

In this study, a proteomic approach links the J-domain chaperone auxilin, which uncoats clathrin-coated vesicles, to the other major coat complexes in the cell (COPII and COPI). Genetic and biochemical studies support the proposal that auxilin facilitates vesicle traffic in the early secretory pathway.
Abstract
Coat protein complexes contain an inner shell that sorts cargo and an outer shell that helps deform the membrane to give the vesicle its shape. There are three major types of coated vesicles in the cell: COPII, COPI, and clathrin. The COPII coat complex facilitates vesicle budding from the endoplasmic reticulum (ER), while the COPI coat complex performs an analogous function in the Golgi. Clathrin-coated vesicles mediate traffic from the cell surface and between the trans-Golgi and endosome. While the assembly and structure of these coat complexes has been extensively studied, the disassembly of COPII and COPI coats from membranes is less well understood. We describe a proteomic and genetic approach that connects the J-domain chaperone auxilin, which uncoats clathrin-coated vesicles, to COPII and COPI coat complexes. Consistent with a functional role for auxilin in the early secretory pathway, auxilin binds to COPII and COPI coat subunits. Furthermore, ER–Golgi and intra-Golgi traffic is delayed at 15°C in swa2Δ mutant cells, which lack auxilin. In the case of COPII vesicles, we link this delay to a defect in vesicle fusion. We propose that auxilin acts as a chaperone and/or uncoating factor for transport vesicles that act in the early secretory pathway.
INTRODUCTION
Vesicular traffic begins when GTPases of the Sar/Arf family recruit protein coat complexes from the cytosol onto membranes. There are three major coat complexes in the cell: COPII, COPI, and clathrin. At the endoplasmic reticulum (ER), the COPII coat complex initiates the formation of ER-derived vesicles (Barlowe and Miller, 2013). This coat consists of an inner shell (Sec23/Sec24) that sorts cargo and an outer shell (Sec13/Sec31) or cage (Gurkan et al., 2006). The inner shell, or coat adaptor (Sec23/Sec24), is recruited to the ER by the activated form of Sar1 (Sar1-GTP) via an interaction with the Sec23 subunit, while the Sec24 subunit directly binds to and captures cargo into the nascent vesicle. Recruitment of the outer shell of the coat, Sec13/Sec31, leads to coat polymerization and vesicle budding (Barlowe and Miller, 2013).
It was initially thought that the vesicle-tethering machinery, which links the vesicle to its target membrane, recognizes and binds to an uncoated vesicle. However, in 2007, we reported the unexpected finding that an ER–Golgi tethering factor, TRAPPI, recognizes a COPII vesicle by binding the coat adaptor that sorts cargo into the vesicle (Cai et al., 2007). This result was surprising, because it was thought at the time that all vesicles uncoat soon after they bud. The COPII coat, in particular, was considered to be unstable and to be released from the vesicle without the aid of uncoating factors (Antonny et al., 2001). Our observation not only implied that the COPII coat adaptor plays a role in targeting a vesicle to its correct intracellular destination, it also suggested there was an active mechanism for uncoating these vesicles. Since we reported these findings, other coat complexes (AP-3 adaptor complex, retromer, and COPI) have also been shown to play a role in vesicle targeting (Angers and Merz, 2009; Wassmer et al., 2009; Zink et al., 2009).
In this paper, we describe a proteomic screen and genetic studies that connect the J-domain chaperone auxilin, which is required to uncoat clathrin-coated vesicles (Lemmon, 2001; Eisenberg and Greene, 2007), to the COPII and COPI coat complexes. In support of the proposal that auxilin may facilitate the uncoating of COPII and COPI vesicles, a proteomic approach has revealed a change in the assembly state of these coats in auxilin-depleted yeast cells. Genetic and biochemical studies confirm that auxilin interacts with subunits of both complexes. Additionally, the analysis of a swa2∆ mutant, which lacks auxilin, leads to a delay in ER–Golgi and intra-Golgi traffic at 15°C. In vitro transport studies link this delay to a defect in COPII vesicle fusion. Together these findings reveal an unexpected role for auxilin in the early secretory pathway.
RESULTS
Proteomic analysis of clathrin-coated vesicle fractions from auxilin-depleted cells links auxilin to COPII and COPI coat structures
To identify additional components in clathrin-coated vesicles (CCVs), including potential cargo and novel adaptor proteins, we fractionated CCVs from yeast (Lemmon et al., 1988) and subjected the peak clathrin column fractions from a Sephacryl S-1000 column to mass spectrometry proteomic analysis (see Materials and Methods for details). As the depletion of auxilin might stabilize CCVs, this analysis was performed with both a wild-type strain (SL1463) and a GAL1p:SWA2 strain (SL4827) (Pishvaee et al., 2000) that was depleted for auxilin by shifting cells from galactose to glucose medium (Supplemental Tables S3 and S4). As expected, major peptide hits from both the Swa2-depleted and wild-type strains included clathrin heavy and light chains, the subunits of the trans-Golgi network (TGN)/endosomal AP-1 adaptor complex, and several membrane proteins known, or likely, to be sorted into TGN/endosomal-associated CCVs (see Supplemental Tables S5 and S6 and Figure 1). Neither endocytic coat proteins or actin-associated endocytic factors were isolated in the wild-type or the auxilin-depleted strain, supporting prior studies indicating that clathrin-mediated endocytic vesicle uncoating is rapid and mediated by other factors, such as synaptojanin Sjl2, Ark1/Prk1 kinases, Arf3/Gts1/Lsb5, and cofilin (Sekiya-Kawasaki et al., 2003; Toret et al., 2008; Boettner et al., 2012).
FIGURE 1:

Major coated vesicle components identified in the proteomic analysis of Sephacryl S-1000 fractions from wild-type and auxilin-depletion strains. Shown are clathrin coats and the TGN/endosome adaptor AP-1, ER/Golgi coats COPI and COPII, and several COP-vesicle membrane proteins. Red, hits identified only in the auxilin-depletion strain; yellow, hits found in both wild type and swa2∆; green, found only in wild type; and white, not detected.
Subtraction of the proteins identified in the wild-type CCV fractions from those found in the auxilin-depleted fractions allowed us to eliminate many proteins specific to clathrin-mediated transport, as well as nonspecific hits, such as metabolic enzymes, viral particles, ribosomal subunits, and proteins involved in translation (Supplemental Tables S5–S7). A number of late Golgi/endosomal/vacuolar membrane cargoes were found in the auxilin-only data set (Supplemental Table S7), but several known to be sorted by clathrin may merely be less abundant or less stable CCV components. Although a retromer subunit (Vps17) and eisosome components (Lsp1 and Pil1) were found in the column fractions from the auxilin-depleted lysate, other subunits from these complexes (Pep8/Vps26 and Sur7, respectively) were also found in the wild-type CCV fractions, suggesting these constituents are not auxilin specific.
To our surprise, instead of identifying novel CCV adaptor proteins and cargo, we found proteins associated with COPII- and COPI-coated vesicles in the auxilin-depleted fractions (see Supplemental Tables S5–S7; Figure 1). These included the subunits of the inner shell of the COPII coat complex, Sec23 and Sec24, and the COPI coat subunits Sec26, Ret2, and Sec27 (Brandizzi and Barlowe, 2013; Faini et al., 2013). Additionally, several membrane proteins present in COPII vesicles, such as p24 family members (Figure 1 and Supplemental Table S7; Schuiki and Volchuk, 2012; Hirata et al., 2013) and the Erv14 and Emp47 ER cargo receptors (Sato and Nakano, 2002; Herzig et al., 2012), were only found in the auxilin-depleted fractions. Together these data suggest that COPII- and COPI-coated vesicles copurify with CCVs in swa2Δ mutant fractions.
