

Abstract
AIM: To explore the association of methylation of the CpG island in the promotor of the P16 tumor suppressor gene with the clinicopathological characteristics of the colorectal cancers.
METHODS: Methylation-specific PCR (MSP) was used to detect P16 methylation of 62 sporadic colorectal cancer specimens.
RESULTS: P16 methylation was detected in 42% of the tumors. Dukes’ staging was associated with P16 methylation status. p16 methylation occurred more frequently in Dukes’ C and D patients (75.9%) than in Dukes’ A and B patients (12.1%).
CONCLUSION: P16 methylation plays a role in the carcinogenes is of a subset of colorectal cancer, and it might be linked to poor prognosis.
Keywords: colorectal neoplasma/pathology; genes, P16; polymerase chain reaction/METHODS; CpG region; methylation; MSP
INTRODUCTION
The alterations of p16 gene, i.e. homozygous deletion, point mutation and promoter methylation, and their consequence, P16 gene inactivation, were frequently observed in many human tumors. In colorectal cancers (CRC), since the first two mechanisms have never been demonstrated[1], the role of P16 gene methylation in carcinogenesis has attracted considerable attention recently[2-4]. Whether P16 methylation is associated with clinicopathological characteristics has not been clear[4,5]. In the present study, we detected the occurrence of p16 methylation in primary CRC using a recently developed methylation specific PCR method (MSP) and explored the relationship between methylated p16 and Dukes’ staging and other clinicopathological characteristics of the patients.
MATERIALS AND METHODS
Tumor specimens
Fresh specimens (n = 62) of surgically resected CRC were collected from Rui Jin Hospital and were immediately frozen at -80 °C after rinsed with phosphate buffered saline.
DNA isolation
Tumor DNA was isolated and purified with Trizol reagent (GIBCO Life Technologies) according to the manufacturer’s instruction.
Bisulfite conversion of DNA samples
Bisulfite conversion of DNA samples for methylation detection was carried out based on the principle that treatment of DNA with bisulfite would result in the conversion of unmethylated cytosine residues into uracil, while methylated cytosine residues remain unchanged. Thus, the DNA sequences of methylated and unmethylated genomic regions following bisulfite conversion would be different and distinguishable by sequence specific PCR primers. CpGenome DNA modification kit (Intergen, New York, NY) was used. One microgram of DNA was treated with sodium bisulfite and the bisulfite-converted DNA was resuspended in a total volume of 30 μL of TE.
Methylation-specific PCR (MSP)
MSP was performed using CpG p16 WIZ amplification kit (Intergen). With a complete chemical modification reaction, U primers amplified only unmethylated DNA, and M primers amplified only methylated DNA in the region of P16 gene promoter. W primers amplified unmodified DNA no matter they were methylated or not. Each chemically modified experimental DNA sample was amplified with primers U, M and W respectively. The PCR mixture contained 1 × PCR buffer (500 mmol•L¯¹ KCl, 100 mmol•L¯¹ Tris-HCl, 10 g•L¯¹ Triton X-100, 15 mmol•L¯¹ MgCl2), dNTPs (each at 0.25 mmol•L¯¹), U or M primers 1.0 μL, Ampli Taq Gold polymerase (Perkin-Elmer, Foster city, CA) 1 U, bisulfite-modified DNA 0.1 μg in a final volume of 30 μL. Reactions were started at 95 °C for 12 min. Amplification was carried out in a thermal cycler (Hybaid, Teddington, England) for 35 cycles (45 s at 95 °C, 45s at 60 °C, and 60 s at 72 °C), followed by a final 10 min extension at 72 °C. U, M and W controls provided by the kit served as validation of the reagents and PCR conditions. PCR products were analyzed by agarose gel electrophoresis and ethidium bromide staining.
The expected PCR products visualized on the gel should be an 154 bp nucleotide for unmethylated P16 and 145 bp nucleotide for methylated p16. If the sample contains unmethylated DNA, U primer will produce an 154 bp products (band U), and if it contains methylated DNA, M primer will produce 145 bp products (band M). The appearance of band W from the sample indicated an incomplete bisulfite conversion, and was considered a sign of unqualified chemical modification of the sample.
