

Abstract
Organizing pneumonia (OP) is a clinicopathological entity that occurs idiopathically or in association with several conditions, but there are few reports about myeloperoxidase anti‐neutrophil cytoplasmic antibody (MPO‐ANCA)‐associated OP. We describe a patient with OP whose clinical conditions improved spontaneously. Moreover, serum MPO‐ANCA titers paralleled the clinical activity of the disease, which disappeared in association with disease quiescence. Based on these findings, a subset of patients may have OP related to MPO‐ANCA. ANCA testing should be considered in the work‐up of patients with suspected OP.
Keywords: Microscopic polyangiitis, myeloperoxidase anti‐neutrophil cytoplasmic antibody (MPO‐ANCA), organizing pneumonia
Introduction
Anti‐neutrophil cytoplasmic antibody (ANCA)‐related small vessel vasculitis is one of the most common causes of pulmonary renal syndromes [1]. Interstitial lung disease that mimics idiopathic pulmonary fibrosis sometimes shows ANCA against myeloperoxidase (MPO) [2]. However, there are few reports of organizing pneumonia (OP) associated with MPO‐ANCA. Here, we describe a patient with MPO‐ANCA‐associated OP.
Case Report
A 46‐year‐old woman was referred to our hospital because of a 10‐day history of fever and cough. Before admission, she visited a general physician and had been treated with levofloxacin for 3 days followed by ceftriaxone for 4 days without benefit. She had never smoked and the only issue in her previous medical history was hypothyroidism. She had no history of asbestos exposure. Physical examination showed a body temperature of 38.3°C, but crackles were not audible on auscultation. There was no cutaneous evidence of vasculitis or stigmata of collagen vascular disease. Chest computed tomography (CT) showed sharply demarcated, multiple nodular lesions and interlobular septal thickening, predominantly in both lower lung fields (Fig. 1). Echocardiography did not show any abnormalities. Laboratory findings revealed a white blood cell count of 12,000/μL (77.3% neutrophils, 17.0% lymphocytes, 0.5% eosinophils and 5.0% monocytes). The serum creatinine level was normal at 0.58 mg/dl and a urine examination showed no active sediment. C‐reactive protein was positive at 1.7 mg/dL. MPO‐ANCA was increased at 88 ELISA Units (EU; normal <10 EU), while proteinase 3 ANCA and other autoantibodies were negative. Blood cultures and erythrocytes sedimentation rate were not examined. Bronchoalveolar lavage (BAL) was negative for malignant cells and pathogenic organisms. Total cell count showed 5 × 104 cells per mL and 90% alveolar macrophages, 6% lymphocytes and 1% neutrophils, and the ratio of CD4 to CD8 T lymphocytes was 1.23. Alveolar hemorrhage was also excluded because BAL did not contain blood. Transbronchial lung biopsy showed OP and fibrinous exudate without any vasculitis, granulomas, or necrosis (Fig. 2). We made the diagnosis of MPO‐ANCA‐associated OP. Her condition improved gradually without therapy. Laboratory data including MPO‐ANCA and imaging findings were normalized. In the 1‐year follow‐up, she did not relapse into OP nor develop features of systemic vasculitis.
Figure 1.
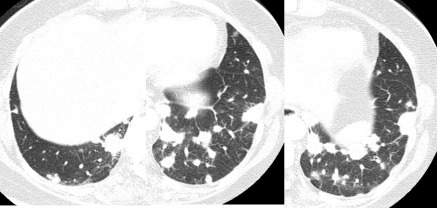
High‐resolution computed tomography scans of the lower lobes show discrete, sharply demarcated, multiple nodular shadows and interlobular septal thickening.
Figure 2.
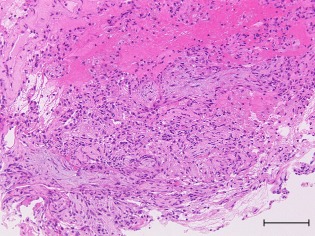
Histological findings of transbronchial biopsy obtained from the left lower lobe nodular shadows. Organizing pneumonia (OP) and fibrinous exudate were found, but vasculitis, granulomas or necrosis were not found. Hematoxylin‐eosin stains. Bar indicates 200 μm.
