

Abstract
The ability to quantify levels of target analytes in biological samples accurately and precisely, in biomonitoring, involves the use of highly sensitive and selective instrumentation such as tandem mass spectrometers and a thorough understanding of highly variable matrix effects. Typically, matrix effects are caused by co-eluting matrix components that alter the ionization of target analytes as well as the chromatographic response of target analytes, leading to reduced or increased sensitivity of the analysis. Thus, before the desired accuracy and precision standards of laboratory data are achieved, these effects must be characterized and controlled. Here we present our review and observations of matrix effects encountered during the validation and implementation of tandem mass spectrometry-based analytical methods. We also provide systematic, comprehensive laboratory strategies needed to control challenges posed by matrix effects in order to ensure delivery of the most accurate data for biomonitoring studies assessing exposure to environmental toxicants.
Keywords: matrix effects, biological analysis, tandem mass-spectrometry, biomonitoring, analytical method development
BACKGROUND
Tandem-mass spectrometry (MS/MS) is a fundamentally powerful analytical technique and is normally used in conjunction with either liquid chromatography (LC) or gas chromatography (GC) for the quantitative analysis of target compounds in biological samples. However, due to its design, it is often vulnerable to matrix effects that may compromise its sensitivity and selectivity thus reduce the accuracy, precision, and robustness of its application (Matuszewski, Constanzer et al. 2003; Antignac, de Wasch et al. 2005; Taylor 2005; Ghosh, Shinde et al. 2012). Generally, the term, “matrix effects,” refers to a difference in mass spectrometric response for an analyte in standard solution versus the response for the same analyte in a biological matrix such as urine, plasma, or serum (Tang and Kebarle 1993). These effects commonly result from endogenous matrix components and preservative agents that can affect chromatographic behavior and the ionization of target compounds, resulting in ion suppression or enhancement (Mei, Hsieh et al. 2003). However, matrix effects vary depending upon ionization type, sample preparation, and biological matrix (Dams, Huestis et al. 2003).
It is important that matrix effects be investigated and managed during the validation and implementation of a method because they can lead to inaccurate measurements of target compounds (Hajslova and Zrostlikova 2003; Chambers, Wagrowski-Diehl et al. 2007; Chiu, Lawi et al. 2010). In their, “Guidance for Industry: Bioanalytical Method Validation,” the U.S. Food and Drug Administration (FDA) states that,
“It may be important to consider the variability of the matrix due to the physiological nature of the sample. In the case of [HP]LC-MS-MS-based procedures, appropriate steps should be taken to ensure the lack of matrix effects throughout the application of the method, especially if the nature of the matrix changes from the matrix used during method validation” (FDA 2001).
According to this recommendation, every laboratory involved in biological analysis should develop procedures that will minimize and manage matrix effects.
In this paper, we present our review, observations, and evaluation of matrix effects during the validation and implementation of tandem-mass-spectrometric-based analytical methods used for the biomonitoring of human exposure to commonly used pesticides such as pyrethroids, organophosphates, and triazine, and commonly used flame retardants such as polybrominated diphenyl ethers (PBDEs). We provide systematic, comprehensive laboratory strategies needed to control existing challenges posed by matrix effects to ensure delivery of the most accurate data on biomonitoring studies. We believe this will help advance existing knowledge on the validation of bioanalytical methods against matrix effects. Additional information regarding matrix effects and their analytical management strategies outside the scope of this review can be found elsewhere (Hewavitharana 2011; Furey, Moriarty et al. 2013).
SOURCES OF MATRIX EFFECTS
Both endogenous and exogenous substances found in biological samples are primary sources of matrix effects associated with either high performance (HP)-LC or GC-MS methods (Mei, Hsieh et al. 2003; Chambers, Wagrowski-Diehl et al. 2007). Endogenous substances include salts, carbohydrates, amines, urea, lipids, peptides, and metabolites (Little, Wempe et al. 2006; Sviridov and Hortin 2009; Ismaiel, Zhang et al. 2010). Exogenous substances contributing to matrix effects include mobile phase additives such as trifluoroacetic acid (TFA) and buffer salts (Garcia 2005) plastic materials such as phthalates, and the commonly used anticoagulant, Li-heparin (Mei, Hsieh et al. 2003; Yu and Xu 2012).
Matrix effects are complex and both compound- and system-specific (Bonfiglio, King et al. 1999; Jewett, Ramaley et al. 1999; Dams, Huestis et al. 2003; Cappiello, Famiglini et al. 2008). Each biological matrix has a unique composition and consequently requires different management strategies (Chiu, Lawi et al. 2010). Additionally, each type of analytical method is affected by matrix components differently (Chiu, Lawi et al. 2010). See Table 1 for the general composition of selected endogenous substances that usually contribute to matrix effects (Ajmani and Rifkind 1998; Picciano 2001; Chiu, Lawi et al. 2010). The main substances found in most matrices include ions, amino acids, and vitamins. In addition, phospholipids such as lysophospholipids are more likely to cause matrix effects in bioanalytical LC/MS/MS methods, but the extent of ionization suppression is analyte-dependent (Xia and Jemal 2009).
Table 1. General composition of selected biological matrices.
| Components | Matrices | ||
|---|---|---|---|
| Plasma/Serum | Urine | Breast Milk | |
| Cell | Epithelial, Leukocytes, Lymphocytes, Macrophages, Neutrophils |
||
| Ions | Na+, K+, Ca2+, Cl−, Mg2+, HCO3−, HPO42−, HSO4− |
Na+, K+, Ca2+, Cl−, Mg2+; NH4+, Sulfates, Phosphates |
Bicarbonate, Calcium Chloride Citrate Magnesium Phosphate Potassium Sodium Sulfate Trace minerals Chromium Cobalt Copper Fluoride Iodine Iron Manganese Molybdenum Nickel Selenium Zinc |
|
Organic
molecules |
Urea, Creatinine, Uric Acid, Amino Acids, Glucose, Bilirubin, Insulin |
Urea, Creatinine, Uric Acid, Citrate, DNA, Amino Acids |
Lactose, Glucose, Nucleotide Sugars, Creatinine, Glucosamine, Urea, Uric Acid, Carotenoids |
| Protein | Albumins, Globulins, Fibrinogen, Clotting factors |
Immunoglobulins, Albumin |
Albumins, Immunoglobulins, Lysozymes, Casiens Thyroxine, Amylaze, Lipase, Glycoproteins |
| Lipid | Phospholipids, Cholesterol, Triglycerides |
Triglycerides, Essential Fatty Acids, Glycolipids, Phospholipids |
|
| Others | Water-soluble vitamins |
Fat-soluble vitamins (A, D, E, K); Water-soluble vitamins, Biotin, Choline, Folate, Inositol, Niacin, Pantothenic acid, Riboflavin, Thiamin |
|
MATRIX EFFECTS IN LIQUID CHROMATOGRAPHY-MASS SPECTROMETRY
In HPLC coupled with either MS or MS/MS, the most commonly observed matrix effect is ion suppression, a loss of ion intensity of the target analyte. Of the two most common atmospheric pressure ionization (API) techniques used, electro-spray ionization (ESI) is more susceptible to ion suppression because of its mechanism of ionization, in comparison to atmospheric pressure chemical ionization (APCI) (Dams, Huestis et al. 2003). In HPLC-ESI-MS, matrix components can suppress the ion intensity of a target analyte by interfering with its ionization during the: (1) addition of a charge to the analyte in the liquid phase and (2) transfer of ions to the gas phase from the droplet surface (Tang and Kebarle 1993; King, Bonfiglio et al. 2000). Additionally, matrix components can compete with the target analyte for the available charges in the liquid phase, causing ion suppression. The presence of interfering compounds at high concentrations increases the viscosity and surface tension of the droplets produced in the ESI interface and can reduce the ability of the analyte to reach the gas phase. Also, co-precipitation of the analytes with nonvolatile material such as macromolecules can limit their transfer into the gas phase. In addition, while in the gas phase, interfering substances can neutralize the analyte ions or impact the stability of the ions produced (King, Bonfiglio et al. 2000). Figure 1 summarizes the mechanisms of matrix effects in ESI.