The CCV purification protocol and proteomics were also applied to a chc1∆ strain expressing auxilin (SWA2). As expected, clathrin and its adaptors were absent in chc1∆ SWA2 Sephacryl S-1000 fractions. Importantly, no significant hits for COPII vesicle ER vesicle membrane proteins or COP coat subunits were obtained. Many of the common/nonspecific hits from the CHC1 SWA2 and the auxilin-depletion strains, however, were also found in the same fractions from the chc1∆ strain (V.A.S. and S.K.L., unpublished observations). These data indicate that the early-stage vesicle transport proteins only significantly accumulate in the absence of auxilin. Thus, in addition to auxilin’s known function for uncoating clathrin-coated vesicles, its modulation of the assembly state of the COPII and COPI coat complexes is also implicated by this analysis.
The loss of SWA2Δ leads to synthetic growth defects when combined with mutations in subunits of the COPII and COPI coat complexes
Concurrent with the proteomic study described above, we used a genetic approach to identify factors that could interact with COPII-coated vesicles. To our surprise, when we crossed the swa2Δ mutant to three different temperature-sensitive (ts) mutants (sec23-1, sec24-1, and sec31-1) that harbor mutations in subunits of the COPII coat complex (Barlowe et al., 1994), the haploid double mutants we obtained (swa2Δ sec23-1, swa2Δ sec24-1, and swa2Δ sec31-1) displayed growth defects that were more pronounced than either of the single mutants alone (Figure 2A). For the swa2Δ sec23-1 mutant, the exaggerated growth defect was most pronounced at 25°C, while the swa2Δ sec24-1 and swa2Δ sec31-1 double mutants were more impaired for growth at 30 and 34°C, respectively. Similar results were obtained when we crossed the swa2Δ mutant to a ts mutant that harbored a mutation in the Sec21 COPI coat subunit (sec21-1), while a double mutant with a mutation in another COPI subunit, Sec27, was only slightly impaired (Figure 2B). Together these findings indicate that SWA2 genetically interacts with genes encoding subunits of the COPII and COPI coat complexes.
FIGURE 2:

Deletion of SWA2Δ exacerbates the ts growth defects of sec mutants that harbor mutations in COPII (A) and COPI (B) coat subunits. Yeast cells were cultured at 25°C in YPD medium to early stationary phase before the cells (∼1 × 108 cells/ml) were serially diluted (10-fold) and spotted onto YPD plates. The plates were incubated at the indicated temperatures for 4–7 d.
The tetratricopeptide repeat domain in auxilin binds directly to the N-terminus of the COPII coat subunit Sec31
The experiments described above prompted us to determine whether auxilin physically interacts with subunits of the COPII and COPI coat complexes. Initial binding studies were performed with the COPII coat, as we can purify sufficient amounts of each subunit of this coat complex from bacteria for direct binding experiments. Genetics linked auxilin to the Sec23, Sec24, and Sec31 subunits (Figure 2), while proteomics linked auxilin to Sec23 and Sec24 (Figure 1). Given that auxilin is known to interact with clathrin, which forms the cage of clathrin-coated vesicles (Ungewickell et al., 1995), we initiated binding studies with the COPII cage subunits Sec31 and Sec13 (Zanetti et al., 2012; Lord et al., 2013). Because full-length Sec31 is a very large protein and not expressed well in bacteria, binding studies were performed with fragments of Sec31 (aa 1–500, aa 501–878, aa 879–1114, and aa 1115–1274) fused to glutathione S-transferase (GST). His6-auxilin bound directly to GST-Sec31 (aa 1–500), and not the other fragments of Sec31 (Figure 3A). Additionally, the binding of His6-Auxilin to GST-Sec31 (aa 1–500) was concentration dependent (Figure 3B). Consistent with our proteomic data, we also observed an interaction with auxilin and GST-Sec23 and GST-Sec24, but not GST or GST-Sec13 (Figure 3A and Supplemental Figure S1, A and B). Thus in vitro binding studies demonstrate that auxilin directly interacts with multiple COPII coat subunits.
FIGURE 3:
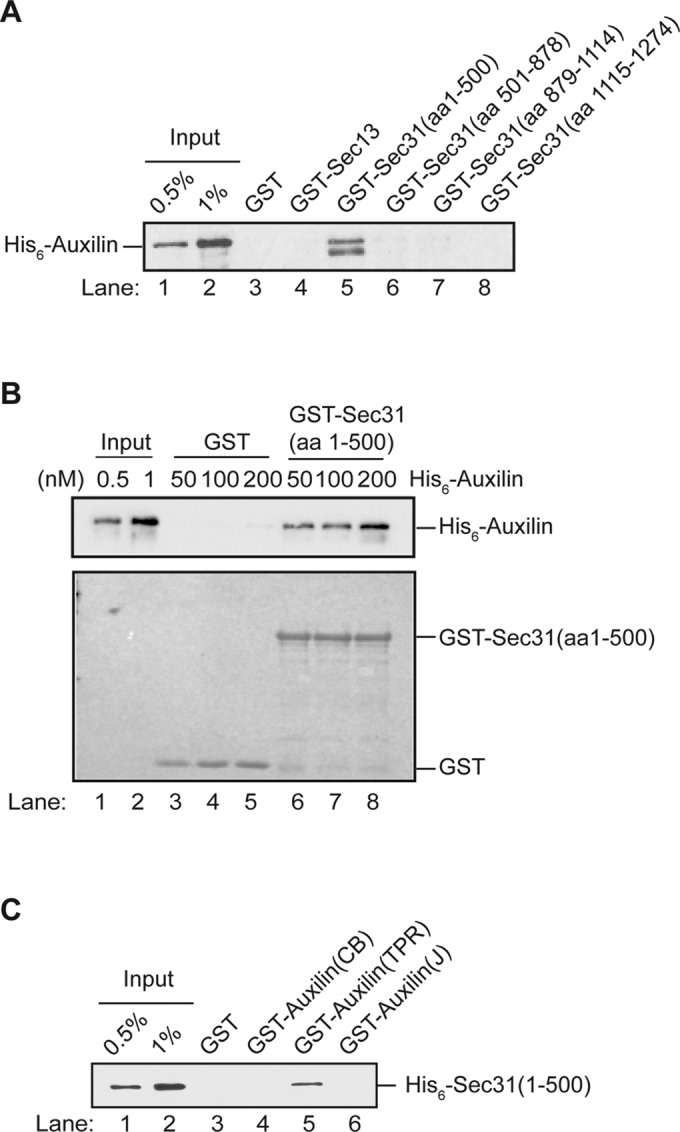
The N-terminus of Sec31 directly binds to the TPR domain of auxilin. (A) Purified recombinant GST, GST-Sec13, and fragments of Sec31 (aa 1–500, 501–878, 879–1114, 1115–1274) fused to GST were prebound to glutathione beads and incubated with His6-auxilin, washed, and analyzed by Western blot analysis using anti-His6 antibody. (B) Top, GST and GST-Sec31 (aa 1–500) were incubated with His6-auxilin at different concentrations and processed for Western blot analysis. Bottom, Ponceau S stain of the binding reactions shows equal loading of GST and GST-Sec31 (aa 1–500). (C) Purified GST and GST fusion proteins were incubated with His6-Sec31 (aa 1–500) and pull-downs with glutathione beads were processed as described in Materials and Methods. The GST fusion proteins include GST-auxilin-CB (clathrin-binding region, aa 1–362), GST-auxilin-TPR domain (aa 363–561), and GST-auxilin-J domain (aa 562–668).
