) at 4 °C for 12 h. Immunocomplexes were incubated with protein G–Sepharose 4 Fast Flow (cat. no. 17-0618-01, Amersham Biosciences/GE Healthcare, Piscataway, NJ) for 3 h, and the Sepharose beads were washed five times with ice-cold PBS. Immunoprecipitated samples were subjected to SDS-PAGE and immunoblotting.
Immunoaffinity isolation of exosomes
The EF was incubated with anti-Tsg101 pAb (1 μg per 100 μg total protein, Abcam) conjugated to protein G–Sepharose (Amersham Biosciences) at 4 °C for 12 h as described26. The Sepharose beads were washed five times with ice-cold PBS, and the affinity-purified samples were subjected to SDS-PAGE and immunoblotting.
Exosome injection into mouse brain
The EF was prepared from microglial CM after sequential treatment with aggregated tau, LPS and ATP or with ATP alone. To fluorescently label exosomes, 2 ml of microglial CM was incubated with 0.5 μl of DiI (2.5 mg ml−1 in DMSO, cat. no. D282, Invitrogen/Molecular Probes) for 3 h. DiI-labeled EF was isolated by sequential ultracentrifugation as described above, and the amount of tau in the EF was calibrated by immunoblotting with purified tau protein. After calibration of tau load, 1 μl of DiI-labeled EF containing 5 ng of hTau was stereotaxically injected into the OML of DG of 4-months-old C57BL/6 mice with the following coordinates: anteroposterior, −2.18 mm; lateral, 1.25; dorsoventral, −1.80. Equivalent amounts of aggregated hTau (5 ng μl−1) or non-tau-containing exosome were also injected into the same brain region in control groups. Animals were sacrificed 3 weeks after the injection and the fixed brain tissues were subjected to immunofluorescence for imaging pTau, DiI and DAPI.
Immunoelectron microscopy of exosomal fraction
The EF collected from microglia CM and PS19 mouse brains were subjected to immunoelectron microscopy as described60. Briefly, 5 μl of the EF were adsorbed for 1 min to a carbon-coated grid that had been made hydrophilic by a 30-s exposure to a glow discharge. Excess liquid was removed with a filter paper (Whatman no. 1, Fisher Scientific), and samples were stained with 0.75% uranyl formate for 30 s. After excess uranyl formate was removed with filter paper, grids were examined in a TecnaiG2 Spirit BioTWIN (Field Emission Inc, Hillsboro, Oregon), and images were recorded with an AMT 2k CCD camera (Advanced Microscopy Techniques, Woburn, MA). For immunogold labeling, pelleted exosomes were fixed with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) and processed for ultrathin cryosectioning. Cell pellets were infiltrated with 2.3 M sucrose in PBS for 15 min, frozen in liquid nitrogen and sectioned at −120 °C. Sections 60–80 nm thick were picked up and transferred to formvar carbon-coated copper grids, and immunogold labeling was carried out at room temperature on a piece of Parafilm. All antibodies were diluted in 1% BSA in PBS. Grids were floated on drops of 1% BSA for 10 min to block nonspecific labeling, transferred to 5-μl drops of primary antibodies (Tsg101, rabbit pAb, 1:20; HT7, mouse mAb, 1:10; or PHF1, mouse mAb, 1:20; kindly provided by P. Davies) and incubated for 30 min, then washed in four drops of PBS (total 10 min) before incubation in 5-nm protein A–gold or, for mouse mAbs, rabbit anti-mouse bridging antibody (1:100, cat. no. 55436, Capell) followed by 5-nm protein A–gold (University Medical Center, Utrecht, the Netherlands) for 20 min. Grids were washed in two drops of PBS followed by four drops of water (total 15 min) and contrasted in a mixture of 0.3% uranyl acetate in 2% methylcellulose for 5 min, then excess liquid was blotted off with filter paper and the grids were examined at 80 kV with a TecnaiG2 Spirit BioTWIN transmission electron microscope. Images were recorded with an AMT 2k CCD camera. The captured images with scale bars were analyzed for the area size of tau+ or tau− vesicles (a total of 368 vesicles) and the average diameter of each vesicle was calculated by the transformation from area size with the following formula:
A histogram was developed from the transformed values and vesicles of 50–100 nm were classified as exosomes.
