

Sixty‐four genomic loci and seven genes that contribute to heritable variation in a model quantitative trait—resistance to oxidative stress—are identified across three yeast strains. The high‐resolution understanding of this phenotype provides new insight into the genetic and molecular basis of quantitative traits.
Abstract
Quantitative traits are often influenced by many loci with small effects. Identifying most of these loci and resolving them to specific genes or genetic variants is challenging. Yet, achieving such a detailed understanding of quantitative traits is important, as it can improve our knowledge of the genetic and molecular basis of heritable phenotypic variation. In this study, we use a genetic mapping strategy that involves recurrent backcrossing with phenotypic selection to obtain new insights into an ecologically, industrially, and medically relevant quantitative trait—tolerance of oxidative stress, as measured based on resistance to hydrogen peroxide. We examine the genetic basis of hydrogen peroxide resistance in three related yeast crosses and detect 64 distinct genomic loci that likely influence the trait. By precisely resolving or cloning a number of these loci, we demonstrate that a broad spectrum of cellular processes contribute to hydrogen peroxide resistance, including DNA repair, scavenging of reactive oxygen species, stress-induced MAPK signaling, translation, and water transport. Consistent with the complex genetic and molecular basis of hydrogen peroxide resistance, we show two examples where multiple distinct causal genetic variants underlie what appears to be a single locus. Our results improve understanding of the genetic and molecular basis of a highly complex, model quantitative trait.
INTRODUCTION
Mapping experiments in model organisms have served and continue to play a crucial role in advancing our understanding of the genetic and molecular basis of quantitative traits (Mackay et al., 2009; Bloom et al., 2013). However, as discussed elsewhere (e.g., Parts et al., 2011; Cubillos et al., 2013), these studies typically involve genetic mapping strategies that are capable of identifying loci at the resolution of broad genomic regions. For quantitative traits that are influenced by a large number of loci, this mapping resolution can make it challenging to detect and localize causal genetic variants, and can also make it difficult to measure accurately the effects of individual loci.
Recent work in Saccharomyces cerevisiae highlights this problem. Studies using large, statistically powerful mapping populations in crosses involving two strains have detected dozens of loci per phenotype (Ehrenreich et al., 2010, 2012; Parts et al., 2011; Bloom et al., 2013; Taylor and Ehrenreich, 2014; Treusch et al., 2015). However, as more strains are considered, the number of loci identified for a given trait typically increases substantially, with loci often showing complicated patterns of detection across genetic backgrounds (e.g., Cubillos et al., 2011; Ehrenreich et al., 2012; Treusch et al., 2015). A variety of biological phenomena, including genetic interactions among loci or close linkage of different genetic variants with phenotypic effects, might explain these observations. Differentiating among these possibilities requires identifying combinations of interacting alleles and precisely delimiting loci to very small genomic intervals, ideally to specific genes and nucleotides.
In this study, we dissect at high resolution the genetic basis of heritable variation in hydrogen peroxide resistance among three budding yeast isolates—the lab strain BY4716, the wine strain RM11-1a, and the oak strain YPS163 (hereafter BY, RM, and YPS, respectively). We chose these strains because they are known to possess genetically complex differences in their tolerances of hydrogen peroxide (Kvitek et al., 2008; Ehrenreich et al., 2012). To determine the genetic basis of this heritable phenotypic variation, we generated multiple backcross populations from each cross of the three strains and implemented additional rounds of backcrossing coupled to stringent phenotypic selections (Figure 1). The rationale behind our approach was to delimit causal loci to discrete genomic intervals that had been introgressed into the backcross parent’s genome. By conducting this process multiple times in parallel, we could then combine information from related backcrosses and map loci even more precisely while retaining high statistical power (Supplemental Note S1). Furthermore, by using such a mapping strategy, we hoped to introgress combinations of alleles that collectively confer resistance, as these alleles could then be tested for additive effects and genetic interactions in a manner similar to Taylor and Ehrenreich (2014, 2015a).
FIGURE 1:
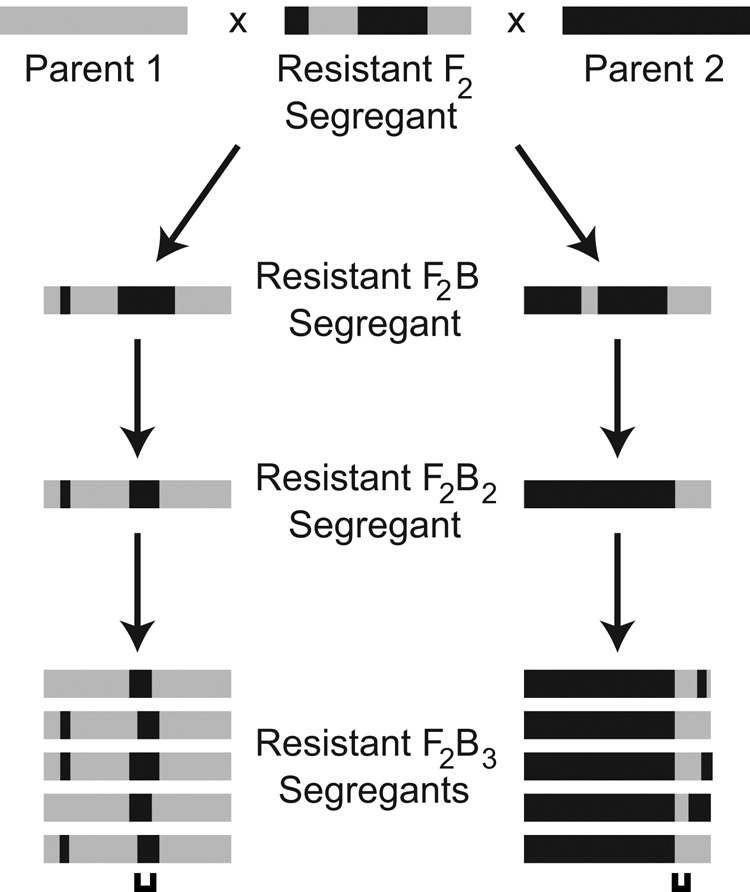
Backcrossing strategy. F2B3 segregants with high hydrogen peroxide resistance were generated through multiple rounds of backcrossing with phenotypic selection. The filled rectangles at each stage represent chromosomes, with gray and black depicting chromosomal regions inherited from Parent 1 and Parent 2, respectively. The brackets shown beneath the chromosomes from resistant F2B3 segregants indicate causal loci. Numbers of individuals screened at each stage of crossing are reported in Materials and Methods.
