

Abstract
The formation of intricately organized aortic and pulmonic valves from primitive endocardial cushions of the outflow tract is a remarkable accomplishment of embryonic development. While not always initially pathologic, developmental semilunar valve (SLV) defects, including bicuspid aortic valve, frequently progress to a disease state in adults requiring valve replacement surgery. Disrupted embryonic growth, differentiation, and patterning events that “trigger” SLV disease are coordinated by gene expression changes in endocardial, myocardial, and cushion mesenchymal cells. We explored roles of chromatin regulation in valve gene regulatory networks by conditional inactivation of the Brg1-associated factor (BAF) chromatin remodeling complex in the endocardial lineage. Endocardial Brg1-deficient mouse embryos develop thickened and disorganized SLV cusps that frequently become bicuspid and myxomatous, including in surviving adults. These SLV disease-like phenotypes originate from deficient endocardial-to-mesenchymal transformation (EMT) in the proximal outflow tract (pOFT) cushions. The missing cells are replaced by compensating neural crest or other non-EMT-derived mesenchyme. However, these cells are incompetent to fully pattern the valve interstitium into distinct regions with specialized extracellular matrices. Transcriptomics reveal genes that may promote growth and patterning of SLVs and/or serve as disease-state biomarkers. Mechanistic studies of SLV disease genes should distinguish between disease origins and progression; the latter may reflect secondary responses to a disrupted developmental system.
Keywords: Brg1, BAF complex, chromatin remodeling, endocardial-to-mesenchymal transformation, endocardial cushions, outflow tract, semilunar valves, bicuspid aortic valve, valve disease
Graphical Abstract

INTRODUCTION
Congenital valve defects affect at least 2% of humans, a remarkable frequency that underlines the urgent need to understand their etiology (Hoffman and Kaplan 2002; Siu and Silversides 2010; Go et al. 2013). Frequently, developmentally abnormal valves do not progress to a pathologic state until late adulthood. For example, many cases of adult aortic valve disease characterized by thickened and myxomatous valve cusps display bicuspid rather than tricuspid arrangements originating from fusions during development (Angelini et al. 1989; Roberts and Ko 2005). The rationale design of diagnostics and therapeutics therefore must distinguish between disrupted developmental processes, and their genetic and environmental influences, that trigger valve defects from mechanisms that drive the progression of an anatomically abnormal valve to a disease state.
Valvulogenesis is a dynamic and multistep process that transforms primitive endocardial cushions (ECs) into highly patterned valves with thin, elongated leaflets or cusps. ECs form at discrete sites of the looping heart tube by the local secretion of a specialized extracellular matrix called the cardiac jelly. The mesenchyme of atrioventricular canal ECs, which generate the mitral and tricuspid valves, originates from an endothelial-to-mesenchymal transition (EMT) of a subset of overlying endocardial cells. The outflow tract contains two pairs of neighboring ECs that together will form the semilunar valves. The proximal outflow tract (pOFT) cushions are primarily populated by EMT-derived mesenchyme. In contrast, the distal outflow tract (dOFT) cushion mesenchyme is largely derived from invading cardiac neural crest cells (NCCs) (Kirby et al. 1983; Waldo et al. 1998; Jiang et al. 2000). Additional cell populations, possibly of second heart field (SHF) origin, may contribute additional mesenchyme to the OFT cushions (Engleka et al. 2012; Sizarov et al. 2012).
EMT begins at embryonic day 9.5 (E9.5) in the AVC and soon after in the pOFT (Camenisch et al. 2002). Signaling factors, notably TGF-beta superfamily and Notch proteins, direct EMT by promoting gene expression program changes that transition endocardial cells from an interconnected epithelial to invasive mesenchymal state (Ramsdell and Markwald 1997; Timmerman et al. 2004; Fischer et al. 2007; Venkatesh et al. 2008; Luna-Zurita et al. 2010; Chang et al. 2011; Wang et al. 2013). Deficient mesenchyme production, including when EMT is disrupted, impedes subsequent steps of valve development and leads to congenitally abnormal valves. A connection between EMT disruption and semilunar valve disease is suggested by Notch1 being one of the few identified human aortic valve disease genes (Garg et al. 2005), although Notch signaling may also have later roles in valve development or homeostasis (Hofmann et al. 2012; Theodoris et al. 2015).
After EMT, the paired OFT ECs undergo a complex rearrangement to form two sets of symmetric tricuspid semilunar valves. By E13.5, the ECs meet and rearrange concomitantly with OFT septation to form separate pulmonic and aortic valves. The three cusps of each semilunar valve elongate and refine into thin tissues that will distend to meet and form a tight seal but have the flexibility to promptly open under pressure. Through postnatal development, the valve interstitium becomes stratified into layers of distinct extracellular matrix (ECM) composition to confer appropriate stiffness/flexibility across the cusps’ short axis (Gross and Kugel 1931; Broom 1978; Hinton et al. 2006). Genetic loss or disruption of several extracellular proteins, such as Versican (Vcan) and Periostin (Postn), or their regulators, notably Adamts-family matrix proteases, produce valve defects (Tkatchenko et al. 2009; Kern et al. 2010; Dupuis et al. 2011; Cheek et al. 2012).
The semilunar valves are also patterned along their proximal-to-distal (long) axis. Each cusp of both aortic and pulmonic valves can be considered as a shallow bisected cup fastened within a cylindrical structure (Fig. S1A–B). The cusps’ base region that connects them to underlying muscle is enriched with Tenascin C (Tnc), which helps confer tissue rigidity (Zhang et al. 1993; Satta et al. 2002; Hinton et al. 2006). In contrast, the flexible distal part of the cusps express high levels of Vcan and Postn (Kruzynska-Frejtag et al. 2001; Norris et al. 2008; Snider et al. 2008; Kern et al. 2010). The junction between the stiff base and flexible distal regions is commonly termed the hinge. The multi-dimensional organization of semilunar valve interstitium suggests that valve patterning requires a highly coordinated series of events that act over an extended period of development. Any of these steps could be perturbed to cause formation of a congenitally abnormal valve.
Human genetics and model organism studies, largely in mice, highlight disrupted regulatory pathways that coalesce on transcriptional changes as primary sources of SLV defects. For example, disrupted Notch signaling produces bicuspid aortic valve (BAV) in both mice and humans (Garg et al. 2005; Jain et al. 2011; Bosse et al. 2013). Additional mouse models of semilunar valve disease implicate Gata5 and Pax3 transcription factors and transcriptional effectors of Tgf-β family signaling (Jain et al. 2011; Laforest et al. 2011; Thomas et al. 2012; Dupuis et al. 2013). The molecular activities of these transcription factors, including an understanding of how they function within a chromatinized landscape to direct specific responses, are poorly understood. A role for chromatin regulation in human SLV disease is supported by the high frequency of de novo variants in histone modifying genes in congenital heart disease (Zaidi et al. 2013). Further the sporadic inheritance patterns in afflicted families (Clementi et al. 1996; Huntington et al. 1997) suggest epigenetic influences that modify penetrance and/or expressivity of disease-causing alleles.
The Brg1-associated factor (BAF) chromatin remodeling complex facilitates activation and repression of genes by ATP-dependent nucleosome repositioning and/or replacement. The core ATPase subunit is predominantly provided by Brg1; in most cases, loss of Brg1 alone inactivates the BAF complex (Kadoch and Crabtree 2013). Coordinately, Brg1-null mice are peri-implantation lethal (Bultman et al. 2000). The BAF complex has been implicated in multiple steps of heart development and disease, including cardiac specification and hypertrophy (Lickert et al. 2004; Hang et al. 2010; Takeuchi et al. 2011). Further, Brg1 is required in endocardial cells to maintain the microenvironment required for the initiation of cardiac trabeculation (Stankunas et al. 2008). Brg1 expression in endocardial and mesenchymal cells of the developing valves suggests additional roles for endocardial lineage BAF complexes during valvulogenesis.
Here, we disrupt endocardial-lineage Brg1 in mice to demonstrate that the BAF chromatin remodeling complex is essential for semilunar valve development. Endocardial Brg1-deficient embryos develop thickened aortic and pulmonic valves that frequently are bicuspid and progress to a myxomatous, disease-like state. We map the origins of these defects to a partial disruption of pOFT EMT, a process an earlier deletion of endocardial Brg1 shows the BAF complex is required for in both the pOFT and AVC. Developing semilunar valves compensate for the deficiency of EMT-derived mesenchyme, but the responding mesenchymal populations are insufficient to fully pattern a complex, multi-origin semilunar valve interstitium. Therefore, an initial deficiency of pOFT EMT can trigger secondary responses that culminate in semilunar valve disease. By RNA-seq analysis of the Brg1-deficient valves, we additionally identify and characterize the expression of novel transcripts that may promote growth and patterning of semilunar valves and/or serve as biomarkers of a diseased state.
MATERIALS AND METHODS
Mice
All mouse husbandry and procedures were approved and monitored by the University of Oregon’s Institutional Animal Care and Use Committee. Transgenic lines are described in Supplemental Materials.
Tissue processing and sectioning
Mouse embryos or dissected adult hearts were fixed overnight in 4% PFA/PBS at 4°C. Fixed tissue was washed thoroughly and then equilibrated overnight with PBS at 4°C. Samples were dehydrated through a graded ethanol series and cleared with xylenes prior to paraffin embedding. 7 µm paraffin sections were transferred to microscope slides for either histological or fluorescent antibody staining. Cryosections were used for RNA in situ hybridizations and occasional antibody staining (see Supplementary Materials). For cryosections, embryos were fixed as described, washed thoroughly with PBS, equilibrated in 30% sucrose overnight at 4°C, embedded in OCT (Tissue-Tek), and sectioned (16 µm).
Histology
Hematoxylin and eosin (H&E) staining used paraffin sections and conventional reagents and methodology (Ricca Chemical Company). Masson’s Trichrome staining on paraffin sections used a reagent kit following manufacturer’s instructions (Electron Microscopy Sciences). Von Kossa stained paraffin sections were counterstained with Alcian Blue (pH 2.5) and Nuclear Fast Red (Vector Laboratories).
Immunostaining
Paraffin-sectioned tissue on slides was de-paraffinized and rehydrated to distilled water using a graded series of ethanol washes. Antigen retrieval was performed as described in Supplemental Materials. The slides were blocked with M.O.M protein diluent (Vector Laboratories) or 10% normal goat serum (NGS) in PBS for 1 hour at RT and then stained overnight with primary antibodies diluted in blocking buffer (sources and dilutions in Supplemental Materials). Alexa dye-conjugated fluorescent secondary antibodies (Life Technologies) were used to detect signal. Nuclei were stained with Hoechst (Life Technologies). Slides were mounted with Vectashield (Vector Laboratories). Immunofluorescent images were acquired using a widefield microscope or an Olympus FV1000 laser-scanning confocal microscope. Confocal images shown are average intensity projections of image stacks.
