

Abstract
We review morphogenesis of the enteric nervous system from migratory neural crest cells, and defects of this process such as Hirschsprung disease, centering on cell motility and assembly, and cell adhesion and extracellular matrix molecules, along with cell proliferation and growth factors. We then review continuum and agent-based (cellular automata) models with rules of cell movement and logistical proliferation. Both movement and proliferation at the individual cell level are modeled with stochastic components from which stereotyped outcomes emerge at the population level. These models reproduced the wave-like colonization of the intestine by enteric neural crest cells, and several new properties emerged, such as colonization by frontal expansion, which were later confirmed biologically. These models predict a surprising level of clonal heterogeneity both in terms of number and distribution of daughter cells. Biologically, migrating cells form stable chains made up of unstable cells, but this is not seen in the initial model. We outline additional rules for cell differentiation into neurons, axon extension, cell-axon and cell-cell adhesions, chemotaxis and repulsion which can reproduce chain migration. After the migration stage, the cells rearrange as a network of ganglia. Changes in cell adhesion molecules parallel this, and we describe additional rules based on Steinberg's Differential Adhesion Hypothesis, reflecting changing levels of adhesion in neural crest cells and neurons. This was able to reproduce enteric ganglionation in a model. Mouse mutants with disturbances of enteric nervous system morphogenesis are discussed, and these suggest future refinement of the models. The modeling suggests a relatively simple set of cell behavioral rules could account for complex patterns of morphogenesis. The model has allowed the proposal that Hirschsprung disease is mostly an enteric neural crest cell proliferation defect, not a defect of cell migration. In addition, the model suggests an explanations for zonal and skip segment variants of Hirschsprung disease, and also gives a novel stochastic explanation for the observed discordancy of Hirschsprung disease in identical twins.
Keywords: Neural crest, enteric nervous system, cell migration, gangliogenesis, mathematical modeling
The Enteric Nervous System: Structure and Diseases
1. Structure and Function of the Enteric Nervous System
The enteric nervous system (ENS) is the largest division of the autonomic nervous system, with numerous neuron types identified by neurochemical coding and neurite and cell body morphology. These control, modulate or mediate almost all aspects of gastrointestinal function, including motility, secretion and adsorption, water and electrolyte balance, chemical sensing, and communication between gut segments and the CNS. The ENS includes motor, sensory and inter-neurons, so that it has a capacity for integrated function (Furness, 2012; Hao and Young, 2009). Despite these similarities to the CNS, the organization of the ENS is quite different and, superficially, relatively simple. The ganglia are small with variable numbers of neurons and glia usually arranged in two layers or plexuses (Fig. 1a). Neurite bundles ensheathed by glial cells connect local ganglia, and extend to nearby gut tissues. This organizational unit is replicated with regional variations (Wattchow et al., 2008) countless times to form an ENS network.
Fig. 1.

Diagrams of the structure and development of the ENS. (a) The ENS forms two 2-dimensional arrays (myenteric and submucosal plexuses) in the wall of the gut, associated with the intestinal smooth muscle. (b) Colonization of the foregut, midgut and hindgut in succession by vagal NC cells derived from the embryonic hindbrain. Sacral NC cells have a minor contribution to hindgut ENS. In Hirschsprung disease, the wave of vagal-derived ENC cells fails to complete the final occupation of the distal hindgut. (c) En face overview of the ENS at an early stage near the wavefront of colonization showing chain migration of ENC cells (grey) in the overall direction of the arrow. In mouse but not chick, some early-differentiating neurons (black circle) extend axons (black line) paralleling the ENC cell chains. (d) The same region after initial ganglionation, showing the geometric arrangement of the ENS ganglia. Neurons (colored circles representing different neuron types) form clusters and glial/ENC cells (grey) tend to surround the neurons. Nerve fibers (colored lines) accompanied by glial cells link the ganglia.
2. Pathologies of the Enteric Nervous System
The ENS is subject to many pathological alterations (Furness, 2008), but research on development of the ENS has been dominated by a desire to understand a single ENS dysmorphology, Hirschsprung disease (congenital aganglionic megacolon). This disease, which occurs in about 1/5000 births, is marked by intractable constipation, due to a lack of peristalsis. This in turn is caused by a regional lack of ENS in the distal intestine but the length affected is highly variable. Mostly the ENS of the sigmoid colon is absent, which may also extend to the descending colon, but much longer defects are known, up to total intestinal agangiolionosis (Solari et al., 2003). Other rare Hirschsprung-like conditions also occur: zonal aganglionosis, where a region of colon lacks ENS but the regions proximal and distal to this are ganglionated, and skip segment, in which a short ganglionated region is flanked proximally and distally by agangliononic colon (O'Donnell and Puri, 2010).
Mutations in about a dozen genes predispose to Hirschsprung disease, but mutations in RET, encoding the GDNF receptor, may underlie most cases (Amiel et al., 2008). These result in full or partial inactivation of RET function. An identical condition has also arisen spontaneously or been engineered into many animal models. Although structurally and functionally Hirschsprung disease could qualify as the world's simplest structural birth defect, its genetics is complex, with a host of modifier genes (Walters et al., 2010), genetic alterations of various types, skewed sex ratio and environmental influences (Fu et al., 2010). Hirschsprung disease shows incomplete penetrance and dramatic differences in expressivity in siblings, as graphically displayed by variations in the length of intestine affected, and even monozygotic twins are frequently discordant (Jung, 1995).
Hypoganglionosis, hyperganglionosis, ganglion cell disorganization and selective loss of one or a few types of nerve cell also occur and these impair intestinal function (De Giorgio and Camilleri, 2004). Some of these are clearly congenital while others have a later onset such as oesophageal achalasia. Reduced ENC cell and ganglion density, size and pattern (hypoganglionosis) occurs in the transition zone in Hirschsprung patients, and it also occurs independently, for example in mice with one functional copy of the GDNF gene (Flynn et al., 2007; Gianino et al., 2003; Shen et al., 2002). RET; with MEN2B mutations resulting in constitutive activation of the receptor, produces ENS hyperganglionation (ganglioneuroma) (Takahashi et al., 1999; Yin et al., 2006), and Zic2 mutant mice also show ENS hyperplasia (Zhang and Niswander, 2013). Larger ganglia have also been reported in the submucosal plexus in the controversial IND type B (Meier-Ruge et al., 2004). In some patients with severe non-Hirschsprung constipation, reduction of Substance-P and VIP nerves relative to nitrergic nerves have been described (King et al., 2010), suggesting a problem in the balance of various ENS nerve cell types. For the most part, however, it is not known what directly leads to these enteric neuropathologies.
Colonization of the Gastrointestinal Tract by Enteric Neural Crest Cells
We review key local cell behaviors and molecular actions in the developing ENS before summarizing how these can be incorporated into the mathematical models.
1. Enteric neural crest cell migration in the intestinal wall
The ENS is derived from the neural crest (NC), with most derived from the hindbrain vagal level (Kuntz, 1922; Le Douarin and Teillet, 1973; Yntema and Hammond, 1954). Vagal NC cells migrate through adjacent tissues to reach the foregut (Kuo and Erickson, 2011), and from there the cells directionally colonize the midgut and hindgut (Epstein et al., 1991; Tucker et al., 1986; Young et al., 1998), eventually reaching the cloaca (O' Donnell et al., 2006). The sacral NC also makes a minor contribution (Fig. 1b). In this review, we will term these cells enteric NC-derived cells (ENC cells). These cells can be detected within the gut mesenchyme by probes to such molecules as p75, HNK-1 and Sox10.
ENC cell migration has been timetabled in chick (Allan and Newgreen, 1980), mouse (Young et al., 1998), rat (Newgreen and Hartley, 1995) and human (Fu et al., 2004) and the colonization is lengthy in terms of distance (several millimeters; mouse, chick) and duration (4 days, mouse; 4 weeks, human). The colonization sequence immediately suggests a cause for Hirschsprung disease: ENC cells fail to complete migration so the distal gut remains aganglionic. Consequently Hirschsprung disease has been regarded as the archetypal defect of cell migration.
The population of ENC cells advances predictably as revealed by observations of labeled ENC cells. This movement is by cell migration in the midgut and hindgut mesenchyme, mostly within a narrow almost 2-dimensional layer beneath the serosa via intersecting chains of cells (Fig. 1c) (Conner et al., 2003; Epstein et al., 1991; Young et al., 2004), which later aggregate as ganglia (Fig. 1d). Time-lapse imaging of this migration in organotypic gut culture has shown that the chains are fairly stable yet the cells forming the chains are in a constant flux with rapid bursts of movement, slower gradual movements, pauses and constant exchange of neighbors, which are unpredictable at the level of each individual cell (Druckenbrod and Epstein, 2005, 2007; Young et al., 2004).
2. Roles of adhesion molecules in ENC cell migration
Extracellular matrix (ECM) and cell-cell adhesion molecules (CAMs) have long been known to be fundamental to NC morphogenesis (Erickson and Perris, 1993).
Extracellular matrix molecules and their receptors
ENC cells have many potential ECM substrates for locomotion, guidance and signaling, such as fibronectin (FN), tenascin-C (TNC), laminins, collagens and proteoglycans (Fujimoto et al., 1989)(Bolcato-Bellemin et al., 2003; Breau et al., 2009; Nagy et al., 2009; Simon-Assmann et al., 1995). These could act as permissive adhesive pathways while others could restrict movement. In vitro studies of mouse ENS cells suggest FN plays the former role and TNC the latter (Breau et al., 2009). ECM components do not seem to define the layer or the chain structure preferred by early migrating ENC cells but the later ganglia occupy a border defined structurally and by CAMs and ECM (Newgreen and Hartley, 1995).
ENC cells express integrin superfamily cell-ECM adhesion receptors especially those with the β1 chain (Bixby et al., 2002; Breau et al., 2006; Kruger et al., 2003). Undifferentiated ENC cells expressed higher levels of β1-integrins compared to neurons and glial cells (Bondurand N and Dufour S, unpublished results). Recently, mice lacking β1-integrins in NC cells have been engineered with Cre-LoxP technology using the human tissue plasminogen activator promoter for lineage targeting (Pietri et al., 2003). These cells colonized the midgut (Fig. 2a, b) similarly to normal, in a low FN and TNC ECM environment (Breau et al., 2009). In normal mouse there is a moderate temporary loss of chain formation as ENC cells enter the cecum (Druckenbrod and Epstein, 2005) but the mutants exhibited a major migratory defect at this point (Breau et al., 2009) leading to aganglionosis of the colon (Fig. 2c, d) resembling Hirschsprung disease. Depletion of β1-integrin in ENC cells disturbed chain formation, decreased the speed, persistence and directionality of movement in the cecum and hindgut where the ECM is enriched in FN and TNC, and led to aggregation. β1-integrins were required to overcome TNC-mediated inhibition of ENC adhesion and stimulate FN-mediated ENC migration (Breau et al., 2009). β1-integrin signaling is structurally and functionally linked to the actin motility machinery, and this is regulated in ENC through Phactr4 and PP1 activities (Zhang and Niswander, 2012).
Fig. 2.