Clinicopathological data
Clinicopathological data were collected after MSP for p16 methylation was accomp lished. The clinicopathological data included age, gender of the patients, anatomical location[2] and Dukes stage of CRC.
Statistical analysis
The χ² test was performed to analyze the relationship between p16 methylation status and each of the clinicopathologic parameters.
RESULTS
MSP analysis of p16 methylation
Of 62 CRC specimens, 26 displayed bands M, 154 bp products, signifying that 42% of the tumors had detectable p16 methylation. Band U, 154 bp product, was visible in all specimens with varied intensity, indicating the existence of unmethylated DNA in these specimens. No band W appeared in all specimens (Figure 1).
Figure 1.
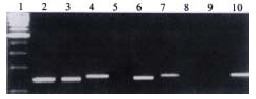
Electrophoresis of MSP products of p16 CpG region. 1. 100 bp marker, 2. W control (142 bp), 3. M control (145 bp), 4. U control (154 bp), 5, 6, 7. The same sample (from patient No.9) reactive with W, M, U primers respec tively, 6. 145 bp product, 7. 154 bp product, indicating p16 was methylated in this sample, 8, 9, 10. Same sample (from patient No.34) reactive with W, M, U primers respectively, 10. 154 bp product, indicating that p16 was unmethylated in this sample.
Clinicopathological correlations with p16 methylation
No correlation was found between p16 methylation and some of clinical factors, including age, gender, tumor location, etc. However, P16 methylation was signif icantly associated with Dukes’ staging. Dukes’ C, D patients were more likely to contain methylated p16 compared with Dukes’ A, B patients (Table 1).
Table 1.
Clinicopathological correlations with p16 methylation
| n | Methylated | Unmethylated | |
| Male | 35 | 18 (51.4) | 17 (48.6) |
| Female | 27 | 8 (29.6) | 19 (70.4) |
| Age (yrs) | |||
| < 50 | 15 | 7 (46.7) | 8 (53.3) |
| > 50 | 47 | 19 (40.4) | 28 (59.6) |
| Tumor location[2] | |||
| Proximal | 21 | 12 (57.1) | 9 (42.9) |
| Distal | 41 | 14 (34.1) | 27 (65.9) |
| Dukes’ staging | |||
| A, B | 33 | 4 (12.1) | 29 (87.9) |
| C, D | 29 | 22 (75.9) | 7 (24.1)b |
P < 0.01, vs A, B.
DISCUSSION
DNA methylation involves addition of a methyl group to the carbon 5 position of the cytosine ring. This reaction is catalyzed by DNA methyltransferase in the context of the sequence 5’-CG-3’, which is also referred to as a CpG dinucleotide[6,7]. The potential contribution of DNA methylation to oncogenesis is mediated by one or more of mechanisms that include DNA hypomethylation, hyper methylation of tumor suppressor gene and chromosomal instability in cancers[8-13]. The methylation of gene, particularly the methylation of CpG-rich promoters, could block transcriptional activation[6]. P16 tumor suppres sor gene plays a monitor role in the passage of cells through the G1 to S phase of the cell cycle by binding to cyclin-dependent kinase 4 and inhibiting its effect on cyclin D1[14-16]. Methylation of cytosine residues at CpG sites in p16 gene promotor, resulting in a silenced p16 expression, has been reported in many cell lines, including CRC, and some primary carcinomas in varied origins, such as colon, brain, breast, bladder, ovary, lung, and myeloma and so on[17-30].
In the previous studies on p16 methylation, most researchers used Southern analysis and digestion of methylation-sensitive enzymes to detect methylation[15,31]. We used MSP, in the present study, for analysis of the methylation status of p16. This method provided significant advantages over previous ones used for assaying methylation. MSP is much more sensitive than Southern analysis, facilitating the detection of low numbers of methylated alleles and the study of DNA from small samples. MSP allows examination of all CpG sites, not just those within sequences recognized by methylation sensitive restriction enzymes. In summary, MSP is a simple, sensitive and specific method for determining the methylation status of CpG rich region[32]. This might account for that the occurrence of p16 methylation detected by MSP (20%-50%)[4,5,33] was usually higher than those by Southern analysis and other METHODS (10%-20%)[15,31]. In our study, methylated p16 in primary CRC was found in a frequency as high as 42%, probably revealing a number more precise than those detected not by MSP.