RNA In situ hybridization
In situ hybridizations were performed using cryosections (16 µm) except for Nptx1, which used paraffin sections (7 µm), as described (Stewart et al. 2014). Briefly, frozen sections were washed with PBS and PBST prior to Proteinase K treatment. Alternatively, paraffin sectioned tissue was first de-paraffinized and rehydrated. Sections were post-fixed with 4% PFA, washed, and incubated with pre-hybridization buffer at 65°C for 4 hours. Digoxigenin (DIG)-labeled RNA probes (see Supplemental Materials) were hybridized overnight at 65°C. Following multiple washes, the tissue was blocked with 1% Roche blocking buffer (Roche) and then stained with alkaline phosphatase-conjugated anti-DIG antibody (Roche) overnight at 4°C. The signal was developed after extensive TBST washes using BCIP/NBT (Promega). Tissue was counterstained with Nuclear Fast Red (Vector Laboratories) before being processed for mounting with Permount (Electron Microscopy Sciences).
Valve morphometrics
Optimally matched H&E stained sections for each studied valve of wildtype and littermate Nfatc1Cre;Brg1F/F embryos were imaged for area and distance measurements using ImageJ (NIH). Length-to-width ratios were determined as described previously (Stankunas et al. 2010). Cell numbers were counted manually. Data from multiple litters was combined by normalizing values to the mean of wildtype embryos within each letter. Statistical significance was determined using two-tailed Student’s t-tests.
Valve 3D reconstructions
7 µm serial paraffin sections covering the entire E16.5 aortic valve were collected, H&E stained and imaged. All images (slices in stack) were imported into ImageJ (NIH) and aligned manually to neighboring valve sections using the Trakem2 plugin (Cardona et al. 2012). An ‘area list’ for each cusp (LCC, RCC, NCC) was generated manually for every layer before being merged across layers to establish 3-D shapes. Adjustments made for the z-space and thickness were standardized for a given litter to allow a direct comparison between wildtype and Nfatc1Cre;Brg1F/F aortic valves. A two-tailed Student’s t-test comparing total aortic valve volumes (all cusps) between wildtype and Nfatc1Cre;Brg1F/F embryos used data normalized to the mean of the wildtype aortic valve volumes within each litter.
In vivo EdU labeling and proliferation assays
Pregnant females were IP injected with 50 mg/kg of 5-ethynyl-2´-deoxyuridine (EdU; Life Technologies) 2–3 hours before embryos were harvested. Paraffin sections were prepared for staining as described. For combined lineage tracing and EdU proliferation studies, slides were incubated for 10’ in a pressure cooker with DAKO antigen retrieval solution (DAKO). The slides were blocked for one hour with 10% NGS/PBS after antigen retrieval followed by a Click-iT reaction (Life Technologies). Subsequently, slides were washed 6× 10’ with PBS before proceeding with anti-GFP antibody staining as described in the Supplemental Materials.
EMT explant assays
Collagen gel EMT explant assays using dissected E9.5 outflow tract tissue were performed as described (Xiong et al. 2011). 1 mg/ml rat tail collagen I (BD Biosciences) matrices were supplemented with 5 µg/ml doxycycline to induce the TRE:H2BGFP reporter used for fluorescent lineage tracing. Explants were incubated at 37°C in 5% O2/5% CO2 for 48 hours before brightfield and fluorescent imaging on an inverted Nikon Eclipse Ti widefield microscope.
Quantitative RT-PCR
qRT-PCR was performed using KAPA SYBR FAST qPCR Master Mix (Kapa Biosystems) and cDNA synthesized using SuperScript III (Life Technologies) from Trizol-isolated RNA prepared from dissected valve regions. Relative mRNA expression was normalized using a control transcript (HPRT) to calculate ΔCTs (threshold cycles). Expression levels between wildtype and Nfatc1Cre;Brg1F/F hearts were compared using a ΔΔCT approach. ΔCT values were used for two-tailed Student’s t-tests with a Bonferroni correction for multiple comparisons to determine statistical significance. Primer sequences are in Supplemental Materials.
Illumina RNA sequencing
The valve-containing region of E14.5 hearts was dissected under a fluorescent stereomicroscope using the guide of an endocardial-lineage nuclear GFP reporter (see Supplemental Materials). RNA was extracted using TRIzol (Life Technologies). RNA-sequencing libraries were prepared from two sets of individual paired control and littermate Nfatc1Cre;Brg1F/F embryos as described in Supplemental Materials. The bioinformatics workflow to determine differentially gene expression is also presented therein.
RESULTS
Endocardial Brg1 is required for semilunar valve development
Prior studies of Brg1 roles in the developing endocardium showed that the BAF complex is required at embryonic day E9.5 to transcriptionally maintain a microenvironment that supports ventricular trabeculation (Stankunas et al. 2008). Persistent endocardial Brg1 expression throughout the embryonic heart suggested additional endocardial BAF complex functions in other heart regions or during later steps of cardiogenesis. The previous use of the Tie2:Cre line to conditionally delete floxed Brg1 alleles precluded studying later roles due to lethality arising from hematopoietic and yolk sac vasculature defects in Tie2:Cre;Brg1F/F embryos (Stankunas et al. 2008). Therefore, we used the Nfatc1Cre line that expresses Cre from the Nfatc1 locus to specifically remove Brg1 from endocardial but not endothelial or hematopoietic cells (Wu et al. 2012).
Nfatc1Cre;Brg1F/F mice rarely survived to adulthood, accounting for 3% rather than an expected 25% of animals generated from Nfatc1Cre;Brg1F/+ X Brg1F/F intercrosses (Fig. S2A, F). A decreased fraction of viable Nfatc1Cre;Brg1F/F embryos was first observed at E16.5 with few animals surviving after birth, demonstrating late embryonic or perinatal lethality. Hearts from the rare viable postnatal day 0 (P0) Nfatc1Cre;Brg1F/F were grossly larger and histologically had a thickened compact myocardium, possibly reflecting endocardial BAF complex roles in trabecular development (Fig. S2B–E). The coronary artery network was reduced (Fig. S2G–J), consistent with known BAF complex functions in vasculogenesis (Griffin et al. 2008; Stankunas et al. 2008) and Nfatc1Cre-driven recombination in coronary vessel endothelium of endocardial origin (Wu et al. 2012). These defects likely contributed to the perinatal lethality. In addition to these phenotypes, hearts from Nfatc1Cre;Brg1F/F newborn mice had histologically thickened aortic and pulmonic valves (Fig. S2K–P).
We examined if the semilunar valve defects seen in Nfatc1Cre;Brg1F/F mice manifested earlier in development by H&E staining of sectioned E16.5 embryos. The semilunar valve cusps of Nfatc1Cre;Brg1F/F embryos were strikingly thickened and poorly elongated. The thinner “hinge” region that demarcates the boundary between the distal and basal regions of each cusp of both aortic and pulmonic valves was absent (Fig. 1A–B, D–E). In contrast, the mitral valve of Nfatc1Cre;Brg1F/F embryos was of normal size and organization (Fig. 1C, F). Quantitatively, the left and right cusps of the pulmonic valve from Nfatc1Cre;Brg1F/F E16.5 embryos had significantly decreased length:width ratios (0.998 vs. 0.581, P < 1.0E-4), were increased in area (0.998 vs. 1.76, P < 6.0E-3), and had a concordant modest increase in the number of interstitial cells per valve section (P < 2.0E-3) (Fig. 1G). Semilunar valve defects in Nfatc1Cre;Brg1F/F embryos first became apparent at E14.5 as a decreased length:width ratio of the forming cusps although without a change in sectional area (Fig. S3B, F). E14.5 Nfatc1Cre;Brg1F/F embryos also occasionally had a small membranous ventricular septal defect, which usually resolved by E16.5 in surviving embryos (> 90%, n= 11) (Fig. S3J–M). E16.5 Nfatc1Cre;Brg1F/F mitral valve leaflets exhibited no significant change in morphology or cell number (Fig. 1H), even though many mitral valve endocardial and mesenchymal cells had lost Brg1 protein by E14.5 (Fig. S4A–F). The divergent effects on semilunar vs. atrioventricular canal valves suggested that the BAF complex has a unique role during semilunar valve development or that the idiosyncrasies of Nfatc1Cre deletion kinetics only perturbed Brg1’s role in a common process in the developing outflow tract.
Figure 1. Endocardial-lineage Brg1 deficient embryos exhibit semilunar valve defects.

(A–F) Hematoxylin and eosin (H&E) stained sections from E16.5 wildtype and Nfatc1Cre;Brg1F/Fembryos showing indicated valves. Arrowheads mark the basal cusp regions of the semilunar valves. (G, H) Morphometric analyses of pulmonic and mitral valves from E16.5 wildtype and Nfatc1Cre;Brg1F/F embryos. Each data point represents one valve, with values normalized to the mean of the wildtype samples from individual litters. Asterisks indicate a significant difference (P < 0.05, Student’s two-tailed t-tests). (I, J) 3-dimensional rendering of serial sections of aortic valves from E16.5 wildtype and Nfatc1Cre;Brg1F/F littermate embryos. AoV: aortic valve; L/W: length-to-width; CC: mesenchymal cell count; NS, not statistically significant; LCC (blue): left coronary cusp; NCC (red): non-coronary cusp; RCC (yellow): right coronary cusp; LCC-NCC (purple): bicuspid fusion between left and non-coronary cusps. Scale bars: 100 µm.
3-D reconstruction of serial sections through the entire aortic valve of E16.5 Nfatc1Cre;Brg1F/F and littermate embryos revealed that Nfatc1Cre;Brg1F/F aortic valves lacked the typical thin elongated cusps and extended aortic sinuses of wildtype littermates (Fig. S5A–D). However, Nfatc1Cre;Brg1F/F embryos retained coronary ostia with normal origins of the coronary arteries (Fig. S5E, F). Three of the six 3-D reconstructed Nfatc1Cre;Brg1F/F aortic valves were bicuspid. In each case, the bicuspid valve originated from a fusion between the left and non-coronary cusps (LCC-NCC), with residual individual cusp attachment points to the surrounding muscle. While several Nfatc1Cre;Brg1F/F aortic valves had larger volumes than those of paired littermates, this change was not statistically significant (Fig. S5G–R). Therefore, area changes seen by sectional analysis likely represented the bulbous shape of the valves rather than increased size, additionally highlighting how 2-D morphometric analyses of valve development can be misleading. Consistent with the valves having a normal volume, the pulmonic valve mesenchyme of E14.5/E15.5 Nfatc1Cre;Brg1F/F mice had no change in overall proliferation (measured by EdU incorporation and phospho-histone H3 staining, Fig. S6A–D, I–J) or apoptosis (measured by TUNEL and anti-cleaved caspase 3, Fig. S6E–H, K). Collectively, these results suggested that the semilunar valve defects in Nfatc1Cre;Brg1F/F mice primarily represent a patterning/morphogenesis defect rather than overall misregulated growth.