Changes in the ECM adhesion receptor repertoire of ENC cells modulate their colonization of the developing gut. ENC are revealed by Xgal staining in blue. In control mice at E16.5, the midgut (a) and distal colon (c) are colonized by ENS cells, whereas when ENC cells lack β1-integrins, the midgut is colonized (b) in a disorganized way and the distal colon is not colonized (d). (Image: M. Breau).
As ENC cells migrate distally, wavefront cells encounter an increasingly mature microenvironment. Experimentally, older intestinal microenvironment slows ENC occupation of the mouse hindgut (Druckenbrod and Epstein, 2009; Hotta et al., 2009) though whether this slowing is due to age-related changes in ECM is unclear.
Cell-cell adhesion molecules
The surfaces of gut mesenchyme cells, endothelial cells and other ENS cells could also act as motile substrates, based on adhesion via CAMs. Mouse ENC cells show an affinity for mesoderm cells, made especially obvious when integrin-based ECM adhesion is suppressed (Breau et al., 2009). The homophilic CAM N-cadherin is expressed by both ENC cells and by ENS neurons (Breau et al., 2006; Gaidar et al., 1998; Hackett-Jones et al., 2011; Nagy et al., 2012). Genetic deletion of N-cadherin specifically from mouse NC cells reduced intercellular adhesion of ENS cells in cell culture assays, but in gut tissue this had surprisingly little effect on ENC migration. However when examined in detail, loss of N-cadherin in ENC cells changed their directionality of movement subtly; the cells tended to move more circumferentially (Broders-Bondon et al., 2012). It could be hypothesized that loss of N-cadherin-mediated cell-cell adhesion means the direction of ENC cell motility becomes dominated by ECM and its increasingly circumferential orientation.
The Ig superfamily homophilic CAM NCAM has important roles in neural and mesoderm cell adhesion. NCAM cannot be detected by antibody labeling in early migratory avian ENC cells (Hackett-Jones et al., 2011), but it appears as neurons differentiate (Gaidar et al., 1998; Mirsky et al., 1986; Newgreen and Hartley, 1995). Gut mesoderm cells of the mouse express NCAM, but this is reported to occur after the initial colonization phase (Faure et al., 2007).
L1CAM (termed Ng-CAM in Aves) is related to NCAM, and the gene is mutated in some Hirschsprung patients with X-linked hydrocephalus (Okamoto et al., 2004), and it acts as a modifier for Hirschsprung-associated genes like Sox10, ednrb and edn3 (Wallace and Anderson, 2011). L1CAM is expressed transiently by early migrating ENC (Anderson et al., 2006; Hackett-Jones et al., 2011; Nagy et al., 2012; Thiery et al., 1985) and later reappears during ENS neuron differentiation and axon extension (Turner et al., 2009). Soluble L1CAM extracellular domain competitively inhibits cell surface L1CAM adhesive interactions. When administered to mouse hindgut organ cultures during early colonization, this reagent decreased ENC intercellular adhesion, as shown by break-up of ENC cell chains, and slowed (but did not totally prevent) migration (Anderson et al., 2006).
Conclusions on the roles of ECM and CAMs for ENC cell migration
It is clear that β1-integrins and their ECM ligands are necessary in a general sense for complete ENC cell migration and ENS formation. The preference of early migrating ENC cells for a narrow layer in the gut mesoderm cannot be assigned to obvious layers of ECM (or CAM) molecules or combination of molecules (Newgreen and Hartley, 1995). The surprising colonization up to the midgut in the absence of these integrins in mouse mutants correlates with low FN (and TNC), and suggests alternative adhesive substrates, possible mesodermal cells themselves, must exist here (Breau et al., 2009). Migrating ENC cells make repeated transient adhesive contacts with other ENC cells, yet regarding the major CAMs (cadherin and NCAM), there is little evidence that they grossly influencing ENC cell migration. However, it was concluded by Anderson et al. (2006) that L1CAM has a role in the transient adhesions between ENC cells in motile chains, and that this chain formation makes movement more efficient, but how this occurs is not yet understood.
3. ENC cell proliferation during colonization of the intestine
Mitosis has been observed in migrating ENC cells (Druckenbrod and Epstein, 2005; Young et al., 2004). In chorio-allantoic membrane grafts of aneural intestinal regions supplied with vagal NC or ENC cells, the final ENS density resembles the norm for that intestinal region, despite differences in progenitor cell number at the start and despite great growth of the intestinal tissue (Zhang et al., 2010). It has been concluded that ENS cell number rises via proliferation up to a cell density set by the intestinal tissue. This phenomenon, termed logistical growth, could result from consumption of any resource vital for the cells.
Typically in the nervous system there is a numerical overshoot of neurons followed by paring back by programmed cell death to match the neuron number with the size of the field to be innervated. A source-and-sink model with competition for trophic resources underlies this (Barde, 1989). However, this tactic is not followed by the ENS; instead proliferation ramps up ENS cell numbers until the capacity level is reached. The absence of cell death in the ENS may simply arise because of the low initiating NC progenitor population and the enormous final population size.
Role of growth factors in ENC cell proliferation
Candidates for a trophic resource are growth factors produced by the surrounding tissues, with ENC cells acting as a sink. GDNF via RET tyrosine kinase receptor is one of the most important signaling pathways in ENS development. Consistent with the logistical growth model i) GDNF is produced by gut mesoderm cells (Suvanto et al., 1996), ii) in GDNF+/− mice the ENS density is reduced at all intestinal regions at all stages (Flynn et al., 2007; Gianino et al., 2003), iii) with mutations that constitutively activate RET (MEN2B-type mutations) there may be enteric hyperganglionosis (Takahashi et al., 1999) and iv) ENS-derived cells, particularly early stage ENS cells, show a proliferative response to GDNF in vitro (Hearn et al., 1998). However sustained mitogenic responses require insulin-like signaling at the same time (Focke et al., 2003), mediated via a PI-3-K pathway (Focke et al., 2001; Srinivasan et al., 2005).
Mathematical models of colonization and their emergent properties
The intrinisic cell properties discussed above are expressed as “rules” which can be implemented in a mathematical formulation. We illustrate how the mathematical models (both continuum and probabilistic cellular automata) can be used to develop ideas on how these local rules combine to control ENS morphogenesis at a system level. As part of this, we will also outline how deviations from these rules may result in dysmorphogeneses of the types described above. These arguments support the view that the ENS is self-organizing at the local level; with no “grand plan”.
1. Deducing rules from intrinsic cell properties: continuum and cellular automata models
Key features from the above observations which must be exhibited in a basic model are the stereotyped directional colonization by the ENC population, contrasting with the apparently random movement at the cell level. The key proliferative feature to be accounted for is logistical growth. We propose two general rules of colonization of the gut by ENC agents. (Note: we use the term agents to denote the modeled cells). In addition, because cells are physical entities, we can include an exclusion processes: put simply, two agents cannot occupy the same space. In the initial model, adhesion is an overall permissive quality and unnecessary to be included explicitly; but adhesion will be introduced later for chain migration and for ganglion formation. We can also approximate the field of movement to two-dimensions, simplifying modeling:
Basic rules for colonization by ENC agents (see
Fig. 3).
|
Fig. 3.

Rules for the occupation of the gut by ENC agents. (a) Continuum model rules in which the change in ENC agent density C with any time increment dt in any region χ along the intestine is governed by the diffusion-like drift (in this case linear) of ENC agents into and out of the region plus the generation of new ENC agents by logistic production (proliferation) at a set rate λ. Note that when the value of C reaches carrying capacity Cmax, all change in C becomes entirely due to diffusion-like drift. (b) CA model on a square lattice in which ENC movement is equiprobable on all four directions as long as the site of placement is not occupied (this has an effect similar to the movement term outlined in red in 3a). At a set rate ENC agents can proliferate with progeny placed adjacently at random, as long as the site of placement is not occupied (this has an effect similar to the production term outlined in green in 3a). The outcome of these models is shown in Fig. 4.
These rules can be encapsulated in two types of mathematical models. One type is a continuous description, whereby the ENC agent density is governed by a partial differential equation that takes into account diffusion-like movement, and logistic growth (Fig. 3a). This gives a population-level description. To obtain an individual-level description, favored by biologists, agent-based models, such as cellular automata (CA) models are most useful (Fig. 3b). Such models assign probabilities to agent movement and proliferation (Simpson et al., 2006; Simpson et al., 2007a).
Progressive invasion wave: an emergent property
The continuum model generates a directional invasion wave (Fig. 4a), as does the CA model (Fig. 4b-d) but the latter has the advantage of revealing details below the population level that can be verified biologically by cell labeling. In CA models it can be seen that the speed and directionality of individual ENC agents is not predictable. The interplay of these simple rules has considerable explanatory power when modeled, and when tested experimentally (Simpson et al., 2007b). Although expected, the directional colonization is an emergent property since directionality was not specifically encoded in the rules. However, a number of other unexpected properties have also emerged which have helped in interpreting the roles of the Hirschsprung disease genes, and indeed led to a re-evaluation of the disease. The formulation of testable but not intuitive predictions is a major advantage in mathematical modeling.
Fig. 4.

Simulations of the occupation of the gut by ENC agents. (a) A continuum model with diffusion-like movement and proliferation of ENC agents (dark blue trace) up to a set carrying capacity Cmax (dark blue arrow). This generates a directional wave (light blue arrow) of colonization. (b-d) CA model of ENC agent (red) occupation of the gut field (yellow). Dark blue line represents the density of ENC agents, and the dark blue arrows indicate the nominal carrying capacity Cmax. ENC agents are initially placed at density below Cmax Colonization of unoccupied gut is negligible until local ENC agent density rises by proliferation to approach Cmax. After this the ENC agents colonize the gut as a directional wave. Stochastic variation of movement and proliferation means that, in a CA simulation, the achieved density C varies continually around Cmax, unlike the agent density in the continuum model. (Images: M. Simpson). (e) Arbitrarily color coding regions of the ENC agent population shows that the front phalanx of ENC agents expands by proliferation to colonize the remaining unoccupied gut field (yellow). Two frontal ENC agents are labeled blue; their descendants form compact clones which in this particular simulation have merged. (Image: B. Binder) (f) In a partially colonized gut field (as for Fig. 4e), two frontal ENC agents are labeled blue (top panel); and migration and proliferation proceed under the basic conditions until the field is fully colonized. With identical stating conditions, stochastic variation in agent motility and proliferation results later in widely varying clonal outcomes. Some clones are moderate sizes (2nd panel) but some huge clones occur (3rd panel), as do clones with minimal or no expansion (bottom panel). This enormous range of clone sizes is an emergent property of this model that has not yet been investigated biologically. (Image: B. Binder). (g) The gut mesenchyme agent field (yellow) can be coded to grow in CA models. Individual mesenchyme agents in each row (g, top) are chosen at random to divide (orange) with daughter cell placed one site distal (to the right). In this way the gut field (g, bottom) elongates by one column per division cycle. (h) ENC agents on this growing field still show frontal expansion (red agents) as in non-growing fields (see Fig. 4e) but there is considerable population expansion behind the wavefront and mixing of ENC agents (green, black ENC agents). In addition, ENC clones (blue agents) do not remain compact when the field is growing. (Image: B. Binder)
Frontal expansion: an emergent property
These rules were tested in continuum and CA models and in biological experiments by labeling the ENC cell population (either the entire population or sub-regions) and tracking the migrating cells/agents. Both models predict firstly that colonization will be inefficient while the ENC agents are below carrying capacity because newly produced agents merely relocate within the original occupied zone (Fig. 4b, c). Attainment of carrying capacity then leads to a disproportionate relocation of agents to previously unoccupied territory as an invasion wave (Fig. 4d). The CA version of the model predicts that there will be stochastic noise around the attainment of carrying capacity (Fig. 4c-d). Both models also predict frontal ENC agents will make the greatest contribution to further colonization. The front ENC cells and their daughter cells will on average advance faster than those behind the front, and they will proliferate more than those behind the front (at least in non-growing gut scenarios). In short, the frontal cell sub-population occupies more space more rapidly than otherwise identical rearguard cells (Fig. 4e).