Though p16 methylation is common in CRC cell lines[4,34,35] and has been suggested to involve also in the primary CRC[2,4,5,33,36-38], the association of p16 methylation with the clinicopathological characteristics of primary CRC has rarely been investigated and concluded with conflicts[2,5,37-39]. The role of p16 methylation in the development and progress of the primary CRC therefore awaits to be elucidated. Wiencke et al[2] found that women were much more likely than men to have p16-methylated tumors and that proximal tumors were more likely to contain methylated p16. They reported that p16 methylation was also associated with poorly differentiated tumors. Liang et al[5] reported that the presence of p16 methylation predicted shorter survivals in colorectal cancer patients. Zhang et al[37,38] found that p16 methylation in CRC was not associated with clinicopathologic data. In our study, no correlation was found between p16 methylation and various clinicopathologic factors including age, gender or tumor location. However, p16 methylation was significantly associated with Dukes’ staging. Dukes C, D patients were more likely (75.9%) to contain methylated p16 as compared with Dukes’ A, B patients (12.1%). Dukes’ staging is a clinical classification for colorectal cancers based on the tumor size, local extent, metastatic status, i.e. lymph node involvement. Hence, Dukes’ staging has been considered a most important prognostic determinant. In the present study, p16 methylation was more likely to occur in Dukes’ C, D patients. This result suggested that p16 methylation might link to a more malignant outcome of CRC.
How does the existence of methylated p16 promoter affect the expression pattern of p16 protein in primary tumors Controversy has emerged over recent years on the relationship of p16 promoter methylation and its gene expression in whole tumors. Although most of authors hold the traditional viewpoint that detectable p16 methylation necessarily link to the inactivation of p16 protein, or transcriptional silencing of p16 gene[3,10,19,31,40], coexistence of p16 methylation and p16 expression in one specimen has been frequently described[2,41-44]. It was noticeable that, in our study, band U, i.e. PCR products amplified by primer U, was visible in each specimen. Similar results were also reported by other investigators[2]. This might first be attributed to the contamination of non-neoplastic cells that naturally harbored unmethylated DNA. However, Guan et al[4] demonstrated that band U appeared even in the samples acquired by microdissection which got rid of the non-neoplastic cells. This indicated the possibility that tumor cells in one sample were virtually the mixture of those contained methylated and unmethylated DNA. In fact, the cell line in which p16 methylation was detected could still express p16 protein, due to a partial methylation[45]. In summary, coexistence of p16 methylation and p16 expression in one sample might reflect the cell heterogeneity, in which, as we speculated, a part of cells contained methylated p16 and loss of p16 expression whereas another part of cells kept expressing or even overexpressing p16 protein. We found that, in a previous immunohistochemical study on 71 archival specimens of CRC, p16 expression-positive tumor cells were seen in roughly 50% specimens, and these p16 expressive specimens were coincident with those with a poorer differentiation grade[46]. Other investigators have also proposed that activation but not inactivation of p16 gene was associated with primary CRC[47]. We will continue our effects to explore the correspondent relation of p16 methylation and p16 expression in the identical CRC specimen, so as to determine how the seemly paradoxical events might unite in one tumor. The elucidation of the relationship between p16 expression and p16 gene methylation in primary tumors will certainly help better understand the role of methylation of tumor suppressor genes in carcinogenesis.
Footnotes
Edited by Ma JY
Supported by the grants from Ministry of Public Health of China, No.98-1-303 and The Educational Committee of Shanghai, No.2000B02.