Bicuspid aortic valve (BAV) disease in endocardial Brg1 deficient mice
The semilunar valve (SLV) defects present at E16.5 indicated that Nfatc1Cre;Brg1F/F mice would provide a mouse model of adult semilunar valve disease if not for the predominant perinatal lethality. Therefore, we conditionally deleted Brg1 with the less efficient parent line, Nfatc1Cre-PGK that retains the hygromycin-resistance cassette used for gene targeting (Wu et al. 2012). As described, the majority of Nfatc1Cre-PGK;Brg1F/F mice survived to adulthood, gradually losing their hair and whiskers due to a failure of hair follicle stem cell renewal (Xiong et al. 2013). While most Nfatc1C re-PGK;Brg1F/F mice had a normal lifespan (monitored for up to two years), their hearts were frequently enlarged with thickened ventricles (Fig. 2B, C, Fig. S7A–D). This cardiomyopathy could represent cardiac hypertrophy arising from pressure overload caused by a partial outflow tract obstruction. We examined hearts from adult Nfatc1Cre-PGK;Brg1F/F mice for semilunar valve defects (Fig. 2D–G) and found that Nfatc1Cre-PGK;Brg1F/F adult mice frequently had substantially thickened aortic valve cusps (21/26; 0/26 control littermates were affected). Nine had a bicuspid aortic valve (six LCC-NCC and three RCC-NCC), recapitulating the E16.5 Nfatc1Cre;Brg1F/F embryonic phenotype. The pulmonic valves were similarly thickened, while the mitral valve appeared qualitatively normal (Fig. S7E–H).
Figure 2. Endocardial-lineage Brg1 deficient adult mice develop aortic valve disease.

(A) Fraction of adult Nfatc1Cre-PGK;Brg1F/F or Nfatc1Cre;Brg1F/F mice with 1) thickened/myxomatous and/or 2) bicuspid aortic valve. 0/26 littermate control animals had abnormal aortic valves. (B, C) Whole mount anterior view of hearts from one year old (1yo) wildtype and Nfatc1Cre-PGK;Brg1F/F littermate mice. (D–G) H&E stained wildtype (D, E) and Nfatc1Cre-PGK;Brg1F/F (F, G) aortic valve sections. (H–K) Adult aortic valve sections stained using Masson’s Trichrome stain (collagen deposition in blue). Wildtype (H, I) and Nfatc1Cre;Brg1F/F (J, K) samples are shown. Black arrows mark mislocalized collagen. (L–O) Von Kossa and Alcian Blue (counterstained with Nuclear Fast Red) stained tissue sections from wildtype (L, M) and Nfatc1Cre;Brg1F/F (N, O) adult aortic valves. Black arrowheads indicate cartilaginous nodules. LCC: left coronary cusp; NCC: non-coronary cusp; RCC: right coronary cusp; LCC/RCC-NCC: bicuspid fusion between left/right and non-coronary cusps. Scale bars: 100 µm.
Trichrome staining indicated that Nfatc1Cre-PGK;Brg1F/F and rare surviving Nfatc1Cre;Brg1F/F mice aortic valves had myxomatous characteristics, with extensive proteoglycan-rich material throughout the valve and intermittent and dispersed collagen deposits rather than the typical enrichment of collagen along the arterial side of the cusps. Irregular chondrocyte-like cells with large nuclei and scattered small-nucleated cells were present, especially at the lateral edges of the cusps where they contact neighboring muscle (Fig. 2H–K). Similar histologic abnormalities are seen in human aortic valve disease and additional mouse models (Satta et al. 2002; Jackson et al. 2003; Kern et al. 2010; Cheek et al. 2012; Hofmann et al. 2012; Krishnamurthy et al. 2012). We assessed if the valves became calcified, as can occur in human disease, using Von Kossa staining. Unexpectedly, we observed little or no change in calcification in adult Nfatc1Cre;Brg1F/F aortic valves irrespective of age or severity of the valve thickening/disorganization phenotype (Fig. 2L–O). Collectively, Nfatc1Cre-PGK;Brg1F/F and rare surviving Nfatc1Cre;Brg1F/F mice establish a unique model of aortic valve disease that 1) has thickened, misorganized and myxomatous cusps, 2) forms bicuspid arrangements usually arising from LCC-NCC fusions and 3) does not become overtly calcified.
Endocardial Brg1 deficient semilunar valves exhibit a loss of interstitial organization
We explored whether the ECM defects seen in adult Nfatc1Cre-PGK;Brg1F/F adult mice manifested developmentally or reflected disease-like progression by antibody staining for ECM proteins expressed in developing valves. Vcan, which is essential for valve maturation and enriched in the medial-distal region of semilunar valve cusps (Kern et al. 2010; Dupuis et al. 2011), became diffusely localized in E16.5 Nfatc1Cre;Brg1F/F embryos with expression extending into the base regions (Fig. 3A–H). Postn expression, another ECM protein essential for valve maturation (Norris et al. 2008; Snider et al. 2008), was both expanded and increased in Nfatc1Cre;Brg1F/F semilunar valve cusps (Fig. 3I–L). In contrast to Vcan and Postn, Tnc was concentrated on the ventricular side and towards the base of wildtype E16.5 semilunar valve cusps (Fig. 3M, O) (Lincoln et al. 2004; Hinton et al. 2006). Reduced Tnc expression in Nfatc1Cre;Brg1F/F embryos was especially apparent in the left cusps of both aortic and pulmonic valves (Fig. 3N, P), correlating with the sites of elevated Postn. Together, the mislocalized ECM indicates a loss of patterning of the semilunar valve cusps into distinct base and distal regions in Nfatc1Cre;Brg1F/F embryos. Mechanistically, the disrupted ECM pattern could reflect the misexpression of locally restricted ECM or ECM-modifying transcripts and/or altered organization of distinct valve interstitial cell populations.
Figure 3. Altered localization and levels of extracellular matrix proteins in Nfatc1Cre;Brg1F/F semilunar valves.

(A–H) Widefield fluorescence images of anti-Versican (Vcan) antibody stained sections of E16.5 wildtype and Nfatc1Cre;Brg1F/F embryos. (I–P) Representative confocal imaged cryosections of E16.5 wildtype and Nfatc1Cre;Brg1F/F semilunar valves fluorescently immunostained for Periostin (Postn) (I–L) or Tenascin C (Tnc) (M–P) (n=3, paired control and Brg1-deficient valves). Pulmonic (PV) or aortic (AoV) valves are shown as indicated. ECM proteins are shown in green/gray scale (A–D) or cyan (I–P). Hoechst-stained nuclei are blue. White arrowheads mark areas of normal protein expression. Yellow arrowheads show regions with broadened (Vcan), broadened/increased (Postn) or decreased (Tnc) levels. Scale bars: 100 µm.
Nfatc1Cre;Brg1F/F semilunar valves have reduced endocardial-derived mesenchyme
Semilunar valve mesenchyme is derived from endocardial, neural crest, and possibly additional lineages that have inconsistently been reported to populate different regions of semilunar valve cusps (Jiang et al. 2000; de Lange et al. 2004; Nakamura et al. 2006; Ma et al. 2008; Jain et al. 2011; Engleka et al. 2012; Phillips et al. 2013). To explore if the aberrant ECM pattern in Nfatc1Cre;Brg1F/F embryos reflected misorganization of distinct mesenchymal sub-populations, we fate mapped endocardial cells and their mesenchymal derivatives in Nfatc1Cre;Brg1F/F hearts using the Rosa26RmTmG Cre reporter line (Muzumdar et al. 2007) while double-staining for Tnc and Postn. In E16.5 wildtype embryos, EMT-derived cells were scattered throughout the right and left pulmonic valve cusps but were enriched towards the Tnc-rich base of the cusp (Fig. 4A, B). In Nfatc1Cre;Brg1F/F embryos, EMT-derived cells were reduced, likely accounting for the loss of the Tnc-high field seen in the base of the left semilunar valve cusps (Fig. 4C, D). The mitral valves of Nfatc1Cre;Brg1F/F embryos had normal Tnc expression in spite of substantial loss of Brg1 protein in mitral valve mesenchyme (Figs. 4E, F and S4C, F). Therefore, the decreased Tnc in Nfatc1Cre;Brg1F/F semilunar valve cusps likely reflected depleted EMT-derived mesenchyme rather than BAF-dependent control of Tnc expression. The high Postn-expressing areas in the pulmonic valve cusps of Nfatc1Cre;Brg1F/F embryos were largely populated by non-labeled cells (Fig. 4G–J). Therefore, the increased Postn originated from a non-cell-autonomous response to endocardial-lineage Brg1 deletion. These results further indicate that, at least for the left cusps, specialized EMT-derived mesenchyme produces the concentrated Tnc in the base of the semilunar valves. The depletion of these cells upon loss of endocardial-lineage Brg1 accounts for the bulbous valves that lack robust basal cusp regions and therefore fail to restrain Postn-expressing cells to the distal cusp.
Figure 4. Disorganized extracellular matrix in endocardial Brg1 deficient semilunar valves reflects an altered organization of distinct mesenchymal lineages.

Immunofluorescence images of E16.5 control (Nfatc1Cre;Brg1F/+;R26RmTmG) or endocardial Brg1-deficient (Nfatc1Cre;Brg1F/F;R26RmTmG) embryo sections simultaneously stained for GFP to monitor Nfatc1Cre-lineage cells and either Tenascin C (Tnc) (A–F) or Periostin (Postn) (G–J). Pulmonic valves (A–D, G–J) and mitral valves (E–F) are shown. All panels are confocal images except (E–F), which are widefield images. Overlay images show Tnc or Postn in red, GFP indicating Nfatc1Cre-lineage cells in green, and Hoechst-stained nuclei in blue. Magnified regions are outlined in dashed boxes. White arrowheads denote normal ECM localization and yellow arrowheads represent either deficient (Tnc) or ectopic (Postn) ECM protein. Scale bars: 100 µm.
We quantitatively assessed the depletion of EMT-derived semilunar valve mesenchyme upon endocardial Brg1 deletion by scoring the fraction of GFP-labeled pulmonic valve mesenchymal cells in Nfatc1Cre;Brg1F/F;Rosa26RmTmG embryos (Fig. 5A, B). At E16.5, there was a significant decrease in the number of Nfatc1Cre-derived mesenchymal cells in both left and right pulmonic cusps (Fig. 5E, F, J). In contrast, the mitral valve retained its full complement of EMT-derived cells (Fig. 5G, H). At E14.5, when the semilunar valve defects first became evident, a similar decrease in the number of EMT-derived cells was observed in the left cusp of Nfatc1Cre;Brg1F/F pulmonic valves (mean 33.3%; P < 0.04) (Fig. 5C, D, I). A comparable trend of reduced EMT-derived cells likewise was seen in the right cusp. Given Nfatc1Cre;Brg1F/F SLVs were of normal volume and cell number, the change in the ratio of mesenchyme of different origins suggested non-EMT derived mesenchyme expanded to replace the scarce EMT-derived population.