We focused on hydrogen peroxide resistance because it is commonly used as a proxy for oxidative stress tolerance, a trait that has potential relevance for both human health and yeast biology. Susceptibility to oxidative stress has been linked to aging (Fabrizio et al., 2003; Braun and Westermann, 2011; Petti et al., 2011; Cui et al., 2012; Longo et al., 2012), as well as to Alzheimer’s disease (Jomova et al., 2010; Greenough et al., 2013; Koppenhofer et al., 2015), diabetes (Varvarovska et al., 2004; Aouacheri et al., 2015), and other disorders. Furthermore, tolerance of oxidative stress has ecological and economic ramifications for yeast, in particular for strains that mainly inhabit aerobic environments or are used in fermentations or industrial applications (Higgins et al., 2003; Sasano et al., 2012; Dhar et al., 2013; Fierro-Risco et al., 2013; Brown et al., 2014; Kitagaki and Takagi, 2014).
By applying our backcrossing strategy to variation in hydrogen peroxide resistance in the BYxRM, BYxYPS, and RMxYPS crosses, we identified 64 distinct genomic loci that likely contribute to the trait. Analysis of allele combinations at certain subsets of these loci suggests that variability in hydrogen peroxide resistance has a genetic basis that is largely additive. Furthermore, identification of a number of these loci at the resolution of small genomic windows, as well as cloning of certain quantitative trait genes and nucleotides, indicates that many distinct molecular processes contribute to hydrogen peroxide resistance. Consistent with the high genetic and molecular complexity of this trait, we show that multiple causal genetic variants can underlie what appears to be a single locus based on genetic mapping data. Our results advance understanding of the genetic and molecular basis of highly complex quantitative traits in yeast and potentially other organisms as well.
RESULTS
Generation of mapping populations using recurrent backcrossing with phenotypic selection
We first determined the minimum inhibitory concentrations (MICs) for hydrogen peroxide of BYxRM, BYxYPS, and RMxYPS segregants (Supplemental Figure S1 and Supplemental Table S1; Materials and Methods). For each cross, 864 haploid recombinants were screened. After this initial phenotyping, each of the five most resistant F2 segregants from the three crosses were individually subjected to several rounds of selective backcrossing to both of their parents (Figure 1; Materials and Methods). This resulted in the generation of two F2B3 families per hydrogen peroxide–resistant F2 segregant. During the iterations of backcrossing, haploid recombinants were frozen to create immortalized stocks and then phenotyped using colony growth assays conducted across a range of hydrogen peroxide doses (Materials and Methods). The most resistant haploid segregant in a given backcross was then used as the founder for the next round of backcrossing (Materials and Methods). To prevent hydrogen peroxide–induced mutations from accumulating during the backcrossing, we performed each round of crossing using fresh cultures from the frozen stocks that had never been exposed to hydrogen peroxide.
From each of the 30 backcross families, 12–15 highly resistant F2B3 progeny were genotyped by low-coverage, whole-genome sequencing and used to detect loci (Materials and Methods). In total, 417 F2B3 segregants were genotyped. However, we found karyotype instability in families derived from one of the BYxYPS F2 segregants and therefore excluded individuals in these families from the study (Supplemental Note S2). Thus, the analyses described here are based on the 392 F2B3 segregants that were generated from the remaining 28 backcross families that did not show aneuploidies.
Identification of loci that contribute to hydrogen peroxide resistance
Loci were identified within individual families based on allele frequency skew among the 12–15 resistant backcross segregants that had been genotyped (Materials and Methods). Given that resistant individuals from the same family were generated from a common diploid progenitor and identified by screening of individual strains (as opposed to pools of segregants), we do not expect bias in our results due to inadvertent selection on other traits, such as sporulation or mating efficiency. Excluding the MAT locus on chromosome III, which is a control marker that we used to generate haploid segregants, we identified 60 loci at a FDR (q) of ≤ 0.05: 10 in BYxRM, 28 in BYxYPS, and 22 in RMxYPS (Figure 2 and Supplemental Figure S2; Materials and Methods). Lowering the threshold to 0.1 or 0.2 resulted in the detection of 79 and 104 total loci, respectively, with crosses involving YPS showing the highest number of detected loci (Figure 2).
FIGURE 2:

Genetic mapping results. Regions of the genome that showed nominal significance (p ≤ 0.05) are plotted with their associated q values. (A–C) Results from advanced backcross families derived from different BYxRM, BYxYPS, and RMxYPS F2 segregants, respectively. Each row represents an F2 segregant from which two advanced backcross families were derived by recurrent backcrossing to each of the strain’s parents.
Resolution of loci within individual families
Because we identified loci using segregants that had been individually genotyped, we were able to delimit loci detected in each family based on recombination breakpoints that were observed in the data (Materials and Methods). Among the 60 loci detected at q ≤ 0.05, this resulted in an average resolution of 54.1 kb (Materials and Methods; Supplemental Table S2). However, 11 of these loci were delimited to genomic intervals of <10 kb (Materials and Methods). Two loci were detected at a resolution of a single gene: YIL177C, a putative Y′ element helicase that was detected on chromosome IX in two RMxYPS families, and PFK2, a subunit of phosphofructokinase involved in glycolysis that was detected on chromosome XIII in one BYxYPS family. An additional three loci were detected at a resolution of two genes (Supplemental Table S2). These occurred on chromosomes I, IX, and XIV in the BYxYPS cross and corresponded respectively to the lectin-like cell wall protein FLO1 and the oxysterol-binding protein SWH1, the vitamin-related transcriptional activator VHR1 and the respiratory induced gene RGI2, and the BLOC1 component SNN1 and the highly pleiotropic gene MKT1. MKT1 and SWH1 are known quantitative trait genes in the BYxRM cross that were identified through mapping studies focused on other phenotypes, as reviewed in Ehrenreich et al. (2009) and recently shown in Wang and Kruglyak (2014), respectively.
Validation and deeper genetic analysis of a subset of loci
When we compared data from families and crosses, we found that detected loci (q ≤ 0.2) could be collapsed into 64 distinct genomic regions (Figure 2; Materials and Methods). For the remainder of the article, we refer to these distinct regions as “loci.” Although 16 of these loci were identified in at least two different families from the same cross, most were not detected in multiple families from the same cross (Figure 2 and Supplemental Figure S3). Some of the loci that were not replicated may be false positives. However, we expect that a large fraction of the detected loci, even those seen only a single time, have biological effects (Supplemental Note S1).
To confirm that most of the identified loci have biological effects, we examined in more detail two RMxYPS families that had been generated by repeated backcrossing to YPS (referred to here as (RMxYPS)xYPS families). None of the loci detected in either of these families were the same (Figure 3A), and some of the loci identified in these families were not seen in mapping data for other RMxYPS families (Figure 2). We generated 96 random F2B3s from each of these families, screened these segregants for their MICs, and genotyped these segregants at the relevant loci (Materials and Methods). We then used the genotype and phenotype data to test whether the loci individually exhibited effects (Materials and Methods). When we did this, six of the eight (75%) tested loci showed significant additive effects (t test, p ≤ 0.045; Figure 3, B and C), a result that is consistent with our false discovery rate (FDR) threshold.
FIGURE 3:

Validation of loci detected in families derived from the RMxYPS cross. (A) Different combinations of alleles detected in RMxYPS backcrosses to YPS. (B, C) The 95% confidence intervals of MIC for alleles tested in larger F2B3 populations. (B) The 96 YPS-backcrossed RMxYPS F2B3 segregants from a family in which a combination of three loci were detected were phenotyped and genotyped at these loci. (C) The 96 YPS-backcrossed RMxYPS F2B3 segregants from a family in which a combination of four alternate loci were detected, as well as a locus shared with only one other YPS-backcrossed RMxYPS family, were similarly phenotyped and genotyped at these loci. (D, E) Additive effects of these alleles across genotypes, with black lines illustrating regression models that only include additive effects.
We also examined whether genetic interactions influenced the effects of any of the eight queried loci. Specifically, we used full factorial analysis of variance (ANOVA) models to assess the relationship between genotype at the loci with significant additive effects and MIC in the two (RMxYPS)xYPS families (Materials and Methods). These models included not only the additive terms corresponding to each individual locus, but also all possible pairwise and higher-order interaction terms among the loci. Although all of the loci continued to show significant additive effects in these models, only one significant interaction term (p < 0.05) was observed, which was between L7-I and L15 (Figure 3A). We also tested for statistically significant genetic interactions between alleles detected in the two families using F2 segregants but did not identify any such interactions (Supplemental Figure S4, A and B). These results suggest that genetic interactions contribute little to heritable variation in hydrogen peroxide resistance (Figure 3, D and E). Thus, even though our approach should be capable of revealing pairwise and higher-order genetic interactions (Taylor and Ehrenreich, 2014, 2015a, b), our results agree with recent work demonstrating that quantitative traits in yeast have a genetic basis that is largely additive (Bloom et al., 2013).
Using detection of loci in multiple families to improve mapping resolution
We attempted to improve our resolution of loci that were detected in multiple families. There were five, five, and six such loci in the BYxRM, BYxYPS, and RMxYPS crosses, respectively (Figure 2). By aggregating data from the families in which these loci were detected at a q-value of ≤0.2 (Figure 2; Materials and Methods), we achieved an average resolution for these loci of 14, 6, and 13 genes in the BYxRM, BYxYPS, and RMxYPS crosses, respectively.
We concentrated on cloning quantitative genes underlying loci detected in multiple families in the BYxYPS cross, as these were identified at the most precise resolution (Figure 2). We first used reciprocal hemizygosity analysis (RH) (Steinmetz et al., 2002) to examine nearly all of the nonessential candidate genes underlying the five loci, which were located on chromosomes V, VII, IX, XIV, and XVI (25 total genes examined; Supplemental Figure S5A and Supplemental Table S3; Materials and Methods). This successfully identified two quantitative trait genes: MKT1 (chromosome XIV; Figure 4A and Supplemental Figure S6) and the aquaporin AQY1 (chromosome XVI; Figure 4B and Supplemental Figure S6). We validated AQY1 and MKT1 by performing allele replacements spanning the entire coding and noncoding regions of these genes in BYxYPS F2B3 segregants that carried the resistance allele at the locus being tested (Supplemental Figure S5B; Materials and Methods).
FIGURE 4:

Localization and cloning of loci detected in multiple BYxYPS advanced backcross families. By combining data from advanced backcross families, we improved our mapping resolution. Gray vertical bars indicate the bounds delimited by aggregated data. Horizontal dashed lines represent the nominal significance cut-off (p ≤ 0.05) for a one-tailed binomial test. The causal gene or variant at each locus is illustrated with a red box or asterisk, respectively. Genes represented with white boxes had no effect on hydrogen peroxide resistance.