Figure 1.

Mechanisms of matrix effects in ESI
APCI, while offering different mechanisms of ionization than ESI, is not free from matrix effects. However, it is known to usually be less susceptible (Jewett, Ramaley et al. 1999; Dams, Huestis et al. 2003; Sangster, Spence et al. 2004; Souverain, Rudaz et al. 2004). In HPLC-APCI-MS, a variety of factors may lead to ion suppression. Theoretically, the transfer of charge in APCI occurs in the gas phase, therefore no competition for charge in the liquid phase or competition to get into the gas phase, should exist. A process possibly leading to ion suppression in APCI, from co-precipitation with non-volatile compounds, is the difference in electron affinity between compounds in the gas phase. The presence of a large number of chargeable species in the gas phase could increase the possibility of suppression by competing with the target analyte to receive the charge (Sangster, Spence et al. 2004). In positive mode, protonated ions will be formed if the proton affinity (PA) of the compound exceeds that of the reagent gas. In negative mode, deprotonated ions will be observed if the gas-phase acidity of the reagent gas exceeds that of the compound.
In comparison with different ionization polarities, the negative mode is generally considered to be more specific and therefore less subject to ion suppression (Antignac, de Wasch et al. 2005). Ghosh et al. (2010) demonstrated this by investigating the role of ionization polarity on matrix effects. They analyzed enalapril and its metabolite enalaprilat in positive and negative polarity by using ESI-LC-MS/MS. Plasma was used as the working matrix. The results showed approximately 30-35% ion suppression in positive polarity for both analytes, but approximately 20% ion suppression for enalapril and 10% ion enhancement for enalaprilat in negative polarity (Ghosh, Shinde et al. 2010). A lesser degree of matrix effects in negative polarity may be explained by the fact that there are a fewer number of compounds forming negative ions in the negative mode, in comparison to in the positive mode, thus reducing the competition among ions to receive charge (Antignac, de Wasch et al. 2005). In positive polarity, a large number of compounds such as proteins, peptides, amino acids, and salts, are able to form positively charged ions, which consequently contribute to a higher degree of ion suppression, as explained above in the mechanism of matrix effects. In addition, use of acidic mobile phase in HPLC with reversed phase chromatography to stabilize the retention time and selectivity of target analytes as well as to reduce peak broadening or peak tailing due to the, “silanol effect,” can promote the number of positively charged ions in the system and therefore cause ion suppression in ESI. The silanol effect results from silanols on the silica surface of the analytical column becoming deprotonated at mid pH and thus interacting with positively charged ions, causing ion-exchange interactions. This is considered a secondary interaction that usually results in peak broadening and tailing of basic compounds in commonly-used silica phase analytical columns. Acidic mobile phase is able to reduce silanol effects by donating a proton to neutralize silanol groups (Agilent 2012).
MATRIX EFFECTS IN GAS CHROMATOGRAPHY-MASS SPECTROMETRY
In GC-based methods, a, “matrix-induced chromatographic response,” (Hajslova and Zrostlikova 2003) is a well-known matrix effect. Erney et al. discussed improved chromatographic peak intensity and shape for certain compounds when samples were injected in the presence of a complex matrix (Erney, Gillespie et al. 1993). Without matrix components during injection, analytes showed broad (or asymmetric) peaks and low response. The enhancement of matrix-induced chromatographic response is caused by a blockage of active sites in the injector by matrix components, which prevents thermal degradation/adsorption of the target analyte. Active sites in the GC injector refer to free silanol groups and metals potentially present in the surface of even high-quality glass injection liners (although commercially available as a deactivated product). In addition, the enhancement of matrix-induced chromatographic response is caused by active sites in the analytical column being blocked by co-eluted matrices that are typically considered the most abundant components of a sample. This results in target analytes being eluted faster and with a sharper peak shape due to reduced interactions between target analytes and active sites in the analytical column, thus leading to fewer losses of intensity (Hajslova and Zrostlikova 2003). However, this often, but not always, leads to low resolution of separated peaks, such as in the case of isomeric peaks observed in most pyrethroid insecticides.
Usually, compounds prone to matrix-induced chromatographic enhancement are either thermally labile or polar analytes capable of hydrogen bonding. For pesticides, these include carbamates and organophosphates, respectively (Schenck and Lehotay 2000; Hajslova and Zrostlikova 2003). Recently, we found that such enhancement can be observed with pyrethroids, which are non-polar compounds, when analyzed in serum and breast milk. Figure 2 shows evidence of matrix-induced chromatographic enhancement of the pyrethroids: permethrin, cypermethrin, cyfluthrin, deltamethrin, and fenvalerate. Sharper peaks with more intensity were observed for these pyrethroids when analyzed together with biological matrices.
Figure 2. An example of matrix-induced chromatographic response observed for pyrethroids when analyzed in breast milk and serum samples.

Note: Pyrethroids in serum and breast milk samples were analyzed using a GC-MS/MS system (7000) from Agilent Technologies(Waldbronn, Germany) coupled with electron impact ionization interface. The GC and MS modules were programmed and controlled using Mass Hunter Software version B.03.01 (Agilent Technologies). A DB-5MS (30 m × 0.250 mm ID × 0.50 μm film thickness) analytical column from Agilent Technologies was used. Serum samples were subjected to protein precipitation using a mixture of water and propanol (85:15 v/v) prior to extraction with C18 solid phase extraction cartridges (6 cc, 500 mg) (JT Baker, Center Valley, PA). The eluents (in hexane/ethyl ether) were further purified using florisil cartridges (6 cc, 500 mg) (Restek Corp, Bellefonte, CA) while breast milk samples were subjected to liquid-liquid extraction (using acetonitrile followed by hexane) and overnight freezing to remove lipids. Extractants were then loaded onto ENVI-Carb-II™/PSA cartridges (6cc, 500/300 mg) (Sigma Aldrich, St. Louis, MO). Final eluents (from both methods) were evaporated to dryness and the residues were reconstituted with 50 μL of an acetonitrile-toluene mixture (66:34 v/v) prior to injection. Theinjection volume was 2 μL under pulsed splitless mode and an injector temperature of 250 °C. A multi-stepwise gradient temperature program (from 70 °C to 310 °C) was used during chromatographic separation. The total run time was 40.2 min. The flow rate of carrier gas helium was at 1.2 mL/min. Quantitative MS/MS transitions were: permethrin m/z183.2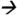 77.0@40V CE; cypermethrin and cyfluthrin m/z163.1
77.0@40V CE; cypermethrin and cyfluthrin m/z163.1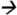 127.2@5V CE; fenvalerate m/z125.2
127.2@5V CE; fenvalerate m/z125.2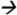 89.0@20V CE; deltamethrin m/z253.1
89.0@20V CE; deltamethrin m/z253.1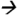 93.2@20V CE; and cypermethrin (13C) m/z170.0
93.2@20V CE; and cypermethrin (13C) m/z170.0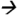 98.0@15V CE.
98.0@15V CE.
The suppression effects induced by co-eluting compounds were also found in GC with either electron impact ionization (EI) or chemical ionization (CI). While the mechanism has not yet been explained, co-eluted matrix components that contribute to such effects include phthalates, caffeine, aliphatic acids, alcohol, aldehydes, and sterols (Yu and Xu 2012). We hypothesize that these matrix components may compete with or suppress the ionization ability of analytes in the gas phase. One plausible mechanism for a loss of intensity in EI is the binding of target ions through opposite electron affinity with other ions present in the matrix.
METHODS FOR EVALUATION OF MATRIX EFFECTS
Flow-based evaluation
Post-column infusion
Bonfiglio et al. proposed a qualitative method to evaluate matrix effects (Bonfiglio, King et al. 1999). In this method, analytes are constantly infused into the system via a T-connector between the column and the MS, and a baseline signal is found. Then, un-spiked matrix that has been extracted and cleaned using the selected sample preparation method is injected into the HPLC-MS. Ionization suppression or enhancement may be observed by comparing changes from baseline across the chromatographic run. Matrix effects may change throughout the chromatographic run, according to what components are eluted into the MS. Matrix effects at the time in which the analyte is expected to elute from the column should be considered most important (Bonfiglio, King et al. 1999).