Immunoelectron microscopy of mouse brain sections
Brains were perfused and fixed with 4% (w/v) PFA, 0.1% glutaraldehyde in 0.1 M sodium phosphate buffer, pH 7.1. Hippocampal dentate gyrus areas were selected from 200-μm-thickness sections, cut into 1-mm squares, placed in 20% BSA, high-pressure frozen (EM PACT2, Leica) and freeze substituted (EM AFS2, Leica) in acetone with 0.1% glutaraldehyde, 0.1% uranyl acetate. Freeze-substituted samples were embedded in Lowicryl (HM20, Polysciences, Inc., Warrington, PA) and ultrathin sections were cut (Ultracut, Leica) and collected on formvar carbon-coated grids (Electron Microscopy Sciences, Fort Washington, PA). Post-immunolabeling was performed at room temperature on a piece of Parafilm: grids were blocked with 1% BSA plus 0.1% Triton X-100 for 30 min and incubated in Iba1 rabbit pAb and PHF1 mouse mAb diluted in 1% BSA, followed by protein A–15-nm gold (University Medical Center, Utrecht, the Netherlands) and donkey anti-mouse 10-nm gold (cat. no. 25129, EMS, Hatfield, PA). Electron microscopy images were acquired using a JEM-1200EX (JEOL, Tokyo, Japan) with a bottom-mounted CCD camera (AMT, Woburn, MA) in the same manner as for the immunoelectron microscopy of exosomal fractions.
Gene expression analysis
Total RNA was isolated by miRNeasy Mini Kit (cat. no. 217004, Qiagen, Valencia, CA) from hippocampal regions of AAV-GFP/tau-injected C57BL/6 mice with or without GW4869 or PLX3397 treatment after transcardial perfusion of DEPC-treated ice-cold PBS (20 ml over 2 min) to eliminate blood cells in the brain. After quantification and evaluation of the integrity of 28S and 18S ribosomal RNA by agarose gel electrophoresis, the samples were subjected to real-time reverse transcription polymerase chain reaction using Rotor-Gene Q (cat. no. 9001630, Qiagen) and specific primer sets for murine Tnfa, Il1b, Il6, Il10 and Tgfb1 genes. The primer sequences used for these genes were previously published61.
Silencing of nSMase2 gene expression by siRNA
Murine neutral sphingomyeli-nase 2 gene (nSMase2, Smpd3)-specific siRNA (sense sequence CCAAAGAAUUGUUGGGUACAUCUU, antisense sequence GAUGUACCCAAVAAUUCUUUGGUU) and scrambled siRNA were purchased from Fisher Thermo Scientific, Inc. Primary cultured murine microglia were transfected with 50 pM Smpd3 siRNA using Lipofectamine 2000 (cat. no. 11668027, Invitrogen) according to the manufacturer’s protocol. Efficient knockdown of Smpd3 was confirmed by real-time RT-PCR using the following primers: 5′-GGCCTATCACTGTTACCCCAATG-3′ and 5′-GTGCTGGTCGAGGAGGTAGATTTC-3′ (ref. 29).
GW4869 treatment of microglia in vitro and mice in vivo
GW4869 (cat. no. D1692, Sigma-Aldrich) was dissolved in tissue culture–grade DMSO at 0.2 mg ml−1. For in vitro experiments, primary cultured murine microglia were treated with 3 μM GW4869 (IC50 = 1 μM) for 24 h before hTau treatment. PS19 mice 3.5 months of age were injected daily intraperitoneally with 1.25 mg kg−1 of GW4869 in 200 μl of 5% DMSO in saline or 200 μl of control vehicle every 4 weeks (28 injections total) as described62. Animals were sacrificed 24 h after the final injection. No obvious behavioral or health problems were observed during the treatment.
Experimental procedure
The investigator in charge of injection of AAV into mouse brain knew the identity of animals receiving the treatments, but the investigators and testers in charge of tissue collection, immunohistochemistry, western blotting and other procedures following euthanasia were blinded to the identity of the animals or samples until the completion of the experiments and assessment of the outcomes. All tested animals and tissue samples were de-identified for data collection (such as immunohistochemistry, image capturing and western blotting) and processing (such as statistical analysis) for testers, and the identity was disclosed at the completion of the experiment.
Statistics
Data are presented as means ± s.e.m. Unpaired t tests were used for comparison of immunostained cell counts in immunohistochemistry and dot-blot intensity between control and drug-treated groups. Multi-group data were analyzed using one-way ANOVA followed by Tukey post hoc test or by two-way ANOVA with multiple comparisons (Prism 6, GraphPad Software, Inc.). The estimate of variance was determined by the s.d. of each group, which was similar between groups for the statistical comparison, and the normality of a parametric test was examined for each experiment using a D’Agostino-Pearson omnibus test (omnibus K2 test, Prism 6) for sample sizes of 6 or higher. Some outliers were excluded after the test. For smaller sample sizes, the data distribution was assumed to be normal but this was not formally tested. Statistical significance was set at P < 0.05. No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those generally employed in the field.