Yeast auxilin has a N-terminal clathrin-binding domain followed by a tetratricopeptide repeat (TPR) domain and a C-terminal J domain that stimulates the ATPase activity of Ssa1 (Pishvaee et al., 2000; Xiao et al., 2006). To determine which domain of auxilin binds to the COPII coat complex, we incubated His6-Sec31 (aa1–500) with GST fusions of each of these regions. His6-Sec31 (aa 1–500) bound to the TPR domain, but not to the other two domains of auxilin (Figure 3C). TPR domains can serve as platforms for protein–protein interactions (D’Andrea and Regan, 2003). Interestingly, deletion of the TPR domain of auxilin but not the N-terminal clathrin-binding region has been reported to result in a growth defect (Xiao et al., 2006). Because disrupting the early secretory pathway impairs growth, the observation that Sec31 binds to a domain in auxilin that is important for growth is consistent with the possibility that auxilin acts on the early secretory pathway.
Auxilin coprecipitates with the COPI coat subunit called COP1
Our proteomics and genetic experiments have also linked auxilin to the COPI coat complex. However, because we were unable to express several of the COPI coat cage subunits in bacteria, we asked whether this coat complex coprecipitates with auxilin. To do this, we precipitated the COPI complex with immunoglobulin G (IgG)–Sepharose beads from a lysate containing tandem affinity purification (TAP)-tagged COP1. Bound protein was eluted, and then Western blot analysis was performed (Figure 4A). Auxilin only coprecipitated with TAP-COP1 from tagged (Figure 4A, top left, lanes 4, 6, and 8) but not untagged lysate (lanes 3, 5, and 7). Furthermore, the COPII coat subunit Sec24, which was used as a specificity control, was not detected in the precipitate (Figure 4A, left bottom panel). As an additional specificity control, we examined another auxilin binding partner, clathrin. When clathrin was precipitated from a lysate containing TAP-tagged clathrin heavy chain (Chc1; Figure 4B), auxilin (Figure 4B, top left) but not the COPI coat subunit Sec28 (Figure 4B, bottom left) was precipitated. Together these findings indicate that the coprecipitation of COP1 with auxilin is specific.
FIGURE 4:

Auxilin coprecipitates with the COPI coat complex. (A) Left, increasing amounts of lysate, prepared from a COP1-TAP–tagged (SFNY1415) or untagged (SFNY 998) strain, were incubated with 30 μl of IgG–Sepharose beads for 2 h at 4°C. The beads were washed, eluted, and then processed for Western blot analysis using anti-auxilin and anti-Sec24 antibodies (1:1000 dilution). Right, anti-coatomer antibody was used to monitor the presence of COP1-TAP in the IgG pull-down. (B) The same as A, but the lysate was prepared from a Chc1-TAP–tagged strain (SFNY1741) and processed for Western blot analysis using anti-auxilin and anti-Sec28 (left) antibodies, or anti-Chc1 antibody (right) at 1:1000 dilution.
The loss of auxilin disrupts secretion
If the interaction of auxilin with the COPII and COPI coat complexes is functionally important, the loss of SWA2Δ should lead to defects in general secretion. While previous studies showed a defect in TGN-mediated pro-α-factor processing in the swa2Δ mutant, no obvious effect on the kinetics of secretion was observed at 30°C (Gall et al., 2000). As trafficking defects in the early secretory pathway impair growth, we reasoned that defects in secretion may only be revealed in swa2Δ cells at a temperature at which growth is dramatically impacted. Consistent with earlier studies, we observed that the swa2Δ mutant grows slowly at several temperatures (Gall et al., 2000; Du et al., 2001) but is most impaired at 15°C (Figure 5A). Therefore we performed our experiments at this lower temperature.
FIGURE 5:

General secretion is delayed in swa2Δ cells at 15°C. (A) The growth of the swa2Δ mutant (SFNY2683) was compared with wild type (SFNY1842) on a YPD plate at 15°C. (B) Secretion analysis: Left, wild type (SFNY1842; lanes 1–3) and the swa2Δ mutant (SFNY2683; lanes 4–6) were incubated for 60 min at 15°C and then pulse labeled at the same temperature with [35S]ProMix for the indicated time points. Proteins secreted into the medium were analyzed as described in Materials and Methods. Secreted p150 is marked. Right, quantification of the percent secretion of p150 into the medium at 15 and 20 min from three independent experiments. Error bars represent SD. *, p < 0.05, Student’s t test.
The swa2Δ mutant and its isogenic wild-type strain were shifted to 15°C for 60 min and then labeled with [35S]ProMix at the same temperature. Proteins secreted into the medium were collected at various times after labeling and analyzed on a 6% SDS polyacrylamide gel. By 15 min, several proteins, including p150, were secreted into the medium in wild-type but not swa2Δ cells (Figure 5B, left, lanes 1 and 4). By 25 min, the observed delay in secretion in the mutant was still pronounced (Figure 5B, compare lanes 3 and 6). Quantification of the p150 band (Figure 5B, right) from three separate experiments revealed a dramatic delay in secretion in the mutant at the 15- and 20-min time points, confirming that the loss of auxilin leads to an obvious delay in general secretion.
The loss of auxilin delays both ER–Golgi and intra-Golgi traffic in vivo
If the delay in secretion in the swa2Δ mutant is a consequence of disrupting COPII and COPI function, ER–Golgi and intra-Golgi traffic should be impaired. To address this possibility, we monitored the trafficking of the vacuolar protease carboxypeptidase Y (CPY). CPY traffics from the ER (p1 form) to the early (p1′) and late Golgi (p2) before it is proteolytically cleaved to the mature form in the vacuole (Stevens et al., 1982). For monitoring the trafficking of CPY in wild type and the swa2Δ mutant, cells were preincubated at 15°C for 1 h, pulse labeled for 10 min, and chased for various times at 15°C (Figure 6A). A delay in the conversion of p1 to p2 CPY was observed in the swa2Δ mutant throughout the chase period (Figure 6A, compare lanes 1–6 with lanes 7–12; see Figure 6B for quantification). Additionally, we also observed that CPY migrated more heterogeneously in the mutant as it was processed from the p1 to the p2 form, suggesting that some CPY may be trapped as p1′ CPY, a form which is difficult to see in wild type (Figure 6A, compare lanes 3–6 with 10–12). Together these findings imply that the loss of auxilin leads to a kinetic delay in the trafficking of CPY between the ER and Golgi and within the Golgi complex. As extensive studies with several different clathrin mutants have shown that traffic through the early secretory pathway occurs normally in the absence of clathrin function (Payne and Schekman, 1985; Payne et al., 1987; Seeger and Payne, 1992a, b; Lemmon et al., 1991), the most likely interpretation of our data is that the observed delay in secretion in the swa2Δ mutant is a consequence of disrupting COPII and COPI function.