Figure 5. A depleted pool of EMT-derived mesenchyme in endocardial Brg1-deficient semilunar valves is replaced by increased proliferation of other mesenchymal lineage(s).

(A–H) Sections of Nfatc1Cre;Brg1F/+;R26RmTmG and Nfatc1Cre;Brg1F/F;R26RmTmG embryos (pulmonic valve except mitral valve in (G, H); genotype and age as indicated) immunostained for GFP (Nfatc1Cre-lineage derived cells, green) and nuclei (Hoechst-stained, purple). Arrowheads indicate EMT-derived mesenchymal cells at the pulmonic valve cusp base near the cushion-myocardium boundary. (I, J) Quantification of the relative contribution of Nfatc1Cre-lineage pulmonic valve mesenchyme in control (Brg1F/+) and endocardial Brg1-deficient embryos (Brg1F/F) at E14.5 (I) and E16.5 (J). Left (L-PV) and right (R-PV) cusps are scored separately. The percentile fraction of Nfatc1Cre-lineage mesenchyme for each embryo is normalized to the mean of corresponding littermate wildtype samples. The mean absolute percentage of labeled and unlabeled cells is shown within individual bars of the chart (n=5). (K–L) E14.5 wildtype (Nfatc1Cre;Brg1F/+;R26RmTmG) (K) and Nfatc1Cre;Brg1F/F;R26RmTmG (L) pulmonic valve sections stained with GFP (Nfatc1Cre-lineage, green), EdU-incorporating proliferating cells (red), and Hoechst (nuclei, blue). White arrows denote EdU+, non-Nfatc1Cre-lineage mesenchymal cells. (M) Quantitation of the proliferation rate of Nfatc1Cre- and other-lineage pulmonic valve mesenchyme in E14.5 control and Nfatc1Cre;Brg1F/F embryos. The mean absolute fraction of EdU-incorporated cells for each condition is shown. For all graphs, error bars show one standard deviation of the mean. Asterisks indicate a significant difference (P < 0.05, two-tailed Student’s t-tests). NS: not significant; GFP+Nfatc1Cre-lineage labeled mesenchyme; Non-LL: non-Nfatc1Cre-lineage labeled mesenchyme. Scale bars: 100 µm.
EMT-derived cells of Nfatc1Cre;Brg1F/F SLVs could be reduced due to a change in their net growth rate and/or deficient EMT when the cells were first generated. To distinguish between these possibilities, we examined proliferation rates by EdU incorporation in EMT-lineage traced wildtype and Nfatc1Cre;Brg1F/F embryos. Residual EMT-derived mesenchyme and endocardial cells proliferated normally (Fig. 5K–M). In contrast, we observed a nearly two-fold increase in EdU-incorporated non-Nfatc1Cre-lineage-labeled mesenchymal cells (Fig. 5M). Therefore, this specialized cushion mesenchyme, likely of NCC and dOFT cushion origins, proliferated to compensate for the deficient EMT-derived pool. Therefore, Brg1 is not inherently required in cushion mesenchyme for cell growth or survival and origins of the semilunar valve defects in Nfatc1Cre;Brg1F/F embryos likely rest earlier in outflow tract development when EMT-derived cells are generated.
Semilunar valve defects originate from deficient pOFT EMT
To determine if outflow tract EMT was disrupted in Nfatc1Cre;Brg1F/F embryos, we examined cushion development at E10.5, less than 24 hours after EMT onset. The pOFT, but not the NCC-populated dOFT cushions of Nfatc1Cre;Brg1F/F embryos contained approximately half the normal number of mesenchymal cells (p < 0.022) (Fig. 6A, B, E). As expected, the AVC cushions were normal (Fig. 6C–E). We used the Rosa26RmTmG reporter line to confirm the deficient pOFT mesenchyme in E10.5 Nfatc1Cre;Brg1F/F embryos represented a loss of EMT-derived cells (Fig. 6F–G). A reduced pOFT with fewer lineage-traced EMT-derived cells persisted at E11.5 in endocardial Brg1-deficient embryos (Fig. 6H–K). We further explored a potential primary pOFT EMT defect in Nfatc1Cre;Brg1F/F embryos using collagen gel explant assays (Runyan and Markwald 1983) and a Nfatc1Cre-driven lineage tracing approach to brightly labeled EMT-derived cell nuclei with GFP. E10.0 Nfatc1Cre;Brg1F/F;Rosa26RrtTA;TRE:H2B–GFP OFT cushion explants produced significantly fewer GFP+ mesenchymal cells (p < 1.0E-4) (Fig. 6L–N). These results confirmed that endocardial deletion of Brg1 driven by Nfatc1Cre produced a modest EMT defect in the pOFT but not AVC cushions.
Figure 6. Reduced EMT-derived semilunar valve mesenchyme i7n Nfatc1Cre;Brg1F/F embryos originates from deficient pOFT EMT, a process that generally requires Brg1 function.
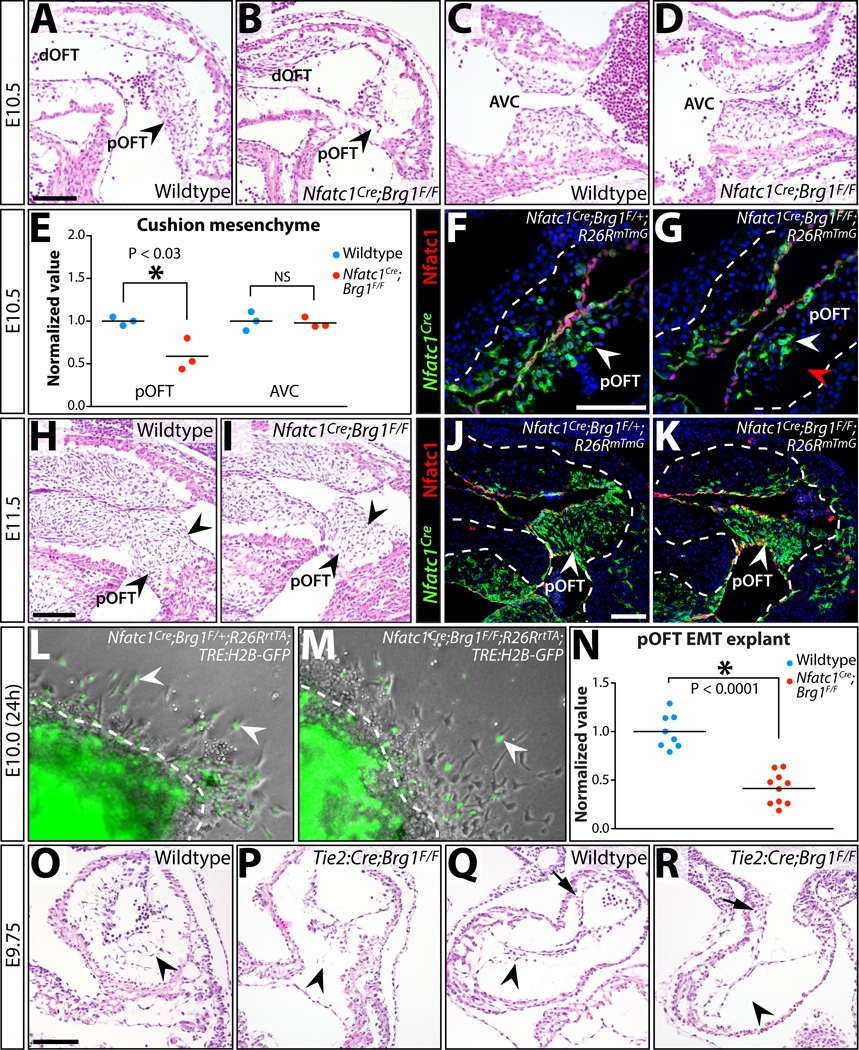
(A–D) H&E stained sagittal sections of E10.5 wildtype and Nfatc1Cre;Brg1F/F embryos. The proximal (pOFT) and distal (dOFT) outflow tract (A, B) and atrioventricular canal (AVC) cushions (C, D) are shown. Arrowheads indicate pOFT mesenchyme. (E) Scoring of mesenchymal cells populating the pOFT or AVC cushions in E10.5 wildtype and Nfatc1Cre;Brg1F/F embryos. Each point shows one embryo and all values are normalized to the mean of littermate wildtype samples. Asterisk denotes a significant difference (two-tailed Student’s t-tests). (F-G, J-K) Widefield fluorescence images of OFT regions from transverse sectioned E10.5 (F, G) or sagittal sectioned E11.5 (J, K) embryos. Tissue is immunostained for GFP (for Nfatc1Cre-lineage tracing, green) and Nfatc1 (endocardium, red). Nuclei are stained with Hoechst (blue). Control (Nfatc1Cre;Brg1F/+;R26RmTmG; F, J) and Nfatc1Cre;Brg1F/F;R26RmTmG (G, K) embryos are shown. (H, I) H&E stained sagittal sections of E11.5 wildtype and Nfatc1Cre;Brg1F/F embryos. Arrowheads indicate pOFT mesenchyme. (L, M) Brightfield and fluorescence overlaid images of collagen gel OFT explants from E10.0 control and Nfatc1Cre;Brg1F/F embryos (24 hours post-dissection). All embryos additionally carry R26RrtTA and TRE:H2B–GFP transgenes and explants are treated with doxycycline to induce H2B–GFP expression (to label Nfatc1Cre-lineage mesenchymal cells, green). White arrowheads denote EMT-derived (GFP+) mesenchymal cells. (N) Quantification of Nfatc1Cre-lineage mesenchymal cells migrating through the collagen matrix. Each point represents one explanted OFT. Values are normalized to the mean of littermate wildtype explants. The asterisk indicates statistical significance (two-tailed Student’s t-test). (O–R) Endocardial Brg1 is required for all cushion EMT. H&E stained sections showing AVC (O,P) and pOFT (Q,R) cushions of wildtype and Tie2:Cre;Brg1F/F E9.75 littermate embryos. Arrowheads denote either the presence or a depleted/absent pool of endocardial-derived cushion mesenchymal cells. Arrows in (Q, R) show neural crest-origin mesenchyme within the dOFT cushions. Scale bars: 100 µm.