We also used allele replacements that spanned the entire coding and noncoding regions of candidate genes to test every gene underlying the chromosome V, VII, and IX loci in resistant BYxYPS F2B3 segregants (Supplemental Figure S5B and Supplemental Table S3; Materials and Methods). By doing this, we identified a single quantitative trait gene at two of the loci: MMS21, an essential SUMO ligase that is involved in DNA repair (chromosome V; Figure 4C), and MRP13, a nuclear-encoded component of the mitochondrial ribosome (chromosome VII; Figure 4D). In addition, we found that the causal variant underlying the chromosome IX locus is a cis regulatory polymorphism in the intergenic region between the mitochondrial porin POR2 and the stress-inducible mitogen-activated protein kinase phosphatase SDP1, which are transcribed in opposite directions. We found this because replacing the entire coding and noncoding regions of either POR2 or SDP1 decreased hydrogen peroxide resistance (Figure 4E), whereas replacing only the coding region of either POR2 or SDP1 had no effect.
One single-nucleotide polymorphism (SNP) differentiates the BY and YPS alleles of the POR2-SDP1 intergenic region. To determine whether this SNP affects the expression of POR2 or SDP1, we used quantitative PCR (qPCR) to measure the transcription of these genes in the presence and absence of hydrogen peroxide (Materials and Methods). We performed qPCR in two genetic backgrounds—a BYxYPS F2B3 segregant that carried the BY allele of the POR2-SDP1 region and a genetically engineered version of the same strain that carried the YPS allele of the region. No expression differences were observed in the absence of hydrogen peroxide (Figure 5A). However, the qPCR experiments revealed that the intergenic SNP affects SDP1 expression specifically in the presence of hydrogen peroxide (Figure 5B).
FIGURE 5:

A cis regulatory polymorphism in the BYxYPS cross causes differential expression of SDP1 in response to hydrogen peroxide. (A, B) qPCR analyses of POR2 and SDP1, respectively. Isogenic strains that only differed in their genotype at the POR2-SDP1 locus were used in these experiments.
The functionally diverse quantitative trait genes (AQY1, MKT1, MMS21, MRP13, and SDP1) that were cloned as a part of this section, as well as the loci that were detected at a resolution of one or two genes (Supplemental Table S2), indicate that variation in hydrogen peroxide resistance is shaped by a broad space of molecular mechanisms and cellular processes.
Linkage among genetic variants strongly influences how loci are detected
Recent studies applying statistically powerful genetic mapping techniques to multiple crosses have shown that loci can exhibit complicated patterns of detection among interrelated mapping populations (e.g., Ehrenreich et al., 2012; Treusch et al., 2015). Arguably, the simplest explanation for this phenomenon is that multiple genetic variants segregate on each chromosome and interference among these genetic variants affects how loci are detected in any given cross.
Our study supports linked genetic polymorphisms as the major cause of loci being detected in complicated patterns in studies involving multiple crosses. For example, in addition to identifying AQY1 as a quantitative trait gene in the BYxYPS cross, we also found that AQY1 is a quantitative trait gene in the RMxYPS cross (Supplemental Figure S7). This was determined through a combination of mapping the chromosome XVI locus spanning AQY1 to two genes using data from four RMxYPS families (Figure 2 and Supplemental Figure S7A), as well as by replacing AQY1RM with AQY1YPS in a relevant RMxYPS F2B3 (Supplemental Figure S7B; Materials and Methods). These results might suggest that BY and RM share an allele of AQY1 that confers hydrogen peroxide resistance and differentiates them from YPS. However, this is not the case; there are no polymorphisms shared between BY and RM in the promoter, coding region, or 3′ untranslated region of AQY1. Instead, AQY1BY and AQY1RM harbor distinct loss-of-function variants (a missense polymorphism in BY and a frameshift in RM), which were originally identified due to their similar effects on freeze–thaw tolerance (Will et al., 2010).
We also found evidence for the presence of closely linked causal genetic variants in different quantitative trait genes. In this case, a locus overlapping the BYxYPS quantitative trait gene MRP13 was also identified in the RMxYPS cross (Figure 6A). However, replacement of MRP13RM with MRP13YPS in a resistant RMxYPS F2B3 did not have an effect on hydrogen peroxide resistance (Figure 6C). To determine the causal gene underlying this locus in the RMxYPS cross, we replaced the RM allele for every gene in this interval with the YPS allele in a relevant RMxYPS F2B3 (Materials and Methods). The only gene in this interval that had an effect in the RMxYPS cross was the cytosolic catalase-encoding gene CTT1, which is located three genes away from MRP13 (Figure 6, B and D). These findings are important because they show how individual genomic loci detected in genetic mapping experiments in yeast can in fact correspond to multiple quantitative trait genes and nucleotides.
FIGURE 6:

Two different quantitative trait genes segregate in the same genomic region. (A) Overlapping peaks on chromosome VII were detected in BYxYPS and RMxYPS backcrosses to YPS, with B and R representing the BY and RM allele frequencies in these crosses, respectively. (B) MRP13 and CTT1 are depicted in bold to emphasize their close physical proximity within this overlapping interval. MRP13BY confers resistance in the BYxYPS cross, whereas in the RMxYPS cross, the causal allele in this interval is instead CTT1RM. (C, D) The 95% confidence intervals of MIC for allele replacement strains, which were generated in a YPS-backcrossed RMxYPS F2B3 segregant with the causal allele at the overlapping locus.
DISCUSSION
Recent studies reveal the very high genetic complexity that can underlie heritable phenotypes in budding yeast (e.g., Ehrenreich et al., 2010, 2012; Cubillos et al., 2011; Lorenz and Cohen, 2012; Bloom et al., 2013; Granek et al., 2013; Wilkening et al., 2014). Much of this work has focused on chemical resistance traits, which can easily be screened in large populations of segregants and multiple crosses. Despite the valuable insights gained from efforts to map the genetic basis of these phenotypes, questions about the molecular mechanisms and statistical genetic architecture underlying these traits remain unanswered, largely due to the limited resolution of current genetic mapping approaches.
In this study, we focused on a single chemical resistance trait—hydrogen peroxide resistance—and used a genetic mapping strategy that resulted in the precise detection of combinations of loci. Our strategy was in part motivated by recent discoveries of higher-order genetic interactions in yeast (Dowell et al., 2010; Taylor and Ehrenreich, 2014, 2015a, b; Wang and Kruglyak, 2014), which were found for binary traits, and was aimed at determining whether such complex genetic interactions also contribute to quantitative traits. Our work suggests that higher-order genetic interactions are unlikely to make a significant contribution to quantitative traits in yeast and instead supports a largely additive genetic basis for such phenotypes in this organism, as was recently argued (Bloom et al., 2013).
In relation to our past work (Ehrenreich et al., 2012; Bloom et al., 2013), we used a different mapping technique to characterize the genetic basis of hydrogen peroxide resistance in the present study. Comparison of results from this article to our other publications or of our past manuscripts to each other suggests that roughly half of the loci detected in one study are replicated in another. For example, analysis of pools of hydrogen peroxide–resistant BYxRM, BYxYPS, and RMxYPS segregants identified 28 loci (Ehrenreich et al., 2012). Of these, 16 (57%) were also detected in the present study. Disparities in the loci identified by these studies may be due to either biological or technical factors and are consistent with the high genetic complexity of hydrogen peroxide resistance.
Of note, the present study is distinguished from our past efforts by the concerted effort we made to clone multiple quantitative trait genes that contribute to hydrogen peroxide resistance. This resulted in the identification of CTT1, MKT1, MMS21, MRP13, SDP1, and two different alleles of AQY1. Our results indicate that many molecular and cellular processes influence hydrogen peroxide resistance. It is possible that this breadth of mechanisms that contribute to the trait may have provided a large mutational target space for the accumulation of functional genetic variation. However, proving such a point is difficult without knowing more of the genes that contribute to hydrogen peroxide resistance in BY, RM, YPS, and other strains, and also determining the causal variants in these genes.
Our findings also provide insights into why certain loci show complicated patterns of detection in studies involving more than two strains. We find that this arises due to the presence of multiple variants with additive effects occurring in the same gene, as well as the presence of distinct additive variants in different genes that are closely linked. Which of these scenarios is more prevalent is unclear from the present work due to the limited number of examples provided by this study, as well as the fact that AQY1 is a common target for environment-specific adaptive mutations and thus may be unusual (Will et al., 2010).
In conclusion, although we focused on hydrogen peroxide resistance, our findings may have general relevance for other chemical resistance phenotypes and highly complex quantitative traits. Given that we examined only three related crosses, we expect that the effects of high genetic complexity and linkage that we described will be more severe in studies that involve a larger number of strains. If this is true, then precisely resolving loci to specific genes and causal variants will be crucial for studies of highly complex, quantitative traits in yeast and other model organisms moving forward. Such resolution will be necessary to maximize the insights that genetic mapping can provide into the genetic and molecular basis of quantitative traits.
MATERIALS AND METHODS
Screening for hydrogen peroxide resistance
Strains were first inoculated into 96 deep-well plates with liquid yeast extract–peptone–dextrose (YPD) medium. These cultures were then incubated for 24 h at 30°C with shaking. After incubation, cells were manually pinned onto YPD agar plates containing different concentrations of hydrogen peroxide. Pinned colonies were incubated at 30°C for 48 h, after which colonies were imaged using a standard digital camera. The MIC was calculated as the lowest concentration of hydrogen peroxide at which a segregant was incapable of growing. Representative images of individuals pinned across a range of doses are shown in Supplemental Figure S8A. Supplemental Figure S8B and Supplemental Table S4 show that, in our experimental setup, there is a low correlation between OD600 readings and MICs. This finding implies that the preculturing steps in our experiments are unlikely to have an effect on our genetic mapping results.
Generation of resistant advanced backcross populations
Standard yeast techniques were used for mating and sporulation. At each stage of crossing, the Synthetic Genetic Array marker system (Tong and Boone, 2006) was used to generate large numbers of MATa recombinant segregants through random spore analysis (Ehrenreich et al., 2010). We started by screening 864 F2 segregants each from the BYxRM, BYxYPS, and RMxYPS crosses. The five most hydrogen peroxide–resistant F2 segregants from each cross were then backcrossed to both of their parents. At the F2B and F2B2 steps, 96 recombinants were screened, with the most resistant individual used for the subsequent backcrossing. Freezer stocks were generated for every segregant before phenotyping for hydrogen peroxide resistance, and all crosses were performed using these freezer stocks. At the F2B3 stage, 672 segregants were phenotyped per backcross family. Of these, 12–15 of the most resistant segregants were genotyped using low-coverage, whole-genome sequencing.
Generation of parental reference genomes
We sequenced whole-genome libraries from each of our parent strains to ∼50× coverage on an Illumina (San Diego, CA) HiSeq and used these data to identify genetic differences between our cross parents and the S288c reference genome. Sequencing reads were mapped to the S288c reference genome using Burrows-Wheeler Aligner (BWA-MEM; Li and Durbin, 2009) using the commands bwa –mem –t 6 ref.fsa read1.fq read2.fq > output.sam. Duplicate reads were removed using the SAMtools rmdup command. Mpileup files were generated in SAMtools (Li et al., 2009) using the commands samtools mpileup –f ref.fsa read.rmdp.srt.bam > output.mp, and SNPs were identified using custom Python scripts. The numbers of SNP and small indel differences observed between our parent strains and the S288c reference genome were 111 for BY, 46,900 for RM, and 65,193 for YPS. We constructed strain-specific reference genomes for each strain by integrating these variants into the S288c reference genome. These strain-specific reference genomes were used in subsequent analyses.
Low-coverage, whole-genome sequencing of resistant backcross segregants
Whole-genome libraries were generated for resistant F2B3 segregants using the Illumina Nextera kit, using DNA that had been extracted with Qiagen (Valencia, CA) DNeasy Blood & Tissue Kits. Each backcross segregant was tagged with a unique barcode identifier. Equimolar fractions of the libraries were mixed together and sequenced at low coverage on an Illumina HiSeq. The average coverage per sequenced segregant was 4.31×. After demultiplexing the sequencing reads, reads were aligned to the reference genome of the parent strain used for backcrossing with BWA-MEM using the same parameters described earlier (Li and Durbin, 2009). This was followed by the generation of mpileup files with SAMtools (Li et al., 2009). Haplotypes were determined by using the fraction of reads from each parental strain as input for hidden Markov models executed in R chromosome by chromosome (Taylor and Ehrenreich, 2014). Aneuploidies were identified by examining coverage throughout the genome, as determined from the mpileup files. All sequencing data are available from the Sequence Read Archive (www.ncbi.nlm.nih.gov/sra) under the BioProject accession identifier PRJNA291876, study accession identifier SRP063000, and BioSample accession identifiers SAMN03998889–SAMN03999280.
Examination of RMxYPS loci
To determine the effects of sets of RM alleles detected in the RMxYPS cross, we genotyped F2B3 segregants at the relevant loci and combined these data with our knowledge of MICs for each F2B3. Genotyping at these loci was conducted using PCR and restriction enzyme digests. The t tests and ANOVAs were conducted using the t.test() and lm() functions in R (https://cran.r-project.org).
Genetic mapping
Loci were identified by one-tailed binomial tests conducted on the F2B3 genotype data for single families, with statistical tests implemented in R. We conducted one-tailed tests because we expected causal alleles from a given parent to be largely fixed and undetectable in repeated backcrosses to that parent. In contrast, causal alleles from the nonbackcross parent should show significant enrichment among F2B3s from the same family, whereas noncausal alleles should segregate at ∼50% frequency. The binomial tests were conducted for all genomic positions at which the nonbackcross parental genotype was detected in a given family. The FDR associated with each p value was determined using the QVALUE package in R (Storey and Tibshirani, 2003). To estimate q values for each test, we combined p values from all families into a single vector and inputted into QVALUE at the same time. This was done under the assumption that data from different families share a common null distribution, which can be more accurately estimated using the whole data set generated in this study. Detected intervals were determined as the regions of maximal significance at a given locus. The size of an identified locus was determined as the region of a chromosome at which a locus showed maximal significance. To map more finely loci that were identified in multiple families from the same cross, we pooled data from each of the families from the same cross in which a locus was nominally significant. We then delimited the locus as the region of the chromosome showing maximal significance in the pooled data. The 64 unique genomic loci described here were identified by combining detected loci from all crosses. In cases in which overlapping loci were detected in multiple families from the same cross or from different crosses, we defined the region of overlap as the unique genomic locus.
Reciprocal hemizygosity analysis
For a given gene, haploid deletion strains were generated in both of the haploid parents of a cross using the CORE cassette (Storici et al., 2001). To facilitate these knockouts, 60–base pair tails were added to the CORE cassette by PCR. The tails were designed so that integration of the cassette into the genome resulted in loss of the entire coding region of that gene in the recipient strain. To generate hemizygotes, haploid deletion strains were mated to a wild-type version of the other cross parent. These hemizygotes were then screened for hydrogen peroxide resistance as described. We considered a gene cloned by RH if a significant effect was detected based on the examination of at least three independently generated hemizygotes per allele (i.e., six total hemizygotes per gene). Significance was assessed using t tests implemented in R.
Allele replacements
Marker-assisted allele replacement was conducted as described in Matsui et al. (2015). Unless stated otherwise, for a given allele replacement, the full donor allele (200–300 base pairs of upstream sequence, the entire coding region, and ∼50 base pairs of downstream sequence) was amplified from the relevant haploid cross parent. At the same time, the hphMX cassette (Goldstein and McCusker, 1999) was amplified with 60–base pair homology tails. One of the hphMX tails was designed to be identical to the 3′ end of the donor allele PCR product. The other hphMX tail was designed to be identical to the region just downstream of the donor sequence in the recipient strain. Selection for hygromycin resistance was used to obtain at least 20 transformants per attempted replacement. Because recombination between the donor allele and the recipient genome can occur anywhere along the length of the donor allele, marked replacements often result in only partial replacement of a gene. Thus we used Sanger sequencing or restriction typing to identify transformants that possessed complete replacement of a gene or that only integrated the hphMX cassette. These two groups of strains served as the allele replacement and control strains described in this article.
Quantitative PCR
A BYxYPS F2B3 segregant with the BY version of the POR2-SDP1 region and a version of this strain that was genetically engineered to carry the YPS allele of this region were cultured overnight in YPD. These cultures were then transferred to fresh YPD the next day and incubated for 3 h at 30°C with shaking (200 rpm). After this setback step, six replicate YPD cultures were generated from each strain, each of which contained ∼2 × 107 cells. For both genotypes, half of the cultures were kept as controls that were never exposed to hydrogen peroxide, and the other half of the cultures were supplemented with hydrogen peroxide to a concentration of 3 mM. After 30 min, the cultures were pelleted by centrifugation. The supernatant was decanted from these pellets, and each pellet was washed with sterile water. After the wash step, the cultures were pelleted again by centrifugation, snap frozen using liquid nitrogen, and stored at −80°C for subsequent total RNA extraction. The Qiagen RNeasy Plant Mini Kit was used for RNA extraction, and cDNA was obtained using the ThermoFisher Scientific (Waltham, MA) SuperScript VILO cDNA Synthesis Kit. qPCRs were then performed on each sample using the Kapa Biosystems (Wilmington, MA) SYBR Fast qPCR kit and Bio-Rad Laboratories (Hercules, CA) DNA Engine Opticon 2. The relative abundances of POR2 and SDP1 in each sample were determined through comparison to the reference gene, ACT1.
