

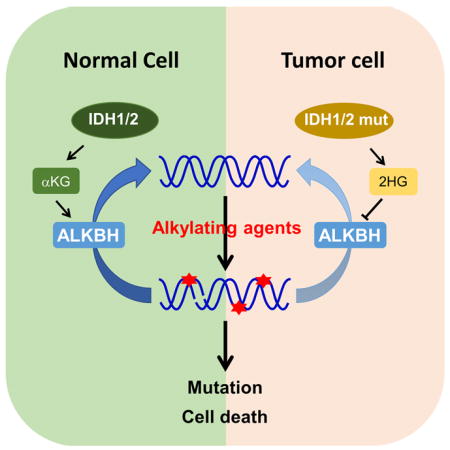
SUMMARY
Chemotherapy of a combination of DNA alkylating agents, procarbazine and lomustine (CCNU), and a microtubule poison, vincristine, offers a significant benefit to a subset of glioma patients. The benefit of this regime, known as PCV, was recently linked to IDH mutation that occurs frequently in glioma and produces D-2-hydroxyglutarate (D-2-HG), a competitive inhibitor of α-ketoglutarate (α-KG). We report here that D-2-HG inhibits the α-KG-dependent alkB homolog (ALKBH) DNA repair enzymes. Cells expressing mutant IDH display reduced repair kinetics, accumulate more DNA damages and are sensitized to alkylating agents. The observed sensitization to alkylating agents requires the catalytic activity of mutant IDH to produce D-2-HG and can be reversed by the deletion of mutant IDH allele or overexpression of ALKBH2 or AKLBH3. Our results suggest that impairment of DNA repair may contribute to tumorigenesis driven by IDH mutations and that alkylating agents may merit exploration for treating IDH-mutated cancer patients.
Graphical abstract
INTRODUCTION
Genes encoding for isocitrate dehydrogenases 1 and 2, IDH1 and IDH2, are frequently mutated in grade II and grade III anaplastic oligodendrogliomas (AO), mixed oligoastrocytomas and astrocytomas, and WHO grade IV secondary glioblastomas (GBM)(>75%) (Parsons et al., 2008) as well as several other types of human cancer, including acute myeloid leukemia (AML, ~20%), cartilaginous tumors (75%), intrahepatic cholangiocarcinomas (10–23%), angioimmunoblastic T-cell lymphoma (AITLs, ~20%) and melanoma (~5%) [Reviewed by (Cairns and Mak, 2013; Yang et al., 2012)]. Tumor-derived IDH1 and IDH2 mutations simultaneously cause loss of its normal activity, the production of α-ketoglutarate (α-KG, also known as 2-oxoglutarate), and gain of a neomorphic activity, the reduction of α-KG to D-2-hydroxyglutarate (D-2-HG) (Dang et al., 2009; Yan et al., 2009; Zhao et al., 2009). D-2-HG is structurally similar to α-KG and acts as an antagonist of α-KG to competitively inhibit multiple α-KG-dependent dioxygenases, including the JmjC domain-containing histone demethylases (KDMs) and the TET (ten-eleven translocation) family of DNA hydroxylases (Chowdhury et al., 2011; Xu et al., 2011). Altered epigenetic regulation is currently considered to be a major mechanism whereby IDH mutation and D-2-HG exert their oncogenic effects.
The unique property of mutant IDH1/2 in producing an oncometabolite that has no known physiological function makes mutant IDH enzymes as obvious potential therapeutic targets for the treatment of IDH-mutated tumors (Rohle et al., 2013; Wang et al., 2013). Clinical studies have also suggested the presence of a sequela target(s) for treating IDH-mutated gliomas. Following an early study showing successful chemotherapy for recurrent malignant oligodendroglioma (Cairncross and Macdonald, 1988), randomized controlled trials were carried out in both the North America (RTOG 9402) and Europe (EORTC 26951). These studies have shown clear benefits for both anaplastic oligodendroglioma (AO) and oligoastrocytoma (AOA) patients who in addition to radiation therapy received chemotherapy of procarbazine, CCNU/lomustine and vincristine (PCV) (Cairncross et al., 2013; Cairncross et al., 2014; Erdem-Eraslan et al., 2013; van den Bent et al., 2013). PCV benefit was recently linked to IDH1 mutations with an overall survival of 9.4 years for IDH-mutated patients vs. 5.7 years for patients with wild-type IDH (Cairncross et al., 2014). Of three agents in PCV regimen, vincristine inhibits microtubule assembly, and CCNU and procarbazine are DNA alkylating agents. The molecular mechanism(s) underlying the therapeutic benefits that are conferred by PCV is not known and is investigated in this study.