FIGURE 6:

ER–Golgi and intra-Golgi traffic are delayed in the swa2Δ mutant. (A) CPY processing: wild type (SFNY1842; lanes 1–6) and the swa2Δ mutant (SFNY2683; lanes 7–12) were shifted to 15°C for 60 min, pulse labeled for 10 min, and chased for the indicated times. CPY was immunoprecipitated from lysates and processed as described in Materials and Methods. The p1 (67 kDa), p2 (69 kDa), and mature (m; 61 kDa) forms of CPY are indicated. (B) Quantification of the ratio of p2CPY/p1CPY from three independent experiments. Error bars represent SD. *, p < 0.05, Student’s t test. (C) Invertase processing and transport: wild-type and swa2∆ cells were shifted to 15°C for 1 h and then incubated at the same temperature for 30 min in low-glucose medium (to induce extracellular invertase expression) in the presence of [35S]ProMix. The cells were converted to spheroplasts and pelleted. Then invertase was immunoprecipitated from the cell pellet fraction (Internal; lanes 1–3) and spheroplast supernatant fractions (External; lanes 4 and 5). Cytoplasmic invertase, glycosylated ER, and Golgi forms of invertase are indicated. Lane 3, a darker exposure of sample shown in lane 2.
To confirm this observation with a second protein that trafficks through the early secretory pathway, we monitored the trafficking of the secreted protein invertase. There are two forms of invertase, a cytoplasmic and a secreted form. The cytoplasmic form is constitutively made, while the secreted form is derepressed in low-glucose-containing medium (Carlson and Botstein, 1982). Secreted invertase is translocated into the lumen of the ER, where it is N-glycosylated to yield an 80-kDa ER form. Before it is secreted into the periplasm, it is further glycosylated in the Golgi to a high-molecular-weight form that migrates heterogeneously on an SDS polyacrylamide gel (Newman and Ferro-Novick, 1987). To determine whether the loss of auxilin disrupts invertase secretion, we shifted wild-type and swa2Δ cells to 15°C for 1 h before derepressing invertase for 30 min at 15°C in YP medium with 0.1% glucose in the presence of [35S]ProMix. At the end of the incubation, the cells were washed, converted to spheroplasts, and pelleted, and invertase was immunoprecipitated from the supernatant (external) and pelleted (internal) fractions. The swa2Δ mutant accumulated more of the ER form of invertase (Figure 6C, compare lanes 1 and 2), indicating a delay in traffic from the ER to the Golgi complex. A higher-molecular-weight Golgi form of invertase (Figure 6C, lanes 2 and 3) was also observed. Some of this form of invertase was secreted (lane 5). The Golgi form of invertase synthesized in the mutant migrated more slowly than the Golgi form in wild-type cells (Figure 6C, compare lanes 3–5), suggesting a delay in Golgi traffic. Together these findings indicate that the loss of auxilin delays vesicle traffic between the ER and Golgi and within the Golgi complex, demonstrating a role for auxilin in the early part of the secretory pathway. We speculate that this function for auxilin was uncovered at 15°C, because cells grow more slowly at this temperature, which may delay the dissociation of the coat and unmask this early secretory defect.
Auxilin is required for COPII vesicle fusion in vitro
Next we wanted to address the question of which step in COPII vesicle traffic requires auxilin. COPII vesicle budding, tethering, and fusion can be differentiated in an in vitro transport assay that reconstitutes vesicle traffic from the ER to the Golgi complex (Ruohola et al., 1988; Lian and Ferro-Novick, 1993). For measuring ER–Golgi traffic in vitro, a radiolabeled cargo marker ([35S]prepro-α-factor) is first translocated into ER membranes that are retained within wild-type or swa2∆ permeabilized yeast cells (PYCs). Subsequent to translocation, the signal sequence of prepro-α-factor is cleaved in the ER, and the protein is N-glycosylated. Glycosylated pro-α-factor, retained within the PYCs, is then incubated with an S1 fraction (cytosol and Golgi membranes) that was obtained from either wild type or the swa2Δ mutant, respectively. Transport of pro-α-factor to the Golgi is monitored with the solid-phase lectin absorbant concanavalin A (ConA)–Sepharose, which binds only glycosylated pro-α-factor. Vesicle budding is measured as the ConA precipitable counts that are released from the PYCs, while membrane fusion is assessed with an anti outer-chain antibody that recognizes the Golgi form of carbohydrate on pro-α-factor. As shown in Figure 7A, COPII vesicle fusion, but not vesicle budding, was decreased in the absence of auxilin.
FIGURE 7:

The swa2Δ mutant is defective in COPII vesicle fusion but not budding or tethering in vitro. (A) Vesicle budding (left) and fusion (right) were measured in fractions prepared from wild type and the swa2Δ mutant as described in Materials and Methods. Error bars represent SEM, n = 3. *, p < 0.05, Student’s t test. (B) Top, fractionation of released transport vesicles: sucrose velocity gradient fractionation pattern of vesicles formed in vitro from wild-type permeabilized donor cells and cytosol. Bottom, vesicle tethering: the transport assay was performed with wild-type donor cells and a wild-type S1 fraction (black line) or swa2Δ mutant donor cells and a swa2Δ mutant S1 fraction (dotted line). At the end of the assay, the permeabilized donor cells were pelleted, and the supernatant was fractionated on a sucrose velocity gradient.
To address whether the observed defect in fusion in the swa2Δ mutant was direct, or an indirect consequence of the inability of the COPII vesicles to bind to the Golgi, we performed a vesicle-tethering assay (Barrowman et al., 2000). Transport reactions were performed as in Figure 7A, except that free vesicles (Figure 7B, top) were separated from vesicles that bound to the Golgi on a sucrose velocity gradient (Figure 7B, bottom). COPII vesicles formed from the swa2Δ mutant or wild-type fractions bound to the Golgi with comparable efficiency (Figure 7B, bottom). The bound vesicles in the swa2Δ mutant, however, were incompetent for fusion (Figure 7A), indicating that the loss of auxilin leads to a specific defect in COPII vesicle fusion.
DISCUSSION
The J-domain protein auxilin is primarily known for its role as a cofactor for Hsc70 in uncoating clathrin-coated vesicles (Lemmon, 2001; Eisenberg and Greene, 2007; Kirchhausen et al., 2014). It binds to CCVs after scission and then recruits Hsc70 to the clathrin lattice via its J domain. Hsc70 interacts with the C-terminal region of the clathrin triskelion tripod for rapid ATP-dependent uncoating of clathrin-coated vesicles (Rapoport et al., 2008), a prerequisite for the fusion of the vesicle with its target membrane. Additional studies suggest that auxilin and Hsc70 have other roles during clathrin-mediated endocytosis (Eisenberg and Greene, 2007). For example, at the cell surface, auxilin plays a role in the formation of CCVs by facilitating the exchange of clathrin during coated pit invagination and bud constriction (Wu et al., 2001; Newmyer et al., 2003; Yim et al., 2005; Sever et al., 2006). Auxilin and Hsc70 are also thought to have a chaperone role for the clathrin triskelion after it dissociates from CCVs, preventing it from aggregating in the cytosol and allowing it to rebind to uncoated membranes (Eisenberg and Greene, 2007).
Auxilin and Hsc70 could have similar roles during COPII and COPI vesicle transport. These proteins may perform a chaperone role for individual subunits of the COPII coat or for the entire COPII complex. As vesicle budding was unaffected in vitro with fractions prepared from swa2∆ cells, the data presented in our studies imply that auxilin is not required for COPII vesicle formation. The finding that auxilin facilitates membrane traffic in the early secretory pathway is consistent with a published fractionation study from our lab showing that auxilin cofractionates with ER membranes on sucrose gradients (Du et al., 2001).