Flow injection analysis
Similar to post column infusion, flow injection analysis may be used to evaluate matrix effects. In this method, a sample is injected into the MS using a sample loop which allows the peak to last for approximately one minute. Then, a number of scans, typically 30 or more, are averaged to calculate the instrument response. Next, a mobile phase additive is added to the mobile phase and the injection is repeated. Again, the same scans are averaged to calculate instrument response. In this way, ionization suppression and/or enhancement due to the additive may be calculated. While this method is useful for evaluating matrix effects from particular compounds, it may not be as useful for biological matrices in which co-elutants are unknown (Holcapek, Volna et al. 2004).
An example of flow-based matrix effects evaluation is shown in Figure 3, where a 3-phenoxybenzoic acid (3-PBA, a common metabolic product of pyrethroids) standard solution was constantly infused into the HPLC system using an automated syringe system and via a T-connector. The baseline intensity of this compound was obtained and then the extracted sample was injected into the column. Matrix effects were observed, as shown by an alteration of intensity. In this case, the baseline intensity drastically dropped. Note that ion suppression occurred mostly after the column void volume was reached and the majority of the polar compounds, mostly non-volatile, were eluted from the column.
Figure 3. Example of chromatogram during a flow-based matrix assessment for 3-PBA in LC-MS/MS analysis.

Note: A standard solution of 3-PBA in MeOH was infused into a QQQ 6490 LC-MS/MS system (Agilent Technologies, Waldbronn, Germany) using a Fusion 100 syringe pump (Chemyx Inc., Stafford, TX) at a flow rate of 0.1 ml/min via T-connector. The LC and MS modules were programmed and controlled using Mass Hunter Software version B.04.01 (Agilent Technologies). An isocratic composition of water (10%) and methanol (90%) (with 0.1% acetic acid) was supplied by the HPLC pump at a flow rate of 0.2 ml/min. The MS/MS parameters were set as follows: ESI (negative mode); 300 °C drying gas temperature; 325 °C sheath gas temperature; 5 L/min drying gas flow; 10 L/min sheath gas flow; 35 psi nebulizer pressure; 3000 V capillary voltage; 500 V nozzle voltage; 250 msec dwell time; and 400 V delta EMV (−). Nitrogen was used as collision gas. Monitoring MS/MS transition for 3-PBA was 213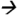 93 @ 16 CE. The injection volume of sample extractant was 5 μL.
93 @ 16 CE. The injection volume of sample extractant was 5 μL.
Post-extraction spike matrix comparison
Matuszewski et al. proposed a comprehensive methodology that relies on the use of spiked matrix comparisons to evaluate matrix effects of biological matrices (Matuszewski, Constanzer et al. 2003). While this method focused on evaluating the matrix effects of plasma samples on the quantification of a drug candidate, it may easily be adapted to accommodate other biological matrices.
In their methodology, three sets of samples were created. The first set was made using neat standard dissolved in a selected solvent (standard solution samples) and the second set was made by fortifying extracted plasma with the compound (post-extraction spiked samples). The third set was made by fortifying plasma with the compound before extraction (pre-extraction spiked samples) and was, on the other hand, used to calculate extraction efficiency. All samples, separated by HPLC, were analyzed using designated ionization interfaces in the MS. First, absolute matrix effects were calculated by comparing the mean peak area of the target analyte in post-extraction spiked samples to the mean peak area of the target analyte in standard solutions. Matrix effects are expressed as a ratio of the mean peak area of an analyte in post-extraction spiked samples to the mean peak area of the same analyte in standard solutions, multiplied by 100. A value greater than 100% indicates ionization enhancement and a value less than 100% indicates ionization suppression. Second, when different lots of biological matrix were obtained, relative matrix effects were assessed via a direct comparison of the mean peak area of the target analyte among post-extraction spiked samples originating from each lot of biological matrix. The variability in these responses, expressed as coefficients of variation (CVs) (%), may be considered a measure of the relative matrix effect for a given analyte. If relative matrix effects are significantly different between groups, the method should not be considered suitable for accurate and precise analyses (Matuszewski, Constanzer et al. 2003).
Alternative method
Another approach to assessing matrix effects in quantitative biological analyses involves adding known amounts of target compounds to biological matrices obtained from at least ten donors. The samples are then prepared and analyzed. Matrix effects are subsequently assessed through calculating the standard deviation (SD) and CV (Olsson, Baker et al. 2004; Baker, Olsson et al. 2005; Montesano, Olsson et al. 2007; Whitehead, Montesano et al. 2010; Panuwet, Nguyen et al. 2012). Based on this method, the observed variation in matrix effects likely represents the differences across samples analyzed. This is very important, particularly for urine sample analysis, because urine concentration (as measured by creatinine concentration or osmolality) can vary drastically across individuals, depending on a variety of factors, including diet and hydration status (WHO 1996; Chiu, Lawi et al. 2010). Urine samples that are more concentrated contain more matrix components that contribute to more matrix effects, in comparison to less concentrated urine samples. An example is shown in a later section.
Also, it is important to note that far more than ten samples may be needed to account fully for the intrinsic variability of the method itself as well as the variability resulting from matrix effects. Simply checking the variation of control samples, which are prepared in the same matrix as the calibration standards (e.g., same dilution, same lot), will not reveal matrix effects observed in the discrete samples (Dewe, De Smet et al. 2007). Also, when pooled samples are used to assess matrix effect variations, they only reveal average results and are unlikely to represent the full spectrum of matrix variations that may occur across all samples. In regard to acceptable variations, it is recommended that whenever variability is greater than 15%, modifications are made to the relevant step(s) in order to reduce the influence of the matrix (Viswanathan, Bansal et al. 2007).
CONTROLLING MATRIX EFFECTS IN AN ANALYTICAL METHOD
While matrix effects cannot be eliminated entirely, they can be minimized or compensated for in an analysis and should be thoroughly investigated as part of any analytical method development and validation. Several measures can be utilized in an attempt to control matrix effects in an analytical method. For example, if co-eluting matrix components and the relation they have with the target analyte are known, adjustment of chromatographic parameters to avoid their co-elutions can minimize effects. Improved chromatography is a straightforward way to separate interfering compounds from analytes (Van Eeckhaut, Lanckmans et al. 2009). However, for a multi-residue analysis, an adjustment of chromatographic parameters can compromise the throughput of the method. Another method used to compensate for matrix effects is the standard addition method (Garrido Frenich, Martinez Vidal et al. 2009). This method allows for internal matrix correction, but is time and resource consuming and often increases the number of analyses necessary by 4-5 times.
Based on our experience, here we present common strategies used to minimize or compensate for matrix effects in an LC- or GC-MS/MS method. A combination of these approaches is recommended to successfully manage matrix effects so that they will not alter method accuracy and precision.
Assessing matrix effects across a dilution range: application for urine analysis
Urine is one of the most complex biological matrices and likely has the widest dilution range. The dilution of urine can be determined using osmolality or estimated using creatinine values. Typically, urine has an osmolality range of 50-1300 mOsmol/kg (Chiu, Lawi et al. 2010). According to a WHO standard testing protocol, urine samples collected from individuals (morning voids or spot samples) can vary in dilution more than 10-fold (creatinine values are <30 ng/dL or >300 ng/dL) (WHO 1996).
For any analyte that is vulnerable to urinary matrix effects, its sensitivity in a given method, as measured by peak intensity, depends upon endogenous concentrations of urinary constituents. Usually, the more concentrated the urine, the more dramatic the observed matrix effects. For example, in our investigation of matrix effects occurring during HPLC-MS/MS analysis of the herbicide atrazine and its metabolites, we observed significant changes in sensitivity when differing amounts of urine were used. Figure 4 summarizes the average sensitivity (n=3) observed for each measured analyte across different volumes of urine. The results demonstrate different degrees of ion suppression or ion enhancement for each measured compound (0% indicated no effect, a negative % value indicated ion suppression, and a positive % value indicated ion enhancement). It is important that matrix effects are investigated in this manner so that an appropriate compensation protocol can be developed. Without knowing the sample dilution ratio, we conclude that analyzing urine samples collected from different individuals, without using appropriate internal standards, is not advised.
