

Abstract
It is well known that CpG dinucleotide steps in DNA, which are highly methylated at the 5-position of cytosine (meC) in human tissues, exhibit a disproportionate number of mutations within certain codons of the p53 gene. There is ample published evidence indicating that the reactivity of guanine with anti-B[a]PDE (a metabolite of the environmental carcinogen benzo[a]pyrene) at CpG mutation hot spots is enhanced by the methylation of the cytosine residue flanking the target guanine residue on the 5′-side. In this work we demonstrate that such a methylation can also dramatically affect the conformational characteristics of an adduct derived from the reaction of one of the two enantiomers of anti-B[a]PDE with the exocyclic amino group of guanine ([BP]G adduct). A detailed NMR study indicates that the 10R (−)-trans-anti-[BP]G adduct undergoes a transition from a minor groove-binding alignment of the aromatic BP ring system in the unmethylated C-[BP]G sequence context, to an intercalative BP alignment with a concomitant displacement of the modified guanine residue into the minor groove in the methylated meC-[BP]G sequence context. By contrast, a minor groove-binding alignment was observed for the stereoisomeric 10S (+)-trans-anti-[BP]G adduct in both the C-[BP]G and meC-[BP]G sequence contexts. This remarkable conformational switch resulting from the presence of a single methyl group at the 5-position of the cytosine residue flanking the lesion on the 5′-side, is attributed to the hydrophobic effect of the methyl group that can stabilize intercalated adduct conformations in an adduct stereochemistry-dependent manner. Such conformational differences in methylated and unmethylated CpG sequences may be significant because of potential alterations in the cellular processing of the [BP]G adducts by DNA transcription, replication, and repair enzymes.
Keywords: conformational switch, cytosine methylation, p53 mutation hot spot, DNA adduct, benzo[a]pyrene, B[a]PDE
Introduction
Benzo[a]pyrene (B[a]P) and structurally related polycyclic aromatic hydrocarbon (PAH) environmental pollutants are metabolically activated in cellular environments to highly reactive and genotoxic diol epoxide derivatives.1 The biologically most important diol epoxide derivative of B[a]P is r7, t8-dihydroxy-t9,10-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene (anti-B[a]PDE) in which the 7-OH and 9,10-epoxide groups are oriented on opposite sides of the planar, pyrene-like aromatic ring system. The major covalent DNA adducts derived from the reactions of B[a]PDE with cellular DNA were first identified more than 25 years ago.2,3 In vitro studies of the reactions of the (+)-7R,8S,9S,10R and the (−)-7S,8R,9R,10S enantiomers of anti-B[a]PDE with native DNA have identified a number of well-defined covalent adducts with G, A, and C in varying proportions.4–6 The dominant mode of covalent adduct formation occurs via reactions of the 10-position of B[a]PDE with the N2-position of guanine, and are designated here as the [BP]G adducts. The most tumorigenic diol epoxide derivative of B[a]P is the (+)-7R,8S,9S,10R enantiomer.7,8 However, both enantiomers are mutagenic,9,10 as shown explicitly by site-specific mutagenesis experiments in vivo with stereochemically defined N2-dG adducts derived from both the (+)-7R,8S,9S,10R and (−)-7S,8R,9R,10S anti-B[a]PDE enantiomers.11–16 In general, the mutagenic properties of the stereo-isomeric [BP]G adducts depends not only on their absolute configurations, but also on the base sequence context and the cell system in which the modified DNA is replicated.12,16
Base sequence context effects are particularly striking in the case of the well-known mutation spectra in the p53 tumor suppressor gene associated with human lung tumors obtained from cigarette smokers.17–22 These mutation hot spots occur at hypermethylated CpG islands at codons 157, 248, and 273 of the p53 gene. It has been proposed that these hot spots arise because the reactivities of B[a]PDE with guanine are known to be greater in methylated meCpG than in unmethylated CpG sequence contexts.17,21,23–26
Another factor that may contribute to the mutagenic properties of [BP]G adducts in methylated meCpG sequences, are potential effects of the methyl group in 5-methylcytosine flanking the lesions on the 5′-side. We have previously shown that two pairs of distinct, stereochemically related adduct conformations are possible when (+)- and (−)-anti-B[a]PDE react with the exocyclic amino group of guanine in DNA.27 Covalent adduct formation can occur by either trans or cis addition of N2-dG to the C10 atom of B[a]PDE. In the cis adducts, the dG moiety is positioned on the same side (relative to the plane of the aromatic pyrene-like ring system) as the 9-OH group, and in the trans adducts it is positioned on the opposite side (Figure 1). The 10S (+)- and 10R (−)-trans-anti adducts are aligned with the bulky pyrenyl ring system in the minor groove, pointing either towards the 5′-end of the modified strand (the (+)-trans-adduct), or towards the 3′-end (the (−)-trans-adduct).28,29 Both the 10S (−)-cis- and the 10R (+)-cis-anti adducts are intercalated, but with the modified guanine residue displaced into the major and minor grooves, respectively, and with the partner dC group in the complementary strand displaced towards the major groove in both cases.30,31 Specifically, we hypothesized earlier that the 10S (+)-trans- and (−)-cis-adduct conformers, on the one hand, and the 10R (−)-trans- and 10R (+)-cis-adduct conformations, on the other, can interconvert because they have the identical absolute configurations about the C10-N2-dG bond, and that the equilibrium between these two conformations could depend on the base sequence context.27 The possibility that a methylated cytosine residue flanking a [BP]G adduct on its 5′-side in a meC-[BP]G sequence context could also influence its conformation or structural characteristics aroused our interest.23,32 Weisenberger & Romano found that methylation of a cytosine residue flanking a 10S (+)-trans-anti-[BP]G adduct changes its electrophoretic mobility and, therefore, its structural properties;23 however, the conformations of the adducts were not determined. Utilizing low-resolution optical spectroscopic techniques, we showed that the 10R (−)-trans-anti-[BP]G adduct (G*) changed from a minor groove adduct conformation32 in the sequence context (5′-CCATCG*CTACC)·(5′-GGTAGCGATGG) to a carcinogen-base stacked conformation, when the C flanking the lesion on the 5′-side was replaced by meC.32 In the present work we used NMR methods to establish that the presence of a single methyl group at the 5-position of cytosine flanking the B[a]PDE-modified guanine, G*, switches the conformation of the 10R (−)-trans-anti-[BP]G adduct from a minor groove to a base-displaced intercalative alignment. In contrast, the stereoisomeric 10S (+)-trans-anti-[BP]G adduct maintains its minor groove conformation in both C-[BP]G-C and meC-[BP]G-C sequence contexts. There are no changes in either of these 10R or 10S adduct conformations when the methyl group is positioned on the 3′-flanking cytosine residue in C-[BP]G-meC sequence contexts. The unique conformational switch in the case of the 10R (−)-trans adduct may be understood in terms of a favorable adduct conformation-dependent hydrophobic interaction of the methyl group in the 5′-flanking 5-methyl-cytosine residue with the pyrenyl aromatic ring system in double-stranded DNA.
Figure 1.

(a)–(d) Absolute configurations of stereoisomeric [BP]G adducts, (e) sequence of the [BP]G·C 11-mer duplex, and (f) structure of the 10R (−)-trans-anti-[BP]-N2-dG adduct ((−)-trans-anti [BP]G). In (e), the arrows point to either of the two cytosine residues that were replaced (singly) by 5-methylcytosine in the 11-mer sequence, the methylated sequences being denoted either by meC-[BP]G-C or by C-[BP]G-meC.
Results
Exchangeable proton spectra
The one-dimensional NMR spectra (7.5–15 ppm) of the 10R (−)-trans-anti-[BP]G adduct in the C-[BP]G-C (a) and the meC-[BP]G-C (b) sequences at the [BP]G·C 11-mer duplex level in H2O buffer solution at 0 °C are depicted in Figure 2. The numbering scheme is defined in Figure 1. The imino proton assignments are shown in (b) and were determined as described for the same sequence context in earlier publications.28–31,33 The partially resolved imino protons resonating between 12.5 ppm and 14 ppm are characteristic of Watson–Crick base-pairing in these 11-mer duplexes. There are dramatic differences in the imino proton spectra on proceeding from the C-[BP]G-C (a) to the meC-[BP]G-C (b) sequence context. The patterns resemble those of the 10R (+)-cis-anti-[BP]G adduct. Three well-resolved imino protons at 12.12 ppm, 11.23 ppm, and 10.78 ppm, assigned to the adduct site, namely [BP]G6 and the flanking G16 and G18, shift upfield following methylation at position 5 of the pyrimidine ring located on the 5′-side of the [BP]G adduct. Such upfield imino proton shifts (b) are characteristic of intercalation of the pyrenyl ring into the helix in the meC-[BP]G-C (b) sequence context. The lack of upfield-shifted imino protons in the unmethylated C-[BP]G-C sequence context (a) is in agreement with the previous results and minor groove structural assignment of the same 10R (−)-trans-anti-[BP]G adduct in the same duplex reported by de los Santos et al.29 However, the presence of the 5-methyl group at the C flanking the same adduct on the 5′-side is unusual and resembles the imino spectrum of the stereoisomeric 10R (+)-cis-adduct in the same unmethylated 11-mer duplex that has a base-displaced intercalated conformation.30
Figure 2.