RESULTS
D-2-HG inhibits ALKBH enzymes in vitro
Endogenous (e.g. S-adenosylmethionine, SAM) and environmental (e.g. nitrosoureas) alkylating agents cause methylated bases in DNA that can be mutagenic and cytotoxic if not repaired. The major enzymes repairing the methylated lesions such as 1-methyladenine (1meA) and 3-methylcytosine (3meC) are the AlkB proteins. Like the JmjC KDMs and TET proteins, AlkB belongs to the Fe(II)- and α-KG-dependent dioxygenases (Falnes et al., 2002), and includes nine distinct genes in human cells (AlkB homolog ALKBH1 to ALKBH8 and FTO) (Sedgwick et al., 2007). The function in repairing DNA alkylation lesion has been demonstrated biochemically in vitro and supported by the genetic analysis of mutant mice for mammalian ALKBH2 and ALKBH3 (Aas et al., 2003; Dango et al., 2011; Duncan et al., 2002; Lee et al., 2005; Ringvoll et al., 2006). We therefore examined the effect of D-2-HG on the activity of ALKBH2 and ALKBH3 using purified recombinant ALKBH2 and ALKBH3 proteins and DNA oligo containing 1-methyldeoxyadenine (1MedA) (Figure S1A). We found that purified ALKBH2 and ALKBH3 rapidly (within 1 min) demethylated (repaired) methylated adenine (Figure S1B). Addition of 0.5 mM D-2-HG resulted in nearly 50% inhibition of ALKBH2 (Figure S1C). This is consistent with a previous observation, showing that D-2-HG inhibits DNA repair enzyme ALKBH2 in vitro, with an IC50 value of 0.424mM (Chowdhury et al., 2011). Similarly, ALKBH3 rapidly (within 1 min) repaired methyl-adenine, a reaction that was also inhibited by D-2-HG (Figure 1A). Although D-2-HG is a relatively weak inhibitor of ALKBH2 and ALKBH3 and may not have significant effect on ALKBH-mediated repair under normal physiological conditions, the high levels of D-2-HG that accumulate in IDH-mutated gliomas [i.e., 5–35 mmol/L in glioma (Dang et al., 2009)] suggest that it could significantly impair the ALKBH function in IDH-mutated cells, like other α-KG-dependent dioxygenases reported to be inhibited by 2-HG (Table S1).
Figure 1. 2-HG inhibits ALKBH2 and ALKBH3 and accumulates DNA damages.

(A) D-2-HG inhibits the activity of ALKBH3 in vitro, with IC50 values being 3.09 mM. Shown are average values of triplicated results with standard deviation (S.D.).
(B) 2-HG accumulation causes decreased DNA adduct repair after MMS treatment. U87-MG cells stably expressing the indicated proteins was treated with 2 mM MMS for 1 hr. After the treatment, the cells were cultured in fresh medium containing no MMS for the indicated time. Genomic DNA was hydrolyzed to nucleotide by enzyme digestion, and was then subjected to LC-MS/MS to determine the concentrations of deoxyadenosine (dA) and 1-methyldeoxyadenosine (1MedA). Shown are average values of triplicated results with standard deviation (S.D.).
(C, D, E) 2-HG accumulation causes increased double strand breaks after MMS treatment. U87-MG and U-373 cells stably expressing the indicated proteins were exposed to increasing concentrations of MMS for 1 hr. After the treatment, the cells were cultured in fresh medium containing no MMS for another 10 hours, and the level of phosphorylated histone variant H2A.X (γ-H2AX) was determined by western blot (C) and immunofluorescence (D). Scale bar:100 μm. The number of γ-H2AX foci was counted from 25 randomly selected cells (E). Shown are average values of triplicated results with standard error of means (SEM).