These mathematical predictions have been validated experimentally in organ cultures of gut with labeled ENC cells (Nishiyama et al., 2012; Simpson et al., 2007b), and have also been found in other NC migrations (Kulesa et al., 2008). This scheme places great weight on ENC proliferation as being the engine of colonization.
Colonization of distal intestine by trans-mesenteric ENC: an emergent property
In the mouse and human, the embryonic midgut and hindgut form a U-shape with a short connecting mesentery. Mouse intestine organ cultures with photoconvertible fluorophore labeling of mouse ENC cells has revealed that as the wavefront advances in the midgut, there is a brief time-window when a few ENC cells behind the front in the midgut stray across the mesentery and enter the uncolonized hindgut. These cells then expand disproportionately in number and spread rostral and caudal, as predicted by the frontal expansion hypothesis, so that much of the colon comes to be occupied by the descendants of these trans-mesenteric cells (Nishiyama et al., 2012). If trans-mesenteric colonization occurs in human embryos, it is possible to imagine how the rare zonal and skip segment variants of Hirschsprung disease (O'Donnell and Puri, 2010) might arise: this is ripe for modeling to investigate its feasibility.
Clonal heterogeneity: an emergent property
An emergent property of the CA model is that, despite a uniformity of outcome at the population level, a few initiating ENC agents make a disproportionately large and spatially unpredictable contribution to the final ENS. This arises because of the stochastic nature of movement and proliferation choices of each cell agent (Fig. 4f). This prediction is testable; it is important to establish how cell division occurs, how variable it is and its symmetric or asymmetric nature because this will place restrictions on how we imagine multi-lineage differentiation must occur in the ENS.
Absence of Hirschsprung disease in silico: a failure of modeling
Varying the parameters of these models never resulted in a failure to complete colonization although the time required to colonize fully could be increased by decreasing the proliferation rate of ENC agents.
2. Models with gut growth: broad similarities and detailed differences
During normal formation of the ENS the gut elongates dramatically (Binder et al., 2008), driven by proliferation of the mesenchyme cells. This was incorporated in CA models:
Rules for gut growth (see
Fig. 4g)
|
Rearguard proliferation and dispersed clones: emergent properties
The same models, but with gut growth (Fig. 4g), predict that frontal expansion is still the main means of colonization (Fig. 4h) but, compared to scenarios without gut growth, there is greater rearguard ENC agent growth. This has been observed biologically (Young et al., 2005). In the CA model, this occurs because of new unoccupied gut-space, due to increased numbers of gut mesenchyme agents. However, since gut mesenchyme cells are the source of ENC cell growth factors (eg. GDNF) (Suvanto et al., 1996), space in the model could be regarded as a surrogate for growth factor availability.
The constant creation of exploitable gut-space means there will be far greater mixing of ENC agents, and looser clones (Fig. 4h), and the unpredictable variation in clone sizes and positions is actually amplified. While not being strictly tracking of clones, biological tracking of the descendants of small numbers of vagal NC cells, which were marked by genome-integrating GFP constructs, has revealed enormous and unpredictable numerical and positional variability of daughter cells in the ENS (Binder et al., 2012).
Hirschsprung disease in silico: an emergent property
This model with gut growth predicts that a subnormal rate of proliferation of ENC agents will lead to incomplete colonization. In effect the ENC agents cannot catch up with the distal end of the gut field as it recedes by elongation. The notion that Hirschsprung disease could be the result of proliferation reduction rather than a defect of cell migration is a unifying hypothesis for most of the known Hirschsprung genes (Landman et al., 2007).
Monozygotic twin discordancy: an emergent property
At some intermediate reduction of ENC agent proliferation rate, running the identical CA model repeatedly gives some outcomes with failure to complete colonization (that is, “Hirschsprung disease”), and other outcomes with full ENS colonization. Thus, a purely stochastic difference between success and failure is an emergent property under certain conditions. We propose that this is a real phenomenon and could contribute to the incomplete penetrance and variable expressivity (length of aganglionosis) in human Hirschsprung disease, where even monozygotic twins may be discordant for the condition (Jung, 1995). We propose that such conditions occur around the cusp of haploinsufficiency for RET.
3. Cellular automata models that reproduce chain migration
There are descriptive deficiencies in this basic model: the rules so far describe the advance of a disorderly mob of ENC agents. This is similar to NC behavior in two-dimensional culture (Newgreen et al., 1979). These formulations never reproduce the elegant chain migration (Fig. 1c) seen in gut tissue (Druckenbrod and Epstein, 2005, 2007; Young et al., 2004). We and others (Cox, 2011; Landman et al., 2011) have introduced several models that reproduce the essential features of the previous model but also produce stable chains made up of unstable cells. This required additional features, but which are consistent with the biological evidence. We discuss two variants here; both rely on many common properties, and they are not mutually exclusive. Chain migration in cranial NC cells has also been modeled recently with many features common to those outlined below (McLennan et al., 2012; Wynn et al., 2012).
Chains based on cell-axon interactions
In addition to the motility and proliferation rules discussed above, ENC agents differentiate into neuron agents which, like biological neurons, have diminished motility and proliferation, and which generate motile axons. These additional events can also be assigned probabilities. In this model there are four agent types: ENC agents, neuron agents, growth cone agents (which are motile) and axon shaft agents (which lengthen but are otherwise immotile). Biological correlates of these events have been described (Hao et al., 2009; Young et al., 2005). This was encoded in CA format as follows:
Rules for differentiation and axon guidance
|
Biologically, CAMs of NC-derived cells and axons (Hackett-Jones et al., 2011; Nagy et al., 2012; Thiery et al., 1985) provide adhesive mechanisms for their association. Guidance of cells by axons for intestinal colonization is not a new idea (Kuntz, 1922) and the intimate relationship of axons and ENC cells at the wavefront in mouse embryo colon is illustrated by Hao and Young (2009). This was encoded into the model:
Rules for ENC movement on axons
|
Chain migration: an emergent property
Application of these rules produced the chain migration pattern which appeared realistic both statically and dynamically, and it still preserved the frontal expansion mode of colonization and the unpredictable paths of ENC agents (Fig. 5a-d). An additional property also emerged: lacunae between ENS chains were increasingly bisected by new chains so the chain network became more dense and complex. This had already been observed in biological examples (Young et al., 2004).
Fig. 5.

CA models with chain migration features. (a,b) Cylindrical scheme of ENC agents (coded green, yellow and blue) advancing in association with axons (red). This reproduced chain migration and preserved the frontal expansion phenomenon. (c,d) Flattened views of ENC agent pathways show that agents (2 groups of 6 color-coded agents indicated by arrows in (c) showed (d) unpredictably variable trajectories. (e) An unrealistic feature that emerged in the initial cell-axon interaction model was the occasional axonal growth cone (white arrows) at the wavefront leading the ENC agents (green). These simulations used a hexagonal lattice. (Images: A. Fernando)
Distal projecting axons: a failure of the model
When observed in detail, at times axons were found projecting distal to the front of ENC agents (Fig. 5e). Discussions with Dr Hideki Enomoto and his group suggested that this feature was unrealistic. In order to bind the growth cone advance to the ENC cell front, we modified this model by coding the following:
Rules uniting ENC agents and growth cone agents
|
Chain migration: an emergent property (modified version)
These rules establish a chemotactic factor gradient across the ENC front, and means that each growth cone's course at the front tends to be directly caudal, up the steepest factor gradient. The caudal direction and relative straightness of early ENS axons had already been noted (Hao et al., 2009; Newgreen and Hartley, 1995). Distal to the ENC agent population front, the chemotactic factor level remains high and uniform, so directionally efficient growth cone movement is compromised. The effect of this is to positionally and directionally tie the growth cone front to the ENC front. This unity of ENC cell and growth cone movement has been demonstrated by experimentally reversing the direction of colonization (Young et al., 2002).
Chains based on cell-cell interactions
Few or no enteric neurons and axons exist at the wavefront in the avian ENS, yet the ENC cells still form dynamic chains (Druckenbrod and Epstein, 2005; Epstein et al., 1991). Likewise in cranial NC cell chains, axons are absent (Kulesa and Fraser, 2000). Thus the above axon-based model cannot be always applied. We therefore proposed rules based on cell interactions:
Rules for ENC movement
|
There are several candidate mechanisms for cells performing a reinforced walk: e.g., cells locally depositing attractive or adhesive cues, locally depleting repulsive cues or altering the ECM. NC cells produce proteases capable of altering their local environment (Cai and Brauer, 2002; Menoud et al., 1989), and changes in ECM have been observed after the passage of NC cells (Brauer et al., 1985). Such a bias, unchecked, eventually produces agent aggregation. Therefore to counter this, a mutually repulsive ENC interaction must also be included. A basis for such a behavior could be contact inhibition of locomotion. Contact inhibition is a subtle phenomenon: it involves initial contact attraction/adhesion, local paralysis at the point of contact and stimulation of locomotion in a direction other than contact (Newgreen, 1990; Thomas and Yamada, 1992). This has been observed in NC-derived cells in vitro (Erickson, 1985; Gooday and Thorogood, 1985; Newgreen et al., 1979) and in vivo (Bard and Hay, 1975; Carmona-Fontaine et al., 2008).
Along with these rules, behind the front we also imposed the rules for ENC movement on axons and for ENC differentiation and axon guidance, as previously described. Absence of axons at the wavefront was achieved by omitting neuronal differentiation, or, more realistically, by imposing it more slowly.
Chain migration: an emergent property of cell interactions
Behind the wavefront, the resulting chain migration patterns are largely indistinguishable from the previous model. However at the wavefront there are significant differences. As intended, there is a scarcity of neuronal and axonal agents at the front in this model, but there are more isolated ENC agents than in the axon-based chain migration model (Landman et al., 2011).
In summary, both models predict an emerging ENS network structure although with subtle differences (Landman et al., 2011). Here we have discussed the two models separately, representing the rodent and avian archetypes. We suggest that the two mechanisms are both active in both animal models, but the apparent difference arises due to the relative difference in timing of neuron differentiation with respect to the ENC wavefront.