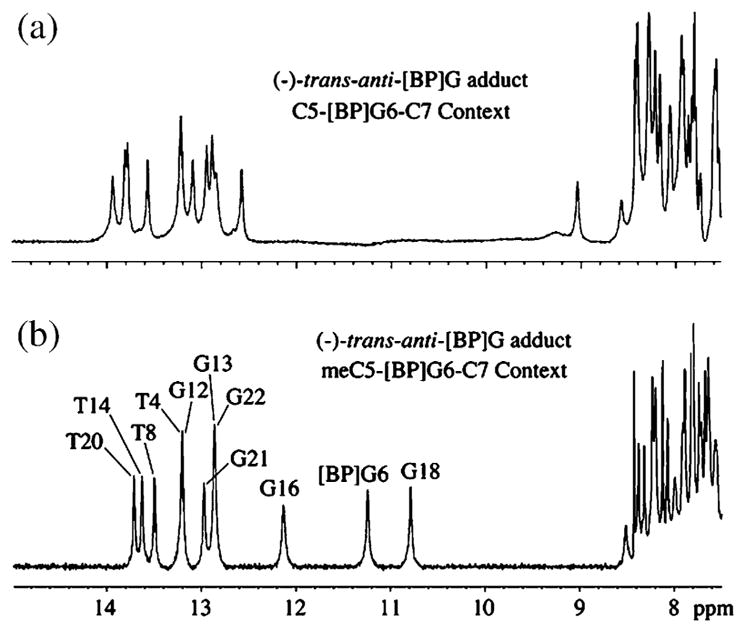
Imino proton NMR spectra (7.5–15 ppm) of the 10R-(−)-trans-anti-[BP]G adduct in (a) the C-[BP]G-C and (b) meC-[BP]G-C sequence contexts at the 11-mer duplex level. Spectra are in 100 mM NaCl, 10 mM phosphate, H2O, pH 6.8 at 0 °C. The imino proton assignments are shown in (b).
Expanded regions of the NOESY contour plot (200 ms mixing time) for the 10R (−)-trans-anti-[BP]G adduct in the meC-[BP]G-C sequence context at the duplex level in H2O buffer at 0 °C are shown in Figure 3. The imino and amino protons have been assigned as described elsewhere.34,35 The NOE patterns between different imino protons are identified in (a) and are labeled A to H, and (b) identifies NOEs between imino protons (10.5–14.0 ppm) and amino and non-exchangeable protons (3.5 ppm to 8.5 ppm). The NOE cross-peaks between DNA protons are labeled A to F, and NOE cross-peaks between aromatic BP protons and DNA protons are labeled 1–11. There are no cross-peaks between the G6 imino and the meC5 methyl protons positioned in the major groove. However, the observation of cross-peak 5 between the G6 imino and the meC5-H1′ protons positioned in the minor groove, indicates that the modified G6 residue is displaced into the minor groove and is aligned towards the 5′-direction of the modified strand. Imino–imino NOE connectivities are observed between all adjacent base-pairs on either side of the adduct (Figure 3). However, NOE connectivities are not observed between the imino proton of the modified dG6 and the imino proton of the adjacent dG16, while only a weak NOE connectivity is observed between the imino proton of dG6 and the imino proton of the adjacent dG18 (G in (a)). In (b), the observed NOE patterns establish Watson–Crick base-pairing at all dA·dT pairs (deoxythymidine imino to deoxyadenosine H2 across the dA·dT pairs) and at all dG·dC pairs (deoxyguanosine imino to deoxycytidine amino across the dG·dC pairs), as shown for the dC5·dG18 and dC7·dG16 pairs (peaks B,B′ and E,E′, respectively), with the exception of the alignment of the [BP]dG6·dC17 pair at the modification site (b). We conclude that hydrogen-bonded base-pairing at [BP]dG6·dC17 does not occur, since, unlike the other paired G·C patterns, there are no cross-peaks between imino protons of [BP]dG6 and amino protons of dC17. Cross-peak A is assigned to the dG6 imino to dG6 amino connectivity, while cross-peaks B,B′ and E,E′ are attributed to the dG18 imino to dC5 amino and the dG16 imino and the dC7 amino cross-peaks, respectively. The dC5 and dC7 amino protons are significantly shifted upfield relative to their normal values, suggesting significant ring current effects due to a nearby aromatic ring system. These results establish formation of stable dC5·dG18 and dC7·dG16 base-pairs on either side of the [BP]dG6 lesion positioned opposite dC. Other cross-peaks shown are those between the G18 imino and G18 amino protons (C), the G18 imino and A19H2 (D), and the G16 imino and A15H2 protons (F).
Figure 3.

Expanded NOESY (200 ms mixing time) contour plots for the 10R-(−)-trans-anti-[BP]G adduct in the meC-[BP]G-C sequence context at the 11-mer duplex level. Spectra are in 100 mM NaCl, 10 mM phosphate, H2O, pH 6.8 at 0 °C. (a) NOEs between imino protons, with the cross-peaks labeled A to H, which are assigned as follows: A, T4(NH3)-G18(NH1); B, T8(NH3)-G16(NH1); C, T14(NH3)-G13(NH1); D, T20(NH3)-G21(NH1); E, T4(NH3)-T20(NH3); F, T8(NH3)-T14(NH3); G, [BP]G6(NH1)-G18(NH1); H, G21(NH1)-G22(NH1). (b) NOEs between imino protons (10.5–14.0 ppm) of dG16, [BP]dG6, and dG18, and amino and non-exchangeable protons (3.5–8.5 ppm), with NOE cross-peaks between DNA protons labeled A to F and NOE cross-peaks between carcinogen and DNA protons labeled 1–11. Cross-peaks A to F are assigned as follows: A, [BP]G6(NH1)-[BP]G6(NH2)/BP(H10); B,B′, G18(NH1)-[Me]C5(NH2); C, G18(NH1)-G18(NH2); D, G18(NH1)-A19(H2); E,E′, G16(NH1)-C7(NH2); F, G16(NH1)-A15(H2). Cross-peaks 1–10 are assigned as follows: 1, G16(NH1)-BP(H4,H5); 2, G16(NH1)-BP(H6); 3, G16(NH1)-BP(H3); 4, G16(NH1)-BP(H1,H2); 5, [BP]G6(NH1)-[Me]C5(H1′); 6, [BP]G6(NH1)-BP(H11); 7, G18(NH1)-[BP]G6(NH2)/BP(H10); 8, G18(NH1)-BP(H4,H5); 9, G18(NH1)-BP(H6); 10, G18(NH1)-BP(H3); 11, [BP]G6(NH1)-BP(H9).
Non-exchangeable protons
The one-dimensional base and sugar H1′ proton NMR spectra (5.0–8.5 ppm) of the 10R (−)-trans-anti-[BP]G adduct in the C-[BP]G-C and meC-[BP]G-C sequence contexts in the 11-mer duplexes in 2H2O phosphate buffer solution at 0 °C are compared in Figure S1 (Supplementary Data). There are dramatic upfield benzo[a]pyrenyl ring proton shifts in the meC-[BP]G-C sequence context that are not observed in the C-[BP]G-C sequence. The upfield benzo[a]pyrenyl ring protons are characteristic of intercalation of the pyrenyl ring into the helix in the meC-[BP]G-C sequence context.
The observed patterns of chemical shifts (Figure S1, Supplementary Data), NOEs, and upfield shifts of the dG18, dG6, and dG16 imino and meC5 and C7 amino protons (Figure 3), are similar to those observed in the case of the stereoisomeric 10R (+)-cis-anti-[BP]G adduct in the identical, but unmethylated 11-mer duplex.30 This adduct is characterized by a base-displaced intercalated conformation. Our observations for the central d(-meC5-[BP]G6-C7-)·d(-G16-C17-G18-) segment of the 11-mer duplex for the 10R (−)-trans-[BP]G adduct suggests that the aromatic ring system of the BP residue is also intercalated between the intact meC5·G18 and the C7·G16 Watson–Crick base-pairs. This is in sharp contrast to the external, minor groove alignment of the aromatic BP ring system of the same 10R (−)-trans-anti-adduct in the unmethylated 11-mer duplex.29
Lack of NOE connectivities at the site of the lesion
Expanded NOESY (250 ms mixing time) contour plots for the 10R-(−)-trans-anti-[BP]G adduct in the meC-[BP]G-C-sequence context at the 11-mer duplex level in 2H2O buffer solution at 0 °C are depicted in Figure S2 (Supplementary Data). From this figure, it is evident that the NOE between the H8 proton of [BP]G6 and the H1′ proton of the 5′-flanking dC5, and the H6 proton of dC7 and the H1′ proton of [BP]G6, are very weak. In the unmodified strand, the NOEs between the H8 proton of G18 and the H1′ proton of C17, and the H6 proton of C17 and the H1′ proton of G16 are also shown to be quite weak. The weak inter-residue connectivities within the meC5-[BP]G6-C7 segment of the modified strand and within the G16-C17-G18 segment of the unmodified strand are consistent with the disruption of stacking of [BP]G6 and C17 with their flanking dC5·dG18 and C7·G16 base-pairs for the 10R-(−)-trans-anti-[BP]G adduct in the meC-[BP]G-C sequence context at the 11-mer duplex level. The base H5 (6.02 ppm), the H6 proton (7.87 ppm), and the H1′ sugar protons of C17 (6.20 ppm) exhibit the greatest downfield shifts among the cytosine residues; this is indicative of a loss of stacking interactions and thus a strong indication of the displacement of C17 out of the duplex. On the other hand, the H5 (4.86 ppm), H6 (7.25 ppm) and H1′(4.47 ppm) protons of C7 and H6 (6.70 ppm), H1′ (5.27 ppm) of meC5 are upfield shifted due to ring current effects, which is consistent with an intercalation of the aromatic pyrenyl ring system between the two base-pairs G18·meC5 and G16·C7 (See Supporting Information for details).