We questioned whether the isolated pOFT EMT defect in Nfatc1Cre;Brg1F/F embryos represented a specific BAF complex role in pOFT endocardium, or if the induction of Cre activity in the Nfatc1Cre line was too late to remove Brg1 in AVC endocardial cells prior to EMT. Supporting this latter possibility, EMT occurs earlier in the AVC than pOFT (Camenisch et al. 2002). Further, Nfatc1Cre;Brg1F/F embryos did not exhibit a widespread loss of endocardial Brg1 protein until E10.5 (Fig. S8, Fig. S9). To achieve an earlier deletion, we used Tie2:Cre with the floxed Brg1 alleles to remove endocardial Brg1 by E9.5 (Stankunas et al. 2008). E9.75 Tie2:Cre;Brg1F/F embryos displayed a near complete absence of EMT in both the pOFT and AVC cushions (Fig. 6O–R). Therefore, endocardial BAF complex-mediated chromatin remodeling is required for both AVC and pOFT EMT. The serendipitous timing of Brg1 deletion at the onset of EMT accounts for the unique and partial pOFT EMT deficiency seen in Nfatc1Cre;Brg1F/F embryos.
To assess potential ongoing endocardial roles for Brg1, we used an additional Cre line, Nfatc1enh:Cre, to delete floxed Brg1 in cushion endocardial cells that undergo minimal or no EMT (Wu et al. 2011). Lineage tracing experiments using Rosa26RmTmG confirmed that Nfatc1enh:Cre was active only in cushion endocardial cells and not valve mesenchyme at E14.5 (Fig. S10A–D). Nfatc1enh:Cre;Brg1F/F embryos exhibited no change in semilunar valve morphology at E14.5 or E16.5 (Fig. S10E–L). The absence of major post-EMT roles for Brg1 in either endocardium or cushion mesenchyme suggests that pOFT EMT is the disrupted process that “triggers” semilunar valve defects in Nfatc1Cre;Brg1F/F embryos. The ultimate manifestation of disorganized ECM and poorly patterned valves thus represents a progression towards a disease-like state, driven by imperfect compensation by non-EMT derived and functionally distinct mesenchymal cells.
Transcriptional profiling of Brg1-deficient cardiac cushions reveals novel regulators
We sought to characterize misexpressed transcripts in Nfatc1Cre;Brg1F/F embryos that could characterize and/or promote the progression of their EMT-deficient SLVs into a diseased state. None amongst a select group of semilunar valve disease-associated transcripts were misexpressed by quantitative qRT-PCR at E14.5 (Fig. S11). Therefore, we used RNA-Seq to identify differences between the E14.5 SLV transcriptomes of wildtype and Nfatc1Cre;Brg1F/F embryos. We identified 43 differentially expressed genes (DEGs) (22 increased, 21 decreased; Table S1) and selected seven for further study (see Supplemental Materials and Methods). We used quantitative RT-PCR on additional sets of dissected tissue to confirm both up-regulated (Nptx1, Clec7a and Cthrc1) and down-regulated (Col23a1, Enpp2, Irf6 and Smoc1) genes were significantly misexpressed at E14.5. At E16.5, Clec7a, Cthrc1, and Nptx1 remained upregulated, suggesting they define a progressively worsening disease-like state (Fig. 7B–C). Conversely, Col23a1, Enpp2 and Smoc1 transcripts were no longer decreased at E16.5, indicating these genes were transiently misexpressed in endocardial-Brg1 deficient embryos at the onset of overt semilunar valve defects.
Figure 7. RNA-seq identifies novel semilunar valve expressed transcripts misexpressed in endocardial lineage Brg1 deficient valves.

(A) Select differentially expressed genes (DEGs) identified from an RNA-seq analysis comparing two paired samples of wildtype and Nfatc1Cre;Brg1F/F dissected E14.5 heart valve tissue. 26 DEGs (green-upregulated; red-downregulated) are listed with fold change and false discovery rate (FDR). Transcripts of particular interest are highlighted. (B,C) Quantitative RT-PCR expression studies of the highlighted DEGs in (A) E14.5 (B) and E16.5 (C) valves. Bar heights represent the log-base-2 fold change in gene expression (linear mean fold change value indicated below bars) between Nfatc1Cre;Brg1F/F and wildtype tissue. Error bars represent one standard deviation with the additional error bars centered along the x-axis showing the variation between wildtype samples. Double asterisks indicate a significant difference including a Bonferroni correction (one-tailed Student’s t-test, n=7, P < 0.007); single asterisk indicates a P < 0.05. (D–K) RNA in situ hybridizations for indicated transcripts on pulmonic valve sections of E13.5 wildtype or littermate Nfatc1Cre;Brg1F/F embryos. Arrowheads highlight areas of highest transcript expression. Scale bar: 100 µm.
To characterize the cell type-specific expression of these potential regulators of SLV development and disease, we performed in situ hybridizations on E14.5 wildtype and Nfatc1Cre;Brg1F/F embryos. Nptx1, a member of the neuronal pentraxin family (Omeis et al. 1996; Boles et al. 2014) was expressed in the most distal endocardial cells of the pulmonic valve and had notably higher levels in endocardial Brg1 mutants (Fig. 7D–E). However, no change in Nptx1 expression was observed in the mitral valve, suggesting that Nptx1 is an OFT endocardial-specific transcript (Fig. S12A–D). In contrast, the collagen triple helix repeat containing 1, Cthrc1 (Pyagay et al. 2005), protein was found only in mesenchymal cells at the very base of the cusps where cushion-derived tissue met adjacent myocardium. As expected, cushion mesenchyme Cthrc1 levels were higher in Nfatc1Cre;Brg1F/F embryos (Fig. 7F–G). Further, Cthrc1 was increased in aortic valve mesenchyme and became ectopically expressed in both valve and ventricular endocardium (Fig. S12E–J). Enpp2, a secreted phospholipase required for lysophosphatidic acid production (Tokumura et al. 2002; Umezu-Goto et al. 2002), and Irf6, an interferon-responsive factor (Kondo et al. 2002), were both specifically expressed in endocardial cells on the arterial side of wildtype pulmonic valves. Enpp2 and Irf6 were largely absent in the endocardium from Nfatc1Cre;Brg1F/F semilunar valves (Fig. 7H–K, Fig. S12K, M, O, R). In contrast, pulmonic arterial endothelial expression of Irf6 was unchanged (Fig. S12Q, T). Given their misexpression in endocardial and/or EMT-derived cells, these four DEGs could represent directly repressed (Nptx1, Cthrc1) or activated (Enpp2, Irf6) targets of BAF complex chromatin remodeling. Further studies are merited to test their potential roles in valve development and disease and/or broader utility as markers of SLV disease progression.
DISCUSSION
Endocardial Brg1 deletion establishes a model of semilunar valve disease with origins in deficient EMT
We describe a new mouse model of semilunar valve disease caused by deletion of Brg1 in the endocardial lineage. Nfatc1Cre;Brg1F/F mice develop thickened, blunted, and occasionally fused valve cusps that become myxomatous in surviving adults. We map the origins of these phenotypes to disrupted pOFT EMT, one of the earliest steps in valve development. OFT mesenchyme of other origins, likely cardiac neural crest, compensates for the deficient EMT-derived cells by relatively increased proliferation but is incompetent to fully generate the complex 3-dimensionally patterned interstitium of a tricuspid semilunar valve. As such, the cellular and extracellular changes that characterize the abnormal semilunar valves represent secondary responses to a disrupted system, rather than reflecting later developmental or physiologic roles of Brg1.
Chromatin remodeling contributes to endocardial cushion EMT
Early deletion of floxed Brg1 in all endocardial cells using the Tie2:Cre driver disrupted EMT in both the AVC and pOFT cushions. These results establish chromatin remodeling as a component of the transcriptional networks that promote EMT. Endocardial Brg1 deletion does not have a significant effect on endocardial cell death or proliferation (Stankunas et al. 2008), suggesting that cushion endocardial BAF complexes have a relatively specific transcriptional role in EMT. A simple explanation is that the BAF complex has a common chromatin remodeling role in all EMT processes, whether in the endocardial cushions or other developmental/disease situations. Consistent with this possibility, the Baf60c subunit of the BAF complex is required for EMT in cancer cell lines (Jordan et al. 2013) and Brg1 interacts with the Zeb1 transcription factor to promote a terminal step in the mesenchymal transition of MCF7 cells (Sánchez-Tilló et al. 2010). Alternatively, effectors of upstream EMT-inducing signaling pathways, such as TGF-beta or Notch signaling, may recruit BAF complex activity to induce EMT. In support of these possibilities, both the Smad2/3 effectors of TGF-beta signaling and activated Notch1, each of which has been implicated in both AVC and pOFT EMT, cooperate with the BAF complex and/or induce chromatin remodeling in other circumstances (Takeuchi et al. 2007; Xi et al. 2008).
The semilunar valve interstitium is organized into regions enriched with mesenchyme of different origins
Multiple lineage tracing studies suggest a complex patterning of multi-origin mesenchymal populations within semilunar valves (Jiang et al. 2000; de Lange et al. 2004; Nakamura et al. 2006; Jain et al. 2011; Phillips et al. 2013). Our simultaneous study of the localization of valve ECM components with Nfatc1-based lineage tracing indicates that the patterned ECM composition of semilunar valves is conferred by the regulated organization of mesenchymal cells with differential competency to produce ECM proteins. For example, EMT-derived cells likely produce the Tnc-enriched base of the left semilunar valve cusps that is depleted in Nfatc1Cre;Brg1F/F embryos. As a corollary, the increased Postn seen in Nfatc1Cre;Brg1F/F embryos could reflect an expanded non-EMT origin (likely neural crest-derived) mesenchyme that continues to fulfill its normal role of secreting Postn. This general concept of semilunar valve patterning provides a framework to understand when and how valve regulators function and comprehend how various mechanisms can generate valve defects.
Interpreting mouse models of semilunar valve disease
A number of other mouse models exhibit either bicuspid aortic valve and/or semilunar valve disease-like phenotypes. When bicuspid, the aortic valves of Nfatc1Cre;Brg1F/F embryos display either a RCC-NCC or, more frequently, what in humans is the rarest LCC-NCC subtype (Fernandes et al. 2004; Fernández et al. 2009; Kang et al. 2013). Grewal et al. proposed that the “type I” subtype showing fusions between RCC-LCC cusps is caused by a deficiency of NCCs (Jain et al. 2011; Phillips et al. 2013; Grewal et al. 2014). In contrast, “type II” fusions (RCC-NCC) are suggested to be of second heart field (SHF) origin, which could encompass pOFT EMT defects given the outflow tract’s SHF origins. Consistent with this idea, the Nos3-deficient mouse model of BAV develops RCC-NCC fusions and may, like the Brg1-deficient model, originate from defective EMT (Fernández et al. 2009). Whether a RCC-NCC or LCC-NCC fusion occurs could reflect the severity of the EMT disruption, reflected by the variable Brg1 deletion in Nfatc1Cre;Brg1F/F embryos.