Ganglionic Morphogenesis of the Enteric Nervous System
1. Cell movement, proliferation and differentiation in ENS gangliogenesis
The formation of multiple small ganglia occurs gradually from the chain migration phase of ENC cells, and simultaneously the ENC cells also differentiate into neurons (Fig. 1c). Neurons can be identified by a number of markers including Hu and PGP9.5, and by simultaneous loss of the Sox10 marker. Axons can be detected by expression of neurofilament, β3-tubulin (Tuj1) and NAPA73 (E/C8). The glial cells can be recognized by expression of GFAP, S100b, BLBP (or BFABP) and Sox10 (Young et al., 2003). There is no shortage of useful biomarkers for the ENS, however distinguishing endogenous ENS progenitor cells from glial cells is still a problem.
Neurons appear at first individually and later in clusters (Hackett-Jones et al., 2011). The proportion of neurons rapidly rises (Flynn et al., 2007): in the mouse midgut this rises from about 4% of all ENS cells at E10.5 to 24% at E12.5 and to 45% at E14.5. In the avian embryo midgut, the proportion of neurons rises rapidly from almost zero E4.5 and by E6 stabilizes to about 55% of all the NC-derived cells despite great increases in both the total number of neural cells and their density of distribution during this period (Zhang, D. unpublished).
In addition to the aggregation of cells into ganglia and the production of new neurons by differentiation of ENC cells, a segregation of the neurons from non-neurons (glial and progenitor cells) evolves. In the avian ENS at midgut level for example, over the period of E5 to E8 the neurons come to lie central in each ganglion with non-neurons as a shell (Hackett-Jones et al., 2011). These ganglia are joined by fibers with attendant cells, presumably glia, connecting adjacent ganglia. The pattern so produced is roughly triangular (Fig. 1d).
In the embryonic rodent midgut there is also segregation of neurons during early morphogenesis but the pattern is somewhat different. From E11.5 the neural cells initially form an increasingly dense lattice on the surface of the future circular muscle layer. By E14.5−15.5 the neurons have begun to coalesce into elongate groups or “ribs ” that align with the future muscle cells and ECM, and intrude centripetally between the muscle cells. By E18.5 the ENS appears as a series of parallel neuron-rich ribs (Figs. 2a, 6) (Lei and Howard, 2011)(Howard, unpublished). This leaves the non-neurons, mostly glial cells, in contact with the neurons but tending to occupy the original more superficial site. Coursing between these elongate ganglia, and at right-angles to them (that is, roughly longitudinal), are bundles of nerve fibers and glial cells, giving the network a distinct rectangular pattern (Fig. 6).
Fig. 6.

ENS cells re-organise to form a geometric array of ganglia and connecting tracts. YFP (green) reveals genetically marked ENS cells in wholemount of the mouse midgut at postnatal day 20. Neurons (Hu, red) and glial cells (BLBP, blue) cells associate in a pattern that is set early in development. (Image: Jun Lei)
Ganglionation occurs in vivo and also in gut grafts to the avian chorio-allantoic membrane (Zhang et al., 2010) and to the rodent renal capsule (Young et al., 1998) where growth of the gut is rapid. Similar ganglionation is also seen in the ENS in organ culture, even though gut growth is minimal (Hearn et al., 1999; Simpson et al., 2007b). It is perhaps surprising that field growth has such little effect on ENS morphogenesis and patterning, and this insensitivity to expansion of the underlying field must be one property that emerges in any attempt at modeling gangliogenesis.
2. Cell-cell and cell-ECM adhesion changes in ENS gangliogenesis
Immunoreactivity to N-cadherin of avian ENS neurons and non-neuronal cells increases with development (Hackett-Jones et al., 2011; Nagy et al., 2012) suggesting a gradual increase in cadherin-based adhesion in all ENS cells in vivo. This may be important in ENS cell aggregation, as it is in sympathetic ganglionation (Kasemeier-Kulesa et al., 2006). The sorting out of avian neuronal and non-neuronal ENS cell lineages in vivo strongly suggests that ENS neurons have greater cohesivity compared to ENS glial and progenitor cells (Hackett-Jones et al., 2011). Consistent with this, NCAM immunoreactivity is markedly higher in avian ENS neurons than in non-neuronal cells and appears slightly earlier than neuronal aggregation. Expression of NCAM is under the control of BMPs expressed by surrounding mesenchyme (Faure et al., 2007; Fu et al., 2006) and promotes aggregation of ENS neurons into ganglia. In functional assays, avian ENS cells isolated by FACS and allowed to aggregate in rotating assays (Takeichi, 1977) form uniform sized aggregates with central neurons and peripheral non-neurons (Fig. 7a,b), reminiscent of avian ENS ganglia in vivo, and cementing the idea that neurons are more cohesive than ENC cells (Rollo B., Zhang, D, unpublished). Modeling of these properties (Fig. 7c,d) is described in more detail later.
Fig. 7.

ENS cells re-organise and aggregate experimentally and in models. (a) Spherical clusters form from dissociated ENS cells from quail embryo gut in standard aggregation assays. (b) Confocal immunofluorescent labeling reveals that in clusters, neurons (HuC/D+ cytoplasm, red) segregate centrally, with ENC cells (SoxE+ nuclei, green) peripherally, suggesting a cell-cell adhesive difference. Nuclei of all cells are labeled blue with DAPI. Scale a,b=50 jim. (Images: D.Zhang, B. Rollo). (c) Simulated sorting behavior of randomly distributed ENC agents (blue) and neurons (red) under different relative and absolute cohesion conditions. Large relative difference between cohesion of neurons (high, 8) and ENC cells (low, 1) leads to neuronal aggregates with continuously motile ENC cells. A lower relative difference of cohesion (N:ENC 2:1) but different absolute levels (6:3 and 8:4) gives varying degrees of aggregation with central neurons and peripheral ENC agents. (d) Ganglion-like aggregation is robust over a range of parameters, shown here by varying the neuron to ENC cell ratio and the total ENS agent density (Hackett-Jones et al., 2011).
At the same time as ENS ganglia commence aggregation, the nearby gut mesenchyme ECM (Newgreen and Hartley, 1995) and cells become increasingly concentrically oriented as smooth muscle layers appear (Duband et al., 1993). In the mouse midgut, the co-alignment of the rib-like ENS ganglia and the ECM is consistent with an influence of ECM on ENS morphogenesis via contact guidance.
Conclusions on the roles of ECM and CAMs for ENS ganglionation
Guilt-by-association and in vitro evidence suggest that changing levels of cell-cell adhesion molecules play an important role in ENS ganglion aggregation and ganglion internal structure, compared to their postulated minor part in ENC cell migration. In contrast, the contributions of cell-ECM adhesions to ganglion aggregation per se may be less dramatic than their role in migration. The level of β1-integrin mRNA in mouse ENS declines (Bondurand N.and Dufour S., unpublished) suggesting a gradual decline in ECM adhesion. The role of cell-ECM adhesion may include sculpting the form of the ENS ganglia and the neural network; this is discussed further in relation to the mutant mice.
3. Variations in the basic ENS ganglionation pattern: lessons from mice
The model outlined above is enacted between ENS cells on a uniform field, but in reality the microenvironment is rich in other cells and ECM. Observations in mice point to important interactions between these and the ENS cells in the process of ganglionation.
General failure of gangliogenesis: NC-Specific deletion of Hand2
Deletion of the basic helix-loop-helix DNA-binding protein Hand2 in enteric neural precursor cells using a Nestin-Cre line of driver mice results in offspring which die around P20 due to gastrointestinal defects (Lei and Howard, 2011). Hand2 is lost in cells that at any stage expressed the neuron precursor molecule Nestin. There is no ENC cell migration defect in these mice but gangliogenesis is impaired, and the ENS network continues to resemble the ENS of an earlier stage. This mutation suggests genetic component(s) to ENS patterning and cell differentiation that is independent of ENC cell migration.
The myenteric ENS network (Fig. 8a) becomes thicker and retains more neurons in the mutants (Fig. 8c), and the deeper coherent rib-like circumferential ganglia (Fig. 8b) fail to form properly (Fig. 8d). Our initial conclusion was that neural network patterning was most affected by cell numbers as there were significant defects in proliferation (Hendershot et al., 2007) which affected both cell numbers and the differentiation of ENC cells into neurons. Interestingly, it appears that not only cell numbers but the distribution of glial cells and expression of CAM and ECM molecules contribute to, or parallel, the neural network patterning defect in Hand2 mutant mice.
Fig. 8.
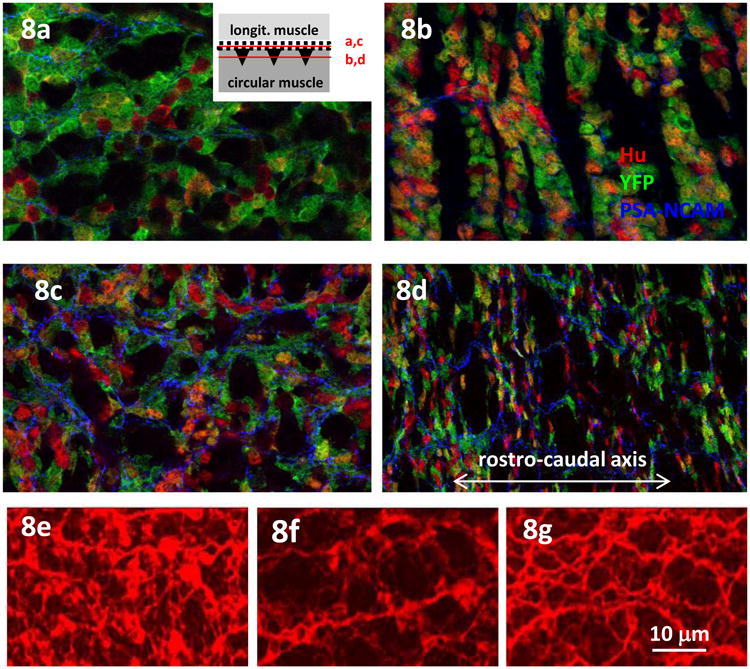
The organization of the ENS is disrupted in mutant mice, shown in mouse embryo gut wholemounts. (a-d) YFP (green) reveals genetically marked Nestin+ ENS cells in myenteric plexus of E18 mouse embryonic colon. In the outer ENS lattice in wild-type mice, the ENS has few neurons (Hu, red). The inset shows the position of confocal section planes in relation to the ENS plexus for a-d). (b) In the same field, neurons are found in the deeper riblike ganglia. (c) In contrast the Hand2 mutant retains many neurons in the outer lattice of ENS. (d) At the deeper layer, the Hand2 mutant ENS cells fail to aggregate into ganglia, suggesting a deficit in cell-cell adhesion. The expression of PSA-NCAM in the ENS is more extensive in the mutant. (Image: Jun Lei). (e-g) The early ENS in mouse E14.5 midgut revealed with anti-β3-tubulin antibody (Tuj1, red). (e) The ENS is a dense network organization in wild-type. (f) In mice with β1-integrin knocked out in ENC cells the ENS is more open. (g) When the ENC cells also lack N-cadherin (double mutant) the ENS organization is partially rescued. (Image: F. Broders-Bondon)
We have postulated above that intercellular adhesion of ENS cells, mediated by CAMS like N-cadherin and/or NCAM, influence ganglionation (Fig. 7). In the Hand2 mutant ENS there is a substantial increase in the level of polysialic acid (PSA)-NCAM (Howard M., unpublished observation) (Fig.8a-d). Addition of PSA reduces cell adhesion (Johnson et al., 2005), and this is not restricted to NCAM homophilic adhesion but also extends to adhesions mediated by other cell and ECM adhesion molecules (Acheson et al., 1991). This could impair the processes needed for ganglion formation (Fu et al., 2006; Faure et al., 2007) and result in the observed looser association between the ENS cells.