Figure 4. Degree(s) of sensitivity alteration of atrazine and its metabolites observed during an LC-MS/MS analysis in urine samples.

Note: Urinary matrices (each with 3 replicates) were acidified with 2 mL of 2% formic acid and extracted using Strata-XC solid phase extraction cartridges (Phenomenex, Torrance, CA). A detailed methoddescription is provided elsewhere (Panuwet, 2010). The extractants were spiked with known amounts of the standard atrazine and its metabolites (10 ppb) and were subjected to separation and analysis using a QQQ 6490 LC-MS/MS system (Agilent Technology, Waldbronn, Germany) coupled with a positive ESI interface. The LC and MS modules were programmed and controlled using Mass Hunter Software version B.04.01 (Agilent Technologies). A gradient composition of water and MeOH (with 0.2% formic acid) was used during chromatographic separation. A flow rate of 0.7 ml/min was used. The analytical column used was C6-Phenyl (3 μm, 4.6 × 100 mm) from Phenomenex (Torrance, CA). The MS/MS parameters were set as follows: 300 °C drying gas temperature; 325 °C sheath gas temperature; 5 L/min drying gas flow; 10 L/min sheath gas flow; 45 psi nebulizer pressure; 3000 V capillary voltage; 500 V nozzle voltage; 120 msec dwell time; and 200 V delta EMV (+). Nitrogen was used as collision gas. Quantitative MS/MS transitions were: DAAM m/z273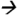 144@9V CE; DACT m/z146
144@9V CE; DACT m/z146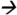 68@25V CE; DEAM m/z315
68@25V CE; DEAM m/z315 186@13V CE; DEA m/z188
186@13V CE; DEA m/z188 146@17V CE; DIAM (13C3)m/z304
146@17V CE; DIAM (13C3)m/z304 175@13V CE; DIA m/z174
175@13V CE; DIA m/z174 68@25V CE; ATZM m/z343
68@25V CE; ATZM m/z343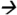 214@13V CE; and ATZ m/z216
214@13V CE; and ATZ m/z216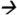 174@13V CE. The sample extractant injection volume was 10 μL and the total run time was 20 mins. An average response of each MS/MS transition was calculated. The degree of matrix effects (%) was calculated using the following equation: 100-[(average response of analyte in matrix eluent/average response of analyte in standard solution)*100]. Negative values represent the degree of suppression (%) whereas positive values represent the degree of enhancement (%).
174@13V CE. The sample extractant injection volume was 10 μL and the total run time was 20 mins. An average response of each MS/MS transition was calculated. The degree of matrix effects (%) was calculated using the following equation: 100-[(average response of analyte in matrix eluent/average response of analyte in standard solution)*100]. Negative values represent the degree of suppression (%) whereas positive values represent the degree of enhancement (%).
Use of dilution or direct injection
The simplest procedure to minimize ion suppression in an analytical method is to introduce less matrix into the analytic system. This can be achieved through dilution or direct-injection of the samples without any pre-concentration steps. It has been noted that these two sample preparation methods result in much less ion suppression in HPLC-ESI-MS/MS (Dams, Huestis et al. 2003). However, the lack of a pre-concentration step in the sample preparation procedure reduces sensitivity and increases the method limit of detection (Dams, Huestis et al. 2003). While both procedures may be applicable in pharmacokinetic studies (i.e. using dosed animals) because these samples usually contain high amounts of analytes, they may not be as useful in biomonitoring studies which aim to measure samples with much lower amounts of target compounds.
Use of solid phase extraction and assessment of its inherent matrix effects
Solid phase extraction (SPE) is commonly used for extracting target analytes from various matrices, including biological samples, due to its robustness. Still, SPE can contribute to the degree of observed matrix effects, particularly ion suppression in HPLC-ESI-MS/MS (Souverain, Rudaz et al. 2004). While matrix clean-up is more extensive with SPE, it allows some interfering compounds that have similar functional groups as the target analytes to be retained and concentrated, thus affecting the ionization of the target analytes (Dams, Huestis et al. 2003).
Typically, the selection of a particular SPE cartridge for sample extraction is made by comparing extraction efficiencies among several candidate cartridges. The cartridge with the highest extraction efficiency, with an eye toward cost, is usually selected. However, we demonstrated through our experiment that the potential matrix effects associated with SPE should be used as an additional criterion for SPE cartridge selection. We compared the degree of matrix effects associated with various types of SPE through the analysis of 3-PBA and 3,5,6-trichloro-2-pyridinol (TCPY), a specific metabolite of chlorpyrifos and chlorpyrifos-methyl, in urine samples. Four cartridges were chosen based on previous reports on extraction recoveries (Olsson, Baker et al. 2004; Nishioka, Mccauley et al. 2006; Le Grand, Dulaurent et al. 2012) or their chemical compatibility with the target analytes. These cartridges include C18 (Silica based C18; JT Baker, Center Valley, PA), HLB (hydrophilic-lipophilic-balanced reversed-phase sorbent; Waters, Milford, MA), Strata-X (reversed-phase sorbent; Phenomenex, Terrance, CA), and Strata-XA (mixed mode reversed-phase/anion exchange; Phenomenex, Terrance, CA). The extraction recoveries of C18 and HLB cartridges for both compounds were more than 80% (Olsson, Baker et al. 2004; Nishioka, Mccauley et al. 2006; Ahn, Lohstroh et al. 2007). We estimated similar results for Strata-X and Strata-XA because they share the same major functional groups as HLB. Additionally, the anion exchange functional group in the Strata-XA provides complete retention of acidic compounds such as 3-PBA and TCPY.
Figure 5 demonstrates the matrix effects associated with each type of SPE on the response of 3-PBA and TCPY. Both ion suppression and enhancement were observed (0% indicated no effect, a negative % indicated ion suppression, and a positive % indicated ion enhancement). Based on the same amount of sorbent mass used, Strata-XA cartridges showed slightly more ion suppression than HLB. In addition, Strata-XA cartridges showed far more degree of suppression than the Strata X despite the fact that less sorbent mass was used. Chambers et al. stated that mixed mode reversed phase/ion exchange cartridges generally provide cleaner samples and therefore reduced ion suppression in the LC-MS/MS system, but our results suggest otherwise (Chambers, Wagrowski-Diehl et al. 2007). A leading candidate responsible for ion suppression in Strata-XA is its retention of urinary salts and acidic compounds. In this experiment, C18 cartridges showed the highest degree of ion suppression. Apart from having the highest amount of sorbent mass to retain target compounds, they may allow large molecules such as peptides, carbohydrates, or vitamins to be co-extracted and thus be present in the final sample to facilitate or cause ion suppression. As previously reported, molecules with a higher mass, such as those mentioned earlier, can suppress the signal of smaller molecules (Sterner, Johnston et al. 2000).
Figure 5. Potential matrix effects associated with type of cartridge used.

Note: Matrix effects were investigated using a post-extraction spike matrix comparison. Experiments began with extracting 1 mL of urine (3 replicates) using 4 different cartridges: Oasis HLB (3 cc, 60 mg) (Waters Corp, Milford, MA); Strata-X (3 cc, 200 mg) (Phenomenex, Torrance, CA); Strata-XA (3 cc, 60 mg) (Phenomenex); and C18 (6 cc, 500 mg) (JT Baker, Center Valley, PA). Generic extraction methods were followed per each manufacturer. Prior to evaporation, the eluents were spiked with standard containing 10 ppb of target compounds. Dried residues were reconstituted with 100 μL of 30:70 MeOH:water (v/v) and 10 μL was injected into a QQQ 6490 LC-MS/MS system (Agilent Technologies, Waldbronn, Germany) coupled with a negative ESI interface. The LC and MS modules were programmed and controlled using Mass Hunter Software version B.04.01 (Agilent Technologies, Waldbronn, Germany). The chromatographic and MS/MS parameters were set the same as those mentioned in Figure 3. An average response of quantitative MS/MS transition for 3-PBA (m/z 213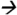 99@15V CE) and quantitative pseudo-MS/MS transition for TCPY (m/z 207
99@15V CE) and quantitative pseudo-MS/MS transition for TCPY (m/z 207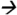 207@1CE) was calculated. The degree of matrix effects (%) was calculated in a manner similar to Figure 3.
207@1CE) was calculated. The degree of matrix effects (%) was calculated in a manner similar to Figure 3.
Our results emphasize that cartridge selection during method development should not only be based on extraction efficiency exclusively, but also on its contribution to matrix effects. Additionally, our results highlight the fact that matrix effects associated with SPE are compound-dependent, in need of thorough investigation, and are usually unpredictable.
Use of isotopically labeled internal standards
Use of the isotopically labeled analogue of the target analyte as an internal standard is the most effective way to rectify or account for matrix effects (Colby and McCaman 1979; Berg and Strand 2011). Theoretically, the same degree of ion suppression or enhancement will be observed for the target analyte and its isotopically labeled analogue. However, the ratio of the two signals should not be affected and correct quantification can still be achieved (Colby and McCaman 1979; Berg and Strand 2011). Unfortunately, isotopically labeled analogues are costly and often unavailable.
Isotopically labeled analogues are compounds for which atoms in the molecule are replaced with their stable isotopes such as 2H, 13C, 15N, or 18O. Generally, the 13C, 15N, and 18O labeled analogues are more similar to their native form than 2H labeled analogues and are therefore expected to behave more similarly in chromatographic separations, resulting in the same retention time (Berg and Strand 2011). The 2H labeled analogue has a stronger binding affinity than the 1H isotope and has twice the atomic weight of its analogue and thus has a slightly different retention time than its native form in GC (Briscoe, Stiles et al. 2007). 2H labeled analogues usually elute several seconds before the native 1H species in GC but because of its poorer resolution, usually coelute in HPLC.
Berg and Strand have investigated both 13C and 2H labeled internal standards for the ultrahigh pressure liquid chromatography (UPLC)-MS/MS analysis of drugs in biological samples, using amphetamine and methamphetamine as example drugs (Berg and Strand 2011). The 13C labeled analogues co-eluted with their native forms under different chromatographic conditions while the 2H labeled analogues and their native forms slightly separated. It was reported that an improved ability to compensate for ion suppression effects, increased linear range, and a better correlation between UPLC-MS/MS analyses and GC-MS analyses of unknown samples, were observed when the 13C labeled analogues were used. The 13C labeled analogues were fully capable of correcting for ion suppression effects up to 70-80% ion suppression. It may be advantageous to use an isotopically labeled standard with few 2H isotopes because increasing the number of 2H substitutes may increase the chromatographic resolution between the native form and its isotopic analogue and therefore not effectively correct for matrix effects (Van Eeckhaut, Lanckmans et al. 2009; Berg and Strand 2011).
Isotopically labeled analogs can also be used as surrogate internal standards for other analytes (Olsson, Baker et al. 2004). However, their ability to compensate for matrix effects other than their respective native forms is usually compromised and needs a thorough assessment. In fact, it needs to be assessed across dilution ranges, different volumes, or different lots, as applicable. In our experience, isotopically labeled standards can help compensate for matrix effects for analytes other than their native forms if they elute closely together. For example, prior to the availability of TCPY (13C3) we used 3-PBA (13C3) as a surrogate internal standard for native TCPY. When TCPY (13C3) was made available, it was incorporated into our analytical method. Although repeat analyses of multiple samples suggested effective correction for matrix effects for TCPY when both isotopic internal standards were used, a better correction was observed when TCPY (13C3) was employed (data not shown).
We notice that, and recommend, if multiple analytes are being measured isocratically or isothermally (depending upon the type of chromatography used), it is necessary to have surrogate internal standard at the beginning, middle, and end of the elution profile to account for matrix effects across the chromatographic run-time. However, when using multi-stepwise gradient separation in either an LC or GC analysis, at least one surrogate internal standard is needed in each step. Additionally, if possible, one analyte can interchangeably be paired with up to two surrogate internal standards (one that eluted before or one that eluted after the target analyte’s peak). Selection in each analytical run is based on the ability of each surrogate standard to compensate for matrix effects occurring for each analyte in the calibration curve or quality control materials.
Use of matrix-matching calibration or pseudo-matrix matching calibration
In the absence of isotopic analogues for use as internal standards, calibration in matrix is useful when dealing with the analysis of target analytes in biological samples. Although matrix-matching calibration cannot correct for problems associated with ion suppression, such as loss of sensitivity and increase of method LODs (Antignac, de Wasch et al. 2005), matrix effects can be accounted for, assuming that every sample (i.e. calibration and unknown samples) are affected to the same extent, thus preventing the generation of biased values in unknown samples. However, the major concern with this approach is that matrix effects can differ substantially across samples (Olsson, Baker et al. 2004). Matrix-matching calibration is also needed to compensate for matrix-induced chromatographic enhancement. This phenomenon impacts quantification if standards in solution were used for calibration. This may lead to an over-estimation of analytical results because at the same concentration or amount injected, target analyte responses in biological samples can be much higher than the responses of the same analytes detected in calibration solutions due to signal enhancement.
Matrix-matching calibration requires the use of adequate matrix pools (i.e. urine, serum, breast milk) collected from donors. To ensure that, “pooled matrix,” contains endogenous components similar to those found in individual samples, samples from multiple donors must be used. To do this properly, a calibration curve for each of the representative samples should be created and compared with the one created from the pooled samples. Differences in calibration characteristics should be noted and a report should be generated to cover the range of matrix effects that are expected. When calibration curves created from a pooled matrix (from a number of donors) differ substantially (>15% difference in CV) from the ones prepared from the samples, a partial within-population calibration curve should be used. A partial within-population calibration curve is prepared using the pool of samples collected from the same population entering the study (Dewe, De Smet et al. 2007).
Whenever we are unable to create a matrix pool similar to the unknown samples or when background concentrations found in a matrix pool are too high, pseudo-matrix matching calibration can be used. For urinary analysis, pseudo-matrix can be a diluted urine pool or synthetic urine. A pseudo-matrix matching calibration curve must have the same slope as the one found in matrix samples. For example, when using diluted urine, the calibration slope must be similar to that found in undiluted urine. The U.S. Centers for Disease Control and Prevention (CDC) has used synthetic urine (CDC 2009) as a matrix for calibration curves during the analysis of several environmental toxicants (e.g., phthalates and bisphenol A) because background levels are found in urine pools and thus interfere with estimates of low standard points. However, synthetic urine is a much simpler matrix than human urine and often is not a suitable substitution for real urine.
In the analysis of some urinary metabolites, 100-fold diluted urine could still produce the same slopes as undiluted urine. On the other hand, we found that, in some matrices, differing dilutions may cause differing slopes for each analyte, regardless of the use of isotopically labeled analogues as internal standards. As seen in Figure 6, a deviation of slope as a result of matrix dilution in rat brain tissue samples was found when analyzed for the 47 PBDE congener. Note that, with this difference in slope, there only would be a few percent error in using any of them to quantify the samples. However, when dealing with trace amounts, these errors can have a large effect on the analytical results.
Figure 6. Varying slope values of PBDE-47 in different matrix dilutions.