2-HG inhibits removal of alkylating agent induced DNA damages in glioma cells
To investigate whether ALKBH2 and ALKBH3 could be inhibited by 2-HG in cells, we established U87-MG glioma stable cell lines expressing wild-type or R132H mutant IDH1 at a level similar to that of endogenous IDH1 (Figure 1B). These stable cells were treated with 2 mM methyl methanesulfonate (MMS), which generates 1-methyldeoxyadenosine (1-MedA). Expression of mutant IDH1 significantly delayed the repair kinetics, resulting a seven-fold increase in the half-life of 1-MedA in genomic DNA from 0.52 hr in cells expressing wild-type IDH1 to 3.67 hr in cells expressing R132H mutant IDH1. Unrepaired DNA adduct would cause DNA double-strand breaks (DSBs). Consistently, when compared with cells expressing wild-type IDH1, both U87-MG and U373-MG cells expressing R132H mutant IDH1 exhibited substantially increased DSBs after MMS treatment, as determined by both western blot and immunofluorescence using an antibody to phosphorylated histone variant H2A.X (γ-H2AX, Figures 1C, 1D and 1E, and Supplementary Figure S1D). Moreover, quantitative reverse transcription (qRT)-PCR analysis showed that multiple genes from different DNA repair pathways were significantly (p<0.05) up-regulated in cells expressing mutant IDH1 when compared to control cells expressing wild-type IDH1 (Figure S1E). The endogenous protein levels of neither ALKBH2 nor ALKBH3 were affected by the expression of either wild-type or mutant IDH1 (Figure S1F). Although IDH1 mutant sensitize cells to alkylating agents, cells expressing wild-type or mutant IDH1 responded to UV and IR similarly (Figure S1G). Together, these results demonstrate that tumor-derived mutant IDH1 inhibits the activity of ALKBH enzymes and results in the accumulation of DNA damages in cells exposure to alkylating agents.
Expression of tumor-derived IDH1 mutant sensitizes cells to alkylating agents
E.coli alkB mutants are sensitive to killing by alkylating agents such as MMS, especially during exponentially doubling (Dinglay et al., 2000; Kataoka et al., 1983). This defect can be rescued by the expression of human ALKBH2 and ALKBH3 (Dinglay et al., 2000; Duncan et al., 2002). The finding that D-2-HG inhibits ALKBH2 and ALKBH3 led us to test whether cultured human cells expressing mutant IDH are sensitized to alkylating agents. We exposed both U87-MG and U373-MG glioblastoma cells stably expressing wild-type or R132H mutant IDH1 to N-methyl-N′-nitro-N-nitrosoguanidine (MNNG) or MMS. Cell death and viability were then assessed by flow cytometry analysis (Figure 2A), MTT (3-(4,5-dimerthylthiazol-2,5- diphenyltetrazolium bromide) assay (Figure 2B), and trypan blue exclusion (Figure 2C). Consistently seen in all three assays, either MNNG or MMS treatment decreased cell viability in a dose-dependent manner in both cell lines, but had more significant (p<0.05) killing effects in cells expressing mutant IDH1 compared to cells expressing wild-type IDH1. FACS analysis revealed that treatments with 4 and 5 mM MMS resulted in 36.1% and 61.4% Annexin-PI double positive cells, respectively, in cells expressing R132H mutant, significantly higher than the cells expressing wild-type IDH1 (20.9% and 42.3%, respectively, Figure S2). Knock down ALKBH2 with two different shRNAs in MMS-treated cells increased the death of U87-MG (wild-type IDH1) cells (Figure 2D), indicating a role of ALKBH2 in protecting U87-MG cells from MMS-induced death. In contrast, depletion of ALKBH2 did not synergistically increase MMS-induced cell death in U87-MG (IDH1R132H) cells. This result is consistent with the notion that the activity of ALKBH2 in protecting MMS-induced cell death requires the α-KG-dependent catalytic function that is inhibited by the high level of D-2-HG accumulated in the U87-MG (IDH1R132H) cells.
Figure 2. Expression of tumor-derived mutant IDH1 sensitizes cells to alkylating agents.

U87-MG and U373-MG cells stably expressing the indicated proteins were exposed to increasing concentrations of MNNG or MMS for 1 hour. The cells were cultured in fresh medium containing no alkylating agents for another 47 hours. After that, cell death was assessed by performing flow cytometry analysis (A), MTT assay (B), and trypan blue staining for viable cell counting (C). Shown are average values of triplicated results with standard deviation (S.D.). *denotes the p < 0.05 for cells expressing mutant IDH1 versus wild-type IDH1; n.s.= not significant. (D). Knockdown ALKBH2 sensitize U87-MG cells expressing wild-type, but not mutant IDH1 to MMS treatment. Cell death was assessed by flow cytometry analysis (right panel).
Alkylating agents sensitizing effect of mutant IDH1 is dependent on 2-HG and can be partially reduced by overexpression of ALKBH2 and ALKBH3
To determine directly whether the sensitization to alkylating agents by IDH1 mutation is dependent on 2-HG, we introduced a second mutation into the D-2-HG-producing IDH1R132H mutant to disrupt its binding to α-KG, which is required for the production of D-2-HG through the NADPH-dependent reduction of α-KG (Dang et al., 2009). Six IDH1 double mutants, R132H/T77A, R132H/S94A, R132H/N96A, R132H/Y139A, R132H/K212Q and R132H/T214A were identified whose catalytic activity to produce D-2-HG was markedly reduced (Figure 3A, 3B). We then established U87-MG cells stably expressing IDH1, IDH1R132H, IDH1R132H/T77A, or IDH1R132H/S94A (Figure S3A) and exposed these stable cells to different concentrations of MMS or MMNG. We observed that the sensitization to MMS and MMNG, conferred by R132H mutant IDH1, was completely abolished by second mutations that eliminated 2-HG production (Figure 3C). Supporting this conclusion, disruption of 2HG-producing activity also hinders the activation of DNA repair genes by the mutant IDH1 (Figure S3B and Figure 3D).
Figure 3. Alkylating agents sensitizing effect of mutant IDH1 is dependent on 2-HG.

(A, B) Characterization of 2-HG producing activity of IDH1 double mutants. The activity of various IDH1 double mutants in producing 2-HG was determined by both the rate of NADPH oxidation (A) and GC-MS analysis (B). Peaks at 19.6 min were identified as bis-TBDMSD derived 2-HG by the mass fragment spectra. The bis-TBDMS 2-HG-specific 433-m/z fragment is shown in the insert. Moreover, the peaks at 18.9 min and 20.3 min were identified as the derivatives of aspartate (Asp) and glutamate (Glu), respectively. ** indicates p < 0.01 by student’s t-test between IDH1R132H and IDH1.
(C) U87-MG cells stably expressing the indicated proteins were exposed to increasing concentrations of MNNG or MMS. Cell death was assessed by performing flow cytometry analysis and trypan blue staining for viable cell counting.
(D) The mRNA expression of selected DNA repair genes was tested in U87-MG cells stably expressing the indicated proteins, as determined by qRT-PCR analysis.
(E) HT1080 (IDH1+/−) cell line was generated by knocking-out the R132C allele in parental HT1080 (IDH1+/R132C) cells using TALEN technique. Metabolites extracted from HT1080 (IDH1+/−) and parental HT1080 (IDH1+/R132C) cells were subjected to GC-MS analysis, showing the loss of 2-HG accumulation in HT1080 (IDH1+/−) cells.
(F) HT1080 (IDH1+/−) and parental HT1080 (IDH1+/R132C) cells were exposed to increased concentrations of MNNG or MMS. Cell death was assessed by performing flow cytometry analysis and trypan blue staining for viable cell counting. Shown are average values of triplicated results with standard deviation (S.D.). *denotes the p < 0.05 for HT1080 (IDH1+/−) versus parental HT1080 (IDH1+/R132C) cells.
(G, H) Parental HT1080 (IDH1+/R132C) and TALEN-edited HT1080 (IDH1+/−) cells stably expressing FLAG tagged ALKBH2 or ALKBH3 were established by retrovirus transduction. Expression of ALKBH was verified by western blot (G). The sensitivity of both IDH1+/R132C and IDH1+/− cells to MMS was determined by MTT assay (H).
HT1080 fibrosarcoma cells carry heterozygous R132C mutation in IDH1 (IDH1+/R132C) (Amary et al., 2011). We deleted the R132C mutant allele from HT1080 by TALEN technique and generated the IDH1+/− cell line (Ma et al., 2015). GC-MS analysis confirmed that 2-HG was produced in the parental IDH1+/R132C cells, but was not detectable in the IDH1+/− cells (Figure 3E). We found that deletion of the R132C mutant allele reduced the sensitivity of HT1080 cells to both MMS and MNNG alkylating agents (Figure 3F). We next stably overexpressed ALKBH2 or ALKBH3 in IDH1+/R132C and IDH1+/− HT1080 cells by retrovirus transduction (Figure 3G). We found that over-expression of either ALKBH partially rescued the sensitivity in parental IDH1+/R132C cells, but not in the IDH1+/− cells, to MMS treatment (Figure 3H). Taken together, we conclude that the ability of mutant IDH1 to sensitize cells to DNA alkylating agents is dependent on its catalytic activity to produce 2-HG.
Mutant IDH1 sensitizes cells to therapeutic alkylating drugs
To explore the potential clinical significance of sensitizing effects by mutant IDH to alkylating agents, we examined the responses of cells expressing either wild-type or mutant IDH1 to busulfan, an alkylator which has commonly been used to treat chronic myeloid leukemia (CML) before the advent of imatinib. We found that U87-MG cells expressing R132H mutant IDH1 were significantly more sensitive to busulfan than control cells expressing wild-type IDH1, and that the enhanced sensitivity was completely abolished by the mutations disrupting D-2-HG production (Figure 4A).
Figure 4. Expression of tumor-derived mutant IDH1 sensitizes cells to clinical alkylating agents.

(A, B) U87-MG cells stably expressing the indicated proteins were exposed to different concentrations of busulfan(A), procabazine, CCNU or vincristine (B) for 48 hrs. Cell viability was assessed by flow cytometry analysis (left), and cells were also stained with trypan blue for viable cell counting (right).
(C) U87-MG cells stably expressing the indicated proteins were treated with different concentrations of CCNU, along with 1 mM procarbazine or 500 nM vincristine for 48 hours. Cell viability was assessed by flow cytometry analysis (upper) and trypan blue exclusion (lower). Shown are average values of triplicated results with standard deviation (S.D.). *denotes the p < 0.05 for cells expressing mutant IDH1 versus wild-type IDH1; n.s.= not significant.
(D) Parental HT1080 (IDH1+/R132C) and TALEN-edited HT1080 (IDH1+/−) cells stably expressing ALKBH2 or ALKBH3 were exposed to different concentrations of CCNU for 72 hr. Cell viability was determined by MTT assay. *denotes the p < 0.05 for HT1080 (IDH1+/−) versus parental HT1080 (IDH1+/R132C) cells.
Of three agents in PCV regimen, vincristine inhibits microtubule assembly, and CCNU and procarbazine are DNA alkylating agents. We found that the expression of R132H mutant IDH1 in U87-MG cells caused significant sensitivity to CCNU compared with the expression of wild-type IDH1 (Figure 4B). In contrast, treatment of cell with vincristine, while effectively reduced cell viability, exhibited indistinguishable effect toward cells expressing either the wild-type or mutant IDH1. Moreover, compared to cells treated with CCNU alone, combined exposure to CCNU and procarbazine or CCNU and vincristine had no additive or synergistic effect on the killing of cells expressing mutant or wild-type IDH1 (Figure 4C). Furthermore, overexpression of either ALKBH2 or ALKBH3 partially reduced the death HT1080 cells exposed to CCNU (Figure 4D), supporting the notion that IDH1 mutation and 2-HG accumulation sensitize HT1080 cells to CCNU by inhibiting the activity of ALKBH. The rescue by overexpressed ALKBH did not reach complete, likely due to the high levels of 2-HG accumulated in the cells. These results provide a plausible molecular explanation for the link between PCV benefit and IDH mutation observed the clinical trials. The interpretation of lack of sensitizing effect by IDH mutation to procarbazine needs to be cautious as procarbazine becomes active only after it is metabolized by cytochrome p450 and monoamine oxidase, mainly in the liver (Weinkam and Shiba, 1978), and works poorly in non-hepatic cells lacking high oxidase activity (Swaffar et al., 1989).
DISCUSSION
This study provides two insights into the IDH mutation. First, our results suggest a mechanism by which IDH mutation contributes to tumorigenesis. There is strong evidence that IDH1/2 mutations alter epigenetic regulation in affected cells (Chowdhury et al., 2011; Figueroa et al., 2010; Noushmehr et al., 2010; Sasaki et al., 2012; Xu et al., 2011). We show in this study that in addition to altering epigenetic control, impairment of DNA repair may also contribute to tumorigenesis driven by IDH mutation. Second, the results presented here implicate a targeted therapy for treating patients with IDH1/2-mutated tumors. As demonstrated in this study, glioma cells engineered to or chondrosarcoma cells endogenously express mutant IDH1 are significantly more sensitive to MNNG, MMS, busulfan, and CCNU, compared to cells expressing only wild-type IDH1, suggesting that the classical alkylating agents may be an appropriate and ‘targeted’ therapy for patients with IDH1/2-mutated cancers. This hypothesis is supported by the results of the aforementioned trials showing that 4–6 cycles of PCV were sufficient with RT to double the survival of patients with AO and AOA (Cairncross et al., 2013; Erdem-Eraslan et al., 2013; van den Bent et al., 2013). The PCV benefit was recently linked to IDH mutation (Cairncross et al., 2014). Our study provides a molecular basis for the PCV benefit linked to IDH mutation. Our finding that IDH mutation and 2HG accumulation sensitize cells to CCNU also suggests that whether future inhibitor targeting mutant IDH and blocking the D-2HG production should be used in combination with PCV need to be investigated.
In addition to glioma, IDH1 and IDH2 mutations also occur at high frequency in several other types of human malignancies. There is no obvious reason that sensitization to DNA alkylating agents conferred by mutant IDH and D-2-HG is unique to glioma. We have demonstrated in this study that chondrosarcoma cells harboring IDH1 mutation are sensitive to alkylating agent in a manner that is dependent on the mutant IDH1. A number of FDA-approved DNA alkylating agents, such as CCNU and busulfan, have long been used in clinical for cancer treatment. They may merit for further exploration for treatment of other IDH-mutated tumors.
EXPERIMENTAL PROCEDURES
Please refer to “Supplemental Experimental Procedures” for more detailed information about procedures for protein expression and purification; In vitro ALKBH2 and ALKBH3 activity assay; cell culture, treatment, transfection, viability assay; Metabolite extraction and LC-MS/MS analysis of nucleosides.
Antibodies, plasmids and chemicals
Antibodies against Flag (ShanghaiGenomics), β-actin (Genescript), H2AX (Sigma-Aldrich), IDH1, ALKBH2, ALKBH3 (Epitomics), and phosph-γ-H2AX (Santa Cruz) were purchased commercially. ALKBH2 and ALKBH3 cDNAs were kind gifts from the Han Jiahuai’s Lab, Xiamen University and subcloned into pSJ3 for the expression and purification in E.coli. Wild-type and mutant IDH1 was constructed into pcDNA3.1 for transient expression or pBABE-puro for stable transduction by retrovirus. ALKBH2 and ALKBH3 were constructed into pBABE-hygro for stable transduction. shRNAs targeting ALKBH2 are cloned into pLKO.1, and the sequence is listed in Table S2. MNNG (Tokyo Chemical Industry, TCI), MMS (Sigma-Aldrich), Temozolomide (TCI), busulfan (TCI), procarbazine/CCNU (Selleckchem) and vincristine (Selleckchem) were purchased commercially.
RNA isolation and qRT-PCR analysis
Total RNA was isolated from cultured cells using Trizol reagent (Invitrogen) following the manufacturer’s instructions. RNA was reversely transcribed with oligo-dT primers and preceded to qRT-PCR with gene-specific primers in the presence of SYBR Premix Ex Taq (TaKaRa). β-actin was used as a housekeeping control. Primer sequences are listed in Table S2.
Supplementary Material
Acknowledgments
We thank the members of the Fudan MCB lab and Xiong lab in UNC for discussion and support throughout this study. Lingchao Chen in Fudan and Dale Ramsden in UNC provided for many valuable suggestions and discussions. This work was supported by the 973 Program (No. 2012CB910303, No. 2012CB910101, No. 2011CB910600, No. 2009CB918401), the NSFC grant (No. 81522033 to DY), and International Postdoctoral Exchange Program of Chinese Postdoctoral Council (to PW). J.W. is supported by K12CA120780-04 and a grant from UNC Neurosurgery Research Fund. This work was supported by NIH grants (CA163834 to YX, R01CA108941 to KLG), and Samuel Waxman Foundation (to YX).
Footnotes
Author contributions
Most of the experiments were done by P.W, and designed by Y.X., D.Y., K.G., P.W., J.W., K.H., M.W. and C.P. The manuscript was written by Y.X., D.Y., K.G., J.W. S.M provided IDH1 knockout HT1080 cells. L.Z and J.Y. did the LC/MS/MS analysis.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aas PA, Otterlei M, Falnes PO, Vagbo CB, Skorpen F, Akbari M, Sundheim O, Bjoras M, Slupphaug G, Seeberg E, et al. Human and bacterial oxidative demethylases repair alkylation damage in both RNA and DNA. Nature. 2003;421:859–863. doi: 10.1038/nature01363. [DOI] [PubMed] [Google Scholar]
- Amary MF, Bacsi K, Maggiani F, Damato S, Halai D, Berisha F, Pollock R, O’Donnell P, Grigoriadis A, Diss T, et al. IDH1 and IDH2 mutations are frequent events in central chondrosarcoma and central and periosteal chondromas but not in other mesenchymal tumours. J Pathol. 2011;224:334–343. doi: 10.1002/path.2913. [DOI] [PubMed] [Google Scholar]
- Cairncross G, Wang M, Shaw E, Jenkins R, Brachman D, Buckner J, Fink K, Souhami L, Laperriere N, Curran W, et al. Phase III trial of chemoradiotherapy for anaplastic oligodendroglioma: long-term results of RTOG 9402. J Clin Oncol. 2013;31:337–343. doi: 10.1200/JCO.2012.43.2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairncross JG, Macdonald DR. Successful chemotherapy for recurrent malignant oligodendroglioma. Ann Neurol. 1988;23:360–364. doi: 10.1002/ana.410230408. [DOI] [PubMed] [Google Scholar]
- Cairncross JG, Wang M, Jenkins RB, Shaw EG, Giannini C, Brachman DG, Buckner JC, Fink KL, Souhami L, Laperriere NJ, et al. Benefit from procarbazine, lomustine, and vincristine in oligodendroglial tumors is associated with mutation of IDH. J Clin Oncol. 2014;32:783–790. doi: 10.1200/JCO.2013.49.3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns RA, Mak TW. Oncogenic isocitrate dehydrogenase mutations: mechanisms, models, and clinical opportunities. Cancer Discov. 2013;3:730–741. doi: 10.1158/2159-8290.CD-13-0083. [DOI] [PubMed] [Google Scholar]
- Chowdhury R, Yeoh KK, Tian YM, Hillringhaus L, Bagg EA, Rose NR, Leung IKH, Li XS, Woon ECY, Yang M. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO reports. 2011;12:463–469. doi: 10.1038/embor.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dango S, Mosammaparast N, Sowa ME, Xiong LJ, Wu F, Park K, Rubin M, Gygi S, Harper JW, Shi Y. DNA unwinding by ASCC3 helicase is coupled to ALKBH3-dependent DNA alkylation repair and cancer cell proliferation. Mol Cell. 2011;44:373–384. doi: 10.1016/j.molcel.2011.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinglay S, Trewick SC, Lindahl T, Sedgwick B. Defective processing of methylated single-stranded DNA by E. coli AlkB mutants. Genes Dev. 2000;14:2097–2105. [PMC free article] [PubMed] [Google Scholar]
- Duncan T, Trewick SC, Koivisto P, Bates PA, Lindahl T, Sedgwick B. Reversal of DNA alkylation damage by two human dioxygenases. Proc Natl Acad Sci U S A. 2002;99:16660–16665. doi: 10.1073/pnas.262589799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdem-Eraslan L, Gravendeel LA, de Rooi J, Eilers PH, Idbaih A, Spliet WG, den Dunnen WF, Teepen JL, Wesseling P, Sillevis Smitt PA, et al. Intrinsic molecular subtypes of glioma are prognostic and predict benefit from adjuvant procarbazine, lomustine, and vincristine chemotherapy in combination with other prognostic factors in anaplastic oligodendroglial brain tumors: a report from EORTC study 26951. J Clin Oncol. 2013;31:328–336. doi: 10.1200/JCO.2012.44.1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falnes PO, Johansen RF, Seeberg E. AlkB-mediated oxidative demethylation reverses DNA damage in Escherichia coli. Nature. 2002;419:178–182. doi: 10.1038/nature01048. [DOI] [PubMed] [Google Scholar]
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka H, Yamamoto Y, Sekiguchi M. A new gene (alkB) of Escherichia coli that controls sensitivity to methyl methane sulfonate. J Bacteriol. 1983;153:1301–1307. doi: 10.1128/jb.153.3.1301-1307.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, Jin SG, Cai S, Chen Y, Pfeifer GP, O’Connor TR. Repair of methylation damage in DNA and RNA by mammalian AlkB homologues. J Biol Chem. 2005;280:39448–39459. doi: 10.1074/jbc.M509881200. [DOI] [PubMed] [Google Scholar]
- Ma S, Jiang B, Deng W, Gu ZK, Wu FZ, Li T, Xia Y, Yang H, Ye D, Xiong Y, et al. D-2-hydroxyglutarate is essential for maintaining oncogenic property of mutant IDH-containing cancer cells but dispensable for cell growth. Oncotarget. 2015;6:8606–8620. doi: 10.18632/oncotarget.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010;17:510–522. doi: 10.1016/j.ccr.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JCH, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. An Integrated Genomic Analysis of Human Glioblastoma Multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringvoll J, Nordstrand LM, Vågbø CB, Talstad V, Reite K, Aas PA, Lauritzen KH, Liabakk NB, Bjørk A, Doughty RW. Repair deficient mice reveal mABH2 as the primary oxidative demethylase for repairing 1meA and 3meC lesions in DNA. The EMBO journal. 2006;25:2189–2198. doi: 10.1038/sj.emboj.7601109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohle D, Popovici-Muller J, Palaskas N, Turcan S, Grommes C, Campos C, Tsoi J, Clark O, Oldrini B, Komisopoulou E, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340:626–630. doi: 10.1126/science.1236062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M, Knobbe CB, Munger JC, Lind EF, Brenner D, Brustle A, Harris IS, Holmes R, Wakeham A, Haight J, et al. IDH1(R132H) mutation increases murine haematopoietic progenitors and alters epigenetics. Nature. 2012;488:656–659. doi: 10.1038/nature11323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedgwick B, Bates PA, Paik J, Jacobs SC, Lindahl T. Repair of alkylated DNA: recent advances. DNA repair. 2007;6:429–442. doi: 10.1016/j.dnarep.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Swaffar DS, Horstman MG, Jaw JY, Thrall BD, Meadows GG, Harker WG, Yost GS. Methylazoxyprocarbazine, the active metabolite responsible for the anticancer activity of procarbazine against L1210 leukemia. Cancer Res. 1989;49:2442–2447. [PubMed] [Google Scholar]
- van den Bent MJ, Brandes AA, Taphoorn MJ, Kros JM, Kouwenhoven MC, Delattre JY, Bernsen HJ, Frenay M, Tijssen CC, Grisold W, et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J Clin Oncol. 2013;31:344–350. doi: 10.1200/JCO.2012.43.2229. [DOI] [PubMed] [Google Scholar]
- Wang F, Travins J, DeLaBarre B, Penard-Lacronique V, Schalm S, Hansen E, Straley K, Kernytsky A, Liu W, Gliser C, et al. Targeted inhibition of mutant IDH2 in leukemia cells induces cellular differentiation. Science. 2013;340:622–626. doi: 10.1126/science.1234769. [DOI] [PubMed] [Google Scholar]
- Weinkam RJ, Shiba DA. Metabolic activation of procarbazine. Life Sci. 1978;22:937–945. doi: 10.1016/0024-3205(78)90358-2. [DOI] [PubMed] [Google Scholar]
- Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Xiao MT. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of [alpha]-Ketoglutarate-Dependent Dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, et al. IDH1andIDH2Mutations in Gliomas. New England Journal of Medicine. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Ye D, Guan KL, Xiong Y. IDH1 and IDH2 mutations in tumorigenesis: mechanistic insights and clinical perspectives. Clin Cancer Res. 2012;18:5562–5571. doi: 10.1158/1078-0432.CCR-12-1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, Yu W, Li Z, Gong L, Peng Y, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009;324:261–265. doi: 10.1126/science.1170944. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.