The ENS of Nestin-targeted Hand2 deletion mice has other defects of organization. Glial cells in the mutant ENS associate primarily with the neuron somata compared to the fiber network (Lei and Howard, 2011). Paralleling the aberrant glial cell distribution, the mutant ENS has an increase in randomly arrayed nerve fibers, so that the rectangular ENS pattern is degraded. It remains unclear whether the disarrayed fiber tracts reflect changes in adhesion (increase: axon-microenvironment) or fasciculation (decrease: axon-axon) cues. However, the increase in PSA-NCAM that we observe in the fibers compared to the neurons may contribute to the lack of fasciculation.
The ENS of Nestin-targeted Hand2 deletion mice lack nitrergic neurons (Lei and Howard, 2011). These are a quantitatively important sub-type (Sang and Young, 1996) and among the first neurons to differentiate. They project the first axons that extend longitudinally (caudally) along the intestine (Sang et al., 1997; Young et al., 2002). It could be proposed that absence of these neurons and hence of their axons may disturb the structuring of the ganglia and the axons generated subsequently, contributing to the ganglion and nerve fiber disarray. In addition, Li et al. (2011) have proposed that early differentiating serotoninergic neurons, via axonal contacts, modulate the differentiation and survival of later-appearing neurons in the same and adjacent ganglia. If this can be generalized to other neuron types, interference with differentiation of nitrergic neurons could have powerful follow-on effects.
The effects of loss of the transcription factor Hand2 expression in a subset of ENS cells has a morphological effect on the ENS that in outline seems a simple failure to progress to higher level of organization, but a molecular understanding of the direct and indirect effects of this initiating genetic alteration will not be simple.
Specific alterations of gangliogenesis: NC-specific deletion of β1-integrin and N-cadherin
Deletion of β1-integrin in all NC cells causes a Hirschsprung-like aganglionosis of the distal gut (Fig. 2d) and also a dysgenesis of ganglion shape and distribution in the midgut (Fig. 2b, 8e). In these mutants the ENS ganglia are bigger, more rounded and more sparsely distributed (Breau et al., 2006), and the rectangular ENS pattern is disturbed. Interestingly, the ENS pattern is not chaotic, but it is different from normal since the mutant ENS ganglia are connected in a rudely triangular network. When assayed in vitro on ECM substrates the β1-integrin-null ENS cells have relatively increased aggregation (Breau et al., 2009). This suggests that the deletion has resulted in the mutant ENS cells having the balance between cell-cell and cell-ECM adhesion altered to favor the former, with organizational cues from the ECM being disregarded.
In mice with NC-specific N-cadherin loss, the ENS ganglionic network is scarcely affected. Surprisingly, in N-cadherin/β1-integrin double mutant mice, morphometry (Broders-Bondon et al., 2012) has shown that the re-organized ganglion network of the β1-integrin-null ENS in vivo is to a degree rescued (Fig. 8g). This rescue is consistent with the previous notion of adhesive balance, and points to the importance of cooperation between these two quite different adhesion systems. It is hard to escape the conclusion that not only the levels of expression and function of adhesion molecules but also the balance between cell-cell and cell-ECM interactions are crucial during ENS morphogenesis (Broders-Bondon et al., 2012). In accord with this, biophysical approaches in other systems have revealed crosstalk between cadherin and integrins to regulate cell adhesion strength and traction force at focal adhesions (Jasaitis et al., 2012; Martinez-Rico et al., 2010; Montero and Heisenberg, 2003; Weber et al., 2011).
Mathematical models of ganglionic morphogenesis and their emergent properties
The general properties that we wish to emerge in CA models of avian ENS ganglion formation include: 1. Ganglia form progressively; 2. Ganglia have both neurons and ENC cells; 3. Ganglionic neurons assemble as a central core with peripheral ENC cells; 4. Ganglia become roughly evenly spaced; 5.Ganglia become roughly evenly sized; 6. Ganglia become relatively stable; 7. Ganglia are similar in non-growing field and growing-field situations.
What would be the input parameters? As in the previously described models, we would include agent motility, proliferation, and differentiation of ENC cell-agents into neuron-agents, the latter having different proliferation properties (modeled for simplicity as loss of proliferation). These would control the final ratio of neurons to ENC agents as well as the final agent density. The additional property to achieve agent aggregation most likely involves cell adhesion (Steinberg, 2007; Townes and Holtfreter, 1955.). We proposed the following aggregation rules:
Rules of ENS cell aggregation
|
Gangliogenesis: an emergent property
Using these few properties, the CA model produced a self-organizing ENS-like array of multiple small stable clusters, with central neuron agents and peripheral ENC agents in each cluster (Fig. 7c). Subtle variations in clustering were induced by altering the ratio of adhesive potential of ENC and neuron agents, and by altering the magnitude of adhesive potential.
Temporal gangliogenesis progression: an emergent property
During assembly of ENS agents, an intermediate stage is marked by clustering of neuron agents with scattered ENC agents; this is observed transiently in avian ENS gangliogenesis (Hackett-Jones et al., 2011).
Stability of ganglia: an emergent property
Ganglion properties altered with parameter settings, so one concern was that this ENS-like state would be unstable, existing only for a small parameter range. In models, dependence on quantitative parameters can be tested by simply sweeping across a range of values. It can easily be imagined that this might result in, for example, at low adhesion values, failure to stably aggregate at all, flipping suddenly to super-aggregation into one or a few huge ganglia as adhesion is increased. Such an unstable situation is unlikely to be realistic; most biological systems are noted for resilience.
In fact the model showed considerable robustness over a range of parameter values (Fig. 7d). In part this was because of an emergent property of lower motility with increasing aggregate size that retarded super-aggregation. Simultaneously, the assembly at the periphery of clusters of ENC agents whose adhesive potential is saturated also limited the size of clusters. The ganglion patterns that emerged were essentially identical in static and growing field scenarios, in accord with biological observations.
Deficiencies of the Biological and Mathematical Models: Future Prospects
1. Colonization phase
Adhesion molecules are clearly of importance in ENC cell colonization, but in our view the current data are little better than lists of ECM and CAMs. Much greater spatial and temporal descriptive detail is required to suggest roles and experiments to test those roles, and to refine models. Experimentally, we summarized progress in targeting the functions of various adhesion molecules in ENS development, but much detail remains to be explored. The β1-integrin knockout mice for example have lost function not in one, but in a sub-family of adhesion receptors. Likewise N-cadherin inactivation in mouse NC cells leaves the role of all other cadherins and all non-cadherin CAMs unexplored.
In the modeling of the colonization of the gut by ENS cells, certain properties were proposed or inferred by comparison with other biological examples and by indirect biological evidence, and then simplified depictions were used. For example the CA models show ENC agents as equipolar (or nonpolar) disks, whereas each ENC cell exhibits a range of complex shapes and polarities which change rapidly. In the models the displacement of each ENC agent per time step was uniform, whereas the speed of each ENC cell varies widely and rapidly. In addition, the ENS was modeled as 2-dimensional, and in vivo it is a narrow layer, but it is not strictly a monolayer. These more complex states can in principal be modeled but at a high cost in input imaging information and output computational requirements. We think that these would not change the model predictions in a fundamental way, but might alter the details of colonization.
Logistical growth is one basis of the ENS colonization model, but it and its molecular controls are not well established. We have proposed that this is exerted through growth factor consumption, but other factors—even physical space in the ENS layer—may be consumed. Moreover we proposed that the growth factor is GDNF, but there may be additional factors. There is no evidence that ENS cells actually consume GDNF, and if they do, whether this consumption would be at a sufficient rate to effect its concentration. This consumption would, we proposed, form a gradient which we invoked to control cell and growth cone directionality. We know of no evidence that unequivocally identifies a gradient of GDNF in the gut mesenchyme, though there is evidence of spatial and temporal mRNA variations. No experiments have been reported in which the level or slope of a putative gradient factor like GDNF is controllably manipulated in gut tissue, to gauge the effect on ENC directionality and speed of movement in a realistic microenvironment.
In one form of the modeling of chain migration, epitomized by the wavefront in the mouse hindgut, axon/ENC agent contacts are ascribed a controlling function. Testing the role of these contacts by prevention of axon extension without collateral damage to the ENC cell locomotion machinery has not been described in mouse ENS.
2. Gangliogenesis phase
At present the modeling of the colonization phase and the ganglionation phase have been presented separately, yet in reality the latter gradually emerges from the former. An aim not yet attempted is to formulate rules and time-dependent variations, possibly quantitative, which will seamlessly allow this progression. A more direct major shortcoming of the basic modeling of ganglionation is that no attempt has yet been made to include axonal growth and connections between forming aggregates (ganglia), yet these axonal connections, with attendant glial cells, are obvious in the real ENS.
Moreover, the role of cell-ECM interactions has been ignored in the basic ganglionation model. The interesting variations in ENS ganglionation in the mouse mutants described strongly suggest that this will need to be factored in. A model encoding changing quantitative adhesivity to ECM, both absolutely, and also relative to adhesivity to cells, will need to be explored. We envisage this as having different ECM and CAM dependencies for ENC agents and neuron agents. Moreover, the enteric ECM clearly has an orientation component that changes with developmental time; this could affect morphogenesis via the well known phenomenon of contact guidance, and could be modeled by gradually introducing an orientation bias. This will need to be included in future models.
As well as this theoretical work, and required for it, much more exact descriptive knowledge of the biomechanical molecules of real systems will also need to be gained. In the mouse mutants for example, the effect of the Hand2 deletion will be genetically very complex; a plethora of downstream genes will be affected. At present the identity of this gene set is not known, let alone that subset which directly affects ENS morphogenesis. The integrin and N-cadherin deletions are obviously much more focused. However. they still experimentally explore only a sub-group of adhesion mechanisms, as mentioned previously.
Summary of Outcomes of Modeling ENS Morphogenesis
Mathematical modelling has provided much insight into the development of the ENS, using local rules for a number of cellular activities (motility, proliferation, differentiation, adhesive capacity) and for how these activities influence each other. From these, stable, resilient patterns of system-wide morphogenesis similar to those observed in the ENS have emerged naturally in the models. It is important to note that only some of these real outcomes (eg. colonization wave) were known and understood (at least intuitively) prior to the modeling. More interestingly, many outcomes emerged in the models. Some of these had been observed biologically but had no credible explanation (eg. stable ENC chains with unstable cells, increasing complexity of the ENS network), and some were only observed in retrospect as a result of predictions based on modeling (eg. frontal expansion, clonal variability). The number of cell behavioral rules used in the models is not large, so in a sense ENS morphogenesis may be simple, although this does not imply that the genetic and molecular underpinnings of the rules are simple.