Benzo[a]pyrenyl proton connectivities and chemical shifts
Expanded NOESY (250 ms mixing time) contour plots for the 10R-(−)-trans-anti-[BP]G adduct in the meC-[BP]G-C sequence context at the 11-mer duplex level are shown in Figure 4. The cross-peaks correspond to NOEs from the numbered benzo[a]pyrenyl protons to methyl and sugar H2′,2′′ protons (a) and to cytosine H5 and sugar H1′ protons (b). The BP proton numbering scheme is shown in Figure 1. The well-resolved NOEs between the benzo[a]pyrenyl protons and DNA protons within the (meC5-[BP]G6-C7)·(G16-C17-G18) segment have been identified and are given in Table 1 (m, medium NOEs; w, weak NOEs). The distribution of these carcinogen–DNA NOEs is sufficient to define the alignment of both the benzo[a]pyrenyl ring and the modified guanine residue centered about the (meC5-[BP]G6-C7)·(G16-C17-G18) segment.
Figure 4.

Expanded NOESY (250 ms mixing time) contour plots for the 10R-(−)-trans-anti-[BP]G adduct in the meC-[BP]G-C sequence context at the 11-mer duplex level. Spectra are in 100 mM NaCl, 10 mM phosphate, 2H2O, pH 6.8 at 0 °C. The numbered cross-peaks correspond to NOEs from benzo[a]pyrenyl protons to methyl and sugar H2′,2′′ protons (a) and to cytosine H5 and sugar H1′ protons (b). In (a), the cross-peaks 1–7 are assigned as follows: 1, BP(H1,H2)-[Me]C5(CH3); 2, BP(H12)-[Me]C5(CH3); 3, BP(H3)-[Me]C5(CH3); 4, BP(H1,H2)-[Me]C5(H2′ ); 5, BP(H12)-[Me]C5(H2′); 6, BP(H11)-[BP]G6(H2′′); 7, BP(H12)-[BP]G6(H2′′). In (b), the cross-peaks 1–9 are assigned as follows: 1, BP(H11)-C7(H1′); 2, BP(H12)-C7(H1′ ); 3, BP(H11)-C7(H5); 4, BP(H1,H2)-C7(H5); 5, BP(H12)-C7(H5); 6, BP(H11)-[Me]C5(H1′); 7, BP(H1,H2)-[Me]C5(H1′ ); 8, BP(H12)-[Me]C5(H1′ ); 9, BP(H6)-G18(H1′).
Table 1.
Intermolecular NOEs in the 10S (−)-trans-anti-[BP]G adduct in the meC-[BP]G-C sequence context in the 11-mer duplex
| 10R (K)-trans-anti-[BP]G | Intermolecular NOEs |
|---|---|
| H1 (7.63 ppm) | MeC5(H6)[m], MeC5(H1′)[w], MeC5(H2′)[m], MeC5(H2′′)[w], MeC5(CH3)[m], G6(H2′′)[w], C7(H5)[m], C7(H6)[w], G16(H1)[m] |
| H2 (7.63 ppm) | MeC5(NH2)[m], MeC5(H6)[m], MeC5(CH3)[m], C7(NH2)[m], G16(H1)[m] |
| H3 (7.40 ppm) | MeC5(NH2)[m], MeC5(CH3)[w], G16(H1)[m], G18(H1)[w] |
| H4 (6.79 ppm) | G16(H1)[m], G18(H1)[w] |
| H5 (6.74 ppm) | G16(H1)[w], G18(H1)[w] |
| H6 (7.32 ppm) | T8(H4′)[m], G16(H1)[w], G18(H1)[m], G18(H1′)[m] |
| H7 (5.14 ppm) | C7(H1′)[m], T8(H4′)[m] |
| H8 (4.36 ppm) | A19(H4′ ′)[m] |
| H9 (4.48 ppm) | G6(H1)[m], G6(H1′)[w] |
| H10 (6.48 ppm) | G18(H1)[w], G6(H1)[m], G6(H2′′), C7(H1′)[m], C7(H4′)[m] |
| H11 (7.96 ppm) | MeC5(H1′)[m], MeC5(H1′′)[m], G6(H1)[m], C7(H5)[w], C7(H1′)[m] |
| H12 (7.48 ppm) | MeC5(H1′)[m], MeC5(H2′)[m], MeC5(H2′′′)[m], MeC5(H6)[m], MeC5(CH3)[w], G6(H2′)[m], G6(H2′′)[m], C7(H6)[m], C7(H5)[m], C7(H1′)[w] |
| NH (11.23 ppm) | MeC5(H1′)[m] |
The chemical shifts of the adduct were recorded at 0 °C in H2O and 2H2O buffered solutions.
The chemical shifts of the aliphatic (H7, H8, H9 and H10) and aromatic (H1, H2, H3, H4, H5, H6, H11 and H12) BP protons between the 10R-(−)-trans-anti-[BP]G adduct in the C-[BP]G-C and the meC-[BP]G-C at the 11-mer duplex level are compared in Figure S3 (Supplementary Data). Analogous chemical shift comparisons between the 10R-(+)-cis-anti-[BP]G adduct in the C-[BP]G-C sequence context and the stereoisomeric 10R-(−)-trans-anti-[BP]G adduct in the meC-[BP]G-C sequence context at the 11-mer duplex level are also compared in Figure S3(a). The aromatic BP protons in the 10R-(−)-trans-anti-[BP]G adduct in the meC-[BP]G-C sequence are shifted upfield by as much as 1.5–1.6 ppm as compared to the same protons in the C-[BP]G-C. These upfield shifts are consistent with intercalation of the benzo[a]pyrenyl ring system into the helix in the meC-[BP]G-C sequence, and groove-binding alignment of the benzo[a]pyrenyl ring system in the C-[BP]G-C sequence. As shown in Figure S3(b), the aromatic BP protons of the 10R-(+)-cis-anti-[BP]G adduct in the C-[BP]G-C sequence context, and the 10R-(−)-trans-anti-[BP]G adduct in the meC-[BP]G-C sequence context are practically superimposable on one another. These similarities in the chemical shifts indicate that the intercalation with base displacement topology defined for the (+)-cis adduct in the unmethylated C-[BP]G-C sequence context, is most likely also adopted by the stereo-isomeric (−)-trans adduct in the methylated meC-[BP]-G-C sequence context.
Stereochemical and 5meC positional effects on adduct conformations
We have evaluated the effects of methylation of C5 in the meC-[BP]G-C sequence context at the 11-mer duplex level in H2O at 0 °C on the conformation of the stereoisomeric 10S (+)-trans-anti-[BP]G adduct. In the identical, but unmethylated sequence, the aromatic BP ring system is positioned in the minor groove and points towards the 5′-end of the modified strand.27 The imino proton NMR spectrum (7.5–15 ppm) of the S-(+)-trans-anti-[BP]G adduct (a) and of the 10R-(−)-trans-anti-[BP]G adduct (b) in the identical meC-[BP]G-C sequence context are compared in Figure 5. There are no upfield-shifted imino proton resonances in the case of the 10S (+)-trans-anti-adduct as there are in the 10R (−)-trans-anti-[BP]G adduct in the same methylated meC-[BP]G-C sequence context. These imino proton spectra clearly establish that intercalation with base displacement is observed only in the case of the 10R-(−)-trans-anti-[BP]G adduct in the meC-[BP]G-C sequence context (Figure 5(b)), while the 10S-(+)-trans-anti-[BP]G adduct in the meC-[BP]G-C sequence context (Figure 5(a)) adopts the groove binding alignment with intact modified base pair alignment.
Figure 5.

Imino proton NMR spectra (7.5–15 ppm) of (a) the 10S-(+)-trans-anti-[BP]G adduct and (b) the 10R-(−)-trans-anti-[BP]G adduct in the meC-[BP]G-C sequence context and (c) the 10S-(+)-trans-anti-[BP]G adduct and (d) the 10R-(−)-trans-anti-[BP]G adduct in the C-[BP]G-meC sequence context at the 11-mer duplex level. Spectra are in 100 mM NaCl, 10 mM phosphate, H2O, pH 6.8 at 0 °C. The assignments of the upfield shifted imino protons are made in (b).
We have also investigated the effect of methylation of cytosine on adduct conformations of the same 10R (−)-trans-anti-[BP]G and 10S (+)-trans-anti-[BP]G adducts in the C-[BP]G-meC sequence context in which the position of the methylated cytosine is switched from the 5′ to the 3′-cytosine flanking the adduct in the modified strand at the 11-mer duplex level. No upfield imino proton shifts are observed in either case, indicating that methylation of the 3′-flanking cytosine residue does not cause a switch from the normal minor groove conformation in the C-[BP]G-C sequence to a base-displaced conformation in the C-[BP]G-meC sequence context in the 11-mer duplexes.
Comparison of NMR characteristics of 10R (−)-trans-anti-[BP]G adduct in the meC-[BP]G-C sequence with 10R (+)-cis-anti-[BP]G adduct in the unmethylated C-[BP]G-C sequence
Similar sequential walking patterns and chemical shifts of the two adducts (Figure S2, Supplementary Data, and Figure 3(a)30) are evidence for their similar base-displaced intercalative alignments. However, chemical shift differences between C5H6 (7.0 ppm) and meC5H6 (6.7 ppm) stem from the presence of the methyl group in the (−)-trans-anti-[BP]G adduct in the meCpG* sequence context. Evidence for the presence of the methyl group in the 10R (−)-trans-anti-[BP]G adduct is also seen from the NOEs between H6/H8 and the methyl proton region, showing connections between the methyl group of meC5 and the methyl group of thymine (Figure S4(a), Supplementary Data). Also, Figure S4(b) shows the through-bond connections between the H5 and H6 protons of all cytosine residues, and that there is no through-bond connection for the meC5 residue.