A Supplementary Methods Checklist is available.
Supplementary Material
Acknowledgments
The authors thank M. Ericsson (Electron Microscopy Facility, Harvard Medical School) for electron microscopic imaging services; P. Davies (Albert Einstein College of Medicine) for providing the MC1, CP13 and PHF1 antibodies; R. Kayed (University of Texas Medical Branch) for providing the T22 polyclonal antibody; Plexxikon, Inc. for providing PLX3397 and control chows; and M. Hasselmo and S. Przedborski for critical reading of the manuscript. This work is supported in part by grants from Alzheimer’s Association (T.I.), Alzheimer’s Art Quilt Initiative (T.I.), Boston University Alzheimer’s Disease Center (P30AG013846, T.I.), BrightFocus Foundation (H.A.) and Coins for Alzheimer’s Research Trust (T.I.).
Footnotes
AUTHOR CONTRIBUTIONS
T.I. and H.A. designed the AAV-GFP/tau injection mouse model and initiated the study. M.M. and J.L. performed field recording electrophysiology and analyses; S.I. and H.A. cultured primary neuronal cells; H.A., S.I. and T.I. purified exosomes for electron microscopy and biochemical analyses; T.H. performed two-photon imaging and laser-scanning confocal microscopy; O.B. generated and provided the Pr2ry12 antibody, performed data analysis and edited the manuscript; B.W. provided tau antibodies, performed data analysis and wrote the manuscript; S.K. generated and provided AAV vectors, performed data analysis and edited the manuscript; and H.A., S.I., J.L., S.T., M.M. and T.I. designed and performed experiments and data analyses and wrote the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Note: Any Supplementary Information and Source Data files are available in the online version of the paper.
References
- 1.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 2.Clavaguera F, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909–913. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahmed Z, et al. A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: the pattern of spread is determined by connectivity, not proximity. Acta Neuropathol. 2014;127:667–683. doi: 10.1007/s00401-014-1254-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boluda S, et al. Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer’s disease or corticobasal degeneration brains. Acta Neuropathol. 2015;129:221–237. doi: 10.1007/s00401-014-1373-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iba M, et al. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J Neurosci. 2013;33:1024–1037. doi: 10.1523/JNEUROSCI.2642-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harris JA, et al. Human P301L-mutant tau expression in mouse entorhinal-hippocampal network causes tau aggregation and presynaptic pathology but no cognitive deficits. PLoS ONE. 2012;7:e45881. doi: 10.1371/journal.pone.0045881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Calignon A, et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron. 2012;73:685–697. doi: 10.1016/j.neuron.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu L, et al. Trans-synaptic spread of tau pathology in vivo. PLoS ONE. 2012;7:e31302. doi: 10.1371/journal.pone.0031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peeraer E, et al. Intracerebral injection of preformed synthetic tau fibrils initiates widespread tauopathy and neuronal loss in the brains of tau transgenic mice. Neurobiol Dis. 2015;73:83–95. doi: 10.1016/j.nbd.2014.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamada K, et al. Neuronal activity regulates extracellular tau in vivo. J Exp Med. 2014;211:387–393. doi: 10.1084/jem.20131685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanders DW, et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. 2014;82:1271–1288. doi: 10.1016/j.neuron.2014.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vingtdeux V, Sergeant N, Buee L. Potential contribution of exosomes to the prion-like propagation of lesions in Alzheimer’s disease. Front Physiol. 2012;3:229. doi: 10.3389/fphys.2012.00229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saman S, et al. Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem. 2012;287:3842–3849. doi: 10.1074/jbc.M111.277061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fiandaca MS, et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: a case-control study. Alzheimers Dement. 2015;11:600–607. doi: 10.1016/j.jalz.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neumann H, Kotter MR, Franklin RJ. Debris clearance by microglia: an essential link between degeneration and regeneration. Brain. 2009;132:288–295. doi: 10.1093/brain/awn109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schafer DP, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.El Andaloussi S, Mager I, Breakefield XO, Wood MJ. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat Rev Drug Discov. 2013;12:347–357. doi: 10.1038/nrd3978. [DOI] [PubMed] [Google Scholar]
- 18.Ghoshal N, et al. Tau-66: evidence for a novel tau conformation in Alzheimer’s disease. J Neurochem. 2001;77:1372–1385. doi: 10.1046/j.1471-4159.2001.00346.x. [DOI] [PubMed] [Google Scholar]
- 19.Serrano-Pozo A, et al. Reactive glia not only associates with plaques but also parallels tangles in Alzheimer’s disease. Am J Pathol. 2011;179:1373–1384. doi: 10.1016/j.ajpath.2011.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shevtsova Z, Malik JM, Michel U, Bahr M, Kugler S. Promoters and serotypes: targeting of adeno-associated virus vectors for gene transfer in the rat central nervous system in vitro and in vivo. Exp Physiol. 2005;90:53–59. doi: 10.1113/expphysiol.2004.028159. [DOI] [PubMed] [Google Scholar]
- 21.Schofield E, Kersaitis C, Shepherd CE, Kril JJ, Halliday GM. Severity of gliosis in Pick’s disease and frontotemporal lobar degeneration: tau-positive glia differentiate these disorders. Brain. 2003;126:827–840. doi: 10.1093/brain/awg085. [DOI] [PubMed] [Google Scholar]
- 22.Fricker M, et al. MFG-E8 mediates primary phagocytosis of viable neurons during neuroinflammation. J Neurosci. 2012;32:2657–2666. doi: 10.1523/JNEUROSCI.4837-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoshiyama Y, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53:337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 24.Elmore MR, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 2014;82:380–397. doi: 10.1016/j.neuron.2014.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qu Y, Franchi L, Nunez G, Dubyak GR. Nonclassical IL-1 beta secretion stimulated by P2X7 receptors is dependent on inflammasome activation and correlated with exosome release in murine macrophages. J Immunol. 2007;179:1913–1925. doi: 10.4049/jimmunol.179.3.1913. [DOI] [PubMed] [Google Scholar]
- 26.Kadiu I, Narayanasamy P, Dash PK, Zhang W, Gendelman HE. Biochemical and biologic characterization of exosomes and microvesicles as facilitators of HIV-1 infection in macrophages. J Immunol. 2012;189:744–754. doi: 10.4049/jimmunol.1102244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perez-Gonzalez R, Gauthier SA, Kumar A, Levy E. The exosome secretory pathway transports amyloid precursor protein carboxyl-terminal fragments from the cell into the brain extracellular space. J Biol Chem. 2012;287:43108–43115. doi: 10.1074/jbc.M112.404467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006 doi: 10.1002/0471143030.cb0322s30. Ch. 3, Unit 3.22. [DOI] [PubMed] [Google Scholar]
- 29.Trajkovic K, et al. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science. 2008;319:1244–1247. doi: 10.1126/science.1153124. [DOI] [PubMed] [Google Scholar]
- 30.Brown GC, Neher JJ. Microglial phagocytosis of live neurons. Nat Rev Neurosci. 2014;15:209–216. doi: 10.1038/nrn3710. [DOI] [PubMed] [Google Scholar]
- 31.Ittner LM, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell. 2010;142:387–397. doi: 10.1016/j.cell.2010.06.036. [DOI] [PubMed] [Google Scholar]
- 32.Clavaguera F, et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci USA. 2013;110:9535–9540. doi: 10.1073/pnas.1301175110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lasagna-Reeves CA, et al. Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci Rep. 2012;2:700. doi: 10.1038/srep00700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Agosta F, et al. Myeloid microvesicles in cerebrospinal fluid are associated with myelin damage and neuronal loss in mild cognitive impairment and Alzheimer disease. Ann Neurol. 2014;76:813–825. doi: 10.1002/ana.24235. [DOI] [PubMed] [Google Scholar]
- 35.Lasagna-Reeves CA, et al. Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. FASEB J. 2012;26:1946–1959. doi: 10.1096/fj.11-199851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Théry C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9:581–593. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- 37.Lu Z, et al. Phagocytic activity of neuronal progenitors regulates adult neurogenesis. Nat Cell Biol. 2011;13:1076–1083. doi: 10.1038/ncb2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Santacruz K, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Castillo-Carranza DL, et al. Passive immunization with Tau oligomer monoclonal antibody reverses tauopathy phenotypes without affecting hyperphosphorylated neurofibrillary tangles. J Neurosci. 2014;34:4260–4272. doi: 10.1523/JNEUROSCI.3192-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Danzer KM, et al. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol Neurodegener. 