Changing the relative levels of the various model components can account for termination of migration and formation of the abnormal ENS distribution and ganglion pattern characteristic of congenital human enteric neuropathies. In particular, the model results and experimental confirmations have important implications for Hirschsprung disease. Arising directly from the modeling, we now view Hirschsprung disease to be more commonly an ENC cell proliferation defect and not, as initially supposed, an ENC cell motility defect. The models also provide a rationale to explain zonal and skip segment aganglionosis. In addition, an emergent feature of the model suggests the novel idea that reduced penetrance of dominant mutation Hirschsprung disease, especially in identical twins, may be in part due to normal stochasticity at the level of behaviour of individual cells which is uncovered by mutation in a Hirschsprung gene.
Highlights.
Reviews the early development of the vertebrate enteric nervous system
Approximates cellular function with probabilistic agent-based rules
Global patterns emerge from modeling interactions of simple local rules
Models provide insight into components of enteric nervous system morphogenesis
Models provide insight into enteric nervous system dysmorphogeneses.
Acknowledgments
The authors would like to thank their colleagues Ben Binder, Nadège Bondurand, Marie Breau, Florence Broders-Bondon, Jeff Craig, Hideki Enomoto, Anthony Fernando, Emily Hackett-Jones, Barry Hughes, Jun Lei, Mick Mariani, Ben Rollo, Johanna Simkin, Matthew Simpson, Lincon Stamp, Heather Young and Dongcheng Zhang for information, published and unpublished, for alerting us to references we had overlooked, and for discussions. Anonymous reviewers also made valuable suggestions and several references. DFN acknowledges that the biomathematical approaches reviewed here owe a 40-year debt to discussions with the late Ian Allan. This work was supported by ARC and NHMRC grants. KAL is an ARC Professorial Fellow. MCRI facilities are supported by the Victorian Government's Operational Infrastructure Support Program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Donald F. Newgreen, The Murdoch Children's Research Institute, Royal Children's Hospital, Parkville 3052, Australia
Sylvie Dufour, Institut Curie/CNRS UMR144, 26 rue d'Ulm, 75005 Paris, France.
Marthe J. Howard, Department of Neurosciences, University of Toledo Health Sciences Campus, Toledo, OH, 43614, USA
Kerry A. Landman, Department of Mathematic and Statistics, University of Melbourne, 3010, Australia
References
- Acheson A, Sunshine JL, Rutishauser U. NCAM polysialic acid can regulate both cell-cell and cell-substrate interactions. The Journal of Cell Biology. 1991;114:143–153. doi: 10.1083/jcb.114.1.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan IJ, Newgreen DF. The origin and differentiation of enteric neurons of the intestine of the fowl embryo. The American journal of anatomy. 1980;157:137–154. doi: 10.1002/aja.1001570203. [DOI] [PubMed] [Google Scholar]
- Amiel J, Sproat-Emison E, Garcia-Barcelo M, Lantieri F, Burzynski G, Borrego S, Pelet A, Arnold S, Miao X, Griseri P, Brooks AS, Antinolo G, de Pontual L, Clement-Ziza M, Munnich A, Kashuk C, West K, Wong KK, Lyonnet S, Chakravarti A, Tam PK, Ceccherini I, Hofstra RM, Fernandez R. Hirschsprung disease, associated syndromes and genetics: a review. J Med Genet. 2008;45:1–14. doi: 10.1136/jmg.2007.053959. [DOI] [PubMed] [Google Scholar]
- Anderson RB, Turner KN, Nikonenko AG, Hemperly J, Schachner M, Young HM. The cell adhesion molecule L1 is required for chain migration of neural crest cells in the developing mouse gut. Gastroenterology. 2006;130:1221–1232. doi: 10.1053/j.gastro.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Bard JB, Hay ED. The behavior of fibroblasts from the developing avian cornea. Morphology and movement in situ and in vitro. J Cell Biol. 1975;67:400–418. doi: 10.1083/jcb.67.2.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barde YA. Trophic factors and neuronal survival. Neuron. 1989;2:1525–1534. doi: 10.1016/0896-6273(89)90040-8. [DOI] [PubMed] [Google Scholar]
- Binder BJ, Landman KA, Newgreen DF, Simkin JE, Takahashi Y, Zhang D. Spatial analysis of multi-species exclusion processes: application to neural crest cell migration in the embryonic gut. Bulletin of mathematical biology. 2012;74:474–490. doi: 10.1007/s11538-011-9703-z. [DOI] [PubMed] [Google Scholar]
- Binder BJ, Landman KA, Simpson MJ, Mariani M, Newgreen DF. Modeling proliferative tissue growth: a general approach and an avian case study. Physical review. 2008;78:031912. doi: 10.1103/PhysRevE.78.031912. [DOI] [PubMed] [Google Scholar]
- Bixby S, Kruger G, Mosher J, Joseph N, Morrison S. Cell-Intrinsic Differences between Stem Cells from Different Regions of the Peripheral Nervous System Regulate the Generation of Neural Diversity. Neuron. 2002;35:643–656. doi: 10.1016/s0896-6273(02)00825-5. [DOI] [PubMed] [Google Scholar]
- Bolcato-Bellemin AL, Lefebvre O, Arnold C, Sorokin L, Miner JH, Kedinger M, Simon-Assmann P. Laminin alpha5 chain is required for intestinal smooth muscle development. Dev Biol. 2003;260:376–390. doi: 10.1016/s0012-1606(03)00254-9. [DOI] [PubMed] [Google Scholar]
- Brauer PR, Bolender DL, Markwald RR. The distribution and spatial organization of the extracellular matrix encountered by mesencephalic neural crest cells. Anat Rec. 1985;211:57–68. doi: 10.1002/ar.1092110110. [DOI] [PubMed] [Google Scholar]
- Breau MA, Dahmani A, Broders-Bondon F, Thiery JP, Dufour S. Beta1 integrins are required for the invasion of the caecum and proximal hindgut by enteric neural crest cells. Development. 2009;136:2791–2801. doi: 10.1242/dev.031419. [DOI] [PubMed] [Google Scholar]
- Breau MA, Pietri T, Eder O, Blanche M, Brakebusch C, Fassler R, Thiery JP, Dufour S. Lack of {beta}1 integrins in enteric neural crest cells leads to a Hirschsprung-like phenotype. Development. 2006;133:1725–734. doi: 10.1242/dev.02346. [DOI] [PubMed] [Google Scholar]
- Broders-Bondon F, Paul-Gilloteaux P, Carlier C, Radice GL, Dufour S. N-cadherin and beta1-integrins cooperate during the development of the enteric nervous system. Dev Biol. 2012;364:178–191. doi: 10.1016/j.ydbio.2012.02.001. [DOI] [PubMed] [Google Scholar]
- Cai DH, Brauer PR. Synthetic matrix metalloproteinase inhibitor decreases early cardiac neural crest migration in chicken embryos. Dev Dyn. 2002;224:441–449. doi: 10.1002/dvdy.10129. [DOI] [PubMed] [Google Scholar]
- Carmona-Fontaine C, Matthews HK, Kuriyama S, Moreno M, Dunn GA, Parsons M, Stern CD, Mayor R. Contact inhibition of locomotion in vivo controls neural crest directional migration. Nature. 2008;456:957–961. doi: 10.1038/nature07441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner PJ, Focke PJ, Noden DM, Epstein ML. Appearance of neurons and glia with respect to the wavefront during colonization of the avian gut by neural crest cells. Dev Dyn. 2003;226:91–98. doi: 10.1002/dvdy.10219. [DOI] [PubMed] [Google Scholar]
- Cox BN. A strain-cue hypothesis for biological network formation. J R Soc Interface. 2011;8:377–394. doi: 10.1098/rsif.2010.0262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Giorgio R, Camilleri M. Human enteric neuropathies: morphology and molecular pathology. Neurogastroenterol Motil. 2004;16:515–531. doi: 10.1111/j.1365-2982.2004.00538.x. [DOI] [PubMed] [Google Scholar]
- Druckenbrod NR, Epstein ML. The pattern of neural crest advance in the cecum and colon. Dev Biol. 2005;287:125–133. doi: 10.1016/j.ydbio.2005.08.040. [DOI] [PubMed] [Google Scholar]
- Druckenbrod NR, Epstein ML. Behavior of enteric neural crest-derived cells varies with respect to the migratory wavefront. Dev Dyn. 2007;236:84–92. doi: 10.1002/dvdy.20974. [DOI] [PubMed] [Google Scholar]
- Druckenbrod NR, Epstein ML. Age-dependent changes in the gut environment restrict the invasion of the hindgut by enteric neural progenitors. Development. 2009;136:3195–3203. doi: 10.1242/dev.031302. [DOI] [PubMed] [Google Scholar]
- Duband JL, Gimona M, Scatena M, Sartore S, Small JV. Calponin and SM 22 as differentiation markers of smooth muscle: spatiotemporal distribution during avian embryonic development. Differentiation. 1993;55:1–11. doi: 10.1111/j.1432-0436.1993.tb00027.x. [DOI] [PubMed] [Google Scholar]
- Epstein ML, Poulsen KT, Thiboldeaux R. Formation of ganglia in the gut of the chick embryo. J Comp Neurol. 1991;307:189–199. doi: 10.1002/cne.903070203. [DOI] [PubMed] [Google Scholar]
- Erickson CA. Control of neural crest cell dispersion in the trunk of the avian embryo. Dev Biol. 1985;111:138–157. doi: 10.1016/0012-1606(85)90442-7. [DOI] [PubMed] [Google Scholar]
- Erickson CA, Perris R. The role of cell-cell and cell-matrix interactions in the morphogenesis of the neural crest. Dev Biol. 1993;159:60–74. doi: 10.1006/dbio.1993.1221. [DOI] [PubMed] [Google Scholar]
- Faure C, Chalazonitis A, Rheaume C, Bouchard G, Sampathkumar SG, Yarema KJ, Gershon MD. Gangliogenesis in the enteric nervous system: roles of the polysialylation of the neural cell adhesion molecule and its regulation by bone morphogenetic protein-4. Dev Dyn. 2007;236:44–59. doi: 10.1002/dvdy.20943. [DOI] [PubMed] [Google Scholar]
- Flynn B, Bergner AJ, Turner KN, Young HM, Anderson RB. Effect of Gdnf haploinsufficiency on rate of migration and number of enteric neural crest-derived cells. Dev Dyn. 2007;236:134–141. doi: 10.1002/dvdy.21013. [DOI] [PubMed] [Google Scholar]
- Focke PJ, Schiltz CA, Jones SE, Watters JJ, Epstein ML. Enteric neuroblasts require the phosphatidylinositol 3-kinase pathway for GDNF-stimulated proliferation. J Neurobiol. 2001;47:306–317. doi: 10.1002/neu.1037. [DOI] [PubMed] [Google Scholar]
- Focke PJ, Swetlik AR, Schilz JL, Epstein ML. GDNF and insulin cooperate to enhance the proliferation and differentiation of enteric crest-derived cells. J Neurobiol. 2003;55:151–164. doi: 10.1002/neu.10204. [DOI] [PubMed] [Google Scholar]