While the NMR characteristics of the (−)-trans-anti-[BP]G adduct in the meC-[BP]G-C sequence context are very similar to those of the stereo-isomeric (+)-cis-anti-[BP]G adduct in the unmethylated C-[BP]G-C sequence context,30 there are also differences. The similarity is not surprising, since the (−)-trans and (+)-cis adducts are identical with one another, with R absolute configurations at the C10 carbon atom, but with opposite configurations of the H atoms at the benzylic carbon atoms C7, C8, and C9 (Figure 1). The model for the (−)-trans-anti-[BP]G adduct in the meC-[BP]G-C sequence context (see below) indicates that the H9 atom should be close to the G6 imino proton, while the H7 proton should be close to the C7H1′ sugar proton (less than 4 Å). Indeed, at short mixing times (50~100 ms), we observe two corresponding NOEs, indicating that the distances between the H9/G6NH1 and H7/C7H1′ protons are 3.9 Å and 3.6 Å, respectively (data not shown). These NOEs are completely consistent with the (−)-trans adduct stereochemistry. Inverting the absolute configurations of the hydrogen atoms at C9 and C7 to yield the (+)-cis adduct would increase the distance between these same protons to distances in excess of ~5.2 Å. Thus, in contrast to our observations, the corresponding NOEs would be extremely weak or unobservable in the case of a (+)-cis adduct stereochemistry.
Molecular mechanics computations
Molecular mechanics methods, followed by distance and intensity-restrained molecular dynamics computations to determine the solution structure of the 10R (−)-trans-anti-[BP]G adduct in the meC-[BP]G-C sequence context at the 11-mer duplex level were performed. The restraints and refinement statistics are listed in Table 2. Stereo views of nine superpositioned intensity-refined structures of the 10R-(−)-trans-anti-[BP]G adduct in the meC-[BP]G-C sequence context at the 11-mer duplex level are shown in Figure 6. These structures are well superimposed on one another (r.m.s.d. 1.37(±0.40) Å for the entire molecule), including the (T4-meC5-[BP]G6-C7-T8)·(A15-G16-C17-G18-A19) segment (r.m.s.d. 0.83(±0.26) Å).
Table 2.
NMR refinement statistics for the 10S (−)-trans-anti-[BP]G adduct in the meC-[BP]G-C sequence context of the 11-mer duplex
| A. NMR distance restraints | |
| Total number of DNA distance restraints | 562 |
| Exchangeable distance restraints | 84 |
| Non-exchangeable distance restraints | 438 |
| Hydrogen bond restraints | 40 |
| Total number of carcinogen intramolecular distance restraints | 21 |
| Total number of carcinogen intermolecular distance restraints | 51 |
| Exchangeable distance restraints | 17 |
| Non-exchangeable distance restraints | 34 |
| B. Structural statistics of the adduct | |
| NOE violations | |
| Number O0.2 Å | 4.3G1.1 |
| Maximum violations (Å) | 0.26G0.14 |
| r.m.s.d. of violations | 0.031G0.002 |
| Deviation from the ideal covalent geometry | |
| Bond lengths (Å) | 0.012G0.001 |
| Bond angles (deg.) | 2.85G0.03 |
| Impropers (deg.) | 0.37G0.04 |
| C. Pair-wise r.m.s.d. (Å) (nine intensity-refined structures) | |
| Full 11-mer adduct duplex | 1.37G0.40 |
| Central 5-mer adduct duplex | 0.83G0.26 |
Figure 6.

Stereo views of nine superpositioned intensity-refined structures of the 10R-(−)-trans-anti-[BP]G adduct in the meC-[BP]G-C sequence context at the 11-mer duplex level. The benzo[a]pyrenyl (BP) ring is in yellow, the modified guanine and its opposing cytosine are in cyan, the methyl group of meC5 is in green.
Stereo views of a representative intensity-refined structure of the 10R-(−)-trans-anti-[BP]G adduct centered within the (meC5-[BP]G6-C7)·(G16-C17-G18) segment are shown in Figure 7. Aview looking into the minor groove is shown in (a), while a view looking down the helix axis is shown in (b). The 10R-(−)-trans-anti-[BP]G adduct centered within the (meC5-[BP]G6-C7)·(G16-C17-G18) segment adopts an intercalation with base displacement architecture centered about the lesion site. The guanine base is positioned in the minor groove approximately parallel with the helix axis and is stacked over the sugar ring of meC5, while C17 is displaced into the major groove (a). The benzo[a]-pyrenyl ring intercalates from the minor groove edge and is sandwiched between the flanking meC5·G18 and C7·G16 pairs (a). The aromatic part of the benzo[a]pyrenyl ring stacks primarily with the meC5 and C7 bases, to a lesser extent with G18, and not at all with G16 (b).
Figure 7.

Stereo views of a representative intensity-refined structure of the 10R (−)-trans-anti-[BP]G adduct centered within the (meC5-[BP]G6-C7)·(G16-C17-G18) segment. A view looking into the minor groove is shown in (a), while a view looking down the helix axis is shown in (b). The benzo[a]pyrene (BP) ring is in yellow, the modified guanine residue and its opposing cytosine residue are in cyan, the methyl group of meC5 is in green, the meC5·G18 pair is in white and the C7·G16 pair is in grey.
Discussion
Our NMR studies establish that the 10R-(−)-trans-anti-[BP]G adduct opposite C in the meC-[BP]G-C sequence context adopts the intercalation with base displacement type alignment, in contrast to a minor groove alignment with an intact modified base-pair for the same adduct in the C-[BP]G-C sequence context. Thus, cytosine methylation in the C-[BP]G-C step of the cytosine residue flanking the adduct on the 5′-side, but not the 3′-side, has a profound effect on the alignment of the covalently attached BP ring. In contrast, the stereo-isomeric 10S (+)-trans-anti-[BP]G adduct maintains its predominant minor groove conformation in both the methylated and unmethylated sequence contexts when one or the other of the two flanking cytosine residues is methylated.
Methylated cytosine residues at CpG dinucleotide steps play an important role in regulating transcription and thus the control of gene expression in mammalian cells.36,37 The impact of the methyl group at the 5-position of cytosine on the characteristics of double-stranded DNA have been studied by a variety of approaches.38–42 Cyclization of ligated mixed-sequence oligonucleotides followed by analysis of electrophoretic mobilities showed that the changes in helical twist or intrinsic flexibility are not altered significantly by cytosine methylation.40 However, in the case of an EcoRI decamer, methylation of the cytosine residue at the CpG dinucleotide steps lowers flexibility by ~30% and causes an underwinding by ~0.5 base-pair per turn.43 The dynamics and local conformational flexibility were also found to be inhibited upon methylation of a CpG step in the HhaI methyltransferase recognition sequence using solid-state NMR methods.44 A molecular dynamics simulation study showed that methylation of CpG indeed reduces the flexibility of double-stranded DNA, attributed to a restriction of the conformational space associated with the bulky and hydrophobic methyl group.45 Sowers et al.38 reported that the major effect of the methyl group in meC is an increase in the polarizability of the pyrimidine that results in increased hydrophobic base-stacking interactions and, in turn, a thermal stabilization of double-stranded DNA. A recent molecular dynamics study of the effects of cytosine methylation in a CpG sequence context suggests that the thermodynamic equilibrium between the BI and BII phosphate backbone conformational states is influenced by the presence of the methyl group, thus thermodynamically leading to an increased base-stacking energy,46 as reported earlier.38 An experimental FTIR study of a different double-stranded CpG oligonucleotide sequence context was interpreted in terms of a different alteration of the BI/BII equilibrium, an enhanced contribution of the C2′-endo sugar conformation, and a change in the glycosidic torsion angle upon methylation.47 X-ray diffraction studies of crystal structures of two identical oligonucleotides differing only by replacement of a single C by meC also concluded that the effects of the methyl group on helical parameters are generally small and are mostly limited to the affected base-pair; the major effect being a displacement of the meC·G base-pair towards the minor groove of the helix.41 Finally, structural analysis by solution NMR and molecular modeling methods shows that replacement of cytosine by meC in a 12-mer oligonucleotide duplex with a single, central CpG dinucleotide step gives rise to a local narrowing of the minor groove, while the major groove becomes more shallow.42 In summary, the reported physical and structural differences in CpG and meCpG sequence contexts do not provide any simple explanations for the unusual conformational switch of the 10R (−)-trans-anti-[BP]G adduct when the cytosine residue flanking the adduct is replaced by 5-methylcytosine in the 11-mer duplex studied in this work.
While the influence of the 5-methyl group in cytosine in a meC-G sequence context on the physical characteristics of double-stranded DNA are not large, it can have a substantial impact on (1) the reactivity of the guanine residue in the meC-G sequence context with anti-B[a]PDE, and (2) a dramatic impact on the mutagenic properties associated with the covalent [BP]G adducts formed. A disproportionate fraction of G→T transversion mutations are observed at methylated CpG sequences in the p53 gene derived from lung tumors of smokers as compared to tumors derived from non-smokers.17,20,48 Different mutation characteristics in this gene have been linked to molecular events arising from the reactions of various classes of carcinogens, including anti-B[a]PDE, with the nucleobases in the p53 gene.49,50 Indeed, the occurrence of mutation hot spots in codons 157, 248, and 273 have been associated with an enhanced reactivity of PAH metabolites such as B[a]PDE.17,20,21,51,52 While this view has been challenged,53,54 there is accumulating in vitro evidence that the methylation of cytosine in CpG dinucleotide steps indeed enhances the reactivity of B[a]PDE with the guanine residue in meCpG sequence contexts.23,24,26,55,56 This effect is most likely associated with the hydrophobicity of the methyl group that enhances both the non-covalent intercalative binding of B[a]PDE to double-stranded poly(dG-medC)·poly(dG-medC) and the subsequent yield of covalent [BP]G adduct formation55,57 as observed in p53 oligonucleotide sequence17,20,21,23,24,26 and other sequence contexts.25
Hydrophobic effects on conformational switching of stereoisomeric (+)- and (−)-trans-anti-[BP]G adduct
While the 10R (−)-trans-anti-[BP]G adduct readily switches from a minor groove alignment in the C-[BP]G-C to a base-displaced intercalative conformation in the meC-[BP]G-C sequence context at the 11-mer duplex level, such a switch was not observed by NMR in the case of the stereoisomeric 10S (+)-trans-anti-[BP]G adduct (Figure 5).
