

Lamin B1 depletion has detrimental effects on brain development. Lamin B1 loss of function affects neuronal development in primary mouse cortical neurons, strongly impairing dendrite length and complexity. This defective dendrite development stems from impaired nuclear ERK signaling and, possibly, mislocalization of nuclear pores.
Abstract
Lamin B1, a key component of the nuclear lamina, plays an important role in brain development and function. A duplication of the human lamin B1 (LMNB1) gene has been linked to adult-onset autosomal dominant leukodystrophy, and mouse and human loss-of-function mutations in lamin B1 are susceptibility factors for neural tube defects. In the mouse, experimental ablation of endogenous lamin B1 (Lmnb1) severely impairs embryonic corticogenesis. Here we report that in primary mouse cortical neurons, LMNB1 overexpression reduces axonal outgrowth, whereas deficiency of endogenous Lmnb1 results in aberrant dendritic development. In the absence of Lmnb1, both the length and complexity of dendrites are reduced, and their growth is unresponsive to KCl stimulation. This defective dendritic outgrowth stems from impaired ERK signaling. In Lmnb1-null neurons, ERK is correctly phosphorylated, but phospho-ERK fails to translocate to the nucleus, possibly due to delocalization of nuclear pore complexes (NPCs) at the nuclear envelope. Taken together, these data highlight a previously unrecognized role of lamin B1 in dendrite development of mouse cortical neurons through regulation of nuclear shuttling of specific signaling molecules and NPC distribution.
INTRODUCTION
Lamin B1 is a major component of the nuclear lamina, and, together with other lamins (e.g., lamin A/C), it plays a role in the structure and function of the nucleus (Zastrow et al., 2006). Over the past few decades, mutations in lamin A and lamin-associated proteins have been shown to cause various human diseases. These diseases, termed laminopathies, include Emery–Dreifuss and limb-girdle muscular dystrophies and Hutchison–Gilford progeria syndrome (Butin-Israeli et al., 2012).
The role of lamin B1 in pathological conditions was discovered relatively recently, when defects in the gene encoding this protein and its regulators were associated with diseases mainly affecting the CNS (Padiath et al., 2006; Brussino et al., 2009; Robinson et al., 2013; Giorgio et al., 2015). Specifically, a duplication of the human lamin B1 gene (LMNB1) has been associated with adult-onset autosomal dominant leukodystrophy (ADLD; Padiath et al., 2006; Brussino et al., 2009), the first discovered laminopathy affecting the CNS (Cortelli et al., 2012). A genomic deletion upstream of LMNB1 that causes up-regulation of its expression has been also identified in one family with an ADLD variant (Giorgio et al., 2015). In addition, loss-of-function mutations in the lamin B1 gene have been linked to neural tube defects as susceptibility factors in mice (De Castro et al., 2012) and humans (Robinson et al., 2013). In mice, Lmnb1 deficiency results in reduced brain size, abnormal layering, and apoptosis of cortical neurons during embryonic corticogenesis (Coffinier et al., 2011; Kim et al., 2011). Conversely, overexpression of murine lamin B1 (Lmnb1) or the human protein (LMNB1) leads to abnormal neuronal activity (e.g., seizures), neuronal degeneration, demyelination, and gliosis accompanied by motor and cognitive impairments (Heng et al., 2013; Rolyan et al., 2015). These pathological phenotypes associated with lamin B1 abnormalities suggest that this protein is essential for proper brain development and function.
The mechanisms by which lamin B1 affects brain development and function are unknown. Although ubiquitous, lamin B1 is highly expressed in rodent brain (Worman et al., 1988; Broers et al., 1997). At the cellular level, lamin B1 defines nuclear mechanics and integrity (Vergnes et al., 2004; Lammerding et al., 2006; Ferrera et al., 2014), regulates nuclear ionic permeability (Ferrera et al., 2014) and signaling through Oct-1 (Columbaro et al., 2013), interacts with chromatin (Malhas et al., 2007; Guelen et al., 2008), and modulates gene expression through mRNA synthesis and processing (Jagatheesan et al., 1999; Malhas et al., 2007; Tang et al., 2008). We demonstrated that in ADLD human fibroblasts and in the brain of Lmnb1-null (Lmnb1Δ/Δ) mice (Vergnes et al., 2004), abnormal levels of lamin B1 are associated with imbalanced expression and splicing of genes involved in neuronal development (Bartoletti-Stella et al., 2015), and lamin B1 finely tunes neuronal versus astrocytic fate commitment during mouse embryonic cortical development (S. Mahajani, C. Giacomini, F. Marinaro, D. DePietri Tonelli, A. Contestabile, and L. Gasparini, unpublished data). However, how abnormal levels of lamin B1 specifically affect the morphological differentiation of postmitotic neurons is unclear.
To address this central question, we examined how lamin B1 regulates axonal and dendritic development of mouse cortical neurons. Here we report that in primary cortical neurons, LMNB1 overexpression reduces axonal outgrowth, whereas deficiency of endogenous Lmnb1 results in aberrant dendritic development. In the absence of Lmnb1, both the length and complexity of dendrites are reduced and their growth is unresponsive to KCl stimulation. This defective dendritic outgrowth stems from impaired extracellular signal–regulated kinase (ERK) 1/2 signaling, likely due to reduced nuclear shuttling of phosphorylated ERK (pERK) and abnormal nuclear pore complex (NPC) distribution.
RESULTS
Lamin B1 expression levels differentially regulate axon and dendrite outgrowth in mouse cortical neurons
In the mouse, Lmnb1 deficiency results in reduced brain size and abnormal layering of cortical neurons (Vergnes et al., 2004; Coffinier et al., 2011). Cortical layers are profoundly disorganized in the Lmnb1Δ/Δ embryonic brain (Coffinier et al., 2011), and βIII-tubulin–positive neurons are abnormally present across the ventricular/subventricular zone (VZ/SVZ; Supplemental Figure S1B), which is usually devoid of mature neurons (Supplemental Figure S1A). In agreement with previous findings (Coffinier et al., 2011), a few apoptotic nuclei are also present in the VZ/SVZ of E17.5 Lmnb1Δ/Δ embryos (Supplemental Figure S1B). These observations suggest that abnormal lamin B1 levels may affect both neuronal survival and differentiation.
To explore whether lamin B1 affects neuronal survival, we analyzed apoptotic nuclei in gain- and loss-of-function conditions in wild-type (WT) cortical neurons nucleofected with pLMNB1–enhanced green fluorescent protein (EGFP) or pEGFP plasmids and in cortical neurons of Lmnb1Δ/Δ embryos and Lmnb1 homozygous littermates (Lmnb1+/+). The number of pyknotic nuclei was comparable in neurons nucleofected with LMNB1 and EGFP or EGFP alone (Supplemental Figure S1C). Conversely, the proportion of apoptotic nuclei in Lmnb1Δ/Δ neuronal cultures was slightly higher than in Lmnb1+/+ (Supplemental Figure S1D), indicating that Lmnb1 deficiency affects the survival of a small subset of neurons. To avoid any confounding effects arising from cell death in subsequent analyses, we considered only neurons with normal chromatin staining.
To test how lamin B1 affects neuronal morphogenesis, we analyzed axonal outgrowth and dendrite development. To measure their length, we immunostained axons and dendrites for Tau (Figures 1, A and B, and 2, A and B) and MAP2 (Figure 2, A and B), respectively. In 7-d-old neurons overexpressing LMNB1 and EGFP, axons were 26% shorter than in neurons expressing EGFP alone (Figure 1C); this reduction occurs early after plating (mean axonal length at 3 d in vitro [DIV]: EGFP, 186 ± 14 μm; LMNB1/EGFP, 113 ± 14 μm; n = 65 neurons/group; p < 0.05, Student’s t test). Neither length nor complexity of dendritic trees was affected by LMNB1 overexpression (Figure 1, D and E). In Lmnb1Δ/Δ neurons, axonal length was reduced at late (7 DIV) but not early (3 DIV) differentiation stages (Figure 2C). Instead, dendrite development was strongly impaired in Lmnb1Δ/Δ neurons at all differentiation stages. The mean total dendrite length was reduced by 63 and 64% at 3 and 7 DIV, respectively (Figure 2D), and the dendritic tree complexity of Lmnb1Δ/Δ neurons was significantly decreased (Figure 2E). To investigate whether the impaired dendritic development also results in altered synapses, we examined the expression of the presynaptic protein synaptophysin and the dendritic spine protein drebrin in Lmnb1Δ/Δ and Lmnb1+/+ neurons at 18 DIV, when synapses reach maturity in primary cortical neurons (Ichikawa et al., 1993). The expression of both synaptophysin and drebrin (Supplemental Figure S2, A and B) was significantly reduced in Lmnb1Δ/Δ neurons, indicating that the effects of Lmnb1 deficiency are still detectable in mature neurons.