Could other mouse models of thickened and disorganized semilunar valves also have origins in pOFT EMT defects? Supporting this hypothesis, several other models disrupt known or potential EMT regulators, including the TGF-β effector Smad2 (combined with Adamts5), TGF-β family receptors (Alk2), Gata-family transcription factors (Gata5), Notch signaling, and BMP signaling (BMP4) (Camenisch et al. 2002; Timmerman et al. 2004; Ma et al. 2005; Wang et al. 2005; McCulley et al. 2008; Laforest et al. 2011; Moskowitz et al. 2011; Thomas et al. 2012; Bosse et al. 2013; Dupuis et al. 2013). Testing this idea would require directly examining these mice for pOFT EMT defects as well as monitoring their valves for a deficient Tnc-expressing base region, as observed in the endocardial Brg1-deficient model. Another set of models, for instance Cxcr7KO, EgfrKO;Ptpnll+/− and Hb-egfKO have clear hyperplastic semilunar valve cusps (Chen et al. 2000; Jackson et al. 2003; Yu et al. 2011), which we did not observe in Nfatc1Cre;Brg1F/F embryos by EdU incorporation and cusp volume measurements. We postulate that this manifestation of a semilunar valve defect likely originates in misregulated proliferative signals rather than deficient EMT. However, our combined EdU incorporation and lineage tracing of Nfatc1Cre;Brg1F/F valves does show how a subset population of semilunar valve mesenchyme can have an increased proliferative rate even while overall mesenchyme proliferation is unchanged. In endocardial Brg1-deficient mice, this effect is non-autonomous, with non-EMT derived mesenchyme increasing their relative rate of proliferation to compensate for the deficient EMT-derived cells. This observation also supports the idea that different valve mesenchymal populations communicate to modulate each other’s growth and organization (Jain et al. 2011; Wu et al. 2011). By extension, neural crest (or other origin) mesenchyme could also be genetically perturbed in semilunar valve disease, as indicated by Rho kinase and Pax3 deficient models (Jain et al. 2011; Phillips et al. 2013).
RNA-Seq of the endocardial Brg1-deficient valves uncovers potential novel regulators of semilunar valve development and disease
Our transcriptomics study of E14.5 Nfatc1Cre;Brg1F/F valve regions identified a number of such potential novel regulators of valve development and/or disease biomarkers. For example, we found misexpressed negative regulators of both BMP signaling (Smoc1, Thomas et al. 2009) and TGF-beta signaling (Mirlet7a-1, Colas et al. 2012), two pathways that can be aberrantly active in mesenchyme of developmentally abnormal valves (reviewed in Conway et al. 2011). Our subsequent RNA ISH analysis revealed that several of the misexpressed genes had highly specific and unique expression patterns, showing the high degree of cellular specialization within semilunar valves. For example, Nptx1 was expressed only in distal tip endocardium of both semilunar valves. Similarly, Enpp2, and Irf6 also are normally expressed in subsets of endocardial cells. In contrast, Cthrc1 transcripts were restricted to mesenchyme at the base of the semilunar valve. Given their altered expression in endocardial or possible EMT-derived cells (Cthrc1) of Nfatc1Cre;Brg1F/F valves, each of these genes may be a direct target of BAF complex activation or repression. Indeed, Brg1 binds near the 5’ end of both Irf6 and Nptx1 genes by ChIP-Seq studies of E11.5 hearts (Li et al. 2013).
Endocardial Brg1-deficient model of semilunar valve disease distinguishes between the origins and progression of semilunar valve disease
Our interpretation of the valve defects in Nfatc1Cre;Brg1F/F embryos provides an explanation for how semilunar valve disease, including thickened and fused cusps, could commonly originate in a sensitive EMT process of early valvulogenesis. Disease progression then represents the “best effort” of robust developmental networks to produce and maintain a functional valve. As such, a genetic understanding of inherited semilunar valve disease may frequently reflect EMT mechanisms, while efforts to therapeutically attenuate disease progression may merit focusing on manipulating compensating cell lineages and/or their ECM products. For the former goal, mutations in BAF component genes themselves are unlikely to be causes of aortic valve disease given the pleiotropic roles of BAF complex driven chromatin remodeling. However, interacting transcription factors and target genes of BAF-direct chromatin regulation during EMT would represent attractive candidate disease genes. For the latter goal, our RNA-Seq analysis of progressively worsening valves in endocardial Brg1-deficient mice has identified new candidate regulators with highly specific expression patterns that may characterize or even drive progression of an anatomically abnormal valve into a disease state.
Supplementary Material
Highlights.
Endocardial-specific deletion of Brg1 establishes a semilunar valve disease model
Brg1-driven chromatin remodeling is required for endocardial cushion EMT
Mesenchyme of distinct origins have unique roles in semilunar valve ECM patterning
Incompetent mesenchymal lineages compensate for deficient outflow tract EMT
RNA-Seq of abnormal semilunar valves uncovers novel valve developmental Genes
ACKNOWLEDGEMENTS
This project originated in the laboratory of Dr. Ching-Pin Chang and benefited from his early technical and intellectual input. Dr. Bin Zhou shared the Nfatc1-based Cre lines in advance of publication. We thank Vidusha Devasthali, Jordan Gessaman, Kelsey Ward, Jackson Corning, Zhi-Yang Tsun, and Gene Ma for technical help and Stankunas lab members for feedback. The UO ACISS computing cluster used for RNA-Seq analyses was NSF-supported (OCI-0960354). B.N.A. was funded by an American Heart Association (AHA) predoctoral fellowship (12PRE11800017). M.L.S. received an AHA summer research award. The March of Dimes (Basil O’Connor Award) and the National Institutes of Health (4R00HL087598 and 1R01HL115294) provided funding (K.S. lab).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
B.N.A. and K.S. designed the experiments. B.N.A. and M.L.S. performed experiments. B.N.A. and K.S. prepared and wrote the manuscript.
REFERENCES
- Angelini A, Ho SY, Anderson RH, Devine WA, Zuberbuhler JR, Becker AE, Davies MJ. The morphology of the normal aortic valve as compared with the aortic valve having two leaflets. J Thorac Cardiovasc Surg. 1989;98:362–367. [PubMed] [Google Scholar]
- Boles NC, Hirsch SE, Le S, Corneo B, Najm F, Minotti AP, Wang Q, Lotz S, Tesar PJ, Fasano CA. NPTX1 Regulates Neural Lineage Specification from Human Pluripotent Stem Cells. Cell Reports. 2014;6:724–736. doi: 10.1016/j.celrep.2014.01.026. [DOI] [PubMed] [Google Scholar]
- Bosse K, Hans CP, Zhao N, Koenig SN, Huang N, Guggilam A, LaHaye S, Tao G, Lucchesi PA, Lincoln J, et al. Endothelial nitric oxide signaling regulates Notch1 in aortic valve disease. Journal of Molecular and Cellular Cardiology. 2013;60:27–35. doi: 10.1016/j.yjmcc.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broom ND. The observation of collagen and elastin structures in wet whole mounts of pulmonary and aortic leaflets. J Thorac Cardiovasc Surg. 1978;75:121–130. [PubMed] [Google Scholar]
- Bultman S, Gebuhr T, Yee D, La Mantia C, Nicholson J, Gilliam A, Randazzo F, Metzger D, Chambon P, Crabtree G, et al. A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Molecular Cell. 2000;6:1287–1295. doi: 10.1016/s1097-2765(00)00127-1. [DOI] [PubMed] [Google Scholar]
- Camenisch TD, Molin DGM, Person A, Runyan RB, Gittenberger-de Groot AC, McDonald JA, Klewer SE. Temporal and Distinct TGFβ Ligand Requirements during Mouse and Avian Endocardial Cushion Morphogenesis. Developmental Biology. 2002;248:170–181. doi: 10.1006/dbio.2002.0731. [DOI] [PubMed] [Google Scholar]
- Cardona A, Saalfeld S, Schindelin J, Arganda-Carreras I, Preibisch S, Longair M, Tomancak P, Hartenstein V, Douglas RJ. TrakEM2 Software for Neural Circuit Reconstruction ed. A. Samuel. PLoS ONE. 2012;7:e38011. doi: 10.1371/journal.pone.0038011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang ACY, Fu Y, Garside VC, Niessen K, Chang L, Fuller M, Setiadi A, Smrz J, Kyle A, Minchinton A, et al. Notch Initiates the Endothelial-to-Mesenchymal Transition in the Atrioventricular Canal through Autocrine Activation of Soluble Guanylyl Cyclase. Developmental Cell. 2011;21:288–300. doi: 10.1016/j.devcel.2011.06.022. [DOI] [PubMed] [Google Scholar]
- Cheek JD, Wirrig EE, Alfieri CM, James JF, Yutzey KE. Differential activation of valvulogenic, chondrogenic, and osteogenic pathways in mouse models of myxomatous and calcific aortic valve disease. Journal of Molecular and Cellular Cardiology. 2012;52:689–700. doi: 10.1016/j.yjmcc.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BB, Bronson RTR, Klaman LDL, Hampton TGT, Wang JFJ, Green PJP, Magnuson TT, Douglas PSP, Morgan JPJ, Neel BGB. Mice mutant for Egfr and Shp2 have defective cardiac semilunar valvulogenesis. Nat Genet. 2000;24:296–299. doi: 10.1038/73528. [DOI] [PubMed] [Google Scholar]
- Clementi M, Notari L, Borghi A, Tenconi R. Familial congenital bicuspid aortic valve: a disorder of uncertain inheritance. Am J Med Genet. 1996;62:336–338. doi: 10.1002/(SICI)1096-8628(19960424)62:4<336::AID-AJMG2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Conway SJ, Doetschman T, Azhar M. The inter-relationship of periostin, TGF beta, and BMP in heart valve development and valvular heart diseases. The Scientific World JOURNAL. 2011;11:1509–1524. doi: 10.1100/tsw.2011.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange FJ, Moorman AFM, Anderson RH, Männer J, Soufan AT, de Gier-de Vries C, Schneider MD, Webb S, van den Hoff MJB, Christoffels VM. Lineage and morphogenetic analysis of the cardiac valves. Circulation Research. 2004;95:645–654. doi: 10.1161/01.RES.0000141429.13560.cb. [DOI] [PubMed] [Google Scholar]