An explanation of these observations can be proposed on the basis of the adduct stereochemistry-dependent opposite orientation of the pyrenyl ring system in the base-displaced intercalative conformations of the stereoisomeric trans-anti-[BP]G adduct pairs with 10R but not 10S absolute configurations. In the 10R (−)-trans-anti-[BP]G adduct, the methyl group of the 5′-neighboring cytosine is positioned in the immediate vicinity of the aromatic pyrenyl ring system. However, in the putative 10S (+)-trans-anti-[BP]G base-displaced intercalated adduct conformation, the methyl group would be positioned away from the aromatic ring system, and instead would be adjacent to the non-aromatic, puckered benzylic ring (Figure 8). Thus, the models suggest that this placement of the methyl group would afford less hydrophobic stabilization of the base-displaced intercalative conformation; consequently, the minor groove orientation continues to be favored in the meC[BP]G sequence context as it is in the C[BP]G sequence context.28
Figure 8.

Base-displaced intercalative conformations. (a) Representative NMR structure of the 10R (−)-trans-anti-[BP]G adduct, and (b) a modeled 10S (+)-trans-anti-[BP]G adduct (see the text), all in the (meC5-[BP]G6-C7)·(G16-C17-G18) sequence context. The selectively displayed protons are highlighted as white spheres, the carbon atoms of benzo[a]pyrene (BP) ring are highlighted as yellow spheres, the modified guanine residue and its opposing cytosine residue are shown as stick models (cyan), the carbon atom of the methyl group of the meC5 residue is shown as a green sphere, the oxygen atoms of the benzylic hydroxyl groups are shown as orange spheres.
The close proximity of the C5 methyl group protons to the pyrenyl aromatic ring system is supported by the observation of NOEs between these –CH3 protons and aromatic ring protons BPH1 and BPH2 (peak 1, Figure 4(a)), BPH12 (peak 2), and BPH3 (peak 3). These NOE cross-peaks suggest that the aromatic, rather than the aliphatic part of the B[a]PDE residue in the 10R (−)-trans adduct is in close proximity to the methyl group in the meC-[BP]G-C sequence context. The structural models (Figures 7(a) and 8(a)) are consistent with this conclusion. The small, but finite chemical shifts of the aromatic ring system protons BPH1, BPH2, and BPH3 in this sequence context relative to the chemical shifts of the 10R (+)-cis-anti-[BP]G adduct in the unmethylated C-[BP]G-C sequence context (Figure S3(b), Supplementary Data), are also consistent with these conclusions.
Finally, while we did not detect any major conformational changes in the case of the 10S (+)-trans-anti-[BP]G adduct, some more subtle conformational changes are nevertheless produced by methylation of the cytosine residue in the meC-[BP]G sequence context. This is indicated by changes in electrophoretic mobilities23 and accessibilities of the aromatic pyrenyl residue to the aqueous environment32 of these adducts in double-stranded oligonucleotides.
Consequences of cytosine methylation in relation to DNA repair
Modulation of adduct repair by cytosine methylation could influence the ultimate mutagenic outcome associated with the [BP]G adduct. Several studies suggest that the susceptibility of [BP]G adducts to removal by DNA repair enzymes is indeed influenced by cytosine methylation. An in vitro nucleotide excision repair NER assay employing human cell extracts revealed stimulation of excision of a 10S (+)-trans-anti-[BP]G adduct in a synthetic oligonucleotide containing the modified p53 codon 273 sequence when the 5′-cytosine residue was methylated as compared to the unmethylated analog.58 Furthermore, Colgate et al.59 have found that the NER efficiency by UvrABC proteins is enhanced by cytosine methylation in the case of both 10S (+) and 10R (−)-trans-anti-[BP]G adducts, and more so for the (−)-trans-adduct, where, as shown here, the methyl group induces a conformational switch. The likelihood that the conformational switch to base-displaced intercalation leads to enhanced NER repair is also supported by findings from the human cell extract NER studies, which revealed that 10R (+) and 10S (−)-cis-anti-[BP]G, which are base-displaced intercalated in solution,30,31 are excised more efficiently than minor groove or classically intercalated adducts.60,61 Similar, though less pronounced differences in the excision of minor groove and base-displaced intercalated [BP]G adducts in unmethylated C[BP]G sequences by UvrABC enzymes from E. coli have been observed as well.62 Thus, the finding of a profound conformational switch in a B[a]PDE adduct induced by cytosine methylation, may impact the mutational hot spot phenomenon, in part through differential repair, and perhaps in part by affecting translesion synthesis during replication. The full details of such effects remain to be elucidated.
Materials and Methods
Oligonucleotides
The unmethylated and methylated oligonucleotides I, I(5′-meC), I(3′-meC) and the complementary strand Ic, were synthesized by standard phosphoramidite methods on a Biosearch Cyclone automated DNA synthesizer (Milligen-Biosearch Corp., San Refael, CA), and were purified and desalted by standard HPLC protocols32,63 on a PRP-1 column (Hamilton) and a Hypersil C-18 column.
Synthesis of site-specifically modified oligonucleotides
The anti-B[a]PDE-modified oligonucleotides synthesized had the following sequences (G* denotes the modified base):
C-[BP]G-C: 5′-d(CCATCG*CTACC)
meC-[BP]G-C: 5′-d(CCAT[meC]G*CTACC)
C-[BP]G-meC: 5′-d(CCATCG*[meC]TACC)
IC: 5′-GGTAGCGATGG (complementary strand)
The unmodified sequences C-G-C, meC-G-C or C-G-meC (G*=G), were reacted with racemic anti-B[a]PDE (dissolved in tetrahydrofuran) as described earlier.63 Caution: benzo[a]pyrene diol epoxides are carcinogenic and mutagenic and should be handled with care, as outlined by National Cancer Institute Guidelines. The modified oligonucleotides were separated from unmodified oligonucleotides, and the modified oligonucleotides containing the single stereoisomeric 10S (+) or 10R (−)-trans-anti-[BP]G adduct (G*, Figure 1) were separated from one another utilizing various reversed-phase HPLC protocols as described in detail elsewhere.63 The stereo-chemical properties of the modified oligonucleotides were established by enzyme digestion of the modified oligonucleotides and HPLC co-elution of the BPDE-modified base with stereochemically defined trans-anti-[BP]G adduct standards.64 The stereochemical properties of each mononucleotide adduct were identified by circular dichroism techniques, and by co-elution with the corresponding mononucleotide adduct standards as described earlier for the same B[a]PDE-modified oligonucleotides.32
NMR methods
Duplexes of the modified oligonucleotides C-[BP]G-C, meC-[BP]G-C and C-[BP]G-meC with the complementary strand IC were prepared for detailed NMR studies as described earlier.30 NMR spectra of the exchangeable protons were collected in aqueous buffer (100 mM NaCl, 10 mM phosphate, pH 6.8) at 0 °C on Varian INOVA and Brucker Avance NMR spectrometers. For the non-exchangeable protons of the same adduct duplexes, NMR spectra were collected in 2H2O at 0 °C. NOESY, COSY and TOCSY data sets were processed using either Varian or Brucker software and analyzed using the FELIX program (Accelrys, Inc.).
Molecular mechanics computations
Two energy minimized starting models for molecular dynamics were created with the torsion space molecular mechanics program DUPLEX.65,66 One was created from the base-displaced-intercalative solution structure of the 10R (+)-cis-anti-[BP]G adduct,30 by remodeling the 10R (+)-cis-anti isomer to the 10R (−)-trans-anti adduct (these two adducts differ from one another only by the absolute configurations at the C7, C8, and C9 carbon atoms, Figure 1), and by adding the methyl group to the 5-position of cytosine residue 5. The second model was created from the NMR solution structure of the minor groove 10R (−)-trans-anti-[BP]G adduct,29 again by addition of the 5-methyl to cytosine residue 5 (Figure 1). A model of the 10S (+)-trans-anti-[BP]G adduct in a base displaced-intercalative conformation in the meC-G-C sequence context was obtained with DUPLEX, by remodeling the NMR solution structure of the 10S (−)-cis-anti-[BP]G adduct,31 with addition of the 5-methyl group to C5 and energy minimization.
Distance restraints
The distances between non-exchangeable inter-protons were obtained from the buildup curves of cross-peak intensities in NOESY spectra at five different mixing times (50, 100, 150, 200 and 250 ms) in 2H2O at 0 °C and given bounds between ±20% and ±30% with distances referenced relative to the cytosine H5–H6 distance of 2.47 Å. Exchangeable proton restraints were based on NOESY data sets at two mixing times (60 ms and 200 ms) in H2O at 0 °C and given bounds of ±30%. Cross-peaks involving exchangeable protons were classified as strong (medium to strong intensity at 60 ms), medium (weak intensity at 60 ms) and weak (observed only at a mixing time of 200 ms) and proton pairs were then restrained respectively to distances of 3.0(±0.9) Å, 4.0(±1.2) Å and 6.0(±1.8) Å.