References
- 1.Ruas M, Peters G. The p16INK4a/CDKN2A tumor suppressor and its relatives. Biochim Biophys Acta. 1998;1378:F115–F177. doi: 10.1016/s0304-419x(98)00017-1. [DOI] [PubMed] [Google Scholar]
- 2.Wiencke JK, Zheng S, Lafuente A, Lafuente MJ, Grudzen C, Wrensch MR, Miike R, Ballesta A, Trias M. Aberrant methylation of p16INK4a in anatomic and gender-specific subtypes of sporadic colorectal cancer. Cancer Epidemiol Biomarkers Prev. 1999;8:501–506. [PubMed] [Google Scholar]
- 3.Baylin SB, Herman JG, Graff JR, Vertino PM, Issa JP. Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv Cancer Res. 1998;72:141–196. [PubMed] [Google Scholar]
- 4.Guan RJ, Fu Y, Holt PR, Pardee AB. Association of K-ras mutations with p16 methylation in human colon cancer. Gastroenterology. 1999;116:1063–1071. doi: 10.1016/s0016-5085(99)70009-0. [DOI] [PubMed] [Google Scholar]
- 5.Liang JT, Chang KJ, Chen JC, Lee CC, Cheng YM, Hsu HC, Wu MS, Wang SM, Lin JT, Cheng AL. Hypermethylation of the p16 gene in sporadic T3N0M0 stage colorectal cancers: association with DNA replication error and shorter survival. Oncology. 1999;57:149–156. doi: 10.1159/000012023. [DOI] [PubMed] [Google Scholar]
- 6.Singal R, Ginder GD. DNA methylation. Blood. 1999;93:4059–4070. [PubMed] [Google Scholar]
- 7.Jones PA, Laird PW. Cancer epigenetics comes of age. Nat Genet. 1999;21:163–167. doi: 10.1038/5947. [DOI] [PubMed] [Google Scholar]
- 8.O'Neill RJ, O'Neill MJ, Graves JA. Undermethylation associated with retroelement activation and chromosome remodelling in an interspecific mammalian hybrid. Nature. 1998;393:68–72. doi: 10.1038/29985. [DOI] [PubMed] [Google Scholar]
- 9.Chen RZ, Pettersson U, Beard C, Jackson-Grusby L, Jaenisch R. DNA hypomethylation leads to elevated mutation rates. Nature. 1998;395:89–93. doi: 10.1038/25779. [DOI] [PubMed] [Google Scholar]
- 10.Schmutte C, Jones PA. Involvement of DNA methylation in human carcinogenesis. Biol Chem. 1998;379:377–388. doi: 10.1515/bchm.1998.379.4-5.377. [DOI] [PubMed] [Google Scholar]
- 11.Lengauer C, Kinzler KW, Vogelstein B. DNA methylation and genetic instability in colorectal cancer cells. Proc Natl Acad Sci USA. 1997;94:2545–2550. doi: 10.1073/pnas.94.6.2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liggett WH, Sidransky D. Role of the p16 tumor suppressor gene in cancer. J Clin Oncol. 1998;16:1197–1206. doi: 10.1200/JCO.1998.16.3.1197. [DOI] [PubMed] [Google Scholar]
- 13.McBurney MW. Gene silencing in the development of cancer. Exp Cell Res. 1999;248:25–29. doi: 10.1006/excr.1999.4454. [DOI] [PubMed] [Google Scholar]
- 14.Wieser RJ, Faust D, Dietrich C, Oesch F. p16INK4 mediates contact-inhibition of growth. Oncogene. 1999;18:277–281. doi: 10.1038/sj.onc.1202270. [DOI] [PubMed] [Google Scholar]
- 15.Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, Baylin SB, Sidransky D. 5' CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med. 1995;1:686–692. doi: 10.1038/nm0795-686. [DOI] [PubMed] [Google Scholar]
- 16.Yu WL, Huang ZH. p16 gene and digestive tract neoplasms. Shijie Huaren Xiaohua Zazhi. 1999;7:1061–1062. [Google Scholar]
- 17.Esteller M, Sanchez-Cespedes M, Rosell R, Sidransky D, Baylin SB, Herman JG. Detection of aberrant promoter hypermethylation of tumor suppressor genes in serum DNA from non-small cell lung cancer patients. Cancer Res. 1999;59:67–70. [PubMed] [Google Scholar]