Discussion
The lungs are among the most common and important organs affected in MPO‐ANCA‐associated vasculitis [1]. Diffuse alveolar hemorrhage and pulmonary fibrosis are the most frequent manifestations [3]. OP, which is a nonspecific pathological finding of lung injury, has been reported in surgical biopsies and autopsies [4], but there are few reports about MPO‐ANCA‐associated OP as a clinical diagnosis. In our case, chest CT findings showed multiple pulmonary nodules, which are reported in cryptogenic OP (COP) [5]. In many patients with COP, rapid improvement is obtained with corticosteroid therapy. However, spontaneous improvement has occasionally been reported [5, 6]. The clinical condition in our case, which spontaneously improved, resembled that of COP. Serum MPO‐ANCA titers decreased concomitantly with the improvement of her clinical symptoms and radiological findings, and there were no clinical findings suggestive of collagen vascular diseases. The clinical course indicates that MPO‐ANCA may be closely related to the pathogenesis of OP.
The mechanisms of MPO‐ANCA‐associated lung diseases are not clear. One prevalent theory suggests that in response to proinflammatory cytokines or microbial products, small quantities of ANCA antigens are translocated to the surface of neutrophils, which activate primed neutrophils directly, injuring localized endothelial cells and alveolar epithelial cells [7, 8]. In our case, the presenting symptoms were fever and cough, which improved gradually without therapy, as did the imaging findings and MPO‐ANCA titers. The clinical course may support the hypothesis that some microbial products triggered the alveolar epithelial injury associated with MPO‐ANCA mentioned above and caused OP. However, we could not detect any pathogens. Further investigations are needed to clarify this hypothesis.
OP can be an accompanying histological finding in cases of ANCA‐associated vasculitis [4, 9]. In patients with granulomatosis with polyangiitis, OP is the major histologic finding in approximately 13% of patients when diagnosed by surgical lung biopsy or autopsy [9]. Transbronchial lung biopsy does not contain sufficient tissue to robustly exclude the possibility of vasculitis in such cases. Based on these facts, the diagnosis of MPO‐ANCA‐associated OP is provisional in the present case. Careful follow‐up is required to confirm this diagnosis.
The prevalence of MPO‐ANCA positivity in patients with OP remains unknown. Our case suggests that a subset of patients have OP related to MPO‐ANCA. It remains to be elucidated whether patients with OP subsequently develop features of systemic vasculitis. ANCA testing should be considered in the work‐up of patients with suspected OP.
Disclosure Statements
No conflict of interest declared.
Appropriate written informed consent was obtained for publication of this case report and accompanying images.
References
- 1. Jennette JC, Falk RJ, Bacon PA, et al. 2013. 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 65:1–11. [DOI] [PubMed] [Google Scholar]
- 2. Tanaka T, Otani K, Egashira R, et al. 2012. Interstitial pneumonia associated with MPO‐ANCA: clinicopathological features of nine patients. Respir. Med. 106:1765–1770. [DOI] [PubMed] [Google Scholar]
- 3. Ando Y, Okada F, Matsumoto S, et al. 2004. Thoracic manifestation of myeloperoxidase‐antineutrophil cytoplasmic antibody (MPO‐ANCA)‐related disease. J. Comput. Assist. Tomogr. 28:710–716. [DOI] [PubMed] [Google Scholar]
- 4. Gaudin PB, Askin FB, Falk RJ, et al. 1995. The pathologic spectrum of pulmonary lesions in patients with anti‐neutrophil cytoplasmic autoantibodies specific for anti‐proteinase 3 and anti‐myeloperoxidase. Am. J. Clin. Pathol. 104:7–16. [DOI] [PubMed] [Google Scholar]
- 5. Akira M, Yamamoto S, and Sakatani M. 1998. Bronchiolitis obliterans organizing pneumonia manifesting as multiple large nodules or masses. AJR 170:291–295. [DOI] [PubMed] [Google Scholar]
- 6. Epler GR, Colby TV, McLoud TC, et al. 1985. Bronchiolitis obliterans organizing pneumonia. N. Engl. J. Med. 312:152–158. [DOI] [PubMed] [Google Scholar]
- 7. Chen M, and Kallenberg CGM. 2009. New advances in the pathogenesis of ANCA‐associated vasculitides. Clin. Exp. Rheumatol. 27(Suppl. 52):S108–S114. [PubMed] [Google Scholar]
- 8. Falk RJ, Terrell RS, Charles LA, et al. 1990. Anti‐neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc. Natl. Acad. Sci. U.S.A. 87:4115–4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Uner AH, Rozum‐Slota B, and Katzenstein ALA. 1996. Bronchiolitis obliterans‐organizing pneumonia (BOOP)‐like variant of Wegener's Granulomatosis. A clinicopathologic study of 16 cases. Am. J. Surg. Pathol. 20:794–801. [DOI] [PubMed] [Google Scholar]