Molecular dynamics computations
The protocol outlined below has been used in our laboratory to determine the solution structures of the 1S-(BPh)G·C and 1R-(BPh)G·C 11-mer duplexes that contain adducts derived from the binding of the r1,t2-dihydroxy-t9,10-epoxy-1,2,3,4-tetrahydrobenzo[c]phenanthrene to N2-dG in DNA.67 The refinement proceeds in two stages where the distance-based refinement is performed first and is then followed by NOE intensity-based refinement. The NMR distance-restrained molecular dynamics (MD) simulations were carried out starting from three initial structures of [BP]G 11-mer duplexes. Two of the initial structures were generated by the DUPLEX program. Both initial structures adopted B-DNA conformations. The first one allowed the carcinogen to intercalate and stack between flanking base-pairs with displacement and breaking of the central [BP]G·C pair as in the 10R (+)-cis-anti-[BP]G adduct.30 The second initial structure embedded the BP into the minor groove of a B-DNA 11-mer duplex without breaking the [BP]G·C base-pairing, as in the 10R (−)-trans-anti-[BP]G adduct.29 The third initial structure also docked the carcinogen into the minor groove, but in an ideal A-DNA 11-mer duplex generated in Insight II (Accelrys, Inc.). X-PLOR68-based restrained MD calculations for each starting structure were carried out with three different sets using the simulated annealing protocol and the CHARM force field. Hydrogen bonding distance restraints were imposed to align the experimentally identified Watson–Crick pairs (±0.10 Å). Only the non-bonded [BP]G6·C17 pair was excluded based on the analysis of NMR spectra. Each cycle of restrained MD simulations was initially carried out at 300 K with a force constant of 1 kcal mol−1 Å−2 on all experimentally obtained distance restraints. The structure was subjected to 2000 cycles of energy minimization and was slowly heated to 1000 K in 7 ps (1 ps per 100 K increase). The force constants on the experimentally obtained distance restraints were slowly scaled up to 32 (non-exchangeable protons), 16 (exchangeable protons) and 64 (hydrogen bonds) kcal mol−1 Å−2, over a period of 18 ps. The system was allowed to evolve for another 20 ps at 1000 K and next cooled gradually to 300 K over 14 ps (0.5 fs time-step) with retention of the full scale of distance restraints and subsequently equilibrated for 12 ps at 300 K. The coordinates were averaged over the last 5 ps and the resulting coordinates subjected to 20 cycles (100 steps each cycle) of conjugate gradient energy minimization. A total of nine distance-refined structures of [BP]G·C 11-mer duplexes were generated and a subset corresponding to the best structures with the least NOE violations, acceptable covalent geometry, and favorable van der Waals energy were subjected to further relaxation matrix refinement.
The resulting nine distance-refined structures were optimized against the NOE cross-peak intensities with the relaxation matrix (relax) refinement routine of the X-PLOR program. The NOE intensities based on five NOESY data sets in 2H2O buffer collected at mixing times of 50, 100, 150, 200 and 250 ms, were incorporated as penalty functions into the relax energy term, in which the exponent 1/6 was used. An isotropic correlation time of 3.75 ns derived from a systematic grid search, along with a cutoff distance of 4.5 Å was used during the relaxation matrix refinement calculations. The distance-refined structures were subjected to 1 ps of molecular dynamics at 1000 K during which the weights for the non-exchangeable NOE intensities were increased from 5 kcal mol−1 to 300 kcal mol−1 while weights for the non-exchangeable distance restraints were reduced from 50 kcal mol−1 Å−2 to 0 kcal mol−1 Å−2. The weights for the hydrogen bonding distances and distance restraints involving exchangeable protons were retained and were the same as those used in the distance-restrained MD refinement. The system was next gradually cooled to 300 K over 2.25 ps (0.5 fs time-step) with retention of the full scale of restraints. This was followed by 5 ps of MD (time-step 1 fs) at 300 K. The coordinates during the last 1.0 ps of dynamics were averaged and these averaged coordinates were subjected to energy minimization.
Protein Data Bank accession number
Coordinates have been deposited with the PDB with accession number 1Y9H.
Supplementary Material
Acknowledgments
Supported by NIH Grants CA-46533 (DJP), CA-099194 (NEG), and CA-28038 (SB). D.J.P. is a member of the New York Structural Biology Center that is supported by NIH/NIGMS grant P41 GM66354. The racemic diol epoxide anti-BP[a]DE was obtained from the National Cancer Institute Carcinogen Reference Standard Repository supported by the U.S. Public Health Service, National Cancer Institute, National Institutes of Health.
Abbreviations used
- B[a]P
benzo[a]pyrene
- B[a]PDE
7,8-dihydroxy-9,10-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene
- anti-B[a]PDE
r7,t8-dihydroxy-t9,10-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene
- (+)-anti-B[a]PDE
(+)-7R,8S,9S,10R B[a]PDE
- (−)-anti-B[a]PDE
(−)-7S,8R,9R,10S B[a]PDE
- [BP]G
modified guanine residue resulting from the reaction of anti-B[a]PDE with N2-dG
- 10S (+)-trans-anti-[BP]G
adduct derived from trans addition of (+)-anti-B[a]PDE to N2-dG
- 10R (−)-trans-anti-[BP]G
adduct derived from trans addition of (−)-anti-B[a]PDE to N2-dG
- 10R (+)-cis-anti-[BP]G
adduct derived from cis addition of (+)-anti-B[a]PDE to N2-dG
- 10S (−)-cis-anti-[BP]G
adduct derived from cis addition of (−)-anti B[a]PDE to N2-dG
- BPh
benzo[c]phenanthrene
- meC
5-methylcytosine
Footnotes
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.jmb.2004.12.027
Tables of chemical shifts of exchangeable and non-exchangeable protons, and Figures (S1, non-exchangeable base and sugar H1′ proton NMR spectra in the 5.0–8.5 ppm region; S2 and S4, expanded NOESY (250 ms mixing time) contour plots; S3, comparative plots of the chemical shifts of the aliphatic and aromatic BP protons).
References
- 1.Conney AH. Induction of microsomal enzymes by foreign chemicals and carcinogenesis by polycyclic aromatic hydrocarbons: G. H. A. Clowes Memorial Lecture. Cancer Res. 1982;42:4875–4917. [PubMed] [Google Scholar]
- 2.Weinstein IB, Jeffrey AM, Jennette KW, Blobstein SH, Harvey RG, Harris C, et al. Benzo[a]pyrene diol epoxides as intermediates in nucleic acid binding in vitro and in vivo. Science. 1976;193:592–595. doi: 10.1126/science.959820. [DOI] [PubMed] [Google Scholar]
- 3.Koreeda M, Moore PD, Wislocki PG, Levin W, Yagi H, Jerina DM. Binding of benzo[a]-pyrene 7,8-diol-9,10-epoxides to DNA, RNA, and protein of mouse skin occurs with high stereo-selectivity. Science. 1978;199:778–781. doi: 10.1126/science.622566. [DOI] [PubMed] [Google Scholar]
- 4.Jerina DM, Chadha A, Cheh AM, Schurdak ME, Wood AW, Sayer JM. Covalent bonding of bay-region diol epoxides to nucleic acids. Advan Expt Med Biol. 1991;283:533–553. doi: 10.1007/978-1-4684-5877-0_70. [DOI] [PubMed] [Google Scholar]
- 5.Wolfe AR, Smith TJ, Meehan T. Benzo[a]pyrene diol epoxide forms covalent adducts with deoxycytidylic acid by alkylation at both exocyclic amino N(4) and ring imino N-3 positions. Chem Res Toxicol. 2004;17:476–491. doi: 10.1021/tx0340201. [DOI] [PubMed] [Google Scholar]
- 6.Cheng SC, Hilton BD, Roman JM, Dipple A. DNA adducts from carcinogenic and non-carcinogenic enantiomers of benzo[a]pyrene dihydrodiol epoxide. Chem Res Toxicol. 1989;2:334–340. doi: 10.1021/tx00011a011. [DOI] [PubMed] [Google Scholar]
- 7.Buening MK, Wislocki PG, Levin W, Yagi H, Thakker DR, Akagi H, et al. Tumorigenicity of the optical enantiomers of the diastereomeric benzo[a]pyrene 7,8-diol-9,10-epoxides in newborn mice: exceptional activity of (+)-7beta,8alpha-dihydroxy-9alpha,10alpha-epoxy-7,8,9,10-tetrahydrobenzo[a]pyrene. Proc Natl Acad Sci USA. 1978;75:5358–5361. doi: 10.1073/pnas.75.11.5358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Slaga TJ, Bracken WJ, Gleason G, Levin W, Yagi H, Jerina DM, Conney AH. Marked differences in the skin tumor-initiating activities of the optical enantiomers of the diastereomeric benzo(a)-pyrene 7,8-diol-9,10-epoxides. Cancer Res. 1979;39:67–71. [PubMed] [Google Scholar]