- Fu M, Sato Y, Lyons-Warren A, Zhang B, Kane MA, Napoli JL, Heuckeroth RO. Vitamin A facilitates enteric nervous system precursor migration by reducing Pten accumulation. Development. 2010;137:631–640. doi: 10.1242/dev.040550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu M, Tam PK, Sham MH, Lui VC. Embryonic development of the ganglion plexuses and the concentric layer structure of human gut: a topographical study. Anat Embryol (Berl) 2004;208:33–41. doi: 10.1007/s00429-003-0371-0. [DOI] [PubMed] [Google Scholar]
- Fu M, Vohra BP, Wind D, Heuckeroth RO. BMP signaling regulates murine enteric nervous system precursor migration, neurite fasciculation, and patterning via altered Ncam1 polysialic acid addition. Dev Biol. 2006;299:137–150. doi: 10.1016/j.ydbio.2006.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto T, Hata J, Yokoyama S, Mitomi T. A study of the extracellular matrix protein as the migration pathway of neural crest cells in the gut: analysis in human embryos with special reference to the pathogenesis of Hirschsprung's disease. J Pediatr Surg. 1989;24:550–556. doi: 10.1016/s0022-3468(89)80504-4. [DOI] [PubMed] [Google Scholar]
- Furness JB. The enteric nervous system: normal functions and enteric neuropathies. Neurogastroenterol Motil. 2008;20(Suppl 1):32–38. doi: 10.1111/j.1365-2982.2008.01094.x. [DOI] [PubMed] [Google Scholar]
- Furness JB. The enteric nervous system and neurogastroenterology. Nature reviews. Gastroenterology & hepatology. 2012;9:286–294. doi: 10.1038/nrgastro.2012.32. [DOI] [PubMed] [Google Scholar]
- Gaidar YA, Lepekhin EA, Sheichetova GA, Witt M. Distribution of N-cadherin and NCAM in neurons and endocrine cells of the human embryonic and fetal gastroenteropancreatic system. Acta Histochem. 1998;100:83–97. doi: 10.1016/S0065-1281(98)80008-1. [DOI] [PubMed] [Google Scholar]
- Gianino S, Grider JR, Cresswell J, Enomoto H, Heuckeroth RO. GDNF availability determines enteric neuron number by controlling precursor proliferation. Development. 2003;130:2187–2198. doi: 10.1242/dev.00433. [DOI] [PubMed] [Google Scholar]
- Gooday D, Thorogood P. Contact behaviour exhibited by migrating neural crest cells in confrontation culture with somitic cells. Cell Tissue Res. 1985;241:165–169. doi: 10.1007/BF00214638. [DOI] [PubMed] [Google Scholar]
- Hackett-Jones EJ, Landman KA, Newgreen DF, Zhang D. On the role of differential adhesion in gangliogenesis in the enteric nervous system. Journal of theoretical biology. 2011;287:148–159. doi: 10.1016/j.jtbi.2011.07.013. [DOI] [PubMed] [Google Scholar]
- Hao MM, Anderson RB, Kobayashi K, Whitington PM, Young HM. The migratory behavior of immature enteric neurons. Dev Neurobiol. 2009;69:22–35. doi: 10.1002/dneu.20683. [DOI] [PubMed] [Google Scholar]
- Hao MM, Young HM. Development of enteric neuron diversity. Journal of cellular and molecular medicine. 2009;13:1193–1210. doi: 10.1111/j.1582-4934.2009.00813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hearn CJ, Murphy M, Newgreen D. GDNF and ET-3 differentially modulate the numbers of avian enteric neural crest cells and enteric neurons in vitro. Developmental biology. 1998;197:93–105. doi: 10.1006/dbio.1998.8876. [DOI] [PubMed] [Google Scholar]
- Hearn CJ, Young HM, Ciampoli D, Lomax AE, Newgreen D. Catenary cultures of embryonic gastrointestinal tract support organ morphogenesis, motility, neural crest cell migration, and cell differentiation. Dev Dyn. 1999;214:239–247. doi: 10.1002/(SICI)1097-0177(199903)214:3<239::AID-AJA7>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Hotta R, Anderson RB, Kobayashi K, Newgreen DF, Young HM. Effects of tissue age, presence of neurones and endothelin-3 on the ability of enteric neurone precursors to colonize recipient gut: implications for cell-based therapies. Neurogastroenterol Motil. 2009 doi: 10.1111/j.1365-2982.2009.01411.x. [DOI] [PubMed] [Google Scholar]
- Jasaitis A, Estevez M, Heysch J, Ladoux B, Dufour S. E-cadherin-dependent stimulation of traction force at focal adhesions via the Src and PI3K signaling pathways. Biophys J. 2012;103:175–184. doi: 10.1016/j.bpj.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson CP, Fujimoto I, Rutishauser U, Leckband DE. Direct evidence that neural cell adhesion molecule (NCAM) polysialylation increases intermembrane repulsion and abrogates adhesion. J Biol Chem. 2005;280:137–145. doi: 10.1074/jbc.M410216200. [DOI] [PubMed] [Google Scholar]
- Jung PM. Hirschsprung's disease: one surgeon's experience in one institution. J Pediatr Surg. 1995;30:646–651. doi: 10.1016/0022-3468(95)90680-0. [DOI] [PubMed] [Google Scholar]
- Kasemeier-Kulesa JC, Bradley R, Pasquale EB, Lefcort F, Kulesa PM. Eph/ephrins and N- cadherin coordinate to control the pattern of sympathetic ganglia 10.1242/dev.02662. Development. 2006;133:4839–4847. doi: 10.1242/dev.02662. [DOI] [PubMed] [Google Scholar]
- King SK, Sutcliffe JR, Ong SY, Lee M, Koh TL, Wong SQ, Farmer PJ, Peck CJ, Stanton MP, Keck J, Cook DJ, Chow CW, Hutson JM, Southwell BR. Substance P and vasoactive intestinal peptide are reduced in right transverse colon in pediatric slow-transit constipation. Neurogastroenterol Motil. 2010;22:883–892. doi: 10.1111/j.1365-2982.2010.01524.x. e234. [DOI] [PubMed] [Google Scholar]
- Kruger GM, Mosher JT, Tsai YH, Yeager KJ, Iwashita T, Gariepy CE, Morrison SJ. Temporally distinct requirements for endothelin receptor B in the generation and migration of gut neural crest stem cells. Neuron. 2003;40:917–929. doi: 10.1016/s0896-6273(03)00727-x. [DOI] [PubMed] [Google Scholar]
- Kulesa PM, Fraser SE. In ovo time-lapse analysis of chick hindbrain neural crest cell migration shows cell interactions during migration to the branchial arches. Development. 2000;127:1161–1172. doi: 10.1242/dev.127.6.1161. [DOI] [PubMed] [Google Scholar]
- Kulesa PM, Teddy JM, Stark DA, Smith SE, McLennan R. Neural crest invasion is a spatially-ordered progression into the head with higher cell proliferation at the migratory front as revealed by the photoactivatable protein, KikGR. Dev Biol. 2008;316:275–287. doi: 10.1016/j.ydbio.2008.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuntz A. Experimental studies on thehistogenesis of the sympathetic nervous system. J Comp Neurol. 1922;34:1–36. [Google Scholar]
- Kuo BR, Erickson CA. Vagal neural crest cell migratory behavior: a transition between the cranial and trunk crest. Dev Dyn. 2011;240:2084–2100. doi: 10.1002/dvdy.22715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landman KA, Fernando AE, Zhang D, Newgreen DF. Building stable chains with motile agents: Insights into the morphology of enteric neural crest cell migration. Journal of theoretical biology. 2011;276:250–268. doi: 10.1016/j.jtbi.2011.01.043. [DOI] [PubMed] [Google Scholar]
- Landman KA, Simpson MJ, Newgreen DF. Mathematical and experimental insights into the development of the enteric nervous system and Hirschsprung's Disease. Development, Growth & Differentiation. 2007;49:277–286. doi: 10.1111/j.1440-169X.2007.00929.x. [DOI] [PubMed] [Google Scholar]
- Le Douarin NM, Teillet MA. The migration of neural crest cells to the wall of the digestive tract in avian embryo. J Embryol Exp Morphol. 1973;30:31–48. [PubMed] [Google Scholar]
- Lei J, Howard MJ. Targeted deletion of Hand2 in enteric neural precursor cells affects its functions in neurogenesis, neurotransmitter specification and gangliogenesis, causing functional aganglionosis. Development. 2011;138:4789–4800. doi: 10.1242/dev.060053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Chalazonitis A, Huang YY, Mann JJ, Margolis KG, Yang QM, Kim DO, Cote F, Mallet J, Gershon MD. Essential roles of enteric neuronal serotonin in gastrointestinal motility and the development/survival of enteric dopaminergic neurons. J Neurosci. 2011;31:8998–9009. doi: 10.1523/JNEUROSCI.6684-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Rico C, Pincet F, Thiery JP, Dufour S. Integrins stimulate E-cadherin-mediated intercellular adhesion by regulating Src-kinase activation and actomyosin contractility. J Cell Sci. 2010;123:712–722. doi: 10.1242/jcs.047878. [DOI] [PubMed] [Google Scholar]
- McLennan R, Dyson L, Prather KW, Morrison JA, Baker RE, Maini PK, Kulesa PM. Multiscale mechanisms of cell migration during development: theory and experiment. Development. 2012;139:2935–2944. doi: 10.1242/dev.081471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier-Ruge WA, Ammann K, Bruder E, Holschneider AM, Scharli AF, Schmittenbecher PP, Stoss F. Updated results on intestinal neuronal dysplasia (IND B) Eur J Pediatr Surg. 2004;14:384–391. doi: 10.1055/s-2004-821120. [DOI] [PubMed] [Google Scholar]
- Menoud PA, Debrot S, Schowing J. Mouse neural crest cells secrete both urokinase-type and tissue-type plasminogen activators in vitro. Development. 1989;106:685–690. doi: 10.1242/dev.106.4.685. [DOI] [PubMed] [Google Scholar]
- Mirsky R, Jessen KR, Schachner M, Goridis C. Distribution of the adhesion molecules N-CAM and L1 on peripheral neurons and glia in adult rats. J Neurocytol. 1986;15:799–815. doi: 10.1007/BF01625196. [DOI] [PubMed] [Google Scholar]
- Montero JA, Heisenberg CP. Adhesive crosstalk in gastrulation. Dev Cell. 2003;5:190–191. doi: 10.1016/s1534-5807(03)00235-1. [DOI] [PubMed] [Google Scholar]
- Nagy N, Burns AJ, Goldstein AM. Immunophenotypic characterization of enteric neural crest cells in the developing avian colorectum. Dev Dyn. 2012;241:842–851. doi: 10.1002/dvdy.23767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy N, Mwizerwa O, Yaniv K, Carmel L, Pieretti-Vanmarcke R, Weinstein BM, Goldstein AM. Endothelial cells promote migration and proliferation of enteric neural crest cells via beta1 integrin signaling. Dev Biol. 2009;330:263–272. doi: 10.1016/j.ydbio.2009.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newgreen DF. Control of the directional migration of mesenchyme cells and neurites. Sem Dev Biol. 1990;1:301–311. [Google Scholar]