2012;7:42. doi: 10.1186/1750-1326-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alvarez-Erviti L, et al. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol Dis. 2011;42:360–367. doi: 10.1016/j.nbd.2011.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arellano-Anaya ZE, et al. Prion strains are differentially released through the exosomal pathway. Cell Mol Life Sci. 2015;72:1185–1196. doi: 10.1007/s00018-014-1735-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Phinney AL, et al. Cerebral amyloid induces aberrant axonal sprouting and ectopic terminal formation in amyloid precursor protein transgenic mice. J Neurosci. 1999;19:8552–8559. doi: 10.1523/JNEUROSCI.19-19-08552.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yamamoto M, Kiyota T, Walsh SM, Ikezu T. Kinetic analysis of aggregated amyloid-beta peptide clearance in adult bone-marrow-derived macrophages from APP and CCL2 transgenic mice. J Neuroimmune Pharmacol. 2007;2:213–221. doi: 10.1007/s11481-006-9049-8. [DOI] [PubMed] [Google Scholar]
- 45.Lewis J, et al. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- 46.Jicha GA, Bowser R, Kazam IG, Davies P. Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res. 1997;48:128–132. doi: 10.1002/(sici)1097-4547(19970415)48:2<128::aid-jnr5>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 47.Otvos L, Jr, et al. Monoclonal antibody PHF-1 recognizes tau protein phosphorylated at serine residues 396 and 404. J Neurosci Res. 1994;39:669–673. doi: 10.1002/jnr.490390607. [DOI] [PubMed] [Google Scholar]
- 48.Butovsky O, et al. Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann Neurol. 2015;77:75–99. doi: 10.1002/ana.24304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Butovsky O, et al. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat Neurosci. 2014;17:131–143. doi: 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vosshall LB, Wong AM, Axel R. An olfactory sensory map in the fly brain. Cell. 2000;102:147–159. doi: 10.1016/s0092-8674(00)00021-0. [DOI] [PubMed] [Google Scholar]
- 51.Hama H, et al. Scale: a chemical approach for fluorescence imaging and reconstruction of transparent mouse brain. Nat Neurosci. 2011;14:1481–1488. doi: 10.1038/nn.2928. [DOI] [PubMed] [Google Scholar]
- 52.Tyler WA, Haydar TF. Multiplex genetic fate mapping reveals a novel route of neocortical neurogenesis, which is altered in the Ts65Dn mouse model of Down syndrome. J Neurosci. 2013;33:5106–5119. doi: 10.1523/JNEUROSCI.5380-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kiyota T, et al. CCL2 accelerates microglia-mediated Aβ oligomer formation and progression of neurocognitive dysfunction. PLoS ONE. 2009;4:e6197. doi: 10.1371/journal.pone.0006197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kiyota T, Ingraham KL, Jacobsen MT, Xiong H, Ikezu T. FGF2 gene transfer restores hippocampal functions in mouse models of Alzheimer’s disease and has therapeutic implications for neurocognitive disorders. Proc Natl Acad Sci USA. 2011;108:E1339–E1348. doi: 10.1073/pnas.1102349108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kiyota T, et al. AAV serotype 2/1-mediated gene delivery of anti-inflammatory interleukin-10 enhances neurogenesis and cognitive function in APP+PS1 mice. Gene Ther. 2012;19:724–733. doi: 10.1038/gt.2011.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Beaudoin GM, III, et al. Culturing pyramidal neurons from the early postnatal mouse hippocampus and cortex. Nat Protoc. 2012;7:1741–1754. doi: 10.1038/nprot.2012.099. [DOI] [PubMed] [Google Scholar]
- 57.Flach K, et al. Tau oligomers impair artificial membrane integrity and cellular viability. J Biol Chem. 2012;287:43223–43233. doi: 10.1074/jbc.M112.396176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Blasi E, Barluzzi R, Bocchini V, Mazzolla R, Bistoni F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J Neuroimmunol. 1990;27:229–237. doi: 10.1016/0165-5728(90)90073-v. [DOI] [PubMed] [Google Scholar]
- 59.Ellman GL, Courtney KD, Andres V, Jr, Feather-Stone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 60.Roccaro AM, et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J Clin Invest. 2013;123:1542–1555. doi: 10.1172/JCI66517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Freilich RW, Woodbury ME, Ikezu T. Integrated expression profiles of mRNA and miRNA in polarized primary murine microglia. PLoS ONE. 2013;8:e79416. doi: 10.1371/journal.pone.0079416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tabatadze N, et al. Inhibition of neutral sphingomyelinase-2 perturbs brain sphingolipid balance and spatial memory in mice. J Neurosci Res. 2010;88:2940–2951. doi: 10.1002/jnr.22438. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.