Supplementary Material
Acknowledgments
We thank Jonathan Lee, Takeshi Matsui, Joann Phan, and Matthew Taylor for critically reviewing a draft of the manuscript. We also thank Charles Nicolet and the USC Epigenome Center staff for their help with Illumina sequencing and Norman Arnheim and Jordan Eboreime for assisting us with qPCR experiments. This work was supported by grants from the National Institutes of Health (R01GM110255 and R21AI108939), National Science Foundation (MCB1330874), Alfred P. Sloan Foundation, and Rose Hills Foundation to I.M.E.
Abbreviations used:
- ANOVA
analysis of variance
- FDR
false discovery rate
- MIC
minimum inhibitory concentration
- RH
reciprocal hemizygosity analysis
- SNP
single-nucleotide polymorphism
- YPD
yeast extract–peptone–dextrose.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-06-0408) on October 28, 2015.
REFERENCES
- Aouacheri O, Saka S, Krim M, Messaadia A, Maidi I. The investigation of the oxidative stress-related parameters in type 2 diabetes mellitus. Can J Diabetes. 2015;39:44–49. doi: 10.1016/j.jcjd.2014.03.002. [DOI] [PubMed] [Google Scholar]
- Bloom JS, Ehrenreich IM, Loo WT, Lite TL, Kruglyak L. Finding the sources of missing heritability in a yeast cross. Nature. 2013;494:234–237. doi: 10.1038/nature11867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun RJ, Westermann B. Mitochondrial dynamics in yeast cell death and aging. Biochem Soc Trans. 2011;39:1520–1526. doi: 10.1042/BST0391520. [DOI] [PubMed] [Google Scholar]
- Brown AJ, Budge S, Kaloriti D, Tillmann A, Jacobsen MD, Yin Z, Ene IV, Bohovych I, Sandai D, Kastora S, et al. Stress adaptation in a pathogenic fungus. J Exp Biol. 2014;217:144–155. doi: 10.1242/jeb.088930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubillos FA, Billi E, Zorgo E, Parts L, Fargier P, Omholt S, Blomberg A, Warringer J, Louis EJ, Liti G. Assessing the complex architecture of polygenic traits in diverged yeast populations. Mol Ecol. 2011;20:1401–1413. doi: 10.1111/j.1365-294X.2011.05005.x. [DOI] [PubMed] [Google Scholar]
- Cubillos FA, Parts L, Salinas F, Bergstrom A, Scovacricchi E, Zia A, Illingworth CJ, Mustonen V, Ibstedt S, Warringer J, et al. High-resolution mapping of complex traits with a four-parent advanced intercross yeast population. Genetics. 2013;195:1141–1155. doi: 10.1534/genetics.113.155515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H, Kong Y, Zhang H. Oxidative stress, mitochondrial dysfunction, and aging. J Signal Transduct. 2012;2012:646354. doi: 10.1155/2012/646354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhar R, Sagesser R, Weikert C, Wagner A. Yeast adapts to a changing stressful environment by evolving cross-protection and anticipatory gene regulation. Mol Biol Evol. 2013;30:573–588. doi: 10.1093/molbev/mss253. [DOI] [PubMed] [Google Scholar]
- Dowell RD, Ryan O, Jansen A, Cheung D, Agarwala S, Danford T, Bernstein DA, Rolfe PA, Heisler LE, Chin B, et al. Genotype to phenotype: a complex problem. Science. 2010;328:469. doi: 10.1126/science.1189015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenreich IM, Bloom J, Torabi N, Wang X, Jia Y, Kruglyak L. Genetic architecture of highly complex chemical resistance traits across four yeast strains. PLoS Genet. 2012;8:e1002570. doi: 10.1371/journal.pgen.1002570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenreich IM, Gerke JP, Kruglyak L. Genetic dissection of complex traits in yeast: insights from studies of gene expression and other phenotypes in the BYxRM cross. Cold Spring Harb Symp Quant Biol. 2009;74:145–153. doi: 10.1101/sqb.2009.74.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenreich IM, Torabi N, Jia Y, Kent J, Martis S, Shapiro JA, Gresham D, Caudy AA, Kruglyak L. Dissection of genetically complex traits with extremely large pools of yeast segregants. Nature. 2010;464:1039–1042. doi: 10.1038/nature08923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizio P, Liou LL, Moy VN, Diaspro A, Valentine JS, Gralla EB, Longo VD. SOD2 functions downstream of Sch9 to extend longevity in yeast. Genetics. 2003;163:35–46. doi: 10.1093/genetics/163.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierro-Risco J, Rincon AM, Benitez T, Codon AC. Overexpression of stress-related genes enhances cell viability and velum formation in Sherry wine yeasts. Appl Microbiol Biotechnol. 2013;97:6867–6881. doi: 10.1007/s00253-013-4850-9. [DOI] [PubMed] [Google Scholar]
- Goldstein AL, McCusker JH. Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast. 1999;15:1541–1553. doi: 10.1002/(SICI)1097-0061(199910)15:14<1541::AID-YEA476>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Granek JA, Murray D, Kayrkci O, Magwene PM. The genetic architecture of biofilm formation in a clinical isolate of Saccharomyces cerevisiae. Genetics. 2013;193:587–600. doi: 10.1534/genetics.112.142067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenough MA, Camakaris J, Bush AI. Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochem Int. 2013;62:540–555. doi: 10.1016/j.neuint.2012.08.014. [DOI] [PubMed] [Google Scholar]
- Higgins VJ, Beckhouse AG, Oliver AD, Rogers PJ, Dawes IW. Yeast genome-wide expression analysis identifies a strong ergosterol and oxidative stress response during the initial stages of an industrial lager fermentation. Appl Environ Microbiol. 2003;69:4777–4787. doi: 10.1128/AEM.69.8.4777-4787.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jomova K, Vondrakova D, Lawson M, Valko M. Metals, oxidative stress and neurodegenerative disorders. Mol Cell Biochem. 2010;345:91–104. doi: 10.1007/s11010-010-0563-x. [DOI] [PubMed] [Google Scholar]