- Newgreen DF, Hartley L. Extracellular matrix and adhesive molecules in the early development of the gut and its innervation in normal and spotting lethal rat embryos. Acta anatomica. 1995;154:243–260. doi: 10.1159/000147776. [DOI] [PubMed] [Google Scholar]
- Newgreen DF, Ritterman M, Peters EA. Morphology and behaviour of neural crest cells of chick embryo in vitro. Cell and tissue research. 1979;203:115–140. doi: 10.1007/BF00234333. [DOI] [PubMed] [Google Scholar]
- Nishiyama C, Uesaka T, Manabe T, Yonekura Y, Nagasawa T, Newgreen DF, Young HM, Enomoto H. Trans-mesenteric neural crest cells are the principal source of the colonic enteric nervous system. Nature neuroscience. 2012;15:1211–1218. doi: 10.1038/nn.3184. [DOI] [PubMed] [Google Scholar]
- O'Donnell A, Bannigan J, Puri P. Vagal neural crest contribution to the chick embryo cloaca. Pediatr Surg Int. 2006;22:983–986. doi: 10.1007/s00383-006-1784-7. [DOI] [PubMed] [Google Scholar]
- O'Donnell AM, Puri P. Skip segment Hirschsprung's disease: a systematic review. Pediatr Surg Int. 2010;26:1065–1069. doi: 10.1007/s00383-010-2692-4. [DOI] [PubMed] [Google Scholar]
- Okamoto N, Del Maestro R, Valero R, Monros E, Poo P, Kanemura Y, Yamasaki M. Hydrocephalus and Hirschsprung's disease with a mutation of L1CAM. Journal of human genetics. 2004;49:334–337. doi: 10.1007/s10038-004-0153-4. [DOI] [PubMed] [Google Scholar]
- Pietri T, Eder O, Blanche M, Thiery JP, Dufour S. The human tissue plasminogen activator- Cre mouse: a new tool for targeting specifically neural crest cells and their derivatives in vivo. Dev Biol. 2003;259:176–187. doi: 10.1016/s0012-1606(03)00175-1. [DOI] [PubMed] [Google Scholar]
- Sang Q, Williamson S, Young HM. Projections of chemically identified myenteric neurons of the small and large intestine of the mouse. J Anat. 1997;190:209–222. doi: 10.1046/j.1469-7580.1997.19020209.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang Q, Young HM. Chemical coding of neurons in the myenteric plexus and external muscle of the small and large intestine of the mouse. Cell Tissue Res. 1996;284:39–53. doi: 10.1007/s004410050565. [DOI] [PubMed] [Google Scholar]
- Shen L, Pichel JG, Mayeli T, Sariola H, Lu B, Westphal H. Gdnf haploinsufficiency causes Hirschsprung-like intestinal obstruction and early-onset lethality in mice. Am J Hum Genet. 2002;70:435–447. doi: 10.1086/338712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon-Assmann P, Kedinger M, De Arcangelis A, Rousseau V, Simo P. Extracellular matrix components in intestinal development. Experientia. 1995;51:883–900. doi: 10.1007/BF01921739. [DOI] [PubMed] [Google Scholar]
- Simpson MJ, Landman KA, Hughes BD, D FN. Looking inside an invasion wave of cells using continuum models: Proliferation is the key. J Theor Biol. 2006;243:343–360. doi: 10.1016/j.jtbi.2006.06.021. [DOI] [PubMed] [Google Scholar]
- Simpson MJ, Merrifield A, Landman KA, Hughes BD. Simulating invasion with cellular automata: connecting cell-scale and population-scale properties. Phys Rev E Stat Nonlin Soft Matter Phys. 2007a;76:021918. doi: 10.1103/PhysRevE.76.021918. [DOI] [PubMed] [Google Scholar]
- Simpson MJ, Zhang DC, Mariani M, Landman KA, Newgreen DF. Cell proliferation drives neural crest cell invasion of the intestine. Developmental biology. 2007b;302:553–568. doi: 10.1016/j.ydbio.2006.10.017. [DOI] [PubMed] [Google Scholar]
- Solari V, Piotrowska AP, Puri P. Histopathological differences between recto-sigmoid Hirschsprung's disease and total colonic aganglionosis. Pediatr Surg Int. 2003;19:349–354. doi: 10.1007/s00383-003-1009-2. [DOI] [PubMed] [Google Scholar]
- Srinivasan S, Anitha M, Mwangi S, Heuckeroth RO. Enteric neuroblasts require the phosphatidylinositol 3-kinase/Akt/Forkhead pathway for GDNF-stimulated survival. Mol Cell Neurosci. 2005;29:107–119. doi: 10.1016/j.mcn.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Steinberg MS. Differential adhesion in morphogenesis: a modern view. Curr Opin Genet Dev. 2007;17:281–286. doi: 10.1016/j.gde.2007.05.002. [DOI] [PubMed] [Google Scholar]
- Suvanto P, Hiltunen JO, Arumae U, Moshnyakov M, Sariola H, Sainio K, Saarma M. Localization of glial cell line-derived neurotrophic factor (GDNF) mRNA in embryonic rat by in situ hybridization. Eur J Neurosci. 1996;8:816–822. doi: 10.1111/j.1460-9568.1996.tb01267.x. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Iwashita T, Santoro M, Lyonnet S, Lenoir GM, Billaud M. Co-segregation of MEN2 and Hirschsprung's disease: the same mutation of RET with both gain and loss-of-function? Hum Mutat. 1999;13:331–336. doi: 10.1002/(SICI)1098-1004(1999)13:4<331::AID-HUMU11>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Takeichi M. Functional correlation between cell adhesive properties and some cell surface proteins. J Cell Biol. 1977;75:464–474. doi: 10.1083/jcb.75.2.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery JP, Delouvee A, Grumet M, Edelman GM. Initial appearance and regional distribution of the neuron-glia cell adhesion molecule in the chick embryo. J Cell Biol. 1985;100:442–456. doi: 10.1083/jcb.100.2.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas LA, Yamada KM. Contact stimulation of cell migration. J Cell Sci. 1992;103(Pt 4):1211–1214. doi: 10.1242/jcs.103.4.1211. [DOI] [PubMed] [Google Scholar]
- Townes P, Holtfreter J. Directed movements and selective adhesion of embryonic amphibian cells. J Exp Zool. 1955;128:53–120. doi: 10.1002/jez.a.114. [DOI] [PubMed] [Google Scholar]
- Tucker GC, Ciment G, Thiery JP. Pathways of avian neural crest cell migration in the developing gut. Dev Biol. 1986;116:439–450. doi: 10.1016/0012-1606(86)90145-4. [DOI] [PubMed] [Google Scholar]
- Turner KN, Schachner M, Anderson RB. Cell adhesion molecule L1 affects the rate of differentiation of enteric neurons in the developing gut. Dev Dyn. 2009;238:708–715. doi: 10.1002/dvdy.21861. [DOI] [PubMed] [Google Scholar]
- Wallace AS, Anderson RB. Genetic interactions and modifier genes in Hirschsprung's disease. World J Gastroenterol. 2011;17:4937–4944. doi: 10.3748/wjg.v17.i45.4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters LC, Cantrell VA, Weller KP, Mosher JT, Southard-Smith EM. Genetic background impacts developmental potential of enteric neural crest-derived progenitors in the Sox10Dom model of Hirschsprung disease. Hum Mol Genet. 2010;19:4353–4372. doi: 10.1093/hmg/ddq357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wattchow D, Brookes S, Murphy E, Carbone S, de Fontgalland D, Costa M. Regional variation in the neurochemical coding of the myenteric plexus of the human colon and changes in patients with slow transit constipation. Neurogastroenterol Motil. 2008;20:1298–1305. doi: 10.1111/j.1365-2982.2008.01165.x. [DOI] [PubMed] [Google Scholar]
- Weber GF, Bjerke MA, DeSimone DW. Integrins and cadherins join forces to form adhesive networks. J Cell Sci. 2011;124:1183–1193. doi: 10.1242/jcs.064618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wynn ML, Kulesa PM, Schnell S. Computational modelling of cell chain migration reveals mechanisms that sustain follow-the-leader behaviour. J R Soc Interface. 2012;9:1576–1588. doi: 10.1098/rsif.2011.0726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin M, King SK, Hutson JM, Chow CW. Multiple endocrine neoplasia type 2B diagnosed on suction rectal biopsy in infancy: a report of 2 cases. Pediatric and Developmental Pathology (Pediatr Dev Pathol) 2006 doi: 10.2350/06-05-0072.1. [DOI] [PubMed] [Google Scholar]
- Yntema CL, Hammond WS. The origin of intrinsic ganglia of trunk viscera from vagal neural crest in the chick embryo. J Comp Neurol. 1954;101:515–541. doi: 10.1002/cne.901010212. [DOI] [PubMed] [Google Scholar]
- Young HM, Bergner AJ, Anderson RB, Enomoto H, Milbrandt J, Newgreen DF, Whitington PM. Dynamics of neural crest-derived cell migration in the embryonic mouse gut. Developmental biology. 2004;270:455–473. doi: 10.1016/j.ydbio.2004.03.015. [DOI] [PubMed] [Google Scholar]
- Young HM, Bergner AJ, Muller T. Acquisition of neuronal and glial markers by neural crest-derived cells in the mouse intestine. J Comp Neurol. 2003;456:1–11. doi: 10.1002/cne.10448. [DOI] [PubMed] [Google Scholar]
- Young HM, Hearn CJ, Ciampoli D, Southwell BR, Brunet JF, Newgreen DF. A single rostrocaudal colonization of the rodent intestine by enteric neuron precursors is revealed by the expression of Phox2b, Ret, and p75 and by explants grown under the kidney capsule or in organ culture. Developmental biology. 1998;202:67–84. doi: 10.1006/dbio.1998.8987. [DOI] [PubMed] [Google Scholar]
- Young HM, Hearn CJ, Farlie PG, Canty AJ, Thomas PQ, Newgreen DF. GDNF is a chemoattractant for enteric neural cells. Developmental biology. 2001;229:503–516. doi: 10.1006/dbio.2000.0100. [DOI] [PubMed] [Google Scholar]
- Young HM, Jones BR, McKeown SJ. The projections of early enteric neurons are influenced by the direction of neural crest cell migration. J Neurosci. 2002;22:6005–6018. doi: 10.1523/JNEUROSCI.22-14-06005.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young HM, Turner KN, Bergner AJ. The location and phenotype of proliferating neural-crest-derived cells in the developing mouse gut. Cell Tissue Res. 2005;320:1–9. doi: 10.1007/s00441-004-1057-5. [DOI] [PubMed] [Google Scholar]
- Zhang D, Brinas IM, Binder BJ, Landman KA, Newgreen DF. Neural crest regionalisation for enteric nervous system formation: implications for Hirschsprung's disease and stem cell therapy. Dev Biol. 2010;339:280–294. doi: 10.1016/j.ydbio.2009.12.014. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Niswander L. Phactr4: a new integrin modulator required for directional migration of enteric neural crest cells. Cell Adh Migr. 2012;6:419–423. doi: 10.4161/cam.21266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Niswander L. Zic2 is required for enteric nervous system development and neurite outgrowth: a mouse model of enteric hyperplasia and dysplasia. Neurogastroenterol Motil. 2013 doi: 10.1111/nmo.12101. [DOI] [PMC free article] [PubMed] [Google Scholar]