

Abstract
The skin irritating principle from Thapsia garganica was isolated, named thapsigargin and the structure elucidated. By inhibiting the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA) thapsigargin provokes apoptosis in almost all cells. By conjugating thapsigargin to peptides, which are only substrates for either prostate specific antigen (PSA) or prostate specific membrane antigen (PSMA) prodrugs were created, which selectively affect prostate cancer cells or neovascular tissue in tumors. One of the prodrug is currently tested in clinical phase II. The prodrug under clinical trial has been named mipsagargin.
Keywords: Thapsigargin, Sarco/endoplasmic reticulum (SERCA), Prodrug, Prostate specific antigen (PSA), Prostate specific membrane antigen (PSMA), Targeting drugs
1. Traditional medicine with Thapsia garganica
Thapsia garganica L. (Apiaceae) is an umbelliferