

Abstract
Leptin is an adipocytokine that circulates in proportion to body fat to signal the repletion of long-term energy stores. Leptin acts via its receptor, LepRb, on specialized neuronal populations in the brain (mainly in the hypothalamus and brainstem) to alter motivation and satiety, as well as to permit energy expenditure and appropriate glucose homeostasis. Decreased leptin, as with prolonged caloric restriction, promotes a powerful orexigenic signal, decreases energy use via a number of neuroendocrine and autonomic axes, and disrupts glucose homeostasis. Here, we review what is known about cellular leptin action and focus on the roles for specific populations of LepRb-expressing neurons for leptin action.
Leptin is secreted primarily from white adipose tissue in approximate proportion to triglyceride content, and signals the adequacy of adipose energy stores (1). Because leptin secretion is constitutive and tied to fat stores, circulating levels fluctuate little over the short term. Prolonged caloric restriction and/or inadequate nutrition to meet chronically high energetic demands decreases adipose fat stores and reduces leptin production, however.
The loss of leptin action provokes an extensive physiologic response, increasing the drive to feed, decreasing satiety, and altering a host of neuroendocrine and autonomic systems to decrease energy use (2). Low leptin inhibits the growth and reproductive axes, as well as decreasing overall metabolic rate by blunting sympathetic tone and thyroid function. Diminished leptin action also promotes increased hepatic glucose production while decreasing glucose uptake into muscle, ensuring the availability of glucose for organs (such as the brain) that require this fuel (3, 4). Additionally, low leptin changes a variety of brain systems that alter mood and behavior, for instance, increasing motivation (to promote food seeking) and anxiety (increasing vigilance, and presumably mitigating the possibility of predation due to riskier food-seeking behaviors). Exogenous leptin blunts or reverses these physiologic and behavioral responses to negative energy balance.
Humans or animal models (eg, Lepob/ob mice) that lack leptin display increased feeding and decreased energy expenditure (and thus obesity), along with infertility, short stature, diabetes, and a variety of other endocrine, autonomic, and behavioral abnormalities (1). Because leptin plays a central role in the control of feeding, energy balance, and metabolism, understanding the cellular and neuronal mechanisms by which it acts may ultimately define targets for therapeutic intervention in a variety of disorders, including obesity and diabetes.
Although leptin reverses the obesity of leptin-deficient animals, most obese individuals or animals display elevated circulating leptin concentrations, and exogenous leptin does not substantially alter feeding, body weight, or other parameters in obesity. Although some have postulated that the failure of elevated or exogenous leptin to promote leanness in most obese individuals results from a failure of leptin action, a number of arguments suggest that such “leptin resistance” is unlikely to underlie most cases of obesity but rather that a ceiling may exist, beyond which additional leptin promotes little additional effect (5). Indeed, antagonizing leptin receptor (LepRb) increases body weight and food intake in both lean and diet induced obese mice, indicating that endogenous leptin continues to suppress feeding, even in hyperleptinemic diet-induced obese mice that fail to suppress food intake in response to exogenous leptin (6). In either case, the biology of leptin lies primarily in the robust response to below-normal leptin, rather than in the minimal response to increased leptin.
Leptin Signaling
Leptin acts via the long form of the leptin receptor (LepRb) (see Figure 1) (7); although other forms of the receptor exist, the classical leptin receptor mutant (Leprdb/db) mouse, which presents a phenotype not different than Lepob/ob mice, lacks only LepRb. Similarly, restoration of LepRb alone in mice lacking all leptin receptor isoforms suffices to normalize physiology (8).
Figure 1.
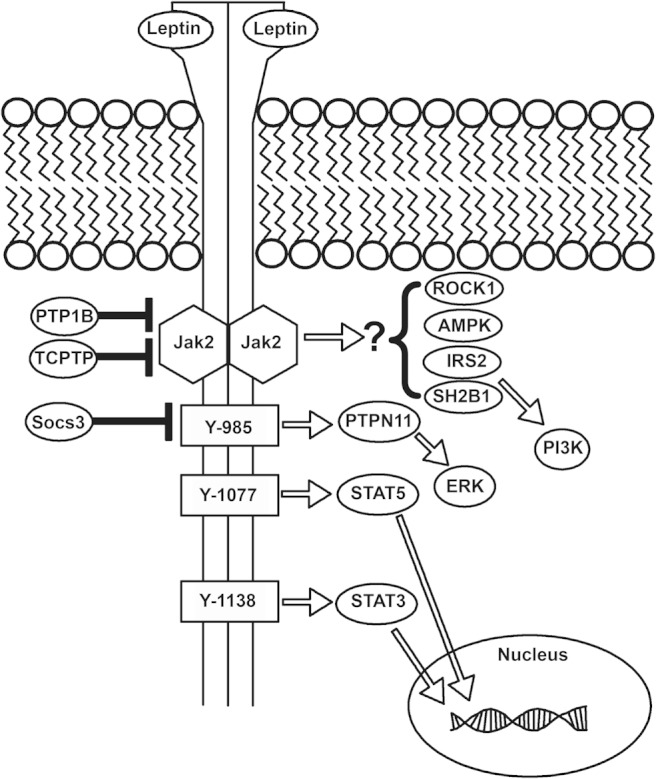
LepRb signaling. Leptin binds to the extracellular domain of LepRb, activating the associated Jak2 tyrosine kinase and promoting the Jak2-mediated phosphorylation of tyrosine residues 985, 1077, and 1138 of LepRb (note, numbering is that of the murine receptor). Phosphorylated Tyr985 recruits PTPN11, which initiates the ERK signaling cascade. Phosphorylated Tyr1077 and Tyr1138 recruit STAT5 and STAT3, respectively, permitting their trafficking to the nucleus to mediate the control of cognate gene expression. In addition, mechanisms that limit LepRb signaling have been defined: Socs3, whose expression is increased by leptin, binds to phosphorylated Tyr985 to blunt LepRb signaling. Also, the tyrosine phosphatases, PTP1B and TCPTP, dephosphorylate Jak2 and/or LepRb to terminate LepRb signaling. In addition, Rho kinase 1, adenosine monophosphate activated protein kinase, insulin receptor substrate (IRS)2, and SH2B1 have been shown to act downstream of LepRb, although the mechanisms by which LepRb controls these pathways have yet to be defined.
LepRb is a type I cytokine receptor of the IL-6 receptor family (9). As for other members of this family, ligand binding activates the associated Janus kinase-2 (JAK2) tyrosine kinase, thereby promoting the tyrosine phosphorylation of LepRb and associated proteins (10, 11). Distinct tyrosine phosphorylation sites on LepRb each recruit different downstream signaling molecules. As for other IL-6 receptor family members, LepRb recruits latent transcription factors of the signal transducers and activators of transcription (STATs) family, promoting their phosphorylation and transcriptional activation. Although STAT3 represents the dominant LepRb-mediated signal, STAT5 and STAT1 are also activated by leptin under some circumstances (10, 12, 13). Although STAT3 is crucial for leptin→LepRb action in vivo (14), STAT5 is dispensable (15, 16), although it contributes to the control of energy balance by other cytokines (17, 18). Potential roles for STAT1 in leptin action have yet to be examined.
Activated LepRb also recruits the tyrosine phosphatase PTPN11 (previously known as SHP2 or SHPTP2), which plays a role in ERK signaling (10). Other tyrosine phosphatases, including PTP1B and TCPTP, dephosphorylate Jak2 and/or LepRb to decrease leptin action (19). Additionally, leptin activates phosphatidylinositol 3′ kinase, at least in part through the combined actions of insulin receptor substrate proteins and SH2B1 (20–24). In addition, Rho kinase-1 and the adenosine monophosphate activated protein kinase are required for important components of leptin action, although the mechanism(s) by which LepRb controls these pathways remain unclear (25–27).
Although no cell type-specific differences in the activation of downstream signaling molecules by leptin→LepRb have been demonstrated, leptin mediates distinct effects on different cell types, depolarizing (activating) some LepRb neurons while hyperpolarizing (inhibiting) others (28–32). The mechanisms by which the same initial LepRb-activated signaling proteins mediate such distinct cell-specific effects remain opaque.
Leptin Acts in the Brain, Especially the Hypothalamus
The intracerebroventricular (icv) injection of leptin in Lepob/ob or normal mice provokes at least as robust a response as does peripheral delivery of the hormone (33, 34), suggesting that leptin mainly acts via the brain. Indeed, not only is LepRb most highly expressed in the brain, but the restoration of LepRb expression in the brain of LepRb-null (Leprdb/db) animals suffices to normalize physiology (8). Consistently, brain-specific deletion of LepRb recapitulates the phenotypes of Leprdb/db (and Lepob/ob) mice (30, 35).
LepRb is not expressed uniformly throughout the brain but is found mainly in subpopulations of neurons in specific areas of the hypothalamus, midbrain and brainstem (see Figure 2) (36). Mice lacking LepRb in specifically in hypothalamic (Nkx2.1-expressing) neurons display phenotypes reminiscent of Leprdb/db mice (37), revealing that leptin action on hypothalamic LepRb neurons is crucial for leptin action.
Figure 2.
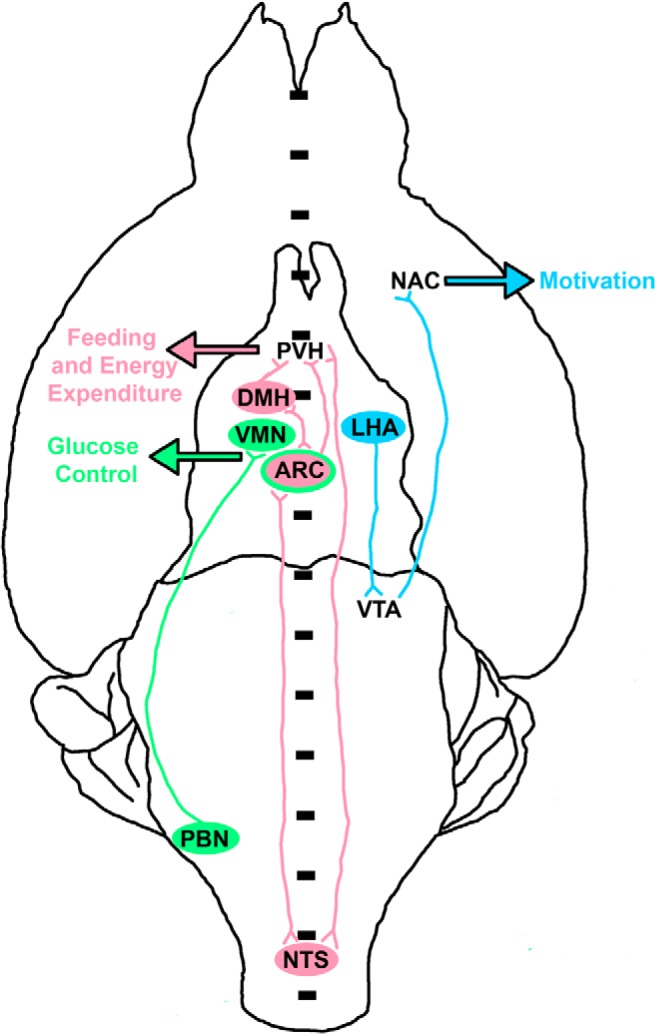
Major leptin-regulated neural systems and their outputs. Shown is a schematic diagram of major leptin-regulate brain systems, in a ventral (from the bottom) view of the rodent brain. Although all structures are bilateral, they are shown on only 1 side of the brain for the sake of simplicity. Note that the ARC, NTS, and PVH are bilateral but straddle the midline. Three major roles for leptin include the control of food intake and energy expenditure (red pathways), glucose homeostasis (green pathways), and motivated behavior (blue pathways). In the hindbrain, leptin action on the NTS increases satiety; these NTS LepRb neurons make reciprocal connections with hypothalamic nuclei (ARC, DMH, and PVH) that control food intake. Leptin also acts directly on the ARC and DMH, which (in addition to their reciprocal connections with each other) share strong reciprocal connections with the major output nucleus of the hypothalamus, the PVH. Leptin acts on these circuits to reduce food intake and increase energy expenditure. Direct leptin action on the ARC and VMH contribute to the suppression of glucose production and the stimulation of glucose disposal. Leptin action on the PBN also contributes to glycemic control, by suppressing glucose production during the CRR; PBN LepRb neurons mediate this effect via projections to the VMH. Leptin acts on LepRb neurons in the LHA to control the mesolimbic DA system. Not only do LHA LepRb neurons project to the VTA but also they modulate the activity of LHA HCRT neurons that project to the VTA. Thus, LHA LepRb neurons control the VTA neurons that project to the NAc to modulate the activity of the mesolimbic DA system and motivated behavior.
Ablation of LepRb in at least 2 large, widely distributed subpopulations of hypothalamic neurons (ie, those that contain vesicular GABA transporter [vGAT] or nitric oxide synthase-1 [NOS1]) also produces a Leprdb/db-like obesity phenotype (30, 35), although LepRb disruption in vGAT cells produces stronger effects on the reproductive and other endocrine axes than does disruption in NOS1 cells. In contrast, disruption of LepRb in several other smaller hypothalamic neural populations produces relatively little effect on body weight (see below). Thus, a network of hypothalamic LepRb neurons with sufficient redundancy to compensate for the lack of LepRb on smaller groups of cells may underlie the control of body weight by leptin. At least some groups of LepRb neurons play specific and nonredundant functions, however. Understanding the roles for specific subsets of LepRb neurons, as well as the neural mechanisms by which they act, represent a crucial areas of inquiry.
Roles for Hypothalamic Arcuate Nucleus (ARC) LepRb Neurons in Leptin Action
The number of distinct regions throughout the brain that contain LepRb neurons (along with the intermingling of LepRb neurons with vastly greater numbers of non-LepRb neurons in each region) present challenges to studying and understanding the function of each group of LepRb cells. Even the ARC, which contains more leptin-responsive cells than any other area of the brain, contains only about 15% of all LepRb neurons in the brain, and LepRb neurons represent the minority of ARC cells (36, 38, 39).
The ARC lies lateral to the base of the third cerebral ventricle and superior to the median eminence, rendering it more rapidly accessible to circulating factors (nutrients, hormones, etc) than areas deeper in the brain. Although other types of ARC neurons contain LepRb, 2 prominent populations of ARC neurons express LepRb: 1) lateral ARC neurons that contain proopiomelanocortin (POMC), and 2) medial ARC neurons that express agouti-related peptide (AgRP) and neuropeptide Y (NPY), along with mediators of inhibitory γ-aminobutyric acid signaling (40). Leptin activates POMC neurons (28), which produce α-melanocyte-stimulating hormone; α-melanocyte-stimulating hormone activates the melanocortin-4 receptor (MC4R) on downstream neurons to promote satiety and increase energy expenditure (41, 42). In contrast, leptin inhibits AgRP neurons, which promote feeding and suppress energy use (28, 43, 44).
POMC and AgRP neurons are each crucial for leptin action and for the control of energy balance. Postnatal deletion of AgRP neurons provokes lethal anorexia (45), even in Lepob/ob animals, whereas animals null for POMC or MC4R are obese and insensitive to leptin (46, 47). In contrast, ablation of LepRb in AgRP and/or POMC neurons produces only mild effects on body weight and food intake (48, 49). Similarly, the loss of STAT3 (which is crucial for LepRb signaling and leptin action) in POMC and AgRP neurons provokes only modest effects on body weight and food intake (50, 51). Thus, although LepRb-expressing POMC and AgRP neurons are crucial for leptin action, LepRb in these neurons is largely dispensable. A number of mechanisms may underlie this apparent paradox. In the case of AgRP/NPY neurons, alterations in these cells (including ablation of AgRP, NPY, or the AgRP/NPY neuron) early in development yields little effect, whereas ablation in adults produces lethal anorexia (45, 52). Thus, developmental compensation may mitigate the phenotype of mice null for LepRb in AgRP cells.
For POMC neurons, because early loss of POMC or MC4R, or the ablation of POMC neurons, promotes dramatic obesity (53, 54), developmental compensation is unlikely to explain the minimal phenotype produced by the POMC-specific loss of LepRb. Rather, leptin may regulate POMC neurons in large part via indirect mechanisms: leptin modulates inhibitory inputs to POMC neurons via vGAT LepRb neurons (30), whereas NOS1 LepRb neurons control Pomc gene expression (POMC neurons contain neither vGAT nor NOS1) (35). Aside from the importance of large groups of hypothalamic LepRb neurons that contain vGAT or NOS1, the molecular fingerprint and location of LepRb neurons crucial for the control of POMC cells and feeding/body weight remains unclear. The dorsomedial hypothalamic nucleus (DMH) represents the main region in which vGAT and NOS1 LepRb neurons overlap, however, suggesting a potential role for DMH LepRb neurons.
Leptin Action in the DMH and Ventromedial Hypothalamic Nucleus (VMN)
Nestled between the rostral aspects of the ARC and DMH lies the VMN; the dorsomedial aspect of the VMN contains a large population of LepRb neurons that express the transcription factor, steroidogenic factor-1 (SF1) (Nr5a1) and the neuropeptide pituitary adenyl cyclase activating protein (PACAP) (Adcyap). Ablation of LepRb in SF1/PACAP neurons provokes little change in energy balance at baseline (31, 55) but increases body weight and adiposity on a palatable high-fat diet, due to a failure to increase energy expenditure with weight gain.
Glutamatergic (vesicular glutamate transporter-2-expressing) LepRb neurons include those in the VMN and ventral premammillary (PMv) nucleus in the hypothalamus, as well as several groups of cells in the brainstem (30). Ablation of LepRb in vesicular glutamate transporter-2 cells provokes only a mild increase in body weight (30), suggesting only minor roles for these regions in the control of feeding and energy balance. Also, deletion of LepRb in prodynorphin-expressing cells (which includes most VMN LepRb cells, a few ARC cells, and about one third of DMH LepRb neurons) yields a phenotype similar to that observed with SF1/PACAP-mediated deletion (56), suggesting that if a set of DMH LepRb neurons play a major role in the control of feeding and energy balance by leptin, it is distinct from those that contain prodynorphin.
Activation of dorsal DMH/dorsal hypothalamic area LepRb neurons promotes energy expenditure, including by increasing energy use for heat generation (57); ablation of LepRb in these dorsal hypothalamic area cells or in prolactin-releasing hormone neurons of the dorsal DMH decreases heat output and energy expenditure in mice (57, 58). Thus, should the DMH contain a population of LepRb neurons focused on the control of feeding and crucial for overall energy balance, they presumably reside in the ventral DMH. However, less is known about the function of LepRb neurons in the more ventral aspects of the DMH.
The Lateral Hypothalamus and the Control of the Mesolimbic Dopamine (DA) System
Food intake is controlled jointly by the brain's motivational circuits (which mediate food seeking and the initiation of feeding) and the systems that modulate satiety to terminate feeding (see below) (59). Food deprivation enhances motivation, increasing locomotor activity and food-seeking behavior, as well as increasing the amount of work animals are willing to expend to attain food. Interestingly, caloric restriction enhances not only the seeking and consumption of food, but also of other rewards (including drugs of abuse), suggesting a general enhancement of motivation during negative energy balance. Importantly, although other hormones and cues contribute to this effect, decreased leptin plays an important role, because exogenous leptin blunts or reverses many of these effects in food-restricted animals (60, 61).
The mesolimbic DA system is crucial for the expression of motivated behaviors (59). At its core, this system contains a set of DA neurons in the midbrain ventral tegmental area (VTA); these VTA DA neurons project widely throughout the brain, including to the nucleus accumbens (NAc), where DA release modulates motivation. A variety of data demonstrate that leptin modulates the mesolimbic DA system, including by the control of VTA tyrosine hydroxylase (Th) (the enzyme that mediates the committed step in DA production) expression and the control of DA reuptake and extracellular DA concentration in the NAc (62, 63).
Although the VTA contains LepRb neurons, most of which are dopaminergic, ablation of LepRb from DA cells in the brain does not alter parameters of motivation, but rather increases anxiety-like behaviors (64). Consistent with this finding, VTA LepRb neurons project to the central amygdala (which plays a major role in anxiety and aversion) and associated structures (65). Interestingly, although deletion of LepRb from DA neurons does not alter motivation (64), short hairpin RNA-mediated suppression of LepRb expression in the VTA does influence hedonic feeding and body weight (66), suggesting a potential role for the (mostly γ-aminobutyric acid-ergic) non-DA VTA LepRb neurons in the control of the mesolimbic DA system.
Because VTA LepRb neurons do not strongly modulate the mesolimbic DA system and motivation, other LepRb neurons must mediate this leptin effect, presumably via projections into the mesolimbic DA system. Although few LepRb neurons project directly to the NAc (65), LepRb neurons in the lateral hypothalamic area (LHA) project densely to the VTA and a few other midbrain structures (32, 62), suggesting a potential role for LHA LepRb neurons in the control of mesolimbic DA function. Consistently, the LHA has long been known to control appetitive behavior: lesioning the LHA diminishes motivation and causes lethal hypophagia, whereas stimulation of the LHA is motivating (67).
In support of a role for LHA LepRb neurons in the control of food intake, body weight, and mesolimbic DA function, intra-LHA leptin administration reduces food intake and body weight gain in Lepob/ob mice and also increases VTA Th expression (62). Furthermore, a subpopulation (∼60%) of LHA LepRb neurons contain neurotensin (NT), and ablation of LepRb in LHA NT neurons increases body weight, adiposity, and food intake while decreasing overall activity and energy expenditure (32). These animals also display decreased mesolimbic DA function as a consequence of increased NAc DA reuptake (32).
NT-containing LHA LepRb neurons also inhibit neighboring hypocretin (HCRT) (also known as Orexin)-containing neurons in response to leptin (32, 68); this represents an important mechanism by which leptin controls the hypothalamus-pituitary-adrenal axis and related stress responses (69). Some NT-containing LHA LepRb neurons also contain the inhibitory neuropeptide, galanin (70), which plays an important role in this inhibition (68). Because HCRT neurons play a role in arousal, including during fasting, the withdrawal of leptin from this circuit increases activity and motivation.
The Brainstem: The Nucleus of the Solitary Tract (NTS) and Satiety
A number of studies have focused on LepRb neurons in the NTS in the caudal brainstem for the control of food intake. The NTS receives important gastrointestinal signals via the vagus nerve, as well as from circulating factors, many of which act via area postrema neuron that project into the NTS (71). These NTS neurons participate in the control of feeding by modulating satiety to promote meal termination, and leptin enhances the response of these NTS cells to gut-derived satiety signals, including cholecystokinin, amylin, and glucagon-like peptide 1 (72–74). Consistently, intra-NTS leptin administration promotes satiety, whereas short hairpin RNA-mediated suppression of NTS LepRb expression increases food intake and body weight gain, especially on a high-fat diet (75, 76).
Other brainstem and midbrain regions also contain significant populations of LepRb neurons, including the parabrachial nucleus (PBN), periaqueductal gray, and dorsal raphe (DR), among others (36, 39). Although roles for leptin action via periaqueductal gray and DR LepRb neurons have not been directly examined (note that the large phenotype previously reported for DR LepRb neurons [77] represents an artifact [78]), PBN LepRb neurons modulate the response to hypoglycemia (see below).
Endocrine Control
Of the neuroendocrine axes, perhaps the most attention has been paid to the control of reproduction by leptin. Reproduction is energetically very costly, especially for females, who bear the burden of nourishing the offspring both before birth and thereafter (via lactation). Thus, the insufficiency of caloric reserves inhibits the reproductive axis, especially in females. Leptin plays a major role in the control of reproduction by nutritional status: Lepob/ob and Leprdb/db mice generally fail to enter puberty (8, 79), as do humans lacking leptin or LepRb (80), and fail to lactate if they become pregnant; leptin administration to Lepob/ob mice restores fertility (79, 81, 82). Furthermore, exogenous leptin restores fertility in females with low adipose mass due to caloric restriction (as in anorexia nervosa) or lipodystrophy (83, 84).
GnRH neurons, which control the reproductive axis, do not contain LepRb, suggesting that leptin must control the reproductive axis indirectly, via LepRb neurons that lie upstream of GnRH cells (85). Roles for several sets of neurons known to lie afferent to GnRH neurons have been investigated, including kisspeptin (KISS1)-expressing neurons of the ARC; very few KISS1 neurons contain LepRb, however, and ablation of LepRb in KISS1 cells does not alter reproductive function (86, 87). The PMv receives and integrates a number of stimuli (including sex steroids and opposite-sex pheromones) to modulate the function of the reproductive system, and contains a large population of LepRb neurons (88–90), suggesting a potential role for PMv LepRb cells in the control of reproductive function. Indeed, PMv LepRb neurons lie in synaptic contact with GnRH neurons and are activated by opposite-sex pheromones (85). Furthermore, lesioning the PMV blocks the ability of leptin to modulate reproductive function and restoration of PMv LepRb expression in a LepRb-null background restores reproductive function (87, 91). PMv LepRb neurons may not be the only LepRb neurons that contribute to the control of the reproductive axis; however, the vast majority of PMv LepRb neurons contain NOS1, but the ablation of LepRb in NOS1 neurons only partially blunts estrus entry and reproductive function in female mice (35). Furthermore, although PMv LepRb neurons do not express vGAT, the deletion of LepRb in vGAT neurons impairs reproduction (92, 93). Additionally, it is not clear whether leptin may control reproductive function indirectly (secondary to changes in adrenal, thyroid, and other endocrine axes), as well as by neural inputs directly onto GnRH neurons.
Fasting or leptin deficiency increases adrenal corticosteroid production while inhibiting the thyroid and growth axes (1). Leptin reverses these effects, demonstrating an important role for leptin in modulating a host of endocrine parameters in line with energy balance (2). There are few LepRb cells in the paraventricular nucleus of the hypothalamus (PVH), where reside the neuroendocrine neurons produce TRH and CRH (which control the thyroid and adrenal axes, respectively) (36, 39). Thus, the control of these endocrine axes by leptin must be mediated indirectly. Indeed, LHA LepRb cells may play an important role in the control of the adrenal axis (69). In contrast, many of the GHRH neurons that control the growth axis contain LepRb (56), suggesting a more direct control of the growth axis by leptin. Although circumscribed sets of LepRb neurons that control the adrenal and thyroid axes have not been identified, the dense projections to the PVH from ARC and DMH LepRb neurons suggest that LepRb neurons in these areas could play a role. Furthermore, leptin action via vGAT LepRb cells is required for the control of both thyroid and adrenal function (30); NOS1 LepRb neurons play a role in the control of thyroid (but not adrenal) function (35). vGAT-containing cells include LHA LepRb neurons, which NOS1 cells do not, consistent with the notion that LHA LepRb neurons may play a crucial role in the control of the adrenal axis by leptin (30, 35, 69).
Glucose Homeostasis
Loss of leptin action also dysregulates glucose homeostasis: Lepob/ob and Leprdb/db mice display marked hyperglycemia, hyperinsulinemia, and glucose intolerance (33, 94, 95). Because caloric restriction/weight loss does not ameliorate these disturbances in glucose homeostasis (33, 96, 97), they are not likely to be caused solely by overnutrition and obesity. Indeed, icv leptin administration in Lepob/ob mice rapidly corrects their defects in glucose homeostasis before the onset of significant weight loss (98), indicating that leptin acts in the brain to control glucose homeostasis. Furthermore, systemic or icv leptin administration in Lepob/ob mice rapidly increases glucose clearance; VMN LepRb neurons may participate in this effect, because intra-VMN leptin injection promotes sympathetic nervous system (SNS) activation and increases glucose uptake into skeletal muscle (99).
Similarly, the loss of leptin action reduces hepatic insulin sensitivity and increases hepatic glucose production, and icv leptin reverses these effects (98, 100, 101). Although it is clear that leptin increases glucose disposal in unperturbed wild-type animals (102), leptin does not appreciably alter hepatic glucose production in animals with normal leptin function, however; this is consistent with the notion that excess leptin mediates little effect, whereas decreased leptin promotes large physiologic changes.
Some studies also report direct leptin action on pancreatic β-cells (103–105), although direct effects of leptin on insulin secretion have not been reported in vivo. In contrast, glucagon, which is elevated in Lepob/ob mice (106), is normalized by leptin administration (107); because icv leptin promotes this response (108, 109), this effect is likely to be mediated by the suppression of SNS activity on α-cells in the pancreatic islets.
A set of very exciting recent studies have revealed the ability of leptin to normalize blood glucose independently of insulin in a model of uncontrolled type 1 diabetes, in which the pancreatic β-cells have been destroyed by treatment with streptozotocin (STZ) (110). As with Lepob/ob mice, these STZ mice exhibit low circulating leptin concentrations before leptin treatment, as a result of the catabolic state produced by uncontrolled insulinopenic diabetes.
A variety of data suggest potential important roles for leptin action via LepRb on POMC neurons for at least some control of glucose homeostasis. Although modestly affecting body weight gain and food intake, the restoration of leptin action in the ARC normalizes hyperinsulinemia and hyperglycemia in Leprdb/db mice (111); similarly, although POMC-specific LepRb restoration in a LepRb-null background does little to alter feeding or other parameters of energy balance, it ameliorates the hyperglycemia and insulin resistance of these animals (112, 113). Furthermore, ablation of LepRb in POMC neurons blunts the ability of leptin to normalize glucose concentrations in STZ mice (114). However, intra-VMN leptin injection also normalizes glucose in STZ rodents (115). Thus, the available evidence suggests that both ARC and VMN LepRb neurons may contribute to the control of glucose homeostasis by leptin; other, untested, sets of neurons may also participate.
Counterregulation
Recently, we demonstrated a role for leptin in the modulation of the response to hypoglycemia (29). Hypoglycemia most often occurs in response to insulin overdose during diabetes therapy, and activates the counterregulatory response (CRR) to attempt to restore blood glucose concentrations to normal. Low glucose concentrations in the brain activate neurons (many of which reside in the VMN) that promote SNS activation, glucagon secretion, and hepatic glucose production as part of the CRR. Because an adequate CRR requires not only adequate energy stores to mobilize, but also adequate SNS activation, one would predict that it is important to augment the CRR in animals with depleted energy stores (as in after a prolonged fast) to compensate for low energy stores and decreased SNS tone. Indeed, we found that mice mount a more pronounced CRR to glucoprivation with 2-deoxyglucose (a competitive inhibitor of glycolysis that mimics hypoglycemia at a cellular level) after a 24-hour fast. Furthermore, leptin blunted this effect of fasting (29), suggesting that low leptin enhances the CRR, presumably to compensate for the low energy stores and decreased SNS tone that accompany negative energy balance.
Recent evidence from our laboratory suggest that leptin inhibits cholecystokinin (CCK)-containing PBN LepRb neurons, which are activated by low glucose and project to the VMN (which plays a crucial role in mediating the CRR to hypoglycemia) (29). CCKCre-mediated leptin receptor ablation enhances the CRR to insulin and 2-deoxyglucose, elevating glucocorticoids, glucagon, catecholamines, and hepatic gluconeogenic enzyme expression. Consistently, pharmacogenetic activation of these cells increases circulating glucagon and glucocorticoids, suppresses insulin, increases hepatic gluconeogenic enzyme expression, and increases blood glucose (29). The response to these cells, and the CRR to glucoprivation, require CCK-mediated neurotransmission and the VMN (29, 116), suggesting that CCK from PBN LepRb neurons acts via a set of CCK-responsive VMN neurons to control the CRR.
Acknowledgments
We thank members of the Myers and Olson labs for helpful discussions.
This work was supported by the Michigan Diabetes Research Center (National Institutes of Health Grant P30 DK020572), the American Diabetes Association, the Marilyn H. Vincent Foundation, and National Institutes of Health Grants DK098853 (to M.G.M.) and DK098833 (to J.N.F.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AgRP
- agouti-related peptide
- ARC
- hypothalamic arcuate nucleus
- CCK
- cholecystokinin
- CRR
- counterregulatory response
- DA
- dopamine
- DMH
- dorsomedial hypothalamic nucleus
- DR
- dorsal raphe
- HCRT
- hypocretin
- icv
- intracerebroventricular
- JAK2
- Janus kinase-2
- KISS1
- kisspeptin
- LepRb
- leptin receptor
- LHA
- lateral hypothalamic area
- MC4R
- melanocortin-4 receptor
- NAc
- nucleus accumbens
- NOS1
- nitric oxide synthase-1
- NPY
- neuropeptide Y
- NT
- neurotensin
- NTS
- nucleus of the solitary tract
- PACAP
- pituitary adenyl cyclase activating protein
- PBN
- parabrachial nucleus
- PMv
- ventral premammillary
- POMC
- proopiomelanocortin
- PVH
- paraventricular nucleus of the hypothalamus
- SF1
- steroidogenic factor-1
- SNS
- sympathetic nervous system
- STAT
- signal transducer and activator of transcription
- STZ
- streptozotocin
- vGAT
- vesicular GABA transporter
- VMN
- ventromedial hypothalamic nucleus
- VTA
- ventral tegmental area.
References
- 1. Myers MG, Leibel RL. Lessons from rodent models of obesity. In: De Groot LJ, Beck-Peccoz P, Chrousos G, et al., eds. Endotext [Internet]. South Dartmouth (MA): MDText.com, Inc.; 2000–2015. February 28. [Google Scholar]
- 2. Ahima RS, Prabakaran D, Mantzoros C, et al. Role of leptin in the neuroendocrine response to fasting. Nature. 1996;382:250–252. [DOI] [PubMed] [Google Scholar]
- 3. Schwartz MW, Seeley RJ, Tschöp MH, et al. Cooperation between brain and islet in glucose homeostasis and diabetes. Nature. 2013;503:59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Myers MG, Jr, Olson DP. Central nervous system control of metabolism. Nature. 2012;491:357–363. [DOI] [PubMed] [Google Scholar]
- 5. Myers MG, Jr, Heymsfield SB, Haft C, et al. Challenges and opportunities of defining clinical leptin resistance. Cell Metab. 2012;15:150–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ottaway N, Mahbod P, Rivero B, et al. Diet-induced obese mice retain endogenous leptin action. Cell Metab. 2015;21:877–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chen H, Charlat O, Tartaglia LA, et al. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell. 1996;84:491–495. [DOI] [PubMed] [Google Scholar]
- 8. de Luca C, Kowalski TJ, Zhang Y, et al. Complete rescue of obesity, diabetes, and infertility in db/db mice by neuron-specific LEPR-B transgenes. J Clin Invest. 2005;115:3484–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baumann H, Morella KK, White DW, et al. The full-length leptin receptor has signaling capabilities of interleukin 6-type cytokine receptors. Proc Natl Acad Sci USA. 1996;93:8374–8378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Banks AS, Davis SM, Bates SH, Myers MG., Jr Activation of downstream signals by the long form of the leptin receptor. J Biol Chem. 2000;275:14563–14572. [DOI] [PubMed] [Google Scholar]
- 11. Kloek C, Haq AK, Dunn SL, Lavery HJ, Banks AS, Myers MG., Jr Regulation of Jak kinases by intracellular leptin receptor sequences. J Biol Chem. 2002;277:41547–41555. [DOI] [PubMed] [Google Scholar]
- 12. Gong Y, Ishida-Takahashi R, Villanueva EC, Fingar DC, Münzberg H, Myers MG., Jr The long form of the leptin receptor regulates STAT5 and ribosomal protein S6 via alternate mechanisms. J Biol Chem. 2007;282:31019–31027. [DOI] [PubMed] [Google Scholar]
- 13. Hekerman P, Zeidler J, Bamberg-Lemper S, et al. Pleiotropy of leptin receptor signalling is defined by distinct roles of the intracellular tyrosines. FEBS J. 2005;272:109–119. [DOI] [PubMed] [Google Scholar]
- 14. Bates SH, Stearns WH, Dundon TA, et al. STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature. 2003;421:856–859. [DOI] [PubMed] [Google Scholar]
- 15. Patterson CM, Villanueva EC, Greenwald-Yarnell M, et al. Leptin action via LepR-b Tyr1077 contributes to the control of energy balance and female reproduction. Mol Metab. 2012;1:61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Singireddy AV, Inglis MA, Zuure WA, Kim JS, Anderson GM. Neither signal transducer and activator of transcription 3 (STAT3) or STAT5 signaling pathways are required for leptin's effects on fertility in mice. Endocrinology. 2013;154:2434–2445. [DOI] [PubMed] [Google Scholar]
- 17. Lee JY, Muenzberg H, Gavrilova O, et al. Loss of cytokine-STAT5 signaling in the CNS and pituitary gland alters energy balance and leads to obesity. PLoS One. 2008;3:e1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Reed JA, Clegg DJ, Smith KB, et al. GM-CSF action in the CNS decreases food intake and body weight. J Clin Invest. 2005;115:3035–3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tsou RC, Bence KK. Central regulation of metabolism by protein tyrosine phosphatases. Front Neurosci. 2012;6:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu AW, Kaelin CB, Takeda K, Akira S, Schwartz MW, Barsh GS. PI3K integrates the action of insulin and leptin on hypothalamic neurons. J Clin Invest. 2005;115:951–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li Z, Zhou Y, Carter-Su C, Myers MG, Jr, Rui L. SH2B1 enhances leptin signaling by both Janus kinase 2 Tyr813 phosphorylation-dependent and -independent mechanisms. Mol Endocrinol. 2007;21:2270–2281. [DOI] [PubMed] [Google Scholar]
- 22. Duan C, Li M, Rui L. SH2-B promotes insulin receptor substrate 1 (IRS1)- and IRS2-mediated activation of the phosphatidylinositol 3-kinase pathway in response to leptin. J Biol Chem. 2004;279:43684–43691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kim YB, Uotani S, Pierroz DD, Flier JS, Kahn BB. In vivo administration of leptin activates signal transduction directly in insulin-sensitive tissues: overlapping but distinct pathways from insulin. Endocrinology. 2000;141:2328–2339. [DOI] [PubMed] [Google Scholar]
- 24. Niswender KD, Morton GJ, Stearns WH, Rhodes CJ, Myers MG, Jr, Schwartz MW. Intracellular signalling. Key enzyme in leptin-induced anorexia. Nature. 2001;413:794–795. [DOI] [PubMed] [Google Scholar]
- 25. Dagon Y, Hur E, Zheng B, Wellenstein K, Cantley LC, Kahn BB. p70S6 kinase phosphorylates AMPK on serine 491 to mediate leptin's effect on food intake. Cell Metab. 2012;16:104–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tanida M, Yamamoto N, Morgan DA, Kurata Y, Shibamoto T, Rahmouni K. Leptin receptor signaling in the hypothalamus regulates hepatic autonomic nerve activity via phosphatidylinositol 3-kinase and AMP-activated protein kinase. J Neurosci. 2015;35:474–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang H, Kong D, Byun KH, et al. Rho-kinase regulates energy balance by targeting hypothalamic leptin receptor signaling. Nat Neurosci. 2012;15:1391–1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cowley MA, Smart JL, Rubinstein M, et al. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–484. [DOI] [PubMed] [Google Scholar]
- 29. Flak JN, Patterson CM, Garfield AS, et al. Leptin-inhibited PBN neurons enhance responses to hypoglycemia in negative energy balance. Nat Neurosci. 2014;17:1744–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vong L, Ye C, Yang Z, Choi B, Chua S, Jr, Lowell BB. Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron. 2011;71:142–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dhillon H, Zigman JM, Ye C, et al. Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron. 2006;49:191–203. [DOI] [PubMed] [Google Scholar]
- 32. Leinninger GM, Opland DM, Jo YH, et al. Leptin action via neurotensin neurons controls orexin, the mesolimbic dopamine system and energy balance. Cell Metab. 2011;14:313–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pelleymounter MA, Cullen MJ, Baker MB, et al. Effects of the obese gene product on body weight regulation in ob/ob mice. Science. 1995;269:540–543. [DOI] [PubMed] [Google Scholar]
- 34. Mistry AM, Swick AG, Romsos DR. Leptin rapidly lowers food intake and elevates metabolic rates in lean and ob/ob mice. J Nutr. 1997;127:2065–2072. [DOI] [PubMed] [Google Scholar]
- 35. Leshan RL, Greenwald-Yarnell M, Patterson CM, Gonzalez IE, Myers MG., Jr Leptin action through hypothalamic nitric oxide synthase-1-expressing neurons controls energy balance. Nat Med. 2012;18:820–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Scott MM, Lachey JL, Sternson SM, et al. Leptin targets in the mouse brain. J Comp Neurol. 2009;514:518–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ring LE, Zeltser LM. Disruption of hypothalamic leptin signaling in mice leads to early-onset obesity, but physiological adaptations in mature animals stabilize adiposity levels. J Clin Invest. 2010;120:2931–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Myers MG, Jr, Münzberg H, Leinninger GM, Leshan RL. The geometry of leptin action in the brain: more complicated than a simple ARC. Cell Metab. 2009;9:117–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Patterson CM, Leshan RL, Jones JC, Myers MG., Jr Molecular mapping of mouse brain regions innervated by leptin receptor-expressing cells. Brain Res. 2011;1378:18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gautron L, Elmquist JK, Williams KW. Neural control of energy balance: translating circuits to therapies. Cell. 2015;161:133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. 2006;443:289–295. [DOI] [PubMed] [Google Scholar]
- 42. Elmquist JK, Coppari R, Balthasar N, Ichinose M, Lowell BB. Identifying hypothalamic pathways controlling food intake, body weight, and glucose homeostasis. J Comp Neurol. 2005;493:63–71. [DOI] [PubMed] [Google Scholar]
- 43. Aponte Y, Atasoy D, Sternson SM. AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nat Neurosci. 2011;14:351–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Krashes MJ, Shah BP, Koda S, Lowell BB. Rapid versus delayed stimulation of feeding by the endogenously released AgRP neuron mediators GABA, NPY, and AgRP. Cell Metab. 2013;18:588–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Luquet S, Perez FA, Hnasko TS, Palmiter RD. NPY/AgRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science. 2005;310:683–685. [DOI] [PubMed] [Google Scholar]
- 46. Marsh DJ, Hollopeter G, Huszar D, et al. Response of melanocortin-4 receptor-deficient mice to anorectic and orexigenic peptides. Nat Genet. 1999;21:119–122. [DOI] [PubMed] [Google Scholar]
- 47. Challis BG, Coll AP, Yeo GS, et al. Mice lacking pro-opiomelanocortin are sensitive to high-fat feeding but respond normally to the acute anorectic effects of peptide-YY(3–36). Proc Natl Acad Sci USA. 2004;101:4695–4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Balthasar N, Coppari R, McMinn J, et al. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron. 2004;42:983–991. [DOI] [PubMed] [Google Scholar]
- 49. van de Wall E, Leshan R, Xu AW, et al. Collective and individual functions of leptin receptor modulated neurons controlling metabolism and ingestion. Endocrinology. 2008;149:1773–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gong L, Yao F, Hockman K, et al. Signal transducer and activator of transcription-3 is required in hypothalamic agouti-related protein/neuropeptide Y neurons for normal energy homeostasis. Endocrinology. 2008;149:3346–3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xu AW, Ste-Marie L, Kaelin CB, Barsh GS. Inactivation of signal transducer and activator of transcription 3 in proopiomelanocortin (Pomc) neurons causes decreased pomc expression, mild obesity, and defects in compensatory refeeding. Endocrinology. 2007;148:72–80. [DOI] [PubMed] [Google Scholar]
- 52. Gropp E, Shanabrough M, Borok E, et al. Agouti-related peptide-expressing neurons are mandatory for feeding. Nat Neurosci. 2005;8:1289–1291. [DOI] [PubMed] [Google Scholar]
- 53. Cone RD. Anatomy and regulation of the central melanocortin system. Nat Neurosci. 2005;8:571–578. [DOI] [PubMed] [Google Scholar]
- 54. Cone RD, Cowley MA, Butler AA, Fan W, Marks DL, Low MJ. The arcuate nucleus as a conduit for diverse signals relevant to energy homeostasis. Int J Obes Relat Metab Disord. 2001;25(suppl 5):S63–S67. [DOI] [PubMed] [Google Scholar]
- 55. Hawke Z, Ivanov TR, Bechtold DA, Dhillon H, Lowell BB, Luckman SM. PACAP neurons in the hypothalamic ventromedial nucleus are targets of central leptin signaling. J Neurosci. 2009;29:14828–14835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Allison MB, Patterson CM, Krashes MJ, Lowell BB, Myers MG, Jr, Olson DP. TRAP-seq defines markers for novel populations of hypothalamic and brainstem LepRb neurons. Mol Metab. 2015;4:299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rezai-Zadeh K, Yu S, Jiang Y, Laque A, et al. Leptin receptor neurons in the dorsomedial hypothalamus are key regulators of energy expenditure and body weight, but not food intake. Mol Metab. 2014;3:681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dodd GT, Worth AA, Nunn N, et al. The thermogenic effect of leptin is dependent on a distinct population of prolactin-releasing peptide neurons in the dorsomedial hypothalamus. Cell Metab. 2014;20:639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Berthoud HR. The neurobiology of food intake in an obesogenic environment. Proc Nutr Soc. 2012;71:478–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Figlewicz DP, Bennett JL, Naleid AM, Davis C, Grimm JW. Intraventricular insulin and leptin decrease sucrose self-administration in rats. Physiol Behav. 2006;89:611–616. [DOI] [PubMed] [Google Scholar]
- 61. Figlewicz DP, Higgins MS, Ng-Evans SB, Havel PJ. Leptin reverses sucrose-conditioned place preference in food-restricted rats. Physiol Behav. 2001;73:229–234. [DOI] [PubMed] [Google Scholar]
- 62. Leinninger GM, Jo YH, Leshan RL, et al. Leptin acts via leptin receptor-expressing lateral hypothalamic neurons to modulate the mesolimbic dopamine system and suppress feeding. Cell Metab. 2009;10:89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fulton S, Pissios P, Manchon RP, et al. Leptin regulation of the mesoaccumbens dopamine pathway. Neuron. 2006;51:811–822. [DOI] [PubMed] [Google Scholar]
- 64. Liu J, Perez SM, Zhang W, Lodge DJ, Lu XY. Selective deletion of the leptin receptor in dopamine neurons produces anxiogenic-like behavior and increases dopaminergic activity in amygdala. Mol Psychiatry. 2011;16:1024–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Leshan RL, Opland DM, Louis GW, et al. Ventral tegmental area leptin receptor neurons specifically project to and regulate cocaine- and amphetamine-regulated transcript neurons of the extended central amygdala. J Neurosci. 2010;30:5713–5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hommel JD, Trinko R, Sears RM, et al. Leptin receptor signaling in midbrain dopamine neurons regulates feeding. Neuron. 2006;51:801–810. [DOI] [PubMed] [Google Scholar]
- 67. Fulton S, Woodside B, Shizgal P. Modulation of brain reward circuitry by leptin. Science. 2000;287:125–128. [DOI] [PubMed] [Google Scholar]
- 68. Goforth PB, Leinninger GM, Patterson CM, Satin LS, Myers MG., Jr Leptin acts via lateral hypothalamic area neurotensin neurons to inhibit orexin neurons by multiple GABA-independent mechanisms. J Neurosci. 2014;34:11405–11415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bonnavion P, Jackson AC, Carter ME, de Lecea L. Antagonistic interplay between hypocretin and leptin in the lateral hypothalamus regulates stress responses. Nat Commun. 2015;6:6266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Laque A, Zhang Y, Gettys S, et al. Leptin receptor neurons in the mouse hypothalamus are colocalized with the neuropeptide galanin and mediate anorexigenic leptin action. Am J Physiol Endocrinol Metab. 2013;304:E999–E1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Norgren R. Projections from the nucleus of the solitary tract in the rat. Neuroscience. 1978;3:207–218. [DOI] [PubMed] [Google Scholar]
- 72. Huo L, Maeng L, Bjørbaek C, Grill HJ. Leptin and the control of food intake: neurons in the nucleus of the solitary tract are activated by both gastric distension and leptin. Endocrinology. 2007;148:2189–2197. [DOI] [PubMed] [Google Scholar]
- 73. Morton GJ, Blevins JE, Williams DL, et al. Leptin action in the forebrain regulates the hindbrain response to satiety signals. J Clin Invest. 2005;115:703–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Williams DL, Baskin DG, Schwartz MW. Leptin regulation of the anorexic response to glucagon-like peptide-1 receptor stimulation. Diabetes. 2006;55:3387–3393. [DOI] [PubMed] [Google Scholar]
- 75. Grill HJ, Schwartz MW, Kaplan JM, Foxhall JS, Breininger J, Baskin DG. Evidence that the caudal brainstem is a target for the inhibitory effect of leptin on food intake. Endocrinology. 2002;143:239–246. [DOI] [PubMed] [Google Scholar]
- 76. Hayes MR, Skibicka KP, Leichner TM, et al. Endogenous leptin signaling in the caudal nucleus tractus solitarius and area postrema is required for energy balance regulation. Cell Metab. 2010;11:77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Yadav VK, Oury F, Suda N, et al. A serotonin-dependent mechanism explains the leptin regulation of bone mass, appetite, and energy expenditure. Cell. 2009;138:976–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lam DD, Leinninger GM, Louis GW, et al. Leptin does not directly affect CNS serotonin neurons to influence appetite. Cell Metab. 2011;13:584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Chehab FF, Lim ME, Lu R. Correction of the sterility defect in homozygous obese female mice by treatment with the human recombinant leptin. Nat Genet. 1996;12:318–320. [DOI] [PubMed] [Google Scholar]
- 80. Montague CT, Farooqi IS, Whitehead JP, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387:903–908. [DOI] [PubMed] [Google Scholar]
- 81. Barash IA, Cheung CC, Weigle DS, et al. Leptin is a metabolic signal to the reproductive system. Endocrinology. 1996;137:3144–3147. [DOI] [PubMed] [Google Scholar]
- 82. Chehab FF, Mounzih K, Lu R, Lim ME. Early onset of reproductive function in normal female mice treated with leptin. Science. 1997;275:88–90. [DOI] [PubMed] [Google Scholar]
- 83. Welt CK, Chan JL, Bullen J, et al. Recombinant human leptin in women with hypothalamic amenorrhea. N Engl J Med. 2004;351:987–997. [DOI] [PubMed] [Google Scholar]