Our previous studies demonstrated that the uncoating of COPII vesicles is highly regulated, occurring after vesicles tether to the Golgi (Cai et al., 2007; Lord et al., 2011). In this paper, we show that, in the absence of auxilin, COPII vesicle tethering to the Golgi is unaffected in vitro, but fusion is blocked. This finding is consistent with a role for auxilin in the uncoating of docked COPII vesicles before fusion. Purified recombinant His6-auxilin was unable to restore fusion activity to swa2∆ mutant fractions (unpublished data), but this may be because recombinant auxilin is not phosphorylated in bacteria. It has been speculated that the activity of auxilin, a known phosphoprotein, is regulated by phosphorylation (Gall et al., 2000; Lemmon, 2001). Phosphorylation of specific sites on auxilin may control the timing or rate at which different classes of vesicles uncoat, providing an additional layer of regulation to membrane fusion. Although our findings are consistent with a role for auxilin in COPII vesicle uncoating, we were unable to directly demonstrate a role for auxilin in this process. COPII cages from yeast coat subunits are not readily formed in vitro, and initial attempts to examine auxilin function in cage assembly/disassembly has not been straightforward (R. Lakshminarayan and E. Miller, personal communication). Nevertheless, our proteomics analysis showing accumulation of both COPII and COPI coat proteins and ER vesicle membrane cargo in the CCV fractions from auxilin-depleted cells implies that auxilin regulates the assembly state of COPII- and COPI-coated structures.
In summary, we have used here a variety of approaches to show that auxilin plays a role in the early secretory pathway in yeast. Further studies are underway to determine whether auxilin plays a similar early secretory role in animal cells. This is of significant interest, because several mutations in DNAJC6, which encodes the nerve specific auxilin, were recently found to cause recessive juvenile parkinsonism. Mutations in DNAJ26, which encodes the ubiquitous auxilin GAK, are implicated in Parkinson’s disease susceptibility (Koutras and Braun, 2014). It is speculated that such auxilin defects affect clathrin-mediated endocytosis at the synapse and thus dopamine receptor recycling. However, based on our findings, it will be important to consider whether early secretory pathway defects may be associated with these clinical manifestations of auxilin deficiency.
MATERIALS AND METHODS
Strains, growth conditions, and antibodies
All yeast and bacterial strains used in this study are listed in Supplemental Tables S1 and S2. Yeast cells were grown in YP (1% yeast extract, 2% peptone) plus 2% dextrose (YPD) or other carbon sources. For in vivo labeling experiments, cells were grown in synthetic complete or minimal medium with the appropriate amino acids and 2% dextrose.
Anti–6x His is from ThermoFisher (Waltham, MA), and anti-coatomer and anti-Sec28 antibodies were a gift from Rainer Duden (University of Lubeck). Anti-rabbit antiserum to auxilin, Sec24, CPY, and invertase were prepared in the Ferro-Novick laboratory.
Anti-Chc1 mouse monoclonal antibodies were prepared in the Lemmon laboratory (Lemmon et al., 1988).
Purification of enriched coated vesicle fractions for proteomics
Wild-type cells (SL1463) were grown in YPD. For the depletion of auxilin, a GAL1p:SWA2 strain (SL4827/GPY2598) was grown to log phase in YP + 2% galactose, and then cultures were shifted to YPD for 15 h. For each strain, clathrin-coated vesicles (CCVs) were purified from 3 l of cells (∼5 × 107/ml) that were harvested and processed for cell fractionation and Sephacryl S-1000 size-exclusion chromatography (1.5 × 100 cm column), as described previously (Lemmon et al., 1988). Peak Chc1 fractions (28–36) were assessed by SDS–PAGE and Coomassie Blue R250 staining of gels, pooled (6–7 fractions of 3 ml each) and concentrated ∼40- to 100-fold in centricon 10 (Millipore, Billerica, MA) or vivaspin 2 (Vivaproducts, Littletown, MA) microconcentrators. Samples were separated on 4–20% polyacrylamide precast Bio-Rad minigels (Bio-Rad Laboratories, Hercules, CA) and stained with Coomassie Blue R250 in 30% methanol and 5% acetic acid. Gel lanes were cut into 10–11 slices, and each slice was trypsinized and analyzed by mass spectrometry by Midwest Bio Services (Overland Park, KS; www.midwestbioservices.com).
Analysis of proteomics
Peptide hits in each gel slice (Supplemental Tables S3 and S4) were compared with the Saccharomyces cerevisiae protein database using the SEQUEST algorithm (performed by Midwest Bio Services) and curated with the Protein Information Resource Peptide Matching analysis tool (http://research.bioinformatics.udel.edu/peptidematch/index.jsp). The data from individual slices were then combined and sorted to generate a composite list of hits for wild-type or the auxilin-depletion vesicle preparation (Supplemental Tables S5 and S6). Finally, to generate a list of protein hits specific to the auxilin-depletion strain, all of the gene products identified in the wild-type strain were subtracted from the hits obtained from the auxilin-depletion strain (Supplemental Table S7). During the purification of coated vesicles, RNase was added to the cell extract to remove polysomes (Lemmon et al., 1988), but ribosomal subunits and translation factors were still a common contaminant in the Sephacryl S-1000 coated-vesicle column fractions. Therefore all proteins involved in translation, including translation factors and ribosomal subunits, were excluded from the auxilin-depletion strain list.
Immunoprecipitation of the COPI coat complex
For the TAP-tag immunoprecipitation experiments described in Figure 4, lysates were prepared from a total of 3000 OD600 units of cells, pelleted, resuspended in 100 ml of spheroplast buffer (1.4 M sorbitol, 100 mM sodium phosphate, pH 7.5, 0.35% β-mercaptoethanol, and 0.5 mg/ml Zymolyase), and incubated for 30 min at 37°C. The spheroplasts were pelleted and washed twice with 150 ml of 1.7 M sorbitol and 20 mM HEPES (pH 7.4), and the spheroplast pellet was resuspended in 8 ml of lysis buffer (20 mM HEPES, pH 7.4, 4 mM EDTA, 1 mM dithiothreitol [DTT]) and Dounce homogenized. The resulting lysate was brought to 0.1 M NaCl, 1% NP-40, and incubated on ice for 20 min and then centrifuged for 30 min at 60,000 × g. The supernatants (4–12 mg protein) were then incubated with 30 μl of IgG–Sepharose beads (GE Healthcare) for 2 h at 4°C. The beads were washed three times with wash buffer (20 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM DTT, 2.5 mM MgCl2, 1% Triton X-100, and a protease inhibitor cocktail that included 1 μg/ml of aprotinin, chymostatin, antipain, pepstatin, and leupeptin) before they were eluted in 20 μl of 3X SDS–PAGE sample buffer and analyzed by Western blotting.
In vitro binding assays with recombinant proteins
The expression of His6-tagged and GST-tagged fusion proteins was induced with 0.5 mM isopropyl β-d-1-thiogalactopyranoside. All fusion proteins were purified as described in the Recombinant Protein Purification handbook from GE Healthcare. Purified His6-tagged fusion proteins (0.2 μM) were incubated with equimolar amounts of GST-fusion proteins or GST (0.2 μM) immobilized on glutathione–Sepharose beads (GE Healthcare) in binding buffer (25 mM Tris, pH 7.5, 300 mM KCL, 2% Triton X-100, 1 mM DTT, 1 mM EDTA, and the protease inhibitor cocktail described above) for 2–4 h at 4°C. The beads were washed three times with binding buffer, eluted in 30 μl of SDS–PAGE sample buffer by heating to 100°C for 5 min, and analyzed by Western blot analysis.
General secretion assay
Cells were grown overnight at 25°C to log phase, and 7.5 OD600 units were resuspended in 2 ml of fresh methionine-free medium containing 0.06 mg/ml bovine serum albumin and then shifted to 15°C for 60 min. After the incubation, the cells were labeled with 500 μCi of [35S]ProMix at 15°C, and aliquots of cells (350 μl) were removed at the end of the indicated time points. Labeled cell suspensions were centrifuged for 1 min at 13,000 × g, and then 300 μl of the medium (extracellular fractions) was transferred to an ice-cold tube containing 30 μl of stop mix (500 mM NaN3, 500 mM NaF). The extracellular fractions were centrifuged again for 1 min at 13,000 × g to remove residual cells, and the supernatant was transferred to a fresh tube containing 20 μl of 100% trichloroacetic acid (TCA) and 1 mg/ml sodium deoxycholate. All samples were incubated on ice for 60 min before the TCA precipitates were pelleted at 13,000 × g for 5 min and washed twice with ice-cold acetone. The acetone-washed pellets were air-dried, resuspended in 40 μl of 1X SDS sample buffer, heated to 100°C for 5 min, and subjected to SDS–PAGE using a 6% gel, followed by autoradiography.
Analysis of the transport and processing of CPY and invertase
For analysis of CPY processing, all strains (20 OD600 units) grown overnight at 25°C were resuspended in 4.5 ml of fresh minimal medium (supplemented with amino acids) and then shifted to 15°C for 60 min before being labeled with 500 μCi of [35S]ProMix for 10 min at 15°C. Cells (700 μl) were removed (0 min time point), and a chase mix (625 μl of 250 mM cysteine and 250 mM methionine) was added to the remaining sample. Additional aliquots (700 μl) were then removed at the indicated time points. Cells were washed twice with cold 10 mM sodium azide and resuspended in 125 μl of spheroplast buffer (1.4 M sorbitol, 50 mM potassium phosphate, pH 7.5, 10 mM sodium azide, 50 mM β-mercaptoethanol, 10 μg Zymolyase-100T/1 OD unit of cells). After a 45-min incubation at 37°C, spheroplasts were pelleted at 450 × g for 3 min, resuspended in 125 μl of 1% SDS, and heated for 5 min at 100°C. The samples were diluted with 900 μl of phosphate-buffered saline (PBS) containing 1% Triton X-100 and centrifuged for 15 min at 13, 000 × g. The supernatant (920 μl) was removed and incubated overnight with 3 μl of anti-CPY antibody (rabbit antiserum; Ferro-Novick laboratory) at 4°C. A 50% slurry of protein A–Sepharose (50 μl) beads was added to each sample, and samples were incubated for 1 h. The beads were then washed twice with urea wash buffer (2 M urea, 200 mM NaCl, 100 mM Tris, pH 7.6, 1% Triton X-100) and twice with 1% β-mercaptoethanol. The contents of the beads were solubilized in 70 μl of Laemmli sample buffer before they were subjected to SDS–PAGE using an 8% gel, followed by autoradiography.
For analysis of invertase secretion, 6 OD600 units of cells grown overnight at 25°C were resuspended in 6 ml of fresh minimal medium with 2% glucose (supplemented with amino acids) and then shifted to 15°C for 60 min. The cells were then transferred into medium containing 0.1% glucose to derepress the synthesis of invertase and labeled with 250 μCi of [35S]ProMix for 30 min at 15°C. After the incubation, the cells were washed twice with cold 10 mM sodium azide and resuspended in 125 μl of spheroplasting buffer for 45 min at 37°C and then centrifuged at 450 × g for 3 min. SDS (5 μl of 20% SDS) was added to 100 μl of the spheroplast supernatant (external), and the spheroplast pellet (internal) was resuspended in 125 μl of 1% SDS. Both samples were then heated for 5 min at 100°C. Aliquots (100 μl) of each sample were diluted with 900 μl of PBS containing 1% Triton X-100 and centrifuged for 15 min at 13,000 × g. The supernatant (920 μl) was removed and incubated overnight with 3.5 μl of anti-invertase antibody (rabbit antiserum; Ferro-Novick laboratory) before a 90-min incubation with 50 μl of a 50% slurry of protein A–Sepharose beads. The beads were then washed and solubilized as described above. Immunoprecipitated invertase was analyzed on a 10% polyacrylamide gel, which was subjected to autoradiography.
In vitro transport assay
The in vitro transport assay was performed as previously described (Groesch et al., 1990; Lian and Ferro-Novick, 1993), using wild-type donor cells and a wild-type S1 fraction (prepared as described in the following section) or swa2∆ donor cells and an swa2∆ S1 fraction. Briefly, permeabilized donor cells were incubated with in vitro translated radiolabeled prepro-α-factor for 50 min at 15°C. Donor cells harboring ER-translocated pro-α-factor were then pelleted, washed, and incubated with an S1 fraction for 90 min at 15°C. At the end of the reaction, the cells were centrifuged at 13,000 × g for 1 min, and the supernatant (containing Golgi and released vesicles) and a cell pellet from each sample were incubated with ConA–Sepharose beads at 4°C for 2 h. The beads were then washed, and samples were eluted in 70 μl of 1X SDS gel sample buffer. Equal amounts (20 μl) of each sample were counted, and the percent of budding was calculated as the counts in the supernatant/supernatant + pellet. For determination of the fusion efficiency, 20 μl of the ConA-treated supernatant fraction was immunoprecipitated with an antibody that recognizes Golgi-specific α1-6 mannose side chains on pro-α-factor. The percent fusion was calculated as the precipitable pro-α-factor counts in the supernatant/total ConA counts in the supernatant and pellet. The budding and fusion of wild type was considered to be maximal (100%).
Preparation of S1 fractions for the transport assay
Cells (1500 OD600 units) grown overnight at 30°C to log phase were resuspended in 1 l of YP medium with 0.01% glucose and incubated at 25°C for 30 min. The cells were pelleted, resuspended in 500 ml of spheroplast buffer (YP, 0.01% glucose, 1.4 M sorbitol, 50 mM potassium phosphate, pH 7.5, 50 mM β-mercaptoethanol, 10 μg Zymolyase-100T/l OD unit of cells), and incubated at 37°C for 30 min. The spheroplasts were then resuspended in recovery medium (YP, 0.01% glucose, 1 M sorbitol) and incubated at 37°C for 90 min. The regenerated spheroplasts were gently lysed in 3.36 ml of 20 mM HEPES (pH 7.2) before being centrifuged at 1000 × g for 10 min. The supernatant was collected, snap frozen, and stored at −80°C.
Vesicle-tethering assay
The supernatants from 10 transport reactions were pooled and loaded onto a sucrose gradient consisting of the following steps of sucrose prepared in TBPS (25 mM HEPES, pH 7.2, 115 mM KOAc, 25 mM Mg(OAc)2, 268 mM sorbitol): 0.5 ml of 50% sucrose (wt/wt), 0.5 ml of 46% sucrose (wt/wt), 1 ml of 42% sucrose (wt/wt), 1.5 ml of 38% sucrose (wt/wt), 1.5 ml of 34% sucrose (wt/wt), 1.5 ml of 30% sucrose (wt/wt), 1.5 ml of 26% sucrose (wt/wt), 1.5 ml of 22% sucrose (wt/wt), and 1.5 ml of 18% sucrose (wt/wt). The gradient was centrifuged for 2 h and 20 min at 173,000 × g. At the end of the centrifugation, twelve 1 ml samples were removed starting from the top of the gradient. Each sample (200 μl) was then treated with ConA–Sepharose beads to affinity purify glycosylated pro-α-factor and processed as described above. The counts from each fraction were graphed to determine the fractionation of pro-α-factor.
The same experiment, as described above, was done to mark where free vesicles migrate on the gradient, but the reactions were performed with cytosol instead of an S1 fraction.
Supplementary Material
Acknowledgments
We thank Yunrui Du for her initial genetic observations with the swa2Δ mutant. We thank G. Payne and R. Duden for reagents and Elizabeth Miller and Douglas Boettner for helpful discussions. Salary support for J.D. and S.F.-N. was provided by the Howard Hughes Medical Institute. V.A.S. was supported in part by National Institutes of Health (NIH) grant T32-HL07188. This work was also supported by NIH grants R01 GM114111 (S. F.-N.) and R01 GM 055796 (S.K.L.).
Abbreviations used:
- CCV
clathrin-coated vesicles
- Chc1
clathrin heavy chain
- ConA
concanavalin A
- CPY
carboxypeptidase Y
- DTT
dithiothreitol
- ER
endoplasmic reticulum
- GST
glutathione S-transferase
- IgG
immunoglobulin G
- PBS
phosphate-buffered saline
- PYC
permeabilized yeast cell
- TCA
trichloroacetic acid
- TGN
trans-Golgi network
- TPR
tetratricopeptide repeat
- ts
temperature-sensitive
- YP
yeast–peptone
- YPD
yeast–peptone–dextrose.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-09-0631) on November 4, 2015.
REFERENCES
- Angers CG, Merz AJ. HOPS interacts with Apl5 at the vacuole membrane and is required for consumption of AP-3 transport vesicles. Mol Biol Cell. 2009;20:4563–4574. doi: 10.1091/mbc.E09-04-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonny B, Madden D, Hamamoto S, Orci L, Schekman R. Dynamics of the COPII coat with GTP and stable analogues. Nat Cell Biol. 2001;3:531–537. doi: 10.1038/35078500. [DOI] [PubMed] [Google Scholar]
- Barlowe C, Orci L, Yeung T, Hosobuchi M, Hamamoto S, Salama N, Rexach MF, Ravazzola M, Amherdt M, Schekman R. COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell. 1994;77:895–907. doi: 10.1016/0092-8674(94)90138-4. [DOI] [PubMed] [Google Scholar]
- Barlowe CK, Miller EA. Secretory protein biogenesis and traffic in the early secretory pathway. Genetics. 2013;193:383–410. doi: 10.1534/genetics.112.142810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrowman J, Sacher M, Ferro-Novick S. TRAPP stably associates with the Golgi and is required for vesicle docking. EMBO J. 2000;19:862–869. doi: 10.1093/emboj/19.5.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettner DR, Chi RJ, Lemmon SK. Lessons from yeast for clathrin-mediated endocytosis. Nat Cell Biol. 2012;14:2–10. doi: 10.1038/ncb2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandizzi F, Barlowe C. Organization of the ER-Golgi interface for membrane traffic control. Nat Rev Mol Cell Biol. 2013;14:382–392. doi: 10.1038/nrm3588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Yu S, Menon S, Cai Y, Lazarova D, Fu C, Reinisch K, Hay JC, Ferro-Novick S. TRAPPI tethers COPII vesicles by binding the coat subunit Sec23. Nature. 2007;445:941–944. doi: 10.1038/nature05527. [DOI] [PubMed] [Google Scholar]
- Carlson M, Botstein D. Two differentially regulated mRNAs with different 5′ ends encode secreted with intracellular forms of yeast invertase. Cell. 1982;28:145–154. doi: 10.1016/0092-8674(82)90384-1. [DOI] [PubMed] [Google Scholar]
- D’Andrea LD, Regan L. TPR proteins: the versatile helix. Trends Biochem Sci. 2003;28:655–662. doi: 10.1016/j.tibs.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Du Y, Pypaert M, Novick P, Ferro-Novick S. Aux1p/Swa2p is required for cortical endoplasmic reticulum inheritance in Saccharomyces cerevisiae. Mol Biol Cell. 2001;12:2614–2628. doi: 10.1091/mbc.12.9.2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg E, Greene LE. Multiple roles of auxilin and hsc70 in clathrin-mediated endocytosis. Traffic. 2007;8:640–646. doi: 10.1111/j.1600-0854.2007.00568.x. [DOI] [PubMed] [Google Scholar]
- Faini M, Beck R, Wieland FT, Briggs JA. Vesicle coats: structure, function, and general principles of assembly. Trends Cell Biol. 2013;23:279–288. doi: 10.1016/j.tcb.2013.01.005. [DOI] [PubMed] [Google Scholar]
- Gall WE, Higginbotham MA, Chen C, Ingram MF, Cyr DM, Graham TR. The auxilin-like phosphoprotein Swa2p is required for clathrin function in yeast. Curr Biol. 2000;10:1349–1358. doi: 10.1016/s0960-9822(00)00771-5. [DOI] [PubMed] [Google Scholar]
- Groesch ME, Ruohola H, Bacon R, Rossi G, Ferro-Novick S. Isolation of a functional vesicular intermediate that mediates ER to Golgi transport in yeast. J Cell Biol. 1990;111:45–53. doi: 10.1083/jcb.111.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurkan C, Stagg SM, Lapointe P, Balch WE. The COPII cage: unifying principles of vesicle coat assembly. Nat Rev Mol Cell Biol. 2006;7:727–738. doi: 10.1038/nrm2025. [DOI] [PubMed] [Google Scholar]
- Herzig Y, Sharpe HJ, Elbaz Y, Munro S, Schuldiner M. A systematic approach to pair secretory cargo receptors with their cargo suggests a mechanism for cargo selection by Erv14. PLoS Biol. 2012;10:e1001329. doi: 10.1371/journal.pbio.1001329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata R, Nihei C, Nakano A. Isoform-selective oligomer formation of Saccharomyces cerevisiae p24 family proteins. J Biol Chem. 2013;288:37057–37070. doi: 10.1074/jbc.M113.518340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhausen T, Owen D, Harrison SC. Molecular structure, function, and dynamics of clathrin-mediated membrane traffic. Cold Spring Harb Perspect Biol. 2014;6:a016725. doi: 10.1101/cshperspect.a016725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koutras C, Braun JE. J protein mutations and resulting proteostasis collapse. Front Cell Neurosci. 2014;8:191. doi: 10.3389/fncel.2014.00191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon S, Lemmon VP, Jones EW. Characterization of yeast clathrin and anticlathrin heavy-chain monoclonal antibodies. J Cell Biochem. 1988;36:329–340. doi: 10.1002/jcb.240360403. [DOI] [PubMed] [Google Scholar]