FIGURE 1:
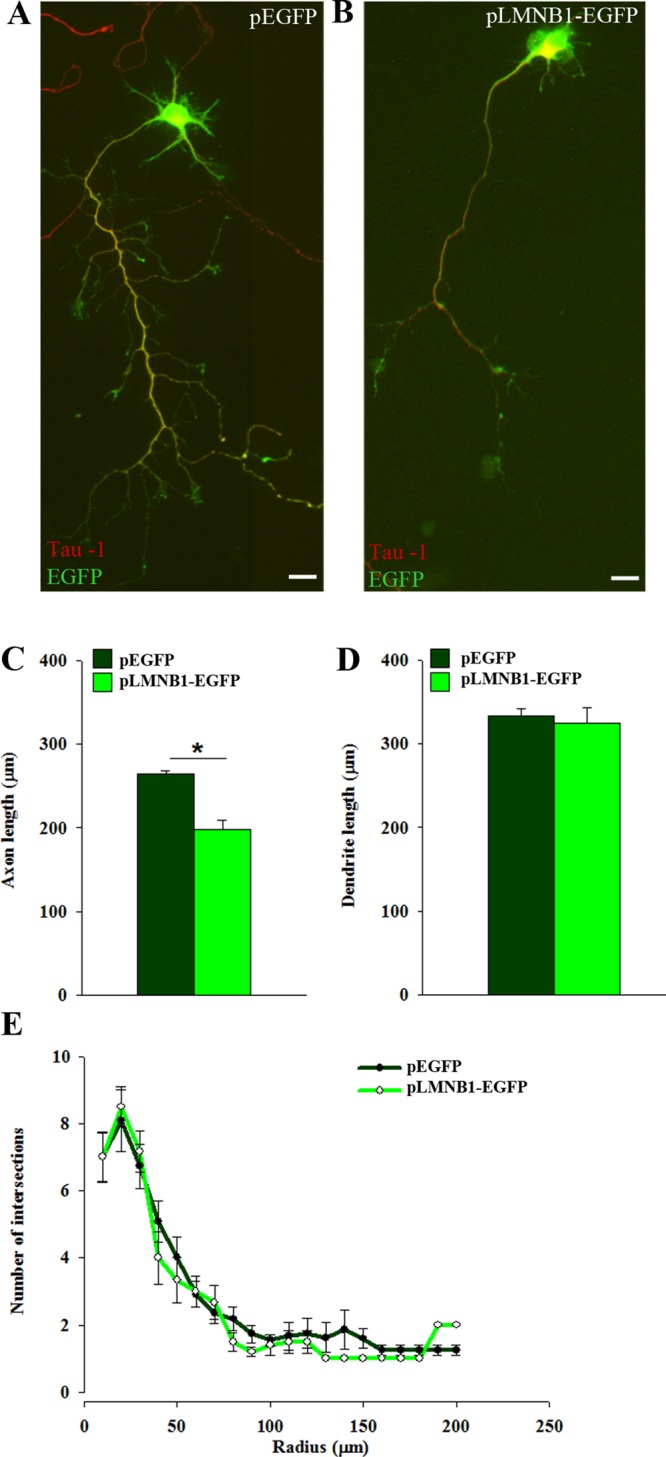
LMNB1 overexpression reduces axon outgrowth in mouse cortical neurons. Primary cortical neurons were transfected before plating by nucleofection with pLMNB1-EGFP or pEGFP. Axonal and dendritic outgrowth was analyzed 7 d later as described in Materials and Methods. (A, B) Representative immunofluorescence images of Tau immunoreactivity (red) in 7 DIV neurons transfected with pEGFP (A) and pLMNB1-EGFP (B). Nuclei are counterstained with DAPI (blue). Scale bars, 20 μm. (C, D) Quantitative analysis of axonal (C) and total dendritic (D) length. Bars represent the mean length ± SEM of 65 neurons/group measured in three independent experiments. *p < 0.05, Student’s t test. (E) Sholl analysis of dendritic tree arborization.
FIGURE 2:

Lmnb1-null cortical neurons develop a deficient dendritic tree. Neuronal morphology was analyzed in Lmnb1+/+ and Lmnb1Δ/Δ primary cortical neurons at 3 and 7 DIV as described in Materials and Methods. (A, B) Representative immunofluorescence images of MAP2 (green) and Tau (red) immunoreactivity in Lmnb1+/+ (A) and Lmnb1Δ/Δ (B) neurons. Scale bars, 20 μm. (C, D) Quantitative analysis of axonal (C) and total dendritic (D) length at 3 and 7 DIV. Bars represent the mean length ± SEM of at least 100 neurons/genotype from four independent experiments. *p < 0.05, **p < 0.01, Student’s t test. (E) Sholl analysis of dendritic tree arborization at 7 DIV. *p < 0.05, two-way ANOVA, followed by multiple comparison with the Holm–Sidak method. (F) Quantitative analysis of total dendrite length in Lmnb1+/+ and Lmnb1Δ/Δ neurons incubated with 18 mM KCl or 12 μm forskolin for 72 h. Data are expressed as mean percentage ± SEM of untreated Lmnb1+/+. At least 100 neurons/genotype for each experimental condition were measured in four independent experiments. *p < 0.05, **p < 0.01, two-way ANOVA, followed by multiple comparison with the Holm–Sidak method.
To investigate whether cognate lamins also contribute to the abnormal morphological differentiation of Lmnb1-null neurons, we analyzed the protein expression of lamin B2 (Lmnb2) and lamin A/C (LmnA/C) by Western blot. In both cultured primary neurons and embryonic brain (Supplemental Figure S3, A and B), the protein levels of Lmnb2 and LmnA/C were similar, ruling out any contribution of cognate lamins to altered dendritic outgrowth.
Taken together, these results indicate that lamin B1 is crucial for morphogenesis of murine cortical neurons and that lamin B1 gain or loss of function has distinct effects on axonal and dendritic compartment development.
Lmnb1 deficiency prevents depolarization-induced dendrite development
Considering that different signaling events regulate axonal and dendritic outgrowth (Shelly et al., 2010, 2011), we next focused on understanding how Lmnb1 loss of function affects dendritic development. The development of dendrites is modulated by neuronal activity (Redmond et al., 2002; Aizawa et al., 2004; Wayman et al., 2006). We thus investigated whether depolarizing stimuli could rescue the dendritic phenotype of Lmnb1Δ/Δ neurons. We treated cortical neurons with or without KCl or forskolin, which induce depolarization through distinct mechanisms—calcium and cAMP elevation, respectively (Impey et al., 1998; Wayman et al., 2006; Mauceri et al., 2011). We started treatment from 5 DIV, a critical time for dendrite elongation and branching (Wayman et al., 2006), and measured the length of dendrites and axons 72 h later. Neither KCl nor forskolin changed axonal length in Lmnb1+/+ (average length of untreated, 238 ± 40 μm; n = 106; KCl, 233 ± 17 μm; n = 96; forskolin, 230 ± 50 μm; n = 95) or Lmnb1Δ/Δ (untreated, 151 ± 20 μm; n = 106; KCl, 182 ± 21 μm; n = 99; forskolin, 174 ± 23 μm; n = 108). Forskolin did not significantly affect dendrite development in either Lmnb1+/+ or Lmnb1Δ/Δ neurons. In contrast, KCl differentially affected dendrite length across genotypes. Dendrite length was increased by 34% with KCl treatment in Lmnb1+/+ neurons but not in Lmnb1Δ/Δ neurons, where it remained similar to that of untreated neurons (Figure 2F). This indicates that in the absence of Lmnb1, dendritic development is insensitive to KCl, suggesting an impaired dendritic outgrowth response to neuronal activity.
Lmnb1 deficiency impairs phospho-ERK nuclear signaling in mouse primary cortical neurons
Activity-induced dendrite outgrowth is regulated through transcriptional programs via cAMP-responsive element–binding protein (CREB), which is activated by phosphorylation on Ser-133 (pCREB) upon nuclear translocation of signaling molecules such as the cAMP-dependent protein kinase A and ERKs (Impey et al., 1998; Wiegert and Bading, 2011). To investigate how Lmnb1 deficiency affects activity-induced PKA and ERK signaling to the nucleus, we investigated CREB activation by examining pCREB in response to depolarizing stimuli.
We stimulated Lmnb1+/+ and Lmnb1Δ/Δ neurons with KCl, which activates CREB via calcium-induced ERK activation (Impey et al., 1998) or forskolin, which activates CREB through cAMP-dependent protein kinase A (Hagiwara et al., 1993; Impey et al., 1998). CREB and pCREB nuclear immunoreactivity showed that total CREB levels were similar in untreated Lmnb1+/+ and Lmnb1Δ/Δ neurons (Figure 3A). When exposed to forskolin, CREB was activated similarly in neurons of both genotypes (Figure 3A). In contrast, stimulation with KCl induced a small but significant increase of pCREB immunoreactivity in Lmnb1+/+ neurons but not in Lmnb1Δ/Δ neurons (Figure 3A). This suggests that in the absence of Lmnb1, the activation of CREB via the cAMP-dependent PKA is preserved, whereas the response mediated by ERK is likely defective.
FIGURE 3:

Lmnb1 deficiency impairs pERK nuclear import in mouse cortical neurons. Lmnb1+/+ and Lmnb1Δ/Δ neurons (7 DIV) were incubated with 50 mM KCl or 72 μM forskolin for 1 h. ERK, pERK, CREB, and pCREB were analyzed as described in Materials and Methods. (A) Quantitative analysis of pCREB immunoreactivity in the nucleus. Bars represent the average intensity ± SEM of nuclear pCREB immunofluorescence normalized to nuclear CREB immunofluorescence. At least 150 neurons/group were analyzed in two independent experiments. *p < 0.01, two-way ANOVA, followed by multiple comparison with the Holm–Sidak method. (B) Representative Western blots of ERK1/2, pERK1/2 and tubulin in neurons incubated with or without KCl. (C) Quantitative analysis of pERK levels. The data are normalized to total ERK. Bars represent the average ratio ± SEM from seven independent experiments. (D, E) Nuclear translocation of pERK in KCl-stimulated neurons. (D) Representative maximal projections of z-stack confocal images of pERK1/2 immunoreactivity (green). (E) Quantitative analysis of pERK immunoreactivity in the nucleus. Bars represent the average intensity ± SEM of nuclear pERK1/2 immunofluorescence normalized to cytosolic pERK1/2 immunofluorescence. At least 65 neurons/group were measured in three independent experiments. *p < 0.01, two-way ANOVA, followed by multiple comparison with the Holm–Sidak method. (F) pERK2-EGFP nuclear translocation. Representative maximal projections of z-stack confocal images of Lmnb1+/+ and Lmnb1Δ/Δ neurons transfected with pERK2-EGFP and incubated with KCl 50 mM for 1 h. (G, H) Representative maximal projections of confocal z-stack images of pHIST3 (green) immunoreactivity in 7DIV Lmnb1+/+ (left panels) and Lmnb1Δ/Δ (right panels) primary cortical neurons incubated in the absence (G) or presence (H) of 50 mM KCl for 1 h. In all fluorescence images, nuclei are counterstained with DAPI (blue). Scale bars, 5 μm.
We next analyzed the expression levels and phosphorylation of ERKs in lysates from Lmnb1Δ/Δ and Lmnb1+/+ cultured neurons incubated with or without KCl for 1 h. In unstimulated neurons from both genotypes, the total protein level of ERKs and their phosphorylation state, indicative of their activation (Wiegert and Bading, 2011), were comparable (Figure 3, B and C). When treated with KCl, pERK levels increased similarly in Lmnb1Δ/Δ and Lmnb1+/+ cortical neurons (Figure 3, B and C), indicating that the absence of Lmnb1 does not affect ERK phosphorylation per se. After phosphorylation, translocation of pERK to the nucleus is required to induce CREB phosphorylation through mitogen and stress-activated kinase 1 (MSK1; Arthur et al., 2004), and it has been shown that lamins regulate pERK nuclear retention in Drosophila cyst stem cells (Chen et al., 2013). To investigate whether Lmnb1 affects pERK nuclear import and signaling, we performed immunocytochemistry in Lmnb1+/+ and Lmnb1Δ/Δ neurons incubated with or without KCl and evaluated pERK subcellular distribution and the activation of the downstream signaling targets MSK1 and histone 3 (HIST3). In untreated neurons of both genotypes, pERK was mainly cytosolic (Figure 3E). When exposed to KCl, pERK was present both in the nucleus and cytoplasm of Lmnb1+/+ neurons but remained largely cytosolic in Lmnb1Δ/Δ (Figure 3, D and E). Indeed, in Lmnb1Δ/Δ nuclei, pERK immunoreactivity inside the nucleus was significantly decreased by 38%, with the vast majority of nuclei (96%) showing nuclear levels of pERKs below the average of Lmnb1+/+ nuclei (Figure 3, D and E). To investigate whether increasing total levels of ERK could rescue its nuclear import, we transiently transfected neurons with EGFP-tagged ERK2 (Zhai et al., 2013) and analyzed its KCl-induced nuclear translocation. In basal conditions, EGFP-ERK2 correctly localized in the cytoplasm of neurons from both genotypes. When stimulated by KCl, EGFP-ERK2 translocated to the nucleus in Lmnb1+/+ neurons but remained cytosolic in Lmnb1Δ/Δ neurons (Figure 3F).
In the nucleus, pERKs activate MSK1, which phosphorylates histone H3 on serine 10 (pHIST3; Soloaga et al., 2003; Brami-Cherrier et al., 2007; Wittmann et al., 2009). To evaluate how the absence of Lmnb1 affects nuclear ERK-mediated signaling, we measured the activated form of MSK1 (phosphorylated on threonine 581) and pHIST3. Consistent with reduced pERK translocation, the immunoreactivity of pHIST3 and active MSK1 was lower in nuclei of Lmnb1Δ/Δ neurons treated with KCl (Figure 3, G and H, and Supplemental Figure S4, A and B) than Lmnb1+/+ nuclei. Therefore, despite unchanged protein levels of pERKs, the absence of Lmnb1 affects their nuclear import and downstream nuclear signaling.
These data indicate that Lmnb1 deficiency selectively impairs nuclear signaling through pERK without affecting cAMP-mediated activation of CREB.
Defective dendritic development in the absence of Lmnb1 stems from impaired pERK signaling
In rat cortical neurons, ERK signaling regulates dendritic complexity (Ha and Redmond, 2008). To investigate the involvement of pERK in the abnormal dendritic outgrowth associated with Lmnb1 loss of function, we incubated mouse cortical neurons with KCl for 72 h with or without the ERK inhibitor FR180204 (Zhai et al., 2013). In WT cortical neurons, this compound inhibits KCl-induced ERK phosphorylation and nuclear translocation (Figure 4, A and B). In Lmnb1+/+ neurons, incubation with FR180204 significantly decreased basal dendrite length and completely blocked KCl-induced dendrite outgrowth (Figure 4C), reducing the dendrite length to levels comparable to those of Lmnb1Δ/Δ neurons. Consistent with defective ERK nuclear signaling, in Lmnb1Δ/Δ neurons, total dendrite length was similar in neurons treated with or without KCl and FR180204 either alone or in combination (Figure 4C). These results indicate that ERK nuclear signaling is required for basal and KCl-induced dendrite outgrowth in mouse cortical neurons and that this signaling is impaired in the absence of Lmnb1.
FIGURE 4:

pERK-induced dendritic outgrowth is impaired in Lmnb1-deficient cortical neurons. pERK nuclear translocation and dendritic outgrowth in primary cortical neurons treated with or without KCl alone or in combination with FR180204. (A, B) Representative maximal projection of confocal z-stack images of pERK immunoreactivity (green) in Lmnb1+/+ primary cortical neurons (7 DIV) treated for 1 h with 50 mM KCl alone (A) or KCl and 36 μM FR180204 (B). Nuclei are counterstained with DAPI. Scale bars, 5 μm. (C) Quantitative analysis of total dendrite length of Lmnb1+/+ and Lmnb1Δ/Δ neurons incubated with or without 50 mM KCl or 36 μm FR180204 alone and in combination for 72 h. Bars represent mean total dendrite length ± SEM from four independent experiments. At least 45 neurons were analyzed for each experimental condition. *p < 0.05, **p < 0.01, two-way ANOVA, followed by multiple comparison with the Holm–Sidak method.
Lamin B1 deficiency affects nuclear morphology in mouse cortical neurons in vitro and in vivo
We next investigated the mechanisms by which Lmnb1 deficiency affects ERK nuclear import. Lmnb1 deficiency causes nuclear abnormalities in Lmnb1Δ/Δ mouse embryonic fibroblasts (Vergnes et al., 2004) and brain (Coffinier et al., 2011). To investigate how Lmnb1 deficiency affects nuclei of postmitotic neurons, we analyzed the nuclear morphology of primary mouse cortical neurons derived from Lmnb1Δ/Δ embryos, using immunofluorescence and transmission electron microscopy (TEM). Nuclei of cultured Lmnb1Δ/Δ neurons were smaller and rounder than those of Lmnb1+/+ embryos (Figure 5, A–F); the area of Lmnb1Δ/Δ nuclei was 43% less than that of Lmnb1+/+ (Figure 5, A–C). With TEM, Lmnb1Δ/Δ nuclei presented a rounder shape than those of Lmnb1+/+ (Figure 5, D and E), as confirmed by increased circularity values (Figure 5F). This phenotype also occurs in vivo: in cortical neurons of Lmnb1Δ/Δ embryos, nuclear area was significantly reduced compared with Lmnb1+/+ littermates (Figure 5, G–I). Taken together, these results indicate that optimal lamin B1 levels are essential to maintain neuronal nuclear size and shape.
FIGURE 5:

Lmnb1 deficiency induces nuclear abnormalities in primary mouse cortical neurons. Nuclei morphology was analyzed in 7 DIV primary cortical neurons or E17.5 embryonic brain from Lmnb1+/+ and Lmnb1Δ/Δ mice as described in Materials and Methods. (A, B) Representative maximal projections of z-stack confocal images of Lmnb1+/+ (A) and Lmnb1Δ/Δ (B) neurons immunostained for βIII-tubulin. Nuclei are counterstained with DAPI (blue). Scale bars, 5 μm. (C) Quantitative analysis of nuclear area. Bars represent the mean area ± SEM of at least 100 neurons/group. (D, E) Representative TEM images of nuclei from Lmnb1+/+ (D) and Lmnb1Δ/Δ (E) primary cortical neurons. Scale bars, 2 μm. (F) Circularity of nuclei determined using TEM images. Lmnb1+/+, n = 28; Lmnb1Δ/Δ, n = 39. (G, H) Representative maximal projections of confocal z-stack images of βIII-tubulin (red) immunoreactivity in the cortical plate of E17.5 Lmnb1+/+ (G) and Lmnb1Δ/Δ (H) embryos. (I) Quantitative analysis of nuclear area. Bars represent the mean area ± SEM. At least 100 neurons from three different embryos/genotype were analyzed. In all graphs, *p < 0.05, **p < 0.01, Student’s t test.
Lamin B1 deficiency affects the distribution and composition of nuclear pore complexes in mouse cortical neurons
Activity-induced dendrite outgrowth is induced by signaling molecules shuttling between cytosol and nucleus (Fainzilber et al., 2011) via NPCs (Raices and D’Angelo, 2012). NPCs are composite structures formed by multiple proteins called nucleoporins (Nups), whose distribution at the nuclear envelope (NE) is regulated by lamins (Chen et al., 2013; Guo et al., 2014). Both A- and B-type lamins bind to Nup153 (Al-Haboubi et al., 2011), which localizes on the nuclear side of NPCs and participates in the nuclear import of several proteins (Walther et al., 2001), including ERK (Vomastek et al., 2008). To investigate whether Lmnb1 deficiency affects NPCs, we analyzed their localization in Lmnb1Δ/Δ and Lmnb1+/+ primary cortical neurons by immunocytochemistry and TEM. The immunoreactivity of NPCs and Nup153 was punctate and evenly distributed on the NE in Lmnb1+/+ neurons both in culture (Figure 6, A and B) and the embryonic brain (Supplemental Figure S5C). In the absence of Lmnb1, NPC and Nup153 signals appeared patchy, with an asymmetric, polarized distribution, covering only discrete regions of the NE in the majority of cortical neurons in vitro (78% of cultured neurons; Figure 6, A and B) and in vivo (Supplemental Figure S5D).
FIGURE 6:

Lmnb1 deficiency alters the distribution of NPCs in mouse cortical neurons. NPCs were analyzed in 7 DIV Lmnb1+/+ and Lmnb1Δ/Δ primary cortical neurons as described in Materials and Methods. (A, B) Representative maximal projections of z-stack confocal images of Lmnb1+/+ and Lmnb1Δ/Δ neurons stained for NPCs (A; green) and Nup153 (B; green). (C–E) TEM images of Lmnb1+/+ (C) and Lmnb1Δ/Δ (D, E) neurons showing NPCs (arrows) in transversal (C, D) and paratangential (E) sections. Asterisks indicate mitochondria. n, nucleus. Scale bars, 200 nm. (F) Top, 3D models of a Lmnb1Δ/Δ nuclear membrane fragment representing the reconstruction of an 800-nm tomogram acquired in HAADF STEM from three semithin serial sections (Supplemental Movie S1). Scale bars, 200 nm. Bottom, single tomogram slices corresponding to sections S1–S3 in the three-dimensional model. White arrows point to NPCs; asterisks indicate mitochondria. n, nucleus. (G) Frequency distribution of interpore distances in nuclei of Lmnb1+/+ (n = 198) and Lmnb1Δ/Δ (n = 557) neurons. The frequency of interpore distances was determined using electron micrographs from >30 transversal sections. (H) Representative Western blot of NPCs and actin in lysates from Lmnb1+/+ and Lmnb1Δ/Δ cultured neurons. (I) Quantitative analysis of NPC expression. The data are normalized to actin. Bars represent the average ratio ± SEM. n = 4 per genotype. *p < 0.05, Student’s t test. (J, K) Lmnb1Δ/Δ neurons were infected with LV syn-tagged-V5-LMNB1 and analyzed by immunocytochemistry. (J) Representative maximal projection of confocal z-stack images of V5 (green) and LMNB1 (red) immunoreactivity in Lmnb1Δ/Δ neurons infected with LV syn-tagged-V5-LMNB1. (K) Representative maximal projection of confocal z-stack images of Tpr (green) and V5 (red) immunoreactivity in Lmnb1Δ/Δ neurons infected with LV syn-tagged-V5-LMNB1. In all fluorescence images, nuclei are counterstained with DAPI (blue). Scale bars, 5 μm.
A similar spatial pattern was found for the nucleoporin Tpr, which is involved in ERK nuclear translocation (Vomastek et al., 2008). In Lmnb1Δ/Δ neurons, Tpr was clustered and asymmetrically distributed across nuclei in vitro (Supplemental Figure S5, A and B) and in vivo (Supplemental Figure S5, C and D). Based on TEM, nuclear tangential and cross sections confirmed that in Lmnb1Δ/Δ, NPCs were distributed irregularly, with some regions of the NE hosting groups of NPCs located close to each other and other areas devoid of NPCs (Figure 6, D and E). High-angle annular dark-field (HAADF) scanning TEM (STEM) tomography performed on serial semithin sections showed that in Lmnb1Δ/Δ neurons, NPCs are distributed in parallel, closely packed rows (Figure 6F and Supplemental Movie S1), whereas in Lmnb1+/+ neurons, NPCs are sparse and distanced (Supplemental Figure S5E and Supplemental Movie S2). Consistently, the interpore distance frequency distribution differed between Lmnb1Δ/Δ and Lmnb1+/+ neurons (Figure 6G). Although the entire distribution covered a broad range (0.1–8 μm) in both genotypes, in Lmnb1Δ/Δ, 66% of interpore distance values were in the range 0.1–1 μm, whereas in Lmnb1+/+, only 37% of values were <1 μm, confirming NPC clustering in the absence of Lmnb1.
To test whether NPC changes specifically relate to Lmnb1 deficiency, we examined whether reinstating levels of lamin B1 by LMNB1 expression restores physiological NPC localization. We transduced primary Lmnb1Δ/Δ cortical neurons with lentiviral particles containing V5-tagged LMNB1 under the synapsin promoter (syn-LMNB1-V5) at 3 DIV. Seven days later, we analyzed the expression and subcellular distribution of LMNB1 and Tpr by immunocytochemistry. In LMNB1-transduced Lmnb1Δ/Δ neurons, the expression of LMNB1 and its localization at the nuclear lamina were partially restored (Figure 6J). In nuclear regions where LMNB1 was correctly expressed and localized, Tpr lost the polarized appearance and was evenly distributed at the nuclear lamina, resuming its normal distribution (Figure 6K). In neighboring nontransduced Lmnb1Δ/Δ neurons, Tpr immunoreactivity remained clustered and asymmetrically distributed across the nucleus (Figure 6K), indicating that lamin B1 is essential for the correct localization of NPCs at the NE.
Nucleocytoplasmic transport of specific molecules through NPCs relies on interactions of transport receptors (e.g., importin and exportin karyopherins) with specific FG-Nups, a subset of nucleoporins harboring repetitive stretches of Phe-Gly residues (Raices and D’Angelo, 2012). To investigate whether changes in Lmnb1 levels affect NPC composition, we examined a subset of FG-Nups in cultured cortical neurons and brain from Lmnb1Δ/Δ and Lmnb1+/+ embryos. FG-Nup protein levels were analyzed by Western blot using the Mab414 antibody, which recognizes several FG-Nups, including Nup62, Nup98, Nup153, Nup214, and Nup358 (Guo et al., 2014). Nup98 was significantly reduced in both Lmnb1Δ/Δ cultured neurons (Figure 6, H and I) and brain (Supplemental Figure S5, F and G), whereas Nup153 was decreased in Lmnb1Δ/Δ brain (Supplemental Figure S5, F and G), but not cultured neurons (Figure 6, H and I). Therefore, abnormal expression of Lmnb1 results in changes in NPC composition and mislocalization, which could underpin the aberrant nuclear shuttling of ERK signaling molecules.
Besides shuttling to the nucleus through direct interaction with NPC, some proteins, including pERK, also bind to karyopherins, which facilitate the transfer of cargoes across NPCs (Tran and Wente, 2006). To investigate whether Lmnb1 deficiency also alters such mechanisms, we examined the protein expression and localization of importin α7 (Imp α7), which mediates alternate pERK nuclear translocation in mammalian (Chuderland et al., 2008) and Drosophila S2 (Lorenzen et al., 2001) cells, and the protein levels of importin β1 (Imp β1), which mediates the import of nuclear localization signal (NLS)–bearing proteins (Soniat and Chook, 2015). Localization of Imp α7 and protein expression of both Imp α7 and Imp β1 were similar in Lmnb1Δ/Δ and Lmnb1+/+ neurons (Supplemental Figure 6, A–D). Further, to investigate potential effects of Lmnb1 on NLS-mediated mechanisms, we transfected Lmnb1Δ/Δ and Lmnb1+/+ neurons with a bicistronic plasmid encoding an NLS-bearing green fluorescent protein (GFP-NLS) and the cytosolic tomato reporter fluorescent protein and examined their subcellular localization. GFP-NLS correctly localized to the nucleus in both Lmnb1Δ/Δ and Lmnb1+/+ neurons, without apparent differences across genotypes (Supplemental Figure S6, E and F). These results indicate that Imp α7-, Imp β1-, and NLS-mediated import mechanisms are preserved in the absence of Lmnb1.
DISCUSSION
Lamin B1 depletion has detrimental effects on brain development (Coffinier et al., 2011; Kim et al., 2011). Here we report that both lamin B1 gain and loss of function affect neuronal development in primary cortical neurons, with distinct outcomes. Whereas LMNB1 overexpression selectively decreases axonal outgrowth, Lmnb1 deficiency strongly impairs dendrite length and complexity. This defective dendritic development likely stems from impaired ERK signaling: in the absence of Lmnb1, ERK is correctly phosphorylated but fails to translocate to the nucleus, likely due to NPC mislocalization.
These results shed new insight into potential mechanisms underlying the deleterious effects of Lmnb1 absence on embryonic cortical development. In the mouse, Lmnb1 deficiency results in reduced brain size (Vergnes et al., 2004), decreased cortical thickness, abnormal cortical layering of neurons, and apoptosis (Coffinier et al., 2011; Kim et al., 2011). The altered lamination of Lmnb1-null cortex has been mainly ascribed to defective migration of cortical newborn neurons (Coffinier et al., 2011). However, other mechanisms may also contribute to the complex phenotype of the Lmnb1-null embryonic brain. Indeed, we recently found that abnormal levels of Lmnb1 during early embryonic development influence the neuroglial commitment of neural progenitors (Mahajani et al., unpublished data). We now show that lamin B1 is required for the morphological development of cortical neurons. Specifically, primary Lmnb1-null neurons have reduced nuclear size, impaired dendrite outgrowth, and reduced synaptic protein content. Further, in the Lmnb1-null brain, neurons are present in the VZ/SVZ area, which is usually devoid of mature neurons. This may be a consequence of several mechanisms, including premature, aberrant differentiation of neurons, changes in neuroglial fate commitment, and/or defective migration of newborn neurons, and may ultimately lead to apoptosis and reduced numbers of neurons (Coffinier et al., 2011; Kim et al., 2011). Further investigation is warranted to elucidate the causal and temporal relationship of all these processes and their mutual interactions in determining the composite effects of Lmnb1 deficiency on brain development.
Lamin B1 regulates neuronal morphological differentiation in a complex manner, with different actions depending on its protein levels. Specifically, when LMNB1 is overexpressed, neurons develop shorter axons. This phenotype appears early during neuronal development (3 DIV), suggesting potential effects during initial axonogenesis. However, in the absence of Lmnb1, axonal length is normal during initial neuronal morphogenesis (up to 3 DIV) but is reduced later (7 DIV). This hints at a role of lamin B1 in axon maintenance and is consistent with previous findings showing that the absence of lamin B induces axonal degeneration in Xenopus retinal ganglion cells (Yoon et al., 2012).
In contrast, dendrite development is unaffected by LMNB1 overexpression but is strongly impaired when Lmnb1 is absent, starting during early neuronal morphogenesis. This indicates that loss of lamin B1 specifically affects the development of the dendritic compartment. We also find that lamin B1 is essential for proper regulation of dendrite development by depolarizing stimuli. Consistent with previous reports (Redmond et al., 2002; Wayman et al., 2006; Mauceri et al., 2011), dendrite elongation and arborization is enhanced by KCl exposure, and ERK signaling is required to sustain dendrite outgrowth in both naive and KCl-stimulated Lmnb1+/+ neurons. However, Lmnb1-deficient neurons show reduced nuclear import of ERKs and are unresponsive to KCl, indicating that ERK nuclear signaling likely mediates the effects of lamin B1 on dendritic outgrowth.
Changes in NPC composition have been associated with altered neuronal differentiation. Under chronic stress, rat hippocampal neurons undergo simplification of their dendritic tree and down-regulate Nup62 (Kinoshita et al., 2014). In the mouse embryo, knockdown of Nup210 blocks the differentiation of stem cells into neuronal progenitors (D’Angelo et al., 2012), whereas Nup133 deficiency impairs the generation of terminally differentiated neurons (Lupu et al., 2008). Our results now indicate that abnormal NPC distribution and composition induced by Lmnb1 deficiency are associated with defective dendritic development and impaired nuclear translocation of pERKs. This is consistent with findings that lamin B promotes nuclear retention of ERK in Drosophila cyst stem cells by controlling the distribution of Nup153 and the Tpr homologue MTOR (Chen et al., 2013) and that, in mammalian cells, Tpr depletion decreases the cytoplasm-to-nucleus translocation of ERK2 (Vomastek et al., 2008). Levels of Imp α7 and Imp β1, subcellular localization of Imp α7, and nuclear import through NLS-mediated mechanisms, as well as signaling via cAMP-dependent protein kinase A, are apparently normal in Lmnb1-null neurons, indicating the preservation of all of these mechanisms in the absence of Lmnb1. Taken together, these findings support a model in which Lmnb1 acts on dendritic development by specifically regulating nuclear pore composition and localization, which in turn regulates pERK transport and thereby signaling.
In the nuclear lamina, lamin A and lamin B1 form independent but interacting networks that assemble at the end of mitosis in a specific sequence of events (Moir et al., 2000), with lamin B1 assembling first, followed by lamin A after assembly of all NE major components, including NPCs (Moir et al., 2000). During this composite process, the lamin B1 network possibly acts as a template spacer for proper distribution of NPCs, which are subsequently stabilized by lamin A (Al-Haboubi et al., 2011). NPCs are anchored to lamins through interaction of the lamin immunoglobulin-fold domain and multiple binding sites on Nup153 (Smythe et al., 2000; Al-Haboubi et al., 2011). The complexity of this interaction translates into distinct phenotypes upon ablation of specific lamins or Nups (Walther et al., 2001; Al-Haboubi et al., 2011). In this study, we report that in mouse primary cortical neurons, absence of Lmnb1 is associated with NPC clustering and reduced levels of Nup98 and to some extent Nup153. We also show that transient overexpression of LMNB1 partially restores normal NPC distribution at the NE, indicating that lamin B1 is required for proper NPC localization.
In conclusion, lamin B1 is required for the maintenance of proper nuclear architecture, and signaling defects arising from its loss of function strongly affect the development of dendrites during differentiation of postmitotic neurons. These actions are possibly due to changes in expression of genes involved in neuronal development (Bartoletti-Stella et al., 2015). Consistently, Lmnb1 deficiency also reduces the expression of Nup98, which shuttles between the NPC and nucleoplasm (Griffis et al., 2002) and regulates the activity of several developmental genes (Capelson et al., 2010; Kalverda et al., 2010). Taken together, these data highlight a previously unrecognized role of lamin B1 in the morphogenesis of cortical neurons. Further studies are warranted to investigate how lamin B1’s influence on neuronal differentiation contributes to neurological diseases associated with abnormalities of the gene encoding this nuclear protein.
MATERIALS AND METHODS
Animals
Time-pregnant females of Lmnb1-null (Lmnb1Δ/Δ; MMRRC Mutant Mouse Regional Resource Center, University of California, Davis, Davis, CA; Vergnes et al., 2004) and C57BL6/J WT (Charles River Italia, Calco, Italy) mice were used for this study. Animal health and comfort were veterinary controlled. The mice were housed in filtered cages in a temperature-controlled room with a 12:12 h dark/light cycle with ad libitum access to water and food. All of the animal experiments were performed in full compliance with the European Community Council directive 86/609/EEC and the revised directive 2010/63/EU and were approved by the Italian Ministry of Health and the Istituto Italiano di Tecnologia Ethics Committee. Offspring genotypes were determined using primers specific for the endogenous Lmnb1 allele (forward, 5′-TCCGTGTCGTGTGGTAGGAGG-3′; reverse, 5′-GCAGGAGGGTTGGGAAAGCC-3′) and for the mutant allele carrying the gene-trap insertion (forward, as described; reverse, 5′-CACTCCAACCTCCGCAAACTC-3′).
Antibodies and reagents
The following primary antibodies were used for immunocytochemistry and Western blot analysis: rabbit polyclonal antibodies against lamin B1, Tpr, importin α7 (Abcam, Cambridge, MA), pHIST3 (serine 10), MAP-2 (Merck Millipore, Darmstadt, Germany), actin, βIII-tubulin (Sigma-Aldrich, Milano, Italy), pERK, pCREB (Cell Signaling, Beverly, MA), lamin A/C (Santa Cruz Biotechnology, Dallas, TX), synaptophysin (SySy, Gottingen, Germany), and drebrin (Enzo Life Sciences, Farmingdale, NY); and mouse monoclonal antibodies against Nup153, lamin B2, CREB (Abcam), nuclear pore complex (mAb414; Covance, Princeton, NJ), Lap2β (BD Biosciences, East Rutherford, NJ), Tau (Tau-1; Merck Millipore), ERK (Cell Signaling), importin β (Sigma-Aldrich), and Alexa 488–conjugated Tuj1 (Covance). For Western blot analysis, horseradish peroxidase (HRP)–conjugated secondary antibodies (Bio-Rad, Hercules, CA) were used for detection. For immunofluorescence analyses, Alexa fluorophore–conjugated anti-mouse, anti-rabbit, and anti-rat antibodies from Invitrogen (Thermo Fisher Scientific, Waltham, MA) were used. Unless otherwise specified, general reagents and chemicals were from Sigma-Aldrich, and reagents for cell cultures were from Invitrogen.
Primary neuronal cultures
Primary cortical neurons were prepared from embryonic day 17–18 mouse embryos of WT, Lmnb1Δ/Δ, and homozygous (Lmnb1+/+) littermates. Neurons were plated onto poly-l-lysine–coated coverslips at (3–8) × 104 cells/coverslip and cultured in Neurobasal medium containing B-27, GlutaMAX, and penicillin/streptomycin at 37°C in a 5% CO2 humidified atmosphere. Such culturing conditions are optimized to sustain viability of rodent embryonic postmitotic neurons only, without significant glial contamination or proliferation (Brewer et al., 1993, 1995; Brewer and Price, 1996). For dendrite growth analysis, neurons at 5 DIV were incubated with or without 18 mM KCl for 72 h (Wayman et al., 2006) in the presence or absence of 36 μM FR180204, which is a specific ERK inhibitor (Zhai et al., 2013). To analyze pERK nuclear translocation and signaling, neurons were incubated with or without 50 mM KCl for 1 h as previously described (Zhai et al., 2013). In selected experiments, neurons were also incubated with 72 μM forskolin for 1 h (Impey et al., 1998).
Plasmids and transfection
The pCAGGS-LMNB1-ires-EGFP (pLMNB1-EGFP) and pCAGGS-ires-EGFP (pEGFP) plasmids were previously described (Ferrera et al., 2014). The pCAGGS-NLS-EGFP-ires-Tomato plasmid was a kind gift from A. Contestabile (Italian Institute of Technology, Genoa, Italy). The pEGFP-ERK2 (#371450) and pCherry-MEK1 (#31880) were from Addgene (Cambridge, MA). The pEGFP-ERK2 and pCherry-MEK1 plasmids were used in a 1:1 ratio, according to the one-to-one stoichiometry of the interaction between EGFP-ERK2 and Cherry-MEK1 required to retain ERK2 in the extranuclear compartment in unstimulated conditions (Zhai et al., 2013). Adherent neurons were transfected by lipofection (Lipofectamine 2000; Invitrogen). To transfect neurons before plating, we used the Amaxa Nucleofector device and the basal Nucleofector kit for primary neurons (Lonza, Basel, Switzerland) using (1.5–4) × 106 neurons/transfection, according to the manufacturer’s protocol. The lentiviral construct p-Syn-tagged-V5-LMNB1 was obtained by cloning the V5-tagged LMNB1 into a lentiviral vector under the control of the synapsin I promoter. Lentiviral particles were obtained as published (Dull et al., 1998).
Immunofluorescence
Primary cortical neurons were fixed with 4% paraformaldehyde (PFA)/4% sucrose in phosphate-buffered saline (PBS), pH 7.4, permeabilized with 0.1% Triton X-100 in PBS for 5 min, and blocked with 12% normal goat serum (NGS; Jackson ImmunoResearch, Baltimore, MD) and 0.25% Triton X-100 in PBS for 30 min. Neurons were sequentially incubated with primary antibodies, followed by Alexa 488 or Alexa 546–conjugated secondary antibodies. Coverslips were mounted using Prolong Gold (Invitrogen) containing 4′,6′-diamidino-2-phenylindole (DAPI) for nuclear staining. Fluorescence images were acquired using an automated Olympus BX51 microscope equipped with a MBF Optonic CX9000 camera and 40× UPLFLN semi-APO fluorite numerical aperture (NA) 0.3 objective and MBF Neurolucida V11 software. Confocal imaging was performed using a Leica TCS SP5 AOBS tandem inverted confocal microscope equipped with HCX PL APO 40×/NA 1.25 oil and HCX PL APO blue 63×/1.4 NA oil objective lenses.
Nuclear morphology was analyzed by an experienced researcher on blinded samples. Pyknotic nuclei displaying chromatin condensation were identified as apoptotic according to previous work (Taylor et al., 2008). To quantify the nuclear area, neuronal nuclei were counterstained with the nuclear dye DAPI or Hoechst 33342, and measurements were performed on images of blind-coded samples using ImageJ software (Schneider et al., 2012).
To analyze axonal and dendrite outgrowth, neurons were stained against the axonal protein Tau and dendritic protein MAP-2. Fluorescence images were acquired, and neurites were traced and measured using MBF Neurolucida V11 software (MBF Bioscience, Williston, VT). Dendritic arborization was analyzed using Sholl analysis (Sholl, 1953) by counting the number of intersections of dendrites with concentric circles centered on the cell body whose radius increased at regular steps of 10 μm.
To quantify protein levels in subcellular compartments, z-stack confocal images of KCl-treated and untreated neurons were acquired side by side in the same confocal session using fixed laser and scanning settings. Using confocal maximal projections, the average immunofluorescence intensity in the nucleus and cytosol was measured using Leica LAS and ImageJ (Schneider et al., 2012). Data are expressed as ratio of nucleus/cytosol immunofluorescence intensity.
Immunohistochemistry
Embryonic brains were fixed in 4% PFA overnight, cryopreserved in 30% sucrose in PBS overnight at 4°C, embedded in Tissue Tek OCT compound, and stored at −80°C. Cryosections (10 μm) were cut on a Leica CM3050S cryostat and kept in PBS with 0.02% sodium azide until used. Immunohistochemistry and fluorescence microscopy were performed as described (Gasparini et al., 2011). Briefly, sections were blocked in 5% NGS in PBS containing 0.25% Triton X-100 and incubated with primary antibodies in blocking solution overnight at 4°C, followed by appropriate Alexa dye–conjugated secondary antibodies. Sections were counterstained with Hoechst 33342 or DAPI to visualize the nuclei. Fluorescence and confocal imaging was performed as described.
Western blot
Primary neurons were lysed in buffer containing 150 mM NaCl, 50 mM Tris, 1% NP-40, and 0.1% SDS supplemented with protease and phosphatase inhibitors. After 5 min of incubation, lysates were sonicated and clarified by centrifugation at 10,000 × g for 10 min. The protein concentration of samples was estimated with the bicinchoninic acid assay (Pierce, Thermo Fisher Scientific). Equal amounts of proteins were separated on 10% Tris-glycine SDS–PAGE gels. Proteins were then blotted onto 0.2-μm nitrocellulose membranes (Whatman, GE Healthcare, Little Chalfont, UK). Membranes were briefly stained with 0.1% Ponceau S, blocked for 1 h in 5% milk in 10 mM Tris, pH 8.0, 150 mM NaCl, and 0.1% Triton X-100, and incubated overnight at 4°C with primary antibodies, followed by HRP-conjugated secondary antibodies (1:5000; Bio-Rad) for 1 h at room temperature. Bands were visualized using the SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific). Chemiluminescence signals were acquired using films (Hyperfilm ECL; GE Healthcare) or an Image Quant LAS 4000 mini (GE Healthcare). Densitometric analysis was performed using ImageJ (Schneider et al., 2012). Protein levels were normalized to actin content.
Electron microscopy
TEM was performed on Lmnb1Δ/Δ and Lmnb1+/+ primary cortical neurons plated on glass coverslips and fixed with 0.66 M cacodylate-buffered 1.25% glutaraldehyde solution for 1 h at room temperature. After extensive rinsing in the same buffer, the samples were postfixed in 1.5% potassium ferrocyanide and 1% osmium tetroxide in 0.1 M cacodylate buffer, stained overnight in the dark in a 0.5% aqueous solution of uranyl acetate, dehydrated in a graded ethanol series, and embedded in EPON resin. Sections of ∼70 nm were cut with a diamond knife (Diatome, Biel, Switzerland) on a Leica EM UC6 ultramicrotome and stained with lead citrate and uranyl acetate. TEM images were collected using a FEI Tecnai F20 equipped with a field-emission gun and recorded with a 2k × 2k–megapixel Gatan BM UltraScan charge-coupled device (CCD) camera. Interpore distances of NPCs and nuclear circularity were determined from electron micrographs of >30 cross sections of Lmnb1Δ/Δ and Lmnb1+/+ primary cortical neurons using ImageJ (Schneider et al., 2012). HAADF STEM tomography was performed using a Tecnai F20 transmission electron microscope operating at 200 kV equipped with a field-emission gun. For the reconstruction of the Lmnb1+/+ nuclear membrane fragment, a 400-nm-thick section was tilted over ±65° with the following tilt scheme: 1° at tilt higher than ±30° and 2° intervals at intermediate tilts. For the reconstruction of the Lmnb1Δ/Δ nuclear membrane fragment, three 350-nm-thick serial sections were tilted over ±62° at 2° intervals. The images were acquired using a HAADF detector at a magnification of 40,000 and 56,000 times, respectively, using a 2k × 2k–megapixel Gatan UltraScan CCD camera, resulting in a pixel size of, respectively, 2.5 and 1.85 nm. Computation of tomogram and serial tomogram joining were done with the IMOD software package (Kremer et al., 1996; Mastronarde, 2005). Segmentation and three-dimensional visualization were done using the Amira package (FEI Visualization Science Group, Bordeaux, France).
Statistical analysis
Statistical analysis of groups with normal distributions was performed using a Student’s t test for two groups or analysis of variance (ANOVA), followed by the Holm–Sidak post hoc test in case of multiple comparisons. Differences among groups were considered statistically significant when p < 0.05. Data throughout the text are reported as average values ± SEM, except when otherwise specified.
Supplementary Material
Acknowledgments
We are grateful to the following colleagues of the Istituto Italiano di Tecnologia: Marina Nanni and Claudia Chiabrera for technical assistance with neuronal cultures and Andrea Contestabile for the kind gift of pCAGGS-NLS-EGFP-ires-Tomato plasmid. This work was funded by Fondazione TELETHON via Grant GGP 10184 to L.G.
Abbreviations used:
- ADLD
autosomal dominant leukodystrophy
- DAPI
4′,6′-diamidino-2-phenylindole
- DIV
days in vitro
- EGFP
enhanced green fluorescent protein
- HAADF
high-angle annular dark-field
- Imp
importin
- LMNB1
human lamin B1 protein
- Lmnb1
mouse lamin B1 protein
- LMNB1
human lamin B1 gene
- Lmnb1
mouse lamin B1 gene
- Lmnb1Δ/Δ
lamin B1–null mouse
- Lmnb1+/+
lamin B1 homozygous mouse
- NE
nuclear envelope
- NLS
nuclear localization signal
- NPC
nuclear pore complex
- Nup
nucleoporin
- PFA
paraformaldehyde
- PBS
phosphate-buffered saline
- STEM
scanning transmission electron microscopy
- SVZ
subventricular zone
- TEM
transmission electron microscopy
- VZ
ventricular zone
- WT
wild type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-05-0307) on October 28, 2015.
REFERENCES
- Aizawa H, Hu SC, Bobb K, Balakrishnan K, Ince G, Gurevich I, Cowan M, Ghosh A. Dendrite development regulated by CREST, a calcium-regulated transcriptional activator. Science. 2004;303:197–202. doi: 10.1126/science.1089845. [DOI] [PubMed] [Google Scholar]
- Al-Haboubi T, Shumaker DK, Koser J, Wehnert M, Fahrenkrog B. Distinct association of the nuclear pore protein Nup153 with A- and B-type lamins. Nucleus. 2011;2:500–509. doi: 10.4161/nucl.2.5.17913. [DOI] [PubMed] [Google Scholar]
- Arthur JS, Fong AL, Dwyer JM, Davare M, Reese E, Obrietan K, Impey S. Mitogen- and stress-activated protein kinase 1 mediates cAMP response element-binding protein phosphorylation and activation by neurotrophins. J Neurosci. 2004;24:4324–4332. doi: 10.1523/JNEUROSCI.5227-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartoletti-Stella A, Gasparini L, Giacomini C, Corrado P, Terlizzi R, Giorgio E, Magini P, Seri M, Baruzzi A, Parchi P, et al. Messenger RNA processing is altered in autosomal dominant leukodystrophy. Hum Mol Genet. 2015;4:2746–2756. doi: 10.1093/hmg/ddv034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brami-Cherrier K, Lavaur J, Pages C, Arthur JS, Caboche J. Glutamate induces histone H3 phosphorylation but not acetylation in striatal neurons: role of mitogen- and stress-activated kinase-1. J Neurochem. 2007;101:697–708. doi: 10.1111/j.1471-4159.2006.04352.x. [DOI] [PubMed] [Google Scholar]
- Brewer GJ. Serum-free B27/neurobasal medium supports differentiated growth of neurons from the striatum, substantia nigra, septum, cerebral cortex, cerebellum, and dentate gyrus. J Neurosci Res. 1995;42:674–683. doi: 10.1002/jnr.490420510. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Price PJ. Viable cultured neurons in ambient carbon dioxide and hibernation storage for a month. Neuroreport. 1996;7:1509–1512. doi: 10.1097/00001756-199606170-00014. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ. Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res. 1993;35:567–576. doi: 10.1002/jnr.490350513. [DOI] [PubMed] [Google Scholar]
- Broers JL, Machiels BM, Kuijpers HJ, Smedts F, van den KR, Raymond Y, Ramaekers FC. A- and B-type lamins are differentially expressed in normal human tissues. Histochem. Cell Biol. 1997;107:505–517. doi: 10.1007/s004180050138. [DOI] [PubMed] [Google Scholar]
- Brussino A, Vaula G, Cagnoli C, Mauro A, Pradotto L, Daniele D, Di GE, Barberis M, Arduino C, Squadrone S, et al. A novel family with Lamin B1 duplication associated with adult-onset leucoencephalopathy. J Neurol Neurosurg Psychiatry. 2009;80:237–240. doi: 10.1136/jnnp.2008.147330. [DOI] [PubMed] [Google Scholar]
- Butin-Israeli V, Adam SA, Goldman AE, Goldman RD. Nuclear lamin functions and disease. Trends Genet. 2012;28:464–471. doi: 10.1016/j.tig.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capelson M, Liang Y, Schulte R, Mair W, Wagner U, Hetzer MW. Chromatin-bound nuclear pore components regulate gene expression in higher eukaryotes. Cell. 2010;140:372–383. doi: 10.1016/j.cell.2009.12.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Chen X, Zheng Y. The nuclear lamina regulates germline stem cell niche organization via modulation of EGFR signaling. Cell Stem Cell. 2013;13:73–86. doi: 10.1016/j.stem.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuderland D, Konson A, Seger R. Identification and characterization of a general nuclear translocation signal in signaling proteins. Mol Cell. 2008;31:850–861. doi: 10.1016/j.molcel.2008.08.007. [DOI] [PubMed] [Google Scholar]
- Coffinier C, Jung HJ, Nobumori C, Chang S, Tu Y, Barnes RH, 2nd, Yoshinaga Y, de Jong PJ, Vergnes L, Reue K, et al. Deficiencies in lamin B1 and lamin B2 cause neurodevelopmental defects and distinct nuclear shape abnormalities in neurons. Mol Biol Cell. 2011;22:4683–4693. doi: 10.1091/mbc.E11-06-0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Columbaro M, Mattiolo E, Maraldi NM, Ortolani M, Gasparini L, D’Apice MR, Postorivo D, Nardone AM, Avnet S, Cortelli P, et al. OCT-1 recruitment to the nuclear envelope in adult-onset autosomal dominant leukodystrophy. Biochim Biophys Acta. 2013;1832:411–420. doi: 10.1016/j.bbadis.2012.12.006. [DOI] [PubMed] [Google Scholar]
- Cortelli P, Terlizzi R, Capellari S, Benarroch E. Nuclear lamins: functions and clinical implications. Neurology. 2012;79:1726–1731. doi: 10.1212/WNL.0b013e31826ea887. [DOI] [PubMed] [Google Scholar]
- D’Angelo MA, Gomez-Cavazos JS, Mei A, Lackner DH, Hetzer MW. A change in nuclear pore complex composition regulates cell differentiation. Dev Cell. 2012;22:446–458. doi: 10.1016/j.devcel.2011.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Castro SC, Malhas A, Leung KY, Gustavsson P, Vaux DJ, Copp AJ, Greene ND. Lamin b1 polymorphism influences morphology of the nuclear envelope, cell cycle progression, and risk of neural tube defects in mice. PLoS Genet. 2012;8:e1003059. doi: 10.1371/journal.pgen.1003059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fainzilber M, Budnik V, Segal RA, Kreutz MR. From synapse to nucleus and back again–communication over distance within neurons. J Neurosci. 2011;31:16045–16048. doi: 10.1523/JNEUROSCI.4006-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrera D, Canale C, Marotta R, Mazzaro N, Gritti M, Mazzanti M, Capellari S, Cortelli P, Gasparini L. Lamin B1 overexpression increases nuclear rigidity in autosomal dominant leukodystrophy fibroblasts. FASEB J. 2014;28:3906–3918. doi: 10.1096/fj.13-247635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparini L, Crowther RA, Martin KR, Berg N, Coleman M, Goedert M, Spillantini MG. Tau inclusions in retinal ganglion cells of human P301S tau transgenic mice: effects on axonal viability. Neurobiol Aging. 2011;32:419–433. doi: 10.1016/j.neurobiolaging.2009.03.002. [DOI] [PubMed] [Google Scholar]
- Giorgio E, Robyr D, Spielmann M, Ferrero E, Di Gregorio E, Imperiale D, Vaula G, Stamoulis G, Santoni F, Atzori C, et al. A large genomic deletion leads to enhancer adoption by the lamin B1 gene: a second path to autosomal dominant leukodystrophy (ADLD) Hum Mol Genet. 2015;24:3143–3154. doi: 10.1093/hmg/ddv065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffis ER, Altan N, Lippincott-Schwartz J, Powers MA. Nup98 is a mobile nucleoporin with transcription-dependent dynamics. Mol Biol Cell. 2002;13:1282–1297. doi: 10.1091/mbc.01-11-0538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, van Steensel B. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008;453:948–951. doi: 10.1038/nature06947. [DOI] [PubMed] [Google Scholar]
- Guo Y, Kim Y, Shimi T, Goldman RD, Zheng Y. Concentration-dependent lamin assembly and its roles in the localization of other nuclear proteins. Mol Biol Cell. 2014;25:1287–1297. doi: 10.1091/mbc.E13-11-0644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha S, Redmond L. ERK mediates activity dependent neuronal complexity via sustained activity and CREB-mediated signaling. Dev Neurobiol. 2008;68:1565–1579. doi: 10.1002/dneu.20682. [DOI] [PubMed] [Google Scholar]
- Hagiwara M, Brindle P, Harootunian A, Armstrong R, Rivier J, Vale W, Tsien R, Montminy MR. Coupling of hormonal stimulation and transcription via the cyclic AMP-responsive factor CREB is rate limited by nuclear entry of protein kinase A. Mol Cell Biol. 1993;13:4852–4859. doi: 10.1128/mcb.13.8.4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng MY, Lin ST, Verret L, Huang Y, Kamiya S, Padiath QS, Tong Y, Palop JJ, Huang EJ, Ptacek LJ, Fu YH. Lamin B1 mediates cell-autonomous neuropathology in a leukodystrophy mouse model. J Clin Invest. 2013;123:2719–2729. doi: 10.1172/JCI66737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichikawa M, Muramoto K, Kobayashi K, Kawahara M, Kuroda Y. Formation and maturation of synapses in primary cultures of rat cerebral cortical cells: an electron microscopic study. Neurosci Res. 1993;16:95–103. doi: 10.1016/0168-0102(93)90076-3. [DOI] [PubMed] [Google Scholar]
- Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron. 1998;21:869–883. doi: 10.1016/s0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- Jagatheesan G, Thanumalayan S, Muralikrishna B, Rangaraj N, Karande AA, Parnaik VK. Colocalization of intranuclear lamin foci with RNA splicing factors. J Cell Sci. 1999;112:4651–4661. doi: 10.1242/jcs.112.24.4651. [DOI] [PubMed] [Google Scholar]
- Kalverda B, Pickersgill H, Shloma VV, Fornerod M. Nucleoporins directly stimulate expression of developmental and cell-cycle genes inside the nucleoplasm. Cell. 2010;140:360–371. doi: 10.1016/j.cell.2010.01.011. [DOI] [PubMed] [Google Scholar]
- Kim Y, Sharov AA, McDole K, Cheng M, Hao H, Fan CM, Gaiano N, Ko MS, Zheng Y. Mouse B-type lamins are required for proper organogenesis but not by embryonic stem cells. Science. 2011;334:1706–1710. doi: 10.1126/science.1211222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita Y, Hunter RG, Gray JD, Mesias R, McEwen BS, Benson DL, Kohtz DS. Role for NUP62 depletion and PYK2 redistribution in dendritic retraction resulting from chronic stress. Proc Natl Acad Sci USA. 2014;111:16130–16135. doi: 10.1073/pnas.1418896111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J Struct Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Lammerding J, Fong LG, Ji JY, Reue K, Stewart CL, Young SG, Lee RT. Lamins A and C but not lamin B1 regulate nuclear mechanics. J Biol Chem. 2006;281:25768–25780. doi: 10.1074/jbc.M513511200. [DOI] [PubMed] [Google Scholar]
- Lorenzen JA, Baker SE, Denhez F, Melnick MB, Brower DL, Perkins LA. Nuclear import of activated D-ERK by DIM-7, an importin family member encoded by the gene moleskin. Development. 2001;128:1403–1414. doi: 10.1242/dev.128.8.1403. [DOI] [PubMed] [Google Scholar]
- Lupu F, Alves A, Anderson K, Doye V, Lacy E. Nuclear pore composition regulates neural stem/progenitor cell differentiation in the mouse embryo. Dev Cell. 2008;14:831–842. doi: 10.1016/j.devcel.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhas A, Lee CF, Sanders R, Saunders NJ, Vaux DJ. Defects in lamin B1 expression or processing affect interphase chromosome position and gene expression. J Cell Biol. 2007;176:593–603. doi: 10.1083/jcb.200607054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN. Automated electron microscope tomography using robust prediction of specimen movements. J Struct Biol. 2005;152:36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Mauceri D, Freitag HE, Oliveira AM, Bengtson CP, Bading H. Nuclear calcium-VEGFD signaling controls maintenance of dendrite arborization necessary for memory formation. Neuron. 2011;71:117–130. doi: 10.1016/j.neuron.2011.04.022. [DOI] [PubMed] [Google Scholar]
- Moir RD, Yoon M, Khuon S, Goldman RD. Nuclear lamins A and B1: different pathways of assembly during nuclear envelope formation in living cells. J Cell Biol. 2000;151:1155–1168. doi: 10.1083/jcb.151.6.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padiath QS, Saigoh K, Schiffmann R, Asahara H, Yamada T, Koeppen A, Hogan K, Ptacek LJ, Fu YH. Lamin B1 duplications cause autosomal dominant leukodystrophy. Nat Genet. 2006;38:1114–1123. doi: 10.1038/ng1872. [DOI] [PubMed] [Google Scholar]
- Raices M, D’Angelo MA. Nuclear pore complex composition: a new regulator of tissue-specific and developmental functions. Nat Rev Mol Cell Biol. 2012;13:687–699. doi: 10.1038/nrm3461. [DOI] [PubMed] [Google Scholar]
- Redmond L, Kashani AH, Ghosh A. Calcium regulation of dendritic growth via CaM kinase IV and CREB-mediated transcription. Neuron. 2002;34:999–1010. doi: 10.1016/s0896-6273(02)00737-7. [DOI] [PubMed] [Google Scholar]
- Robinson A, Partridge D, Malhas A, De Castro SC, Gustavsson P, Thompson DN, Vaux DJ, Copp AJ, Stanier P, Bassuk AG, Greene ND. Is LMNB1 a susceptibility gene for neural tube defects in humans? Birth Defects Res. A Clin Mol Teratol. 2013;97:398–402. doi: 10.1002/bdra.23141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolyan H, Tyurina YY, Hernandez M, Amoscato AA, Sparvero LJ, Nmezi BC, Lu Y, Estecio MR, Lin K, Chen J, et al. Defects of lipid synthesis are linked to the age-dependent demyelination caused by lamin b1 overexpression. J Neurosci. 2015;35:12002–12017. doi: 10.1523/JNEUROSCI.1668-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelly M, Cancedda L, Lim BK, Popescu AT, Cheng PL, Gao H, Poo MM. Semaphorin3A regulates neuronal polarization by suppressing axon formation and promoting dendrite growth. Neuron. 2011;71:433–446. doi: 10.1016/j.neuron.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelly M, Lim BK, Cancedda L, Heilshorn SC, Gao H, Poo MM. Local and long-range reciprocal regulation of cAMP and cGMP in axon/dendrite formation. Science. 2010;327:547–552. doi: 10.1126/science.1179735. [DOI] [PubMed] [Google Scholar]
- Sholl DA. Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat. 1953;87:387–406. [PMC free article] [PubMed] [Google Scholar]
- Smythe C, Jenkins HE, Hutchison CJ. Incorporation of the nuclear pore basket protein nup153 into nuclear pore structures is dependent upon lamina assembly: evidence from cell-free extracts of Xenopus eggs. EMBO J. 2000;19:3918–3931. doi: 10.1093/emboj/19.15.3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soloaga A, Thomson S, Wiggin GR, Rampersaud N, Dyson MH, Hazzalin CA, Mahadevan LC, Arthur JS. MSK2 and MSK1 mediate the mitogen- and stress-induced phosphorylation of histone H3 and HMG-14. EMBO J. 2003;22:2788–2797. doi: 10.1093/emboj/cdg273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soniat M, Chook YM. Nuclear localization signals for four distinct karyopherin-beta nuclear import systems. Biochem J. 2015;468:353–362. doi: 10.1042/BJ20150368. [DOI] [PubMed] [Google Scholar]
- Tang CW, Maya-Mendoza A, Martin C, Zeng K, Chen S, Feret D, Wilson SA, Jackson DA. The integrity of a lamin-B1-dependent nucleoskeleton is a fundamental determinant of RNA synthesis in human cells. J Cell Sci. 2008;121:1014–1024. doi: 10.1242/jcs.020982. [DOI] [PubMed] [Google Scholar]
- Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9:231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- Tran EJ, Wente SR. Dynamic nuclear pore complexes: life on the edge. Cell. 2006;125:1041–1053. doi: 10.1016/j.cell.2006.05.027. [DOI] [PubMed] [Google Scholar]
- Vergnes L, Peterfy M, Bergo MO, Young SG, Reue K. Lamin B1 is required for mouse development and nuclear integrity. Proc Natl Acad Sci USA. 2004;101:10428–10433. doi: 10.1073/pnas.0401424101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vomastek T, Iwanicki MP, Burack WR, Tiwari D, Kumar D, Parsons JT, Weber MJ, Nandicoori VK. Extracellular signal-regulated kinase 2 (ERK2) phosphorylation sites and docking domain on the nuclear pore complex protein Tpr cooperatively regulate ERK2-Tpr interaction. Mol Cell Biol. 2008;28:6954–6966. doi: 10.1128/MCB.00925-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walther TC, Fornerod M, Pickersgill H, Goldberg M, Allen TD, Mattaj IW. The nucleoporin Nup153 is required for nuclear pore basket formation, nuclear pore complex anchoring and import of a subset of nuclear proteins. EMBO J. 2001;20:5703–5714. doi: 10.1093/emboj/20.20.5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Impey S, Marks D, Saneyoshi T, Grant WF, Derkach V, Soderling TR. Activity-dependent dendritic arborization mediated by CaM-kinase I activation and enhanced CREB-dependent transcription of Wnt-2. Neuron. 2006;50:897–909. doi: 10.1016/j.neuron.2006.05.008. [DOI] [PubMed] [Google Scholar]
- Wiegert JS, Bading H. Activity-dependent calcium signaling and ERK-MAP kinases in neurons: a link to structural plasticity of the nucleus and gene transcription regulation. Cell Calcium. 2011;49:296–305. doi: 10.1016/j.ceca.2010.11.009. [DOI] [PubMed] [Google Scholar]
- Wittmann M, Queisser G, Eder A, Wiegert JS, Bengtson CP, Hellwig A, Wittum G, Bading H. Synaptic activity induces dramatic changes in the geometry of the cell nucleus: interplay between nuclear structure, histone H3 phosphorylation, and nuclear calcium signaling. J Neurosci. 2009;29:14687–14700. doi: 10.1523/JNEUROSCI.1160-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worman HJ, Lazaridis I, Georgatos SD. Nuclear lamina heterogeneity in mammalian cells. Differential expression of the major lamins and variations in lamin B phosphorylation. J Biol Chem. 1988;263:12135–12141. [PubMed] [Google Scholar]
- Yoon BC, Jung H, Dwivedy A, O’Hare CM, Zivraj KH, Holt CE. Local translation of extranuclear lamin B promotes axon maintenance. Cell. 2012;148:752–764. doi: 10.1016/j.cell.2011.11.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zastrow MS, Flaherty DB, Benian GM, Wilson KL. Nuclear titin interacts with A- and B-type lamins in vitro and in vivo. J Cell Sci. 2006;119:239–249. doi: 10.1242/jcs.02728. [DOI] [PubMed] [Google Scholar]
- Zhai S, Ark ED, Parra-Bueno P, Yasuda R. Long-distance integration of nuclear ERK signaling triggered by activation of a few dendritic spines. Science. 2013;342:1107–1111. doi: 10.1126/science.1245622. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.