Note: Extraction of PBDE-47 from brain tissue was done while homogenizing the tissue sample with a mixture of hexane and acetone (1:1 ratio). The extractant was later purified using Florisil cartridges (6 cc, 500 mg) (Restek Corp, Bellefonte, CA). Target analytes were eluted from the cartridge using hexane. Chromatographic separation and analysis was performed by an Agilent 7000 GC-MS/MS with electron impact ionization interface (Agilent Technologies, Waldbronn, Germany). The system was fitted with a deactivated silica guard column (0.250 mm internal diameter (ID)) (Agilent Technologies, Santa Clara, CA USA) connected to a HP-5MS analytical column (15 m × 0.250 ID × 0.25 μm film thickness, Agilent Technologies, Santa Clara, CA USA). One milliliter of extractant was injected into the system under pulsed splitless mode and with an injector temperature of 250°C. The helium carrier gas flow rate was 1.8 mL/min. Gradient temperature program (from 70 °C to 315°C) was used during chromatographic separation. The interface, source, and quadrupole temperatures were set to 315°C, 315°C, and 150°C, respectively. The total run time was 16 minutes. Quantitative MS/MS transition for PBDE-47 was m/z485.6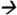 326.0@25V CE while the MS/MS transition for isotopic PBDE-47 analouge was m/z497.5
326.0@25V CE while the MS/MS transition for isotopic PBDE-47 analouge was m/z497.5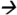 338.0@25V CE.
338.0@25V CE.
The FDA recommends that a calibration curve be generated for each analytical run (FDA 2001). Because matrix effects are system-specific as well as compound- and matrix-dependent, they cause day-to-day variation in calibration slopes. Due to this variation, calibration curves are needed in each analytical run. For example, we have observed changes in slope across days when 3-PBA was analyzed in urine (see Table 2). Based on our data, slope values vary up to 2-fold over the course of one week. Note that, while we were unable to ascertain whether or not these observed alterations result from changes in matrix components of the sample (as well as the mobile phases) or from the amount of matrix component depositing on the surface of the source-interface affecting the efficiency of ionization, we recommend inclusion of a calibration curve in each analytical run.
Table 2. Slope and intercept values observed in linear regression equations resulting from different days of routine analysis of TCPY and 3-PBA.
| Analysis Day | Linear Regression Equation |
|
|---|---|---|
| 3-PBA | TCPY | |
| Day 1 | y = 2.727 * x − 0.010 | y = 2.522 * x − 0.014 |
| Day 2 | y = 2.668 * x − 0.009 | y = 2.498 * x − 0.000 |
| Day 3 | y = 2.552 * x − 0.009 | y = 2.646 * x + 0.007 |
| Day 4 | y = 3.014 * x − 0.013 | y = 3.657 * x + 0.012 |
| Day 5 | y = 2.959 * x − 0.013 | y = 4.105 * x − 0.006 |
| Day 6 | y = 2.792 * x − 0.012 | y = 4.526 * x − 0.009 |
Note: Calibration curves were plotted using relative response values (y) and relative concentrations (x). Relative response is calculated by dividing the response of analyte by the response of its assigned internal standard. The relative concentration is calculated by dividing the expected concentration by the internal standard concentration contained in each level of standard.
Use of nano-flow rate in ESI
A lower solution flow rate is known to reduce the size of the charged droplets in ESI. Because the initial droplets are so small, they require fewer droplet fission reactions and less solvent evaporation prior to the generation of single ions in the gas phase. Thus, a larger portion of the analyte molecules present in the primary droplets may transfer into the MS. Usefulness of a nano-flow LC was demonstrated by Schmidt et al. as they found that, at the minimal flow rates of a few nL/min, signal suppression effects have totally disappeared for the disaccharide, while at a flow rate above 50 nL/min the suppression amounts to about a factor of five (Schmidt, Karas et al. 2003).
Selection of quality control materials
Quality control (QC) materials are an integral component of any analytical run; they ensure the quality of specific samples or batches of samples. During the analysis of biological samples, the selection of matrix for quality control materials is crucial. The matrix used in the preparation of these materials, especially spiked samples, must be similar to the unknown samples. In urine analyses, for example, they should account for different ranges of dilution. They should be prepared to control for matrix effects for at least two different scenarios (e.g. undiluted samples and diluted samples). In serum analyses, they should account for different lots of pooled serum. In these ways, it can be assumed that matrix effects occurring in unknown samples are occurring to the same extent in QC materials and the standard accuracy of the method can be met. We recommend that new lots or batches of matrices used for QC materials be acquired periodically.
Other
Mass spectrometers can sustain their sensitivity if well-maintained and cleaned. Matrix residue from samples, such as precipitated salts or peptides, can accumulate or deposit within the interior parts, particularly at the interface of the mass spectrometer. Deposited matrix components may stay charged and can interfere with the ionization of analytes in the mass spectrometer. This residue can also neutralize ionized compounds or attract target ions, therefore reducing the amount of ions reaching the mass spectrometers via capillary ion transfer tubes, or along the skimmers or optic lenses. This would subsequently reduce the intensity of measured analytes.
To prevent loss of instrumental sensitivity, the instrument should be subject to preventive maintenance more often than recommended by manufacturers (i.e. manufacturers usually recommend performing preventive maintenance every six months). When using an LC-MS/MS, according to our experience, switching to an opposite polarity mode of MS/MS and flushing the LC system for at least two hours with mixtures of water and organic solvents after each analytical run can help maintain instrument sensitivity for considerably longer periods of time. Additionally, daily, weekly, or monthly preventive maintenance, especially those designed to sustain the efficiency of ionization at the interface, must be performed.
MATRIX EFFECTS IN CURRENT BIOANALYTICAL GUIDANCE
To date, there remains a lack of consensus amongst regulatory authorities in terms of guidance for consideration and management of matrix effects in bioanalytical method development and validation (Gonzalez, Blanco et al. 2014). Although all major guidance documents address matrix effects, they vary greatly. For instance, while the FDA draft guidance states that matrix effects should be addressed (FDA 2013), the EMA guidance (EMA 2011) gives study design recommendations including sample number and concentration ranges as well as matrix-specific direction. Figure 7 includes the majority of regulatory guidance requiring matrix effects to be investigated and managed during bioanalytical method development and validation (FDA 2001; Viswanathan, Bansal et al. 2007; EMA 2011; ANVISA 2012; FDA 2013; MHLW 2013).
Figure 7.

Current bioanalytical method development and validation guidance in relation to matrix effects
Note that AAPS/FDA 2007 (USA) refers to Viswanathan et al.
CONCLUSION
Matrix effects are systematic, system-specific, and compound- and matrix-dependent. While they cannot be eliminated entirely, they can be minimized or compensated for using a combination of various measures. Matrix effects in any biological analysis must be thoroughly investigated, taking into account the unique properties of each biological matrix, including inter-sample variation, during method development and validation. During quantification, matrix calibration is needed in order to compensate for matrix effects. The use of isotopically labeled internal standard or surrogate internal standard can minimize the impact of biological matrix effects on quantification. Biological matrix effects can interfere with instrument performance. Therefore, frequent preventive maintenance is necessary. Because matrix effects can differ across bioanalytical settings (e.g. instrumentation, biological matrices, target compounds), additional information regarding the control of ion suppression can be found in the review article published by Furey et al. (2013) (Furey, Moriarty et al. 2013).
ACKNOWLEDGMENTS
The laboratory results presented here were drawn from various projects completed in the Analytical Exposure Science and Environmental Health Laboratory (AESEHL). We wish to acknowledge the contributions from AESEHL laboratory staff members: Albert S. Lee, Christina R. Brosius, Elizabeth K. George, Emma V. Preston, and Grace E. Lee. This work was supported in part by the American Recovery and Reinvestment Act of 2009 under NIH grant 5RC1ES01829902, the National Children’s Study under contract number HHSN267200700007C, PBDE Body Burdens, House Dust Concentrations, and Associations with Thyroid Hormones under NIH grant 1R21ES019697-01, the Emory Parkinson’s disease Collaborative Environmental Research Center under NIEHS grant P01 ES016731, and HERCULES: Health and Exposome Research Center at Emory under NIEHS grant 1P30ES019776-01A1. Samthana A. Radford was supported by Graduate and Postdoctoral Training in Toxicology NIH grant T32 ES012870.
Footnotes
Publisher's Disclaimer: Disclaimer: This is a version of an unedited manuscript that has been accepted for publication. As a service to authors and researchers we are providing this version of the accepted manuscript (AM). Copyediting, typesetting, and review of the resulting proof will be undertaken on this manuscript before final publication of the Version of Record (VoR). During production and pre-press, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal relate to this version also.
REFERENCES
- Agilent T. Control pH During Method Development for Better Chromatography. 2012. [Google Scholar]
- Ahn KC, Lohstroh P, et al. High-throughput automated luminescent magnetic particle-based immunoassay to monitor human exposure to pyrethroid insecticides. Analytical Chemistry. 2007;79(23):8883–8890. doi: 10.1021/ac070675l. [DOI] [PubMed] [Google Scholar]
- Ajmani RS, Rifkind JM. Hemorheological changes during human aging. Gerontology. 1998;44(2):111–120. doi: 10.1159/000021993. [DOI] [PubMed] [Google Scholar]
- Antignac JP, de Wasch K, et al. The ion suppression phenomenon in liquid chromatography-mass spectrometry and its consequences in the field of residue. Analytica Chimica Acta. 2005;529(1-2):129–136. [Google Scholar]
- ANVISA Dispõe sobre os req-uisitos mínimos para a validaç ão de métodos bioanalíticos empregados emestudos com fins de registro e pós-registro de medicamentos, Agência Nacional de Vigilancia Sanitária. 2012;RDC 27 [Google Scholar]
- Baker SE, Olsson AO, et al. High-performance liquid chromatography-tandem mass spectrometry method for quantifying sulfonylurea herbicides in human urine: reconsidering the validation process. Analytical and Bioanalytical Chemistry. 2005;383(6):963–976. doi: 10.1007/s00216-005-0099-1. [DOI] [PubMed] [Google Scholar]
- Berg T, Strand DH. C-13 labelled internal standards-A solution to minimize ion suppression effects in liquid chromatography-tandem mass spectrometry analyses of drugs in biological samples? Journal of Chromatography A. 2011;1218(52):9366–9374. doi: 10.1016/j.chroma.2011.10.081. [DOI] [PubMed] [Google Scholar]
- Bonfiglio R, King RC, et al. The effects of sample preparation methods on the variability of the electrospray ionization response for model drug compounds. Rapid Commun Mass Spectrom. 1999;13(12):1175–1185. doi: 10.1002/(SICI)1097-0231(19990630)13:12<1175::AID-RCM639>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Briscoe CJ, Stiles MR, et al. System suitability in bioanalytical LC/MS/MS. J Pharm Biomed Anal. 2007;44(2):484–491. doi: 10.1016/j.jpba.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Cappiello A, Famiglini G, et al. Overcoming matrix effects in liquid chromatography-mass spectrometry. Analytical Chemistry. 2008;80(23):9343–9348. doi: 10.1021/ac8018312. [DOI] [PubMed] [Google Scholar]
- CDC . Laboratory Procedure Manual: Bisphenol A and other environmental phenols in urine. 2009. [Google Scholar]
- Chambers E, Wagrowski-Diehl DM, et al. Systematic and comprehensive strategy for reducing matrix effects in LC/MS/MS analyses. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;852(1-2):22–34. doi: 10.1016/j.jchromb.2006.12.030. [DOI] [PubMed] [Google Scholar]
- Chiu ML, Lawi W, et al. Matrix Effects-A Challenge Toward Automation of Molecular Analysis. Jala. 2010;15(3):233–242. [Google Scholar]
- Colby BN, McCaman MW. A comparison of calculation procedures for isotope dilution determinations using gas chromatography mass spectrometry. Biomed Mass Spectrom. 1979;6(6):225–230. doi: 10.1002/bms.1200060602. [DOI] [PubMed] [Google Scholar]
- Dams R, Huestis MA, et al. Matrix effect in bio-analysis of illicit drugs with LC-MS/MS: Influence of ionization type, sample preparation, and biofluid. Journal of the American Society for Mass Spectrometry. 2003;14(11):1290–1294. doi: 10.1016/S1044-0305(03)00574-9. [DOI] [PubMed] [Google Scholar]
- Dewe W, De Smet M, et al. Partial within-animal calibration: a new calibration approach in lead optimisation high-throughput bioanalysis. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;854(1-2):183–191. doi: 10.1016/j.jchromb.2007.04.018. [DOI] [PubMed] [Google Scholar]
- EMA, E. M. A. Guideline on bioanalytical method validation. London: 2011. [Google Scholar]
- Erney DR, Gillespie AM, et al. Explanation of the Matrix-Induced Chromatographic Response Enhancement of Organophosphorus Pesticides during Open-Tubular Column Gas-Chromatography with Splitless or Hot on-Column Injection and Flame Photometric Detection. Journal of Chromatography. 1993;638(1):57–63. [Google Scholar]
- FDA. U. F. a. D. A. Guidance for Industry: Bioanalytical Method Validation. U.S. Department of Health and Human Services; 2001. p. 25. [Google Scholar]
- FDA. U. F. a. D. A. Guidance for Industry: Bioanalytical Method Validation. Draft Guidance. U.S. Department of Health and Human Services; 2013. [Google Scholar]
- Furey A, Moriarty M, et al. Ion suppression; a critical review on causes, evaluation, prevention and applications. Talanta. 2013;115:104–122. doi: 10.1016/j.talanta.2013.03.048. [DOI] [PubMed] [Google Scholar]
- Garcia MC. The effect of the mobile phase additives on sensitivity in the analysis of peptides and proteins by high-performance liquid chromatography-electrospray mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;825(2):111–123. doi: 10.1016/j.jchromb.2005.03.041. [DOI] [PubMed] [Google Scholar]
- Garrido Frenich A, Martinez Vidal JL, et al. Compensation for matrix effects in gas chromatography-tandem mass spectrometry using a single point standard addition. Journal of Chromatography A. 2009;1216(23):4798–4808. doi: 10.1016/j.chroma.2009.04.018. [DOI] [PubMed] [Google Scholar]
- Ghosh C, Shinde C, et al. Ionization Polarity as a Cause of Matrix Effects, its Removal and Estimation in ESI-LC-MS/MS Bio-analysis. J Anal Bioanal Tech. 2010;1(106) [Google Scholar]
- Ghosh C, Shinde CP, et al. Influence of ionization source design on matrix effects during LC-ESI-MS/MS analysis. Journal of Chromatography B-Analytical Technologies in the Biomedical and Life Sciences. 2012;893:193–200. doi: 10.1016/j.jchromb.2012.03.012. [DOI] [PubMed] [Google Scholar]
- Gonzalez O, Blanco ME, et al. Bioanalytical chromatographic method validation according to current regulations, with a special focus on the non-well defined parameters limit of quantification, robustness and matrix effect. Journal of Chromatography A. 2014;1353:10–27. doi: 10.1016/j.chroma.2014.03.077. [DOI] [PubMed] [Google Scholar]
- Hajslova J, Zrostlikova J. Matrix effects in (ultra)trace analysis of pesticide residues in food and biotic matrices. Journal of Chromatography A. 2003;1000(1-2):181–197. doi: 10.1016/s0021-9673(03)00539-9. [DOI] [PubMed] [Google Scholar]
- Hewavitharana AK. Matrix matching in liquid chromatography-mass spectrometry with stable isotope labelled internal standards--is it necessary? Journal of Chromatography A. 2011;1218(2):359–361. doi: 10.1016/j.chroma.2010.11.047. [DOI] [PubMed] [Google Scholar]
- Holcapek M, Volna K, et al. Effects of ion-pairing reagents on the electrospray signal suppression of sulphonated dyes and intermediates. J Mass Spectrom. 2004;39(1):43–50. doi: 10.1002/jms.551. [DOI] [PubMed] [Google Scholar]