- 9.Wood AW, Chang RL, Levin W, Yagi H, Thakker DR, Jerina DM, Conney AH. Differences in mutagenicity of the optical enantiomers of the diastereomeric benzo[a]pyrene 7,8-diol-9,10-epoxides. Biochem Biophys Res Commun. 1977;77:1389–1396. doi: 10.1016/s0006-291x(77)80133-2. [DOI] [PubMed] [Google Scholar]
- 10.Wei SJ, Chang RL, Hennig E, Cui XX, Merkler KA, Wong CQ, et al. Mutagenic selectivity at the HPRT locus in V-79 cells: comparison of mutations caused by bay-region benzo[a]pyrene 7,8-diol-9,-10-epoxide enantiomers with high and low carcinogenic activity. Carcinogenesis. 1994;15:1729–1735. doi: 10.1093/carcin/15.8.1729. [DOI] [PubMed] [Google Scholar]
- 11.Moriya M, Spiegel S, Fernandes A, Amin S, Liu T, Geacintov N, Grollman AP. Fidelity of translesional synthesis past benzo[a]pyrene diol epoxide-2′-deoxyguanosine DNA adducts: marked effects of host cell, sequence context, and chirality. Biochemistry. 1996;35:16646–16651. doi: 10.1021/bi9608875. [DOI] [PubMed] [Google Scholar]
- 12.Fernandes A, Liu T, Amin S, Geacintov NE, Grollman AP, Moriya M. Mutagenic potential of stereoisomeric bay region (+)- and (−)-cis-anti-benzo[a]pyrene diol epoxide-N2-2′-deoxyguanosine adducts in Escherichia coli and simian kidney cells. Biochemistry. 1998;37:10164–10172. doi: 10.1021/bi980401f. [DOI] [PubMed] [Google Scholar]
- 13.Shukla R, Liu T, Geacintov NE, Loechler EL. The major, N2-dG adduct of (+)-anti-B[a]PDE shows a dramatically different mutagenic specificity (predominantly, G to A) in a 5′-CGT-3′ sequence context. Biochemistry. 1997;36:10256–10261. doi: 10.1021/bi970541+. [DOI] [PubMed] [Google Scholar]
- 14.Shukla R, Jelinsky S, Liu T, Geacintov NE, Loechler EL. How stereochemistry affects mutagenesis by N2-deoxyguanosine adducts of 7,8-dihydroxy-9,10-epoxy-7,8,9,10-tetrahydrobenzo[a]-pyrene: configuration of the adduct bond is more important than those of the hydroxyl groups. Biochemistry. 1997;36:13263–13269. doi: 10.1021/bi971195z. [DOI] [PubMed] [Google Scholar]
- 15.Hanrahan CJ, Bacolod MD, Vyas RR, Liu T, Geacintov NE, Loechler EL, Basu AK. Sequence specific mutagenesis of the major (+)-anti-benzo[a]pyrene diol epoxide-DNA adduct at a mutational hot spot in vitro and in Escherichia coli cells. Chem Res Toxicol. 1997;10:369–377. doi: 10.1021/tx9601925. [DOI] [PubMed] [Google Scholar]
- 16.Page JE, Zajc B, Oh-hara T, Lakshman MK, Sayer JM, Jerina DM, Dipple A. Sequence context profoundly influences the mutagenic potency of trans-opened benzo[a]pyrene 7,8-diol 9,10-epoxide-purine nucleoside adducts in site-specific mutation studies. Biochemistry. 1998;37:9127–9137. doi: 10.1021/bi980273v. [DOI] [PubMed] [Google Scholar]
- 17.Denissenko MF, Pao A, Tang M, Pfeifer GP. Preferential formation of benzo[a]pyrene adducts at lung cancer mutational hotspots in P53. Science. 1996;274:430–432. doi: 10.1126/science.274.5286.430. [DOI] [PubMed] [Google Scholar]
- 18.Harris CC. p53 tumor suppressor gene: from the basic research laboratory to the clinic—an abridged historical perspective. Carcinogenesis. 1996;17:1187–1198. doi: 10.1093/carcin/17.6.1187. [DOI] [PubMed] [Google Scholar]
- 19.Hollstein M, Shomer B, Greenblatt M, Soussi T, Hovig E, Montesano R, Harris CC. Somatic point mutations in the p53 gene of human tumors and cell lines: updated compilation. Nucl Acids Res. 1996;24:141–146. doi: 10.1093/nar/24.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Denissenko MF, Chen JX, Tang MS, Pfeifer GP. Cytosine methylation determines hot spots of DNA damage in the human P53 gene. Proc Natl Acad Sci USA. 1997;94:3893–3898. doi: 10.1073/pnas.94.8.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen JX, Zheng Y, West M, Tang MS. Carcinogens preferentially bind at methylated CpG in the p53 mutational hot spots. Cancer Res. 1998;58:2070–2075. [PubMed] [Google Scholar]
- 22.Pfeifer GP, Tang M, Denissenko MF. Mutation hotspots and DNA methylation. Curr Top Microbiol Immunol. 2000;249:1–19. doi: 10.1007/978-3-642-59696-4_1. [DOI] [PubMed] [Google Scholar]
- 23.Weisenberger DJ, Romano LJ. Cytosine methylation in a CpG sequence leads to enhanced reactivity with benzo[a]pyrene diol epoxide that correlates with a conformational change. J Biol Chem. 1999;274:23948–23955. doi: 10.1074/jbc.274.34.23948. [DOI] [PubMed] [Google Scholar]
- 24.Pradhan P, Gräslund A, Seidel A, Jernström B. Implications of cytosine methylation on (+)-anti-benzo[a]pyrene 7, 8-dihydrodiol 9,10-epoxide N2-dG adduct formation in 5′-d(CGT), 5′-d(CGA), and 5′-d(CGC) sequence contexts of single- and double-stranded oligonucleotides. Chem Res Toxicol. 1999;12:816–821. doi: 10.1021/tx980230f. [DOI] [PubMed] [Google Scholar]
- 25.Hu W, Feng Z, Tang MS. Preferential carcinogen–DNA adduct formation at codons 12 and 14 in the human K-ras gene and their possible mechanisms. Biochemistry. 2003;42:10012–10023. doi: 10.1021/bi034631s. [DOI] [PubMed] [Google Scholar]
- 26.Matter B, Wang G, Jones R, Tretyakova N. Formation of diastereomeric benzo[a]pyrene diol epoxide-guanine adducts in p53 gene-derived DNA sequences. Chem Res Toxicol. 2004;17:731–741. doi: 10.1021/tx049974l. [DOI] [PubMed] [Google Scholar]
- 27.Geacintov NE, Cosman M, Hingerty BE, Amin S, Broyde S, Patel DJ. NMR solution structures of stereoisometric covalent polycyclic aromatic carcinogen–DNA adduct: principles, patterns, and diversity. Chem Res Toxicol. 1997;10:111–146. doi: 10.1021/tx9601418. [DOI] [PubMed] [Google Scholar]
- 28.Cosman M, de los Santos C, Fiala R, Hingerty BE, Singh SB, Ibanez V, et al. Solution conformation of the major adduct between the carcinogen (+)-anti-benzo[a]pyrene diol epoxide and DNA. Proc Natl Acad Sci USA. 1992;89:1914–1918. doi: 10.1073/pnas.89.5.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de los Santos C, Cosman M, Hingerty BE, Ibanez V, Margulis LA, Geacintov NE, et al. Influence of benzo[a]pyrene diol epoxide chirality on solution conformations of DNA covalent adducts: the (−)-trans-anti-[BP]G·C adduct structure and comparison with the (+)-trans-anti-[BP]G·C enantiomer. Biochemistry. 1992;31:5245–5252. doi: 10.1021/bi00138a002. [DOI] [PubMed] [Google Scholar]
- 30.Cosman M, de los Santos C, Fiala R, Hingerty BE, Ibanez V, Luna E, et al. Solution conformation of the (+)-cis-anti-[BP]dG adduct in a DNA duplex: intercalation of the covalently attached benzo[a]pyrenyl ring into the helix and displacement of the modified deoxyguanosine. Biochemistry. 1993;32:4145–4155. doi: 10.1021/bi00067a001. [DOI] [PubMed] [Google Scholar]
- 31.Cosman M, Hingerty BE, Luneva N, Amin S, Geacintov NE, Broyde S, Patel DJ. Solution conformation of the (−)-cis-anti-benzo[a]-pyrenyl-dG adduct opposite dC in a DNA duplex: intercalation of the covalently attached BP ring into the helix with base displacement of the modified deoxyguanosine into the major groove. Biochemistry. 1996;35:9850–9863. doi: 10.1021/bi9605346. [DOI] [PubMed] [Google Scholar]
- 32.Huang X, Colgate KC, Kolbanovskiy A, Amin S, Geacintov NE. Conformational changes of a benzo[a]pyrene diol epoxide-N2-dG adduct induced by a 5′-flanking 5-methyl-substituted cytosine in a meCG double-stranded oligonucleotide sequence context. Chem Res Toxicol. 2002;15:438–444. doi: 10.1021/tx015588h. [DOI] [PubMed] [Google Scholar]
- 33.Cosman M, Fiala R, Hingerty BE, Amin S, Geacintov NE, Broyde S, Patel DJ. Solution conformation of the (+)-cis-anti-[BP]dG adduct opposite a deletion site in a DNA duplex: intercalation of the covalently attached benzo[a]-pyrene into the helix with base displacement of the modified deoxyguanosine into the minor groove. Biochemistry. 1994;33:11518–11527. doi: 10.1021/bi00204a014. [DOI] [PubMed] [Google Scholar]
- 34.Patel DJ, Shapiro L, Hare D. DNA and RNA: NMR studies of conformations and dynamics in solution. Quart Rev Biophys. 1987;20:35–112. doi: 10.1017/s0033583500004224. [DOI] [PubMed] [Google Scholar]
- 35.Van de Ven FJ, Hilbers CW. Nucleic acids and nuclear magnetic resonance. Eur J Biochem. 1988;178:1–38. doi: 10.1111/j.1432-1033.1988.tb14425.x. [DOI] [PubMed] [Google Scholar]