- 18.Nakamura M, Sugita K, Inukai T, Goi K, Iijima K, Tezuka T, Kojika S, Shiraishi K, Miyamoto N, Karakida N, et al. p16/MTS1/INK4A gene is frequently inactivated by hypermethylation in childhood acute lymphoblastic leukemia with 11q23 translocation. Leukemia. 1999;13:884–890. doi: 10.1038/sj.leu.2401437. [DOI] [PubMed] [Google Scholar]
- 19.Matsuda Y, Ichida T, Matsuzawa J, Sugimura K, Asakura H. p16 (INK4) is inactivated by extensive CpG methylation in human hepatocellular carcinoma. Gastroenterology. 1999;116:394–400. doi: 10.1016/s0016-5085(99)70137-x. [DOI] [PubMed] [Google Scholar]
- 20.Zöchbauer-Müller S, Fong KM, Virmani AK, Geradts J, Gazdar AF, Minna JD. Aberrant promoter methylation of multiple genes in non-small cell lung cancers. Cancer Res. 2001;61:249–255. [PubMed] [Google Scholar]
- 21.Esteller M, González S, Risques RA, Marcuello E, Mangues R, Germà JR, Herman JG, Capellà G, Peinado MA. K-ras and p16 aberrations confer poor prognosis in human colorectal cancer. J Clin Oncol. 2001;19:299–304. doi: 10.1200/JCO.2001.19.2.299. [DOI] [PubMed] [Google Scholar]
- 22.Nakahara Y, Shintani S, Mihara M, Ueyama Y, Matsumura T. High frequency of homozygous deletion and methylation of p16 (INK4A) gene in oral squamous cell carcinomas. Cancer Lett. 2001;163:221–228. doi: 10.1016/s0304-3835(00)00699-6. [DOI] [PubMed] [Google Scholar]
- 23.Nielsen NH, Roos G, Emdin SO, Landberg G. Methylation of the p16 (Ink4a) tumor suppressor gene 5'-CpG island in breast cancer. Cancer Lett. 2001;163:59–69. doi: 10.1016/s0304-3835(00)00674-1. [DOI] [PubMed] [Google Scholar]
- 24.Tannapfel A, Benicke M, Katalinic A, Uhlmann D, Köckerling F, Hauss J, Wittekind C. Frequency of p16 (INK4A) alterations and K-ras mutations in intrahepatic cholangiocarcinoma of the liver. Gut. 2000;47:721–727. doi: 10.1136/gut.47.5.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trzeciak L, Hennig E, Kolodziejski J, Nowacki M, Ostrowski J. Mutations, methylation and expression of CDKN2a/p16 gene in colorectal cancer and normal colonic mucosa. Cancer Lett. 2001;163:17–23. doi: 10.1016/s0304-3835(00)00652-2. [DOI] [PubMed] [Google Scholar]
- 26.Muto S, Horie S, Takahashi S, Tomita K, Kitamura T. Genetic and epigenetic alterations in normal bladder epithelium in patients with metachronous bladder cancer. Cancer Res. 2000;60:4021–4025. [PubMed] [Google Scholar]
- 27.Sanchez-Cespedes M, Esteller M, Wu L, Nawroz-Danish H, Yoo GH, Koch WM, Jen J, Herman JG, Sidransky D. Gene promoter hypermethylation in tumors and serum of head and neck cancer patients. Cancer Res. 2000;60:892–895. [PubMed] [Google Scholar]
- 28.Guo SX, Taki T, Ohnishi H, Piao HY, Tabuchi K, Bessho F, Hanada R, Yanagisawa M, Hayashi Y. Hypermethylation of p16 and p15 genes and RB protein expression in acute leukemia. Leuk Res. 2000;24:39–46. doi: 10.1016/s0145-2126(99)00158-7. [DOI] [PubMed] [Google Scholar]
- 29.Zhu ZY, Tian X, Wang X, Yang YL. P16 gene and APC gene mutation in gastrinomas. Shijie Huaren Xiaohua Zazhi. 2000;8:1418–1419. [Google Scholar]
- 30.Yakoob J, Fan XG, Hu GL, Zhang Z. DNA methylation and carcinogenesis in digestive neoplasms. World J Gastroenterol. 1998;4:174–177. doi: 10.3748/wjg.v4.i2.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gonzalez-Zulueta M, Bender CM, Yang AS, Nguyen T, Beart RW, Van Tornout JM, Jones PA. Methylation of the 5' CpG island of the p16/CDKN2 tumor suppressor gene in normal and transformed human tissues correlates with gene silencing. Cancer Res. 1995;55:4531–4535. [PubMed] [Google Scholar]
- 32.Herman JG, Graff JR, Myöhänen S, Nelkin BD, Baylin SB. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci USA. 1996;93:9821–9826. doi: 10.1073/pnas.93.18.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Esteller M, Tortola S, Toyota M, Capella G, Peinado MA, Baylin SB, Herman JG. Hypermethylation-associated inactivation of p14 (ARF) is independent of p16 (INK4a) methylation and p53 mutational status. Cancer Res. 2000;60:129–133. [PubMed] [Google Scholar]
- 34.Ramchandani S, Bhattacharya SK, Cervoni N, Szyf M. DNA methylation is a reversible biological signal. Proc Natl Acad Sci USA. 1999;96:6107–6112. doi: 10.1073/pnas.96.11.6107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lai DN, Xie YF, Bian L, Yao X. Study on P16 gene aberrant methylation of colon cancer cell lines. Shijie Huaren Xiaohua Zazhi. 1999;7:676–678. [Google Scholar]
- 36.Eads CA, Danenberg KD, Kawakami K, Saltz LB, Danenberg PV, Laird PW. CpG island hypermethylation in human colorectal tumors is not associated with DNA methyltransferase overexpression. Cancer Res. 1999;59:2302–2306. [PubMed] [Google Scholar]
- 37.Zhang J, Lai MD, Chen J. Methylation status of p16 gene in colorectal carcinoma and normal colonic mucosa. World J Gastroenterol. 1999;5:451–454. doi: 10.3748/wjg.v5.i5.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang J, Lai MD, Chen J. Methylation status of p16 gene in colorectal carcinoma and normal colonic mucosa. Zhonghua Binglixue Zazhi. 2000;29:95–98. [PubMed] [Google Scholar]
- 39.Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA. 1999;96:8681–8686. doi: 10.1073/pnas.96.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shim YH, Kang GH, Ro JY. Correlation of p16 hypermethylation with p16 protein loss in sporadic gastric carcinomas. Lab Invest. 2000;80:689–695. doi: 10.1038/labinvest.3780072. [DOI] [PubMed] [Google Scholar]
- 41.Simpson DJ, Bicknell JE, McNicol AM, Clayton RN, Farrell WE. Hypermethylation of the p16/CDKN2A/MTSI gene and loss of protein expression is associated with nonfunctional pituitary adenomas but not somatotrophinomas. Genes Chromosomes Cancer. 1999;24:328–336. [PubMed] [Google Scholar]
- 42.Ng MH, Chung YF, Lo KW, Wickham NW, Lee JC, Huang DP. Frequent hypermethylation of p16 and p15 genes in multiple myeloma. Blood. 1997;89:2500–2506. [PubMed] [Google Scholar]
- 43.Zingg JM, Jones PA. Genetic and epigenetic aspects of DNA methylation on genome expression, evolution, mutation and carcinogenesis. Carcinogenesis. 1997;18:869–882. doi: 10.1093/carcin/18.5.869. [DOI] [PubMed] [Google Scholar]
- 44.Salvesen HB, Das S, Akslen LA. Loss of nuclear p16 protein expression is not associated with promoter methylation but defines a subgroup of aggressive endometrial carcinomas with poor prognosis. Clin Cancer Res. 2000;6:153–159. [PubMed] [Google Scholar]
- 45.Lo YM, Wong IH, Zhang J, Tein MS, Ng MH, Hjelm NM. Quantitative analysis of aberrant p16 methylation using real-time quantitative methylation-specific polymerase chain reaction. Cancer Res. 1999;59:3899–3903. [PubMed] [Google Scholar]
- 46.Chen YY, Yi J, Yang XM, Wu PP. Immunohistochemical study of p16 gene alteration in colorectal cancer. Weichang Bingxue. 2000;5:39–41. [Google Scholar]
- 47.Ohhara M, Esumi M, Kurosu Y. Activation but not inactivation of the MTS1 gene is associated with primary colorectal carcinomas. Biochem Biophys Res Commun. 1996;226:791–795. doi: 10.1006/bbrc.1996.1430. [DOI] [PubMed] [Google Scholar]