- Ismaiel OA, Zhang TY, et al. Investigation of endogenous blood plasma phospholipids, cholesterol and glycerides that contribute to matrix effects in bioanalysis by liquid chromatography/mass spectrometry. Journal of Chromatography B-Analytical Technologies in the Biomedical and Life Sciences. 2010;878(31):3303–3316. doi: 10.1016/j.jchromb.2010.10.012. [DOI] [PubMed] [Google Scholar]
- Jewett BN, Ramaley L, et al. Atmospheric pressure ionization mass spectrometry techniques for the analysis of alkyl ethoxysulfate mixtures. Journal of the American Society for Mass Spectrometry. 1999;10(6):529–536. doi: 10.1016/S1044-0305(99)00017-3. [DOI] [PubMed] [Google Scholar]
- King R, Bonfiglio R, et al. Mechanistic investigation of ionization suppression in electrospray ionization. J Am Soc Mass Spectrom. 2000;11(11):942–950. doi: 10.1016/S1044-0305(00)00163-X. [DOI] [PubMed] [Google Scholar]
- Le Grand R, Dulaurent S, et al. Simultaneous determination of five synthetic pyrethroid metabolites in urine by liquid chromatography-tandem mass spectrometry: application to 39 persons without known exposure to pyrethroids. Toxicology Letters. 2012;210(2):248–253. doi: 10.1016/j.toxlet.2011.08.016. [DOI] [PubMed] [Google Scholar]
- Little JL, Wempe MF, et al. Liquid chromatography-mass spectrometry/mass spectrometry method development for drug metabolism studies: Examining lipid matrix ionization effects in plasma. Journal of Chromatography B-Analytical Technologies in the Biomedical and Life Sciences. 2006;833(2):219–230. doi: 10.1016/j.jchromb.2006.02.011. [DOI] [PubMed] [Google Scholar]
- Matuszewski BK, Constanzer ML, et al. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Analytical Chemistry. 2003;75(13):3019–3030. doi: 10.1021/ac020361s. [DOI] [PubMed] [Google Scholar]
- Mei H, Hsieh YS, et al. Investigation of matrix effects in bioanalytical high-performance liquid chromatography/tandem mass spectrometric assays: application to drug discovery. Rapid Communications in Mass Spectrometry. 2003;17(1):97–103. doi: 10.1002/rcm.876. [DOI] [PubMed] [Google Scholar]
- MHLW . Guideline on Bioanalytical Method Validation in Pharmaceutical Development. Ministry of Health, Labour and Welfare; Japan: 2013. [Google Scholar]
- Montesano MA, Olsson AO, et al. Method for determination of acephate, methamidophos, omethoate, dimethoate, ethylenethiourea and propylenethiourea in human urine using high-performance liquid chromatography-atmospheric pressure chemical ionization tandem mass spectrometry. J Expo Sci Environ Epidemiol. 2007;17(4):321–330. doi: 10.1038/sj.jes.7500550. [DOI] [PubMed] [Google Scholar]
- Nishioka M, Mccauley M, et al. Development, validation, and field use of a novel method for extracting and analyzing organophosphate (OP) and pyrethroid pesticide metabolites and creatinine from commercial disposable diapers. Epidemiology. 2006;17(6):S176–S176. [Google Scholar]
- Olsson AO, Baker SE, et al. A liquid chromatography--tandem mass spectrometry multiresidue method for quantification of specific metabolites of organophosphorus pesticides, synthetic pyrethroids, selected herbicides, and deet in human urine. Analytical Chemistry. 2004;76(9):2453–2461. doi: 10.1021/ac0355404. [DOI] [PubMed] [Google Scholar]
- Panuwet P, Nguyen JV, et al. Quantification of melamine in human urine using cation-exchange based high performance liquid chromatography tandem mass spectrometry. Journal of Chromatography B-Analytical Technologies in the Biomedical and Life Sciences. 2012;887:48–54. doi: 10.1016/j.jchromb.2012.01.007. [DOI] [PubMed] [Google Scholar]
- Picciano MF. Nutrient composition of human milk. Pediatr Clin North Am. 2001;48(1):53–67. doi: 10.1016/s0031-3955(05)70285-6. [DOI] [PubMed] [Google Scholar]
- Sangster T, Spence M, et al. Unexpected observation of ion suppression in a liquid chromatography/atmospheric pressure chemical ionization mass spectrometric bioanalytical method. Rapid Communications in Mass Spectrometry. 2004;18(12):1361–1364. doi: 10.1002/rcm.1477. [DOI] [PubMed] [Google Scholar]
- Schenck FJ, Lehotay SJ. Does further clean-up reduce the matrix enhancement effect in gas chromatographic analysis of pesticide residues in food? Journal of Chromatography A. 2000;868(1):51–61. doi: 10.1016/s0021-9673(99)01137-1. [DOI] [PubMed] [Google Scholar]
- Schmidt A, Karas M, et al. Effect of different solution flow rates on analyte ion signals in nano-ESI MS, or: when does ESI turn into nano-ESI? J Am Soc Mass Spectrom. 2003;14(5):492–500. doi: 10.1016/S1044-0305(03)00128-4. [DOI] [PubMed] [Google Scholar]
- Souverain S, Rudaz S, et al. Matrix effect in LC-ESI-MS and LC-APCI-MS with offline and on-line extraction procedures. Journal of Chromatography A. 2004;1058(1-2):61–66. [PubMed] [Google Scholar]
- Sterner JL, Johnston MV, et al. Signal suppression in electrospray ionization Fourier transform mass spectrometry of multi-component samples. J Mass Spectrom. 2000;35(3):385–391. doi: 10.1002/(SICI)1096-9888(200003)35:3<385::AID-JMS947>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Sviridov D, Hortin GL. Urine albumin measurement: Effects of urine matrix constituents. Clinica Chimica Acta. 2009;404(2):140–143. doi: 10.1016/j.cca.2009.03.034. [DOI] [PubMed] [Google Scholar]
- Tang L, Kebarle P. Dependence of Ion Intensity in Electrospray Mass-Spectrometry on the Concentration of the Analytes in the Electrosprayed Solution. Analytical Chemistry. 1993;65(24):3654–3668. [Google Scholar]
- Taylor PJ. Matrix effects: The Achilles heel of quantitative high-performance liquid chromatography-electrospray-tandem mass spectrometry. Clinical Biochemistry. 2005;38(4):328–334. doi: 10.1016/j.clinbiochem.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Van Eeckhaut A, Lanckmans K, et al. Validation of bioanalytical LC-MS/MS assays: Evaluation of matrix effects. Journal of Chromatography B-Analytical Technologies in the Biomedical and Life Sciences. 2009;877(23):2198–2207. doi: 10.1016/j.jchromb.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Viswanathan CT, Bansal S, et al. Quantitative bioanalytical methods validation and implementation: best practices for chromatographic and ligand binding assays. Pharmaceutical Research. 2007;24(10):1962–1973. doi: 10.1007/s11095-007-9291-7. [DOI] [PubMed] [Google Scholar]
- Whitehead RD, Jr., Montesano MA, et al. Method for measurement of the quaternary amine compounds paraquat and diquat in human urine using high-performance liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878(27):2548–2553. doi: 10.1016/j.jchromb.2009.09.029. [DOI] [PubMed] [Google Scholar]
- WHO . Biological Monitoring of Chemical Exposure in the Workplace Guidelines. Contribution to the International Programme on Chemical Safety (IPCS) Vol. 1. Geneva: 1996. [Google Scholar]
- Xia YQ, Jemal M. Phospholipids in liquid chromatography/mass spectrometry bioanalysis: comparison of three tandem mass spectrometric techniques for monitoring plasma phospholipids, the effect of mobile phase composition on phospholipids elution and the association of phospholipids with matrix effects. Rapid Commun Mass Spectrom. 2009;23(14):2125–2138. doi: 10.1002/rcm.4121. [DOI] [PubMed] [Google Scholar]
- Yu S, Xu XM. Study of matrix-induced effects in multi-residue determination of pesticides by online gel permeation chromatography-gas chromatography/mass spectrometry. Rapid Commun Mass Spectrom. 2012;26(8):963–977. doi: 10.1002/rcm.6193. [DOI] [PubMed] [Google Scholar]