- 36.Hergersberg M. Biological aspects of cytosine methylation in eukaryotic cells. Experientia. 1991;47:1171–1185. doi: 10.1007/BF01918381. [DOI] [PubMed] [Google Scholar]
- 37.Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell. 1980;20:85–93. doi: 10.1016/0092-8674(80)90237-8. [DOI] [PubMed] [Google Scholar]
- 38.Sowers LC, Shaw BR, Sedwick WD. Base stacking and molecular polarizability: effect of a methyl group in the 5-position of pyrimidines. Biochem Biophys Res Commun. 1987;148:790–794. doi: 10.1016/0006-291x(87)90945-4. [DOI] [PubMed] [Google Scholar]
- 39.Hodges-Garcia Y, Hagerman PJ. Cytosine methylation can induce local distortions in the structure of duplex DNA. Biochemistry. 1992;31:7595–7599. doi: 10.1021/bi00148a022. [DOI] [PubMed] [Google Scholar]
- 40.Hodges-Garcia Y, Hagerman PJ. Investigation of the influence of cytosine methylation on DNA flexibility. J Biol Chem. 1995;270:197–201. doi: 10.1074/jbc.270.1.197. [DOI] [PubMed] [Google Scholar]
- 41.Heinemann U, Hahn M. C-C-A-G-G-C-m5C-T-G-G. Helical fine structure, hydration, and comparison with C-C-A-G-G-C-C-T-G-G. J Biol Chem. 1992;267:7332–7341. [PubMed] [Google Scholar]
- 42.Marcourt L, Cordier C, Couesnon T, Dodin G. Impact of C5-cytosine methylation on the solution structure of d(GAAAACGTTTTC)2. An NMR and molecular modelling investigation. Eur J Biochem. 1999;265:1032–1042. doi: 10.1046/j.1432-1327.1999.00819.x. [DOI] [PubMed] [Google Scholar]
- 43.Nathan D, Crothers DM. Bending and flexibility of methylated and unmethylated EcoRI DNA. J Mol Biol. 2002;316:7–17. doi: 10.1006/jmbi.2001.5247. [DOI] [PubMed] [Google Scholar]
- 44.Meints GA, Drobny GP. Dynamic impact of methylation at the M.HhaI target site: a solid-state deuterium NMR study. Biochemistry. 2001;40:12436–12443. doi: 10.1021/bi0102555. [DOI] [PubMed] [Google Scholar]
- 45.Derreumaux S, Chaoui M, Tevanian G, Fermandjian S. Impact of CpG methylation on structure, dynamics and solvation of cAMP DNA responsive element. Nucl Acids Res. 2001;29:2314–2326. doi: 10.1093/nar/29.11.2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rauch C, Trieb M, Wellenzohn B, Loferer M, Voegele A, Wibowo FR, Liedl KR. C5-methylation of cytosine in B-DNA thermodynamically and kinetically stabilizes BI. J Am Chem Soc. 2003;125:14990–14991. doi: 10.1021/ja037218g. [DOI] [PubMed] [Google Scholar]
- 47.Banyay M, Gräslund A. Structural effects of cytosine methylation on DNA sugar pucker studied by FTIR. J Mol Biol. 2002;324:667–676. doi: 10.1016/s0022-2836(02)01104-x. [DOI] [PubMed] [Google Scholar]
- 48.Pfeifer GP. p53 mutational spectra and the role of methylated CpG sequences. Mutat Res. 2000;450:155–166. doi: 10.1016/s0027-5107(00)00022-1. [DOI] [PubMed] [Google Scholar]
- 49.Hussain SP, Harris CC. p53 mutation spectrum and load: the generation of hypotheses linking the exposure of endogenous or exogenous carcinogens to human cancer. Mutat Res. 1999;428:23–32. doi: 10.1016/s1383-5742(99)00028-9. [DOI] [PubMed] [Google Scholar]
- 50.Bennett WP, Hussain SP, Vahakangas KH, Khan MA, Shields PG, Harris CC. Molecular epidemiology of human cancer risk: gene-environment interactions and p53 mutation spectrum in human lung cancer. J Pathol. 1999;187:8–18. doi: 10.1002/(SICI)1096-9896(199901)187:1<8::AID-PATH232>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 51.Yoon JH, Smith LE, Feng Z, Tang M, Lee CS, Pfeifer GP. Methylated CpG dinucleotides are the preferential targets for G-to-T transversion mutations induced by benzo[a]pyrene diol epoxide in mammalian cells: similarities with the p53 mutation spectrum in smoking-associated lung cancers. Cancer Res. 2001;61:7110–7117. [PubMed] [Google Scholar]
- 52.Pfeifer GP, Denissenko MF, Olivier M, Tretyakova N, Hecht SS, Hainaut P. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene. 2002;21:7435–7451. doi: 10.1038/sj.onc.1205803. [DOI] [PubMed] [Google Scholar]
- 53.Rodin SN, Rodin AS. On the origin of p53 G:C to T:A transversions in lung cancers. Mutat Res. 2002;508:1–19. doi: 10.1016/s0027-5107(02)00106-9. [DOI] [PubMed] [Google Scholar]
- 54.Pfeifer GP, Hainaut P. On the origin of G to T transversions in lung cancer. Mutat Res. 2003;526:39–43. doi: 10.1016/s0027-5107(03)00013-7. [DOI] [PubMed] [Google Scholar]
- 55.Geacintov NE, Shahbaz M, Ibanez V, Moussaoui K, Harvey RG. Base-sequence dependence of noncovalent complex formation and reactivity of benzo[a]pyrene diol epoxide with polynucleotides. Biochemistry. 1988;27:8380–8387. doi: 10.1021/bi00422a013. [DOI] [PubMed] [Google Scholar]
- 56.Tretyakova N, Matter B, Jones R, Shallop A. Formation of benzo[a]pyrene diol epoxide–DNA adducts at specific guanines within K-ras and p53 gene sequences: stable isotope-labeling mass spectrometry approach. Biochemistry. 2002;41:9535–9544. doi: 10.1021/bi025540i. [DOI] [PubMed] [Google Scholar]
- 57.Shahbaz M, Geacintov NE, Harvey RG. Noncovalent intercalative complex formation and kinetic flow linear dichroism of racemic syn- and anti-benzo[a]pyrenediol epoxide–DNA solutions. Biochemistry. 1986;25:3290–3296. doi: 10.1021/bi00359a031. [DOI] [PubMed] [Google Scholar]
- 58.Muheim R, Buterin T, Colgate KC, Kolbanovsiy A, Geacintov NE, Naegeli H. Modulation of human nucleotide excision repair by 5-methylcytosines. Biochemistry. 2003;42:3247–3254. doi: 10.1021/bi0268504. [DOI] [PubMed] [Google Scholar]
- 59.Colgate KC, Huang X, Kolbanovskiy A, Skorvaga M, Van Houten B, Amin S, Geacintov NE. Effects of methylation and temperature on nucleotide excision repair of stereo-isomeric benzo[a]pyrene diol epoxide-N2-dG adducts in a 5-meCG*p53 codon 273 sequence context catalyzed by UvrABC proteins from B-caldotenax. Chem Res Toxicol. 2003;16:1676–1677. [Google Scholar]
- 60.Hess MT, Gunz D, Luneva N, Geacintov NE, Naegeli H. Base pair conformation-dependent excision of benzo[a]pyrene diol epoxide-guanine adducts by human nucleotide excision repair enzymes. Mol Cell Biol. 1997;17:7069–7076. doi: 10.1128/mcb.17.12.7069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Buterin T, Hess MT, Luneva N, Geacintov NE, Amin S, Kroth H, et al. Unrepaired fjord region polycyclic aromatic hydrocarbon–DNA adducts in ras codon 61 mutational hot spots. Cancer Res. 2000;60:1849–1856. [PubMed] [Google Scholar]
- 62.Zou Y, Liu TM, Geacintov NE, Van Houten B. Interaction of the UvrABC nuclease system with a DNA duplex containing a single stereoisomer of dG-(+)- or dG-(−)-anti-BPDE. Biochemistry. 1995;34:13582–13593. doi: 10.1021/bi00041a038. [DOI] [PubMed] [Google Scholar]
- 63.Pirogov N, Shafirovich V, Kolbanovskiy A, Solntsev K, Courtney SA, Amin S, Geacintov NE. Role of hydrophobic effects in the reaction of a polynuclear aromatic diol epoxide with oligodeoxynucleotides in aqueous solutions. Chem Res Toxicol. 1998;11:381–388. doi: 10.1021/tx980006q. [DOI] [PubMed] [Google Scholar]
- 64.Geacintov NE, Cosman M, Mao B, Alfano A, Ibanez V, Harvey RG. Spectroscopic characteristics and site I/site II classification of cis and trans benzo[a]pyrene diolepoxide enantiomer-guanosine adducts in oligonucleotides and poly-nucleotides. Carcinogenesis. 1991;12:2099–2108. doi: 10.1093/carcin/12.11.2099. [DOI] [PubMed] [Google Scholar]
- 65.Hingerty BE, Figueroa S, Hayden TL, Broyde S. Prediction of DNA structure from sequence: a build-up technique. Biopolymers. 1989;28:1195–1222. doi: 10.1002/bip.360280703. [DOI] [PubMed] [Google Scholar]
- 66.Wang L, Hingerty BE, Srinivasan AR, Olson WK, Broyde S. Accurate representation of B-DNA double helical structure with implicit solvent and counterions. Biophys J. 2002;83:382–406. doi: 10.1016/S0006-3495(02)75177-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lin CH, Huang X, Kolbanovskiy A, Hingerty BE, Amin S, Broyde S, et al. Molecular topology of polycyclic aromatic carcinogens determines DNA adduct conformation: a link to tumorigenic activity. J Mol Biol. 2001;306:1059–1080. doi: 10.1006/jmbi.2001.4425. [DOI] [PubMed] [Google Scholar]
- 68.Brunger AT. X-PLOR: A System for X-ray Crystallography and NMR. Yale University Press; New Haven, CT: 1992. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.