- 84. Oral EA, Ruiz E, Andewelt A, et al. Effect of leptin replacement on pituitary hormone regulation in patients with severe lipodystrophy. J Clin Endocrinol Metab. 2002;87:3110–3117. [DOI] [PubMed] [Google Scholar]
- 85. Leshan RL, Louis GW, Jo YH, Rhodes CJ, Münzberg H, Myers MG., Jr Direct innervation of GnRH neurons by metabolic- and sexual odorant-sensing leptin receptor neurons in the hypothalamic ventral premammillary nucleus. J Neurosci. 2009;29:3138–3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Cravo RM, Frazao R, Perello M, et al. Leptin signaling in Kiss1 neurons arises after pubertal development. PLoS One. 2013;8:e58698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Donato J, Jr, Cravo RM, Frazão R, et al. Leptin's effect on puberty in mice is relayed by the ventral premammillary nucleus and does not require signaling in Kiss1 neurons. J Clin Invest. 2011;121:355–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Elias CF, Kelly JF, Lee CE, et al. Chemical characterization of leptin-activated neurons in the rat brain. J Comp Neurol. 2000;423:261–281. [PubMed] [Google Scholar]
- 89. Beltramino C, Taleisnik S. Release of LH in the female rat by olfactory stimuli. Effect of the removal of the vomeronasal organs or lesioning of the accessory olfactory bulbs. Neuroendocrinology. 1983;36:53–58. [DOI] [PubMed] [Google Scholar]
- 90. Beltramino C, Taleisnik S. Ventral premammillary nuclei mediate pheromonal-induced LH release stimuli in the rat. Neuroendocrinology. 1985;41:119–124. [DOI] [PubMed] [Google Scholar]
- 91. Donato J, Jr, Silva RJ, Sita LV, et al. The ventral premammillary nucleus links fasting-induced changes in leptin levels and coordinated luteinizing hormone secretion. J Neurosci. 2009;29:5240–5250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Martin C, Navarro VM, Simavli S, et al. Leptin-responsive GABAergic neurons regulate fertility through pathways that result in reduced kisspeptinergic tone. J Neurosci. 2014;34:6047–6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Zuure WA, Roberts AL, Quennell JH, Anderson GM. Leptin signaling in GABA neurons, but not glutamate neurons, is required for reproductive function. J Neurosci. 2013;33:17874–17883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Halaas JL, Gajiwala KS, Maffei M, et al. Weight-reducing effects of the plasma protein encoded by the obese gene. Science. 1995;269:543–546. [DOI] [PubMed] [Google Scholar]
- 95. Coleman DL. Obese and diabetes: two mutant genes causing diabetes-obesity syndromes in mice. Diabetologia. 1978;14:141–148. [DOI] [PubMed] [Google Scholar]
- 96. Wyse BM, Dulin WE. The influence of age and dietary conditions on diabetes in the db mouse. Diabetologia. 1970;6:268–273. [DOI] [PubMed] [Google Scholar]
- 97. Schwartz MW, Baskin DG, Bukowski TR, et al. Specificity of leptin action on elevated blood glucose levels and hypothalamic neuropeptide Y gene expression in ob/ob mice. Diabetes. 1996;45:531–535. [DOI] [PubMed] [Google Scholar]
- 98. Asilmaz E, Cohen P, Miyazaki M, et al. Site and mechanism of leptin action in a rodent form of congenital lipodystrophy. J Clin Invest. 2004;113:414–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Minokoshi Y, Haque MS, Shimazu T. Microinjection of leptin into the ventromedial hypothalamus increases glucose uptake in peripheral tissues in rats. Diabetes. 1999;48:287–291. [DOI] [PubMed] [Google Scholar]
- 100. Liu L, Karkanias GB, Morales JC, et al. Intracerebroventricular leptin regulates hepatic but not peripheral glucose fluxes. J Biol Chem. 1998;273:31160–31167. [DOI] [PubMed] [Google Scholar]
- 101. van den Hoek AM, Teusink B, Voshol PJ, Havekes LM, Romijn JA, Pijl H. Leptin deficiency per se dictates body composition and insulin action in ob/ob mice. J Neuroendocrinol. 2008;20:120–127. [DOI] [PubMed] [Google Scholar]
- 102. Kamohara S, Burcelin R, Halaas JL, Friedman JM, Charron MJ. Acute stimulation of glucose metabolism in mice by leptin treatment. Nature. 1997;389:374–377. [DOI] [PubMed] [Google Scholar]
- 103. Emilsson V, Liu YL, Cawthorne MA, Morton NM, Davenport M. Expression of the functional leptin receptor mRNA in pancreatic islets and direct inhibitory action of leptin on insulin secretion. Diabetes. 1997;46:313–316. [DOI] [PubMed] [Google Scholar]
- 104. Laubner K, Kieffer TJ, Lam NT, Niu X, Jakob F, Seufert J. Inhibition of preproinsulin gene expression by leptin induction of suppressor of cytokine signaling 3 in pancreatic β-cells. Diabetes. 2005;54:3410–3417. [DOI] [PubMed] [Google Scholar]
- 105. Kieffer TJ, Heller RS, Leech CA, Holz GG, Habener JF. Leptin suppression of insulin secretion by the activation of ATP-sensitive K+ channels in pancreatic β-cells. Diabetes. 1997;46:1087–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Dubuc PU, Mobley PW, Mahler RJ, Ensinck JW. Immunoreactive glucagon levels in obese-hyperglycemic (ob/ob) mice. Diabetes. 1977;26:841–846. [DOI] [PubMed] [Google Scholar]
- 107. Della-Fera MA, Choi YH, Hartzell DL, Duan J, Hamrick M, Baile CA. Sensitivity of ob/ob mice to leptin-induced adipose tissue apoptosis. Obes Res. 2005;13:1540–1547. [DOI] [PubMed] [Google Scholar]
- 108. Fujikawa T, Chuang JC, Sakata I, Ramadori G, Coppari R. Leptin therapy improves insulin-deficient type 1 diabetes by CNS-dependent mechanisms in mice. Proc Natl Acad Sci USA. 2010;107:17391–17396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. German JP, Thaler JP, Wisse BE, et al. Leptin activates a novel CNS mechanism for insulin-independent normalization of severe diabetic hyperglycemia. Endocrinology. 2011;152:394–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Wang MY, Chen L, Clark GO, et al. Leptin therapy in insulin-deficient type I diabetes. Proc Natl Acad Sci USA. 2010;107:4813–4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Coppari R, Ichinose M, Lee CE, et al. The hypothalamic arcuate nucleus: a key site for mediating leptin's effects on glucose homeostasis and locomotor activity. Cell Metab. 2005;1:63–72. [DOI] [PubMed] [Google Scholar]
- 112. Berglund ED, Vianna CR, Donato J, Jr, et al. Direct leptin action on POMC neurons regulates glucose homeostasis and hepatic insulin sensitivity in mice. J Clin Invest. 2012;122:1000–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Huo L, Gamber K, Greeley S, et al. Leptin-dependent control of glucose balance and locomotor activity by POMC neurons. Cell Metab. 2009;9:537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Fujikawa T, Berglund ED, Patel VR, et al. Leptin engages a hypothalamic neurocircuitry to permit survival in the absence of insulin. Cell Metab. 2013;18:431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Meek TH, Matsen ME, Dorfman MD, et al. Leptin action in the ventromedial hypothalamic nucleus is sufficient, but not necessary, to normalize diabetic hyperglycemia. Endocrinology. 2013;154:3067–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Garfield AS, Shah BP, Madara JC, et al. A parabrachial-hypothalamic cholecystokinin neurocircuit controls counterregulatory responses to hypoglycemia. Cell Metab. 2014;20:1030–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]