- Dupuis LE, McCulloch DR, McGarity JD, Bahan A, Wessels A, Weber D, Diminich AM, Nelson CM, Apte SS, Kern CB. Altered versican cleavage in ADAMTS5 deficient mice; A novel etiology of myxomatous. Developmental Biology. 2011;357:152–164. doi: 10.1016/j.ydbio.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis LE, Osinska H, Weinstein MB, Hinton RB, Kern CB. Insufficient versican cleavage and Smad2 phosphorylation results in bicuspid aortic and pulmonary valves. Journal of Molecular and Cellular Cardiology. 2013;60:50–59. doi: 10.1016/j.yjmcc.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engleka KA, Manderfield LJ, Brust RD, Li L, Cohen A, Dymecki SM, Epstein JA. Islet1 derivatives in the heart are of both neural crest and second heart field origin. Circulation Research. 2012;110:922–926. doi: 10.1161/CIRCRESAHA.112.266510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes SM, Sanders SP, Khairy P, Jenkins KJ, Gauvreau K, Lang P, Simonds H, Colan SD. Morphology of bicuspid aortic valve in children and adolescents. Journal of the American College of Cardiology. 2004;44:1648–1651. doi: 10.1016/j.jacc.2004.05.063. [DOI] [PubMed] [Google Scholar]
- Fernández B, Durán AC, Fernández-Gallego T, Fernández MC, Such M, Arqué JM, Sans-Coma V. Bicuspid Aortic ValvesWith Different Spatial Orientations ofthe Leaflets Are Distinct Etiological Entities. JAC. 2009;54:2312–2318. doi: 10.1016/j.jacc.2009.07.044. [DOI] [PubMed] [Google Scholar]
- Fischer A, Steidl C, Wagner TU, Lang E, Jakob PM, Friedl P, Knobeloch KP, Gessler M. Combined Loss of Hey1 and HeyL Causes Congenital Heart Defects Because of Impaired Epithelial to Mesenchymal Transition. Circulation Research. 2007;100:856–863. doi: 10.1161/01.RES.0000260913.95642.3b. [DOI] [PubMed] [Google Scholar]
- Garg V, Muth AN, Ransom JF, Schluterman MK, Barnes R, King IN, Grossfeld PD, Srivastava D. Mutations in NOTCH1 cause aortic valve disease. Nature. 2005;437:270–274. doi: 10.1038/nature03940. [DOI] [PubMed] [Google Scholar]
- Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, et al. Heart Disease and Stroke Statistics--2013 Update: A Report From the American Heart Association. Circulation. 2013;127:e6–e245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grewal N, DeRuiter MC, Jongbloed MRM, Goumans MJ, Klautz RJM, Poelmann RE, Gittenberger-de Groot AC. Normal and abnormal development of the aortic wall and valve: correlation with clinical entities. Neth Heart J. 2014;22:363–369. doi: 10.1007/s12471-014-0576-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin CT, Brennan J, Magnuson T. The chromatin-remodeling enzyme BRG1 plays an essential role in primitive erythropoiesis and vascular development. Development. 2008;135:493–500. doi: 10.1242/dev.010090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross L, Kugel MA. Topographic Anatomy and Histology of the Valves in the Human Heart. Am J Pathol. 1931;7:445–474. 7. [PMC free article] [PubMed] [Google Scholar]
- Hang CT, Yang J, Han P, Cheng H-L, Shang C, Ashley E, Bin Zhou, Chang C-P. Chromatin regulation by Brg1 underlies heart muscle development and disease. Nature. 2010;466:62–67. doi: 10.1038/nature09130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton RB, Lincoln J, Deutsch G, Osinska H, Manning P, Benson DW, Yutzey KE. Extracellular Matrix Remodeling and Organization in Developing and Diseased Aortic Valves. Circulation Research. 2006;98:1431–1438. doi: 10.1161/01.RES.0000224114.65109.4e. [DOI] [PubMed] [Google Scholar]
- Hoffman JIE, Kaplan S. The incidence of congenital heart disease. JAC. 2002;39:1890–1900. doi: 10.1016/s0735-1097(02)01886-7. [DOI] [PubMed] [Google Scholar]
- Hofmann JJ, Briot A, Enciso J, Zovein AC, Ren S, Zhang ZW, Radtke F, Simons M, Wang Y, Iruela-Arispe ML. Endothelial deletion of murine Jag1 leads to valve calcification and congenital heart defects associated with Alagille syndrome. Development. 2012;139:4449–4460. doi: 10.1242/dev.084871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntington K, Hunter AGW, Chan K-L. A Prospective Study to Assess the Frequency of Familial Clustering of Congenital Bicuspid Aortic Valve. JAC. 1997;30:1809–1812. doi: 10.1016/s0735-1097(97)00372-0. [DOI] [PubMed] [Google Scholar]
- Jackson LF, Qiu TH, Sunnarborg SW, Chang A, Zhang C, Patterson C, Lee DC. Defective valvulogenesis in HB-EGF and TACE-null mice is associated with aberrant BMP signaling. EMBO J. 2003;22:2704–2716. doi: 10.1093/emboj/cdg264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain R, Engleka KA, Rentschler SL, Manderfield LJ, Li L, Yuan L, Epstein JA. Cardiac neural crest orchestrates remodeling and functional maturation of mouse semilunar valves. J Clin Invest. 2011;121:422–430. doi: 10.1172/JCI44244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. 2000;127:1607–1616. doi: 10.1242/dev.127.8.1607. [DOI] [PubMed] [Google Scholar]
- Jordan NV, Prat A, Abell AN, Zawistowski JS, Sciaky N, Karginova OA, Zhou B, Golitz BT, Perou CM, Johnson GL. SWI/SNF Chromatin-Remodeling Factor Smarcd3/Baf60c Controls Epithelial-Mesenchymal Transition by Inducing Wnt5a Signaling. Molecular and Cellular Biology. 2013;33:3011–3025. doi: 10.1128/MCB.01443-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadoch C, Crabtree GR. Reversible Disruption of mSWI/SNF (BAF) Complexes by the SS18-SSX Oncogenic Fusion in Synovial Sarcoma. Cell. 2013;153:71–85. doi: 10.1016/j.cell.2013.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JW, Song HG, Yang DH, Baek S, Kim DH, Song JM, Kang DH, Lim TH, Song JK. Association Between Bicuspid Aortic Valve Phenotype and Patterns of Valvular Dysfunction and Bicuspid Aortopathy. JCMG. 2013;6:150–161. doi: 10.1016/j.jcmg.2012.11.007. [DOI] [PubMed] [Google Scholar]
- Kern CB, Wessels A, McGarity J, Dixon LJ, Alston E, Argraves WS, Geeting D, Nelson CM, Menick DR, Apte SS. Reduced versican cleaved due to Adamts9 haploinsufficiency is associated with cardiac and aortic anomalies. Matrix Biology. 2010;29:304–316. doi: 10.1016/j.matbio.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby ML, Gale TF, Stewart DE. Neural crest cells contribute to normal aorticopulmonary septation. Science. 1983;220:1059–1061. doi: 10.1126/science.6844926. [DOI] [PubMed] [Google Scholar]
- Kondo S, Schutte BC, Richardson RJ, Bjork BC, Knight AS, Watanabe Y, Howard E, Ferreira de Lima RLL, Daack-Hirsch S, Sander A, et al. Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat Genet. 2002;32:285–289. doi: 10.1038/ng985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy VK, Opoka AM, Kern CB, Guilak F, Narmoneva DA, Hinton RB. Maladaptive matrix remodeling and regional biomechanical dysfunction in a mouse model of aortic valve disease. Matrix Biol. 2012;31:197–205. doi: 10.1016/j.matbio.2012.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruzynska-Frejtag A, Machnicki M, Rogers R, Markwald RR, Conway SJ. Periostin (an osteoblast-specific factor) is expressed within the embryonic mouse heart during valve formation. Mechanisms of Development. 2001;103:183–188. doi: 10.1016/s0925-4773(01)00356-2. [DOI] [PubMed] [Google Scholar]
- Laforest B, Andelfinger G, Nemer M. Loss of Gata5 in mice leads to bicuspid aortic valve. J Clin Invest. 2011;121:2876–2887. doi: 10.1172/JCI44555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Xiong Y, Shang C, Twu KY, Hang CT, Yang J, Han P, Lin C-Y, Lin C-J, Tsai F-C, et al. Brg1 governs distinct pathways to direct multiple aspects of mammalian neural crest cell development. Proc Natl Acad Sci USA. 2013;110:1738–1743. doi: 10.1073/pnas.1218072110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lickert H, Takeuchi JK, Both Von I, Walls JR, McAuliffe F, Adamson SL, Henkelman RM, Wrana JL, Rossant J, Bruneau BG. Baf60c is essential for function of BAF chromatin remodelling complexes in heart development. Nature. 2004;432:107–112. doi: 10.1038/nature03071. [DOI] [PubMed] [Google Scholar]
- Lincoln J, Alfieri CM, Yutzey KE. Development of heart valve leaflets and supporting apparatus in chicken and mouse embryos. Dev Dyn. 2004;230:239–250. doi: 10.1002/dvdy.20051. [DOI] [PubMed] [Google Scholar]
- Luna-Zurita L, Prados B, Grego-Bessa J, Luxán G, del Monte G, Benguría A, Adams RH, Perez-Pomares JM, la Pompa de JL. Integration of a Notch-dependent mesenchymal gene program and Bmp2-driven cell invasiveness regulates murine cardiac valve formation. J Clin Invest. 2010;120:3493–3507. doi: 10.1172/JCI42666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Lu M-F, Schwartz RJ, Martin JF. Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development. 2005;132:5601–5611. doi: 10.1242/dev.02156. [DOI] [PubMed] [Google Scholar]
- Ma Q, Zhou B, Pu WT. Reassessment of Isl1 and Nkx2–5 cardiac fate maps using a Gata4-based reporter of Cre activity. Developmental Biology. 2008;323:98–104. doi: 10.1016/j.ydbio.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCulley DJ, Kang J-O, Martin JF, Black BL. BMP4 is required in the anterior heart field and its derivatives for endocardial cushion remodeling, outflow tract septation, and semilunar valve development. Dev Dyn. 2008;237:3200–3209. doi: 10.1002/dvdy.21743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskowitz IP, Wang J, Peterson MA, Pu WT, Mackinnon AC, Oxburgh L, Chu GC, Sarkar M, Berul C, Smoot L, et al. Transcription factor genes Smad4 and Gata4 cooperatively regulate cardiac valve development. Proceedings of the National Academy of Sciences. 2011;108:4006–4011. doi: 10.1073/pnas.1019025108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. genesis. 2007;45:593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Colbert M, Robbins J. Neural Crest Cells Retain Multipotential Characteristics in the Developing Valves and Label the Cardiac Conduction System. Circulation Research. 2006;98:1547–1554. doi: 10.1161/01.RES.0000227505.19472.69. [DOI] [PubMed] [Google Scholar]