- Kitagaki H, Takagi H. Mitochondrial metabolism and stress response of yeast: applications in fermentation technologies. J Biosci Bioeng. 2014;117:383–393. doi: 10.1016/j.jbiosc.2013.09.011. [DOI] [PubMed] [Google Scholar]
- Koppenhofer D, Kettenbaum F, Susloparova A, Law JK, Vu XT, Schwab T, Schafer KH, Ingebrandt S. Neurodegeneration through oxidative stress: monitoring hydrogen peroxide induced apoptosis in primary cells from the subventricular zone of BALB/c mice using field-effect transistors. Biosens Bioelectron. 2015;67:490–496. doi: 10.1016/j.bios.2014.09.012. [DOI] [PubMed] [Google Scholar]
- Kvitek DJ, Will JL, Gasch AP. Variations in stress sensitivity and genomic expression in diverse S. cerevisiae isolates. PLoS Genet. 2008;4:e1000223. doi: 10.1371/journal.pgen.1000223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo VD, Shadel GS, Kaeberlein M, Kennedy B. Replicative and chronological aging in Saccharomyces cerevisiae. Cell Metab. 2012;16:18–31. doi: 10.1016/j.cmet.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz K, Cohen BA. Small- and large-effect quantitative trait locus interactions underlie variation in yeast sporulation efficiency. Genetics. 2012;192:1123–1132. doi: 10.1534/genetics.112.143107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay TF, Stone EA, Ayroles JF. The genetics of quantitative traits: challenges and prospects. Nat Rev Genet. 2009;10:565–577. doi: 10.1038/nrg2612. [DOI] [PubMed] [Google Scholar]
- Matsui T, Linder R, Phan J, Seidl F, Ehrenreich IM. Regulatory rewiring in a cross causes extensive genetic heterogeneity. Genetics. 2015;201:769–777. doi: 10.1534/genetics.115.180661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parts L, Cubillos FA, Warringer J, Jain K, Salinas F, Bumpstead SJ, Molin M, Zia A, Simpson JT, Quail MA, et al. Revealing the genetic structure of a trait by sequencing a population under selection. Genome Res. 2011;21:1131–1138. doi: 10.1101/gr.116731.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petti AA, Crutchfield CA, Rabinowitz JD, Botstein D. Survival of starving yeast is correlated with oxidative stress response and nonrespiratory mitochondrial function. Proc Natl Acad Sci USA. 2011;108:E1089–E1098. doi: 10.1073/pnas.1101494108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasano Y, Haitani Y, Hashida K, Ohtsu I, Shima J, Takagi H. Enhancement of the proline and nitric oxide synthetic pathway improves fermentation ability under multiple baking-associated stress conditions in industrial baker’s yeast. Microbial Cell Fact. 2012;11:40. doi: 10.1186/1475-2859-11-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz LM, Sinha H, Richards DR, Spiegelman JI, Oefner PJ, McCusker JH, Davis RW. Dissecting the architecture of a quantitative trait locus in yeast. Nature. 2002;416:326–330. doi: 10.1038/416326a. [DOI] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storici F, Lewis LK, Resnick MA. In vivo site-directed mutagenesis using oligonucleotides. Nature Biotechnol. 2001;19:773–776. doi: 10.1038/90837. [DOI] [PubMed] [Google Scholar]
- Taylor MB, Ehrenreich IM. Genetic interactions involving five or more genes contribute to a complex trait in yeast. PLoS Genet. 2014;10:e1004324. doi: 10.1371/journal.pgen.1004324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MB, Ehrenreich IM. Transcriptional derepression uncovers cryptic higher-order genetic interactions. PLoS Genet. 2015a;11:e1005606. doi: 10.1371/journal.pgen.1005606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MB, Ehrenreich IM. Higher-order genetic interactions and their contribution to complex traits. Trends Genet. 2015b;31:34–40. doi: 10.1016/j.tig.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong AH, Boone C. Synthetic genetic array analysis in Saccharomyces cerevisiae. Methods Mol Biol. 2006;313:171–192. doi: 10.1385/1-59259-958-3:171. [DOI] [PubMed] [Google Scholar]
- Treusch S, Albert FW, Bloom JS, Kotenko IE, Kruglyak L. Genetic mapping of MAPK-mediated complex traits across S. cerevisiae. PLoS Genet. 2015;11:e1004913. doi: 10.1371/journal.pgen.1004913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varvarovska J, Racek J, Stetina R, Sykora J, Pomahacova R, Rusavy Z, Lacigova S, Trefil L, Siala K, Stozicky F. Aspects of oxidative stress in children with type 1 diabetes mellitus. Biomed Pharmacother. 2004;58:539–545. doi: 10.1016/j.biopha.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Wang X, Kruglyak L. Genetic basis of haloperidol resistance in Saccharomyces cerevisiae is complex and dose dependent. PLoS Genet. 2014;10:e1004894. doi: 10.1371/journal.pgen.1004894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkening S, Lin G, Fritsch ES, Tekkedil MM, Anders S, Kuehn R, Nguyen M, Aiyar RS, Proctor M, Sakhanenko NA, et al. An evaluation of high-throughput approaches to QTL mapping in Saccharomyces cerevisiae. Genetics. 2014;196:853–865. doi: 10.1534/genetics.113.160291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will JL, Kim HS, Clarke J, Painter JC, Fay JC, Gasch AP. Incipient balancing selection through adaptive loss of aquaporins in natural Saccharomyces cerevisiae populations. PLoS Genet. 2010;6:e1000893. doi: 10.1371/journal.pgen.1000893. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.