- Lemmon SK. Clathrin uncoating: auxilin comes to life. Curr Biol. 2001;11:R49–R52. doi: 10.1016/s0960-9822(01)00010-0. [DOI] [PubMed] [Google Scholar]
- Lemmon SK, Pellicena-Palle A, Conley K, Freund CL. Sequence of the clathrin heavy chain from Saccharomyces cerevisiae and requirement of the COOH terminus for clathrin function. J Cell Biol. 1991;112:65–80. doi: 10.1083/jcb.112.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian JP, Ferro-Novick S. Bos1p, an integral membrane protein of the endoplasmic reticulum to Golgi transport vesicles, is required for their fusion competence. Cell. 1993;73:735–745. doi: 10.1016/0092-8674(93)90253-m. [DOI] [PubMed] [Google Scholar]
- Lord C, Bhandari D, Menon S, Ghassemian M, Nycz D, Hay J, Ghosh P, Ferro-Novick S. Sequential interactions with Sec23 control the direction of vesicle traffic. Nature. 2011;473:181–186. doi: 10.1038/nature09969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord C, Ferro-Novick S, Miller EA. The highly conserved COPII coat complex sorts cargo from the endoplasmic reticulum and targets it to the Golgi. Cold Spring Harb Perspect Biol. 2013;5:a013367. doi: 10.1101/cshperspect.a013367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AP, Ferro-Novick S. Characterization of new mutants in the early part of the yeast secretory pathway isolated by a [3H]mannose suicide selection. J Cell Biol. 1987;105:1587–1594. doi: 10.1083/jcb.105.4.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newmyer SL, Christensen A, Sever S. Auxilin-dynamin interactions link the uncoating ATPase chaperone machinery with vesicle formation. Dev Cell. 2003;4:929–940. doi: 10.1016/s1534-5807(03)00157-6. [DOI] [PubMed] [Google Scholar]
- Payne GS, Hasson TB, Hasson MS, Schekman R. Genetic and biochemical characterization of clathrin-deficient Saccharomyces cerevisiae. Mol Cell Biol. 1987;7:3888–3898. doi: 10.1128/mcb.7.11.3888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne GS, Schekman R. A test of clathrin function in protein secretion and cell growth. Science. 1985;230:1009–1014. doi: 10.1126/science.2865811. [DOI] [PubMed] [Google Scholar]
- Pishvaee B, Costaguta G, Yeung BG, Ryazantsev S, Greener T, Greene LE, Eisenberg E, McCaffery JM, Payne GS. A yeast DNA J protein required for uncoating of clathrin-coated vesicles in vivo. Nat Cell Biol. 2000;2:958–963. doi: 10.1038/35046619. [DOI] [PubMed] [Google Scholar]
- Rapoport I, Boll W, Yu A, Bocking T, Kirchhausen T. A motif in the clathrin heavy chain required for the Hsc70/auxilin uncoating reaction. Mol Biol Cell. 2008;19:405–413. doi: 10.1091/mbc.E07-09-0870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruohola H, Kabcenell AK, Ferro-Novick S. Reconstitution of protein transport from the endoplasmic reticulum to the Golgi complex in yeast: the acceptor Golgi compartment is defective in the sec23 mutant. J Cell Biol. 1988;107:1465–1476. doi: 10.1083/jcb.107.4.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato K, Nakano A. Emp47p and its close homolog Emp46p have a tyrosine-containing endoplasmic reticulum exit signal and function in glycoprotein secretion in Saccharomyces cerevisiae. Mol Biol Cell. 2002;13:2518–2532. doi: 10.1091/mbc.E02-01-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuiki I, Volchuk A. Diverse roles for the p24 family of proteins in eukaryotic cells. Biomol Concepts. 2012;3:561–570. doi: 10.1515/bmc-2012-0028. [DOI] [PubMed] [Google Scholar]
- Seeger M, Payne GS. A role for clathrin in the sorting of vacuolar proteins in the Golgi complex of yeast. EMBO J. 1992a;11:2811–2818. doi: 10.1002/j.1460-2075.1992.tb05348.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger M, Payne GS. Selective and immediate effects of clathrin heavy chain mutations on Golgi membrane protein retention in Saccharomyces cerevisiae. J Cell Biol. 1992b;118:531–540. doi: 10.1083/jcb.118.3.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiya-Kawasaki M, Groen AC, Cope MJ, Kaksonen M, Watson HA, Zhang C, Shokat KM, Wendland B, McDonald KL, McCaffery JM, Drubin DG. Dynamic phosphoregulation of the cortical actin cytoskeleton and endocytic machinery revealed by real-time chemical genetic analysis. J Cell Biol. 2003;162:765–772. doi: 10.1083/jcb.200305077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sever S, Skoch J, Newmyer S, Ramachandran R, Ko D, McKee M, Bouley R, Ausiello D, Hyman BT, Bacskai BJ. Physical and functional connection between auxilin and dynamin during endocytosis. EMBO J. 2006;25:4163–4174. doi: 10.1038/sj.emboj.7601298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens T, Esmon B, Schekman R. Early stages in the yeast secretory pathway are required for transport of carboxypeptidase Y to the vacuole. Cell. 1982;30:439–448. doi: 10.1016/0092-8674(82)90241-0. [DOI] [PubMed] [Google Scholar]
- Toret CP, Lee L, Sekiya-Kawasaki M, Drubin DG. Multiple pathways regulate endocytic coat disassembly in Saccharomyces cerevisiae for optimal downstream trafficking. Traffic. 2008;9:848–859. doi: 10.1111/j.1600-0854.2008.00726.x. [DOI] [PubMed] [Google Scholar]
- Ungewickell E, Ungewickell H, Holstein SE, Lindner R, Prasad K, Barouch W, Martin B, Greene LE, Eisenberg E. Role of auxilin in uncoating clathrin-coated vesicles. Nature. 1995;378:632–635. doi: 10.1038/378632a0. [DOI] [PubMed] [Google Scholar]
- Wassmer T, Attar N, Harterink M, van Weering JR, Traer CJ, Oakley J, Goud B, Stephens DJ, Verkade P, Korswagen HC, Cullen PJ. The retromer coat complex coordinates endosomal sorting and dynein-mediated transport, with carrier recognition by the trans-Golgi network. Dev Cell. 2009;17:110–122. doi: 10.1016/j.devcel.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Zhao X, Baylor L, Kaushal S, Eisenberg E, Greene LE. Clathrin exchange during clathrin-mediated endocytosis. J Cell Biol. 2001;155:291–300. doi: 10.1083/jcb.200104085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J, Kim LS, Graham TR. Dissection of Swa2p/auxilin domain requirements for cochaperoning Hsp70 clathrin-uncoating activity in vivo. Mol Biol Cell. 2006;17:3281–3290. doi: 10.1091/mbc.E06-02-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim YI, Scarselletta S, Zang F, Wu X, Lee DW, Kang YS, Eisenberg E, Greene LE. Exchange of clathrin, AP2 and epsin on clathrin-coated pits in permeabilized tissue culture cells. J Cell Sci. 2005;118:2405–2413. doi: 10.1242/jcs.02356. [DOI] [PubMed] [Google Scholar]
- Zanetti G, Pahuja KB, Studer S, Shim S, Schekman R. COPII and the regulation of protein sorting in mammals. Nat Cell Biol. 2012;14:20–28. doi: 10.1038/ncb2390. [DOI] [PubMed] [Google Scholar]
- Zink S, Wenzel D, Wurm CA, Schmitt HD. A link between ER tethering and COP-I vesicle uncoating. Dev Cell. 2009;17:403–416. doi: 10.1016/j.devcel.2009.07.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.