- Norris RA, Moreno-Rodriguez RA, Sugi Y, Hoffman S, Amos J, Hart MM, Potts JD, Goodwin RL, Markwald RR. Periostin regulates atrioventricular valve maturation. Developmental Biology. 2008;316:200–213. doi: 10.1016/j.ydbio.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omeis IA, Hsu YC, Perin MS. Mouse and human neuronal pentraxin 1 (NPTX1): conservation, genomic structure, and chromosomal localization. Genomics. 1996;36:543–545. doi: 10.1006/geno.1996.0503. [DOI] [PubMed] [Google Scholar]
- Phillips HM, Mahendran P, Singh E, Anderson RH, Chaudhry B, Henderson DJ. Neural crest cells are required for correct positioning of the developing outflow cushions and pattern the arterial valve leaflets. Cardiovascular Research. 2013;99:452–460. doi: 10.1093/cvr/cvt132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyagay P, Heroult M, Wang Q, Lehnert W, Belden J, Liaw L, Friesel R, Lindner V. Collagen Triple Helix Repeat Containing 1, a Novel Secreted Protein in Injured and Diseased Arteries, Inhibits Collagen Expression and Promotes Cell Migration. Circulation Research. 2005;96:261–268. doi: 10.1161/01.RES.0000154262.07264.12. [DOI] [PubMed] [Google Scholar]
- Ramsdell AF, Markwald RR. Induction of Endocardial Cushion Tissue in the Avian Heart is Regulated, in Part, by TGFβ-3-Mediated Autocrine Signaling. Developmental Biology. 1997;188:64–74. doi: 10.1006/dbio.1997.8637. [DOI] [PubMed] [Google Scholar]
- Roberts WC, Ko JM. Frequency by Decades of Unicuspid, Bicuspid, and Tricuspid Aortic Valves in Adults Having Isolated Aortic Valve Replacement for Aortic Stenosis, With or Without Associated Aortic Regurgitation. Circulation. 2005;111:920–925. doi: 10.1161/01.CIR.0000155623.48408.C5. [DOI] [PubMed] [Google Scholar]
- Runyan RB, Markwald RR. Invasion of mesenchyme into three-dimensional collagen gels: a regional and temporal analysis of interaction in embryonic heart tissue. Developmental Biology. 1983;95:108–114. doi: 10.1016/0012-1606(83)90010-6. [DOI] [PubMed] [Google Scholar]
- Satta J, Melkko J, Pöllänen R, Tuukkanen J, Pääkkö P, Ohtonen P, Mennander A, Soini Y. Progression of human aortic valve stenosis is associated with tenascin-C expression. JAC. 2002;39:96–101. doi: 10.1016/s0735-1097(01)01705-3. [DOI] [PubMed] [Google Scholar]
- Sánchez-Tilló E, Lázaro A, Torrent R, Cuatrecasas M, Vaquero EC, Castells A, Engel P, Postigo A. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene. 2010;29:3490–3500. doi: 10.1038/onc.2010.102. [DOI] [PubMed] [Google Scholar]
- Siu SC, Silversides CK. Bicuspid Aortic Valve Disease. JAC. 2010;55:2789–2800. doi: 10.1016/j.jacc.2009.12.068. [DOI] [PubMed] [Google Scholar]
- Sizarov A, Lamers WH, Mohun TJ, Brown NA, Anderson RH, Moorman AFM. Three-dimensional and molecular analysis of the arterial pole of the developing human heart. Journal of Anatomy. 2012;220:336–349. doi: 10.1111/j.1469-7580.2012.01474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider P, Hinton RB, Moreno-Rodriguez RA, Wang J, Rogers R, Lindsley A, Li F, Ingram DA, Menick D, Field L, et al. Periostin Is Required for Maturation and Extracellular Matrix Stabilization of Noncardiomyocyte Lineages of the Heart. Circulation. 2008;102:752–760. doi: 10.1161/CIRCRESAHA.107.159517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankunas K, Hang CT, Tsun Z-Y, Chen H, Lee NV, Wu JI, Shang C, Bayle JH, Shou W, Iruela-Arispe ML, et al. Endocardial Brg1 Represses ADAMTS1 to Maintain the Microenvironment for Myocardial Morphogenesis. Developmental Cell. 2008;14:298–311. doi: 10.1016/j.devcel.2007.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankunas K, Ma GK, Kuhnert FJ, Kuo CJ, Chang C-P. VEGF signaling has distinct spatiotemporal roles during heart valve development. Developmental Biology. 2010;347:325–336. doi: 10.1016/j.ydbio.2010.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart S, Gomez AW, Armstrong BE, Henner A, Stankunas K. Sequential and Opposing Activities of Wnt and BMP Coordinate Zebrafish Bone Regeneration. Cell Reports. 2014;6:482–498. doi: 10.1016/j.celrep.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi JK, Lickert H, Bisgrove BW, Sun X, Yamamoto M, Chawengsaksophak K, Hamada H, Yost HJ, Rossant J, Bruneau BG. Baf60c is a nuclear Notch signaling component required for the establishment of left-right asymmetry. Proc Natl Acad Sci USA. 2007;104:846–851. doi: 10.1073/pnas.0608118104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi JK, Lou X, Alexander JM, Sugizaki H, n PD-OI, Holloway AK, Mori AD, Wylie JN, Munson C, Zhu Y, et al. Chromatin remodelling complex dosage modulates transcription factor function in heart development. Nature Communications. 2011;2 doi: 10.1038/ncomms1187. 187-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodoris CV, Li M, White MP, Liu L, He D, Pollard KS, Bruneau BG, Srivastava D. Human Disease Modeling Reveals Integrated Transcriptional and Epigenetic Mechanisms of NOTCH1 Haploinsufficiency. Cell. 2015;160:1072–1086. doi: 10.1016/j.cell.2015.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas PS, Sridurongrit S, Ruiz-Lozano P, Kaartinen V. Deficient Signaling via Alk2 (Acvr1) Leads to Bicuspid Aortic Valve Development ed. S. Bellusci. PLoS ONE. 2012;7:e35539. doi: 10.1371/journal.pone.0035539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman LA, Grego-Bessa J, Raya A, Bertran E, Perez-Pomares JM, Diez J, Aranda S, Palomo S, McCormick F, Izpisua-Belmonte JC, et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes & Development. 2004;18:99–115. doi: 10.1101/gad.276304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkatchenko TV, Moreno-Rodriguez RA, Conway SJ, Molkentin JD, Markwald RR, Tkatchenko AV. Lack of periostin leads to suppression of Notch1 signaling and calcific aortic valve disease. Physiological Genomics. 2009;39:160–168. doi: 10.1152/physiolgenomics.00078.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokumura A, Majima E, Kariya Y, Tominaga K, Kogure K, Yasuda K, Fukuzawa K. Identification of Human Plasma Lysophospholipase D, a Lysophosphatidic Acid-producing Enzyme, as Autotaxin, a Multifunctional Phosphodiesterase. Journal of Biological Chemistry. 2002;277:39436–39442. doi: 10.1074/jbc.M205623200. [DOI] [PubMed] [Google Scholar]
- Umezu-Goto M, Kishi Y, Taira A, Hama K, Dohmae N, Takio K, Tamori T, Mills GB, Inoue K, Aoki J, et al. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. The Journal of Cell Biology. 2002;158:227–233. doi: 10.1083/jcb.200204026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh DA, Park KS, Harrington A, Miceli-Libby L, Yoon JK, Liaw L. Cardiovascular and Hematopoietic Defects Associated With Notch1 Activation in Embryonic Tie2-Expressing Populations. Circulation Research. 2008;103:423–431. doi: 10.1161/CIRCRESAHA.108.177808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldo K, Miyagawa-Tomita S, Kumiski D, Kirby ML. Cardiac neural crest cells provide new insight into septation of the cardiac outflow tract: aortic sac to ventricular septal closure. Developmental Biology. 1998;196:129–144. doi: 10.1006/dbio.1998.8860. [DOI] [PubMed] [Google Scholar]
- Wang J, Sridurongrit S, Dudas M, Thomas P, Nagy A, Schneider MD, Epstein JA, Kaartinen V. Atrioventricular cushion transformation is mediated by ALK2 in the developing mouse heart. Developmental Biology. 2005;286:299–310. doi: 10.1016/j.ydbio.2005.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wu B, Chamberlain AA, Lui W, Koirala P, Susztak K, Klein D, Taylor V, Zhou B. Katoh M, editor. Endocardial to Myocardial Notch-Wnt-Bmp Axis Regulates Early Heart Valve Development. PLoS ONE. 2013;8:e60244. doi: 10.1371/journal.pone.0060244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Wang Y, Lui W, Langworthy M, Tompkins KL, Hatzopoulos AK, Baldwin HS, Zhou B. Nfatc1 Coordinates Valve Endocardial Cell Lineage Development Required for Heart Valve Formation. Circulation Research. 2011;109:183–192. doi: 10.1161/CIRCRESAHA.111.245035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Zhang Z, Lui W, Chen X, Wang Y, Chamberlain AA, Moreno-Rodriguez RA, Markwald RR, O’Rourke BP, Sharp DJ, et al. Endocardial Cells Form the Coronary Arteries by Angiogenesis through Myocardial-Endocardial VEGF Signaling. Cell. 2012;151:1083–1096. doi: 10.1016/j.cell.2012.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Q, He W, Zhang XH-F, Le H-V, Massagué J. Genome-wide impact of the BRG1 SWI/SNF chromatin remodeler on the transforming growth factor beta transcriptional program. J Biol Chem. 2008;283:1146–1155. doi: 10.1074/jbc.M707479200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Li W, Shang C, Chen RM, Han P, Yang J, Stankunas K, Wu B, Pan M, Zhou B, et al. Brg1 Governs a Positive Feedback Circuit in the Hair Follicle for Tissue Regeneration and Repair. Developmental Cell. 2013;25:169–181. doi: 10.1016/j.devcel.2013.03.015. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Zhou B, Chang C-P. Methods in Molecular Biology, Vol. 843 of Methods in Molecular Biology. Totowa, NJ: Humana Press; 2011. Analysis of the Endocardial-to-Mesenchymal Transformation of Heart Valve Development by Collagen Gel Culture Assay; pp. 101–109. [DOI] [PubMed] [Google Scholar]
- Yu S, Crawford D, Tsuchihashi T, Behrens TW, Srivastava D. The chemokine receptor CXCR7 functions to regulate cardiac valve remodeling. Dev Dyn. 2011;240:384–393. doi: 10.1002/dvdy.22549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi S, Choi M, Wakimoto H, Ma L, Jiang J, Overton JD, Romano-Adesman A, Bjornson RD, Breitbart RE, Brown KK, et al. De novo mutations in histone-modifying genes in congenital heart disease. Nature. 2013;498:220–223. doi: 10.1038/nature12141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H-Y, Kluge M, Timpl R, Chu M-L, Ekblom P. The extracellular matrix glycoproteins BM-90 and tenascin are expressed in the mesenchyme at sites of endothelial-mesenchymal conversion in the embryonic mouse heart. Differentiation. 1993;52:211–220. doi: 10.1111/j.1432-0436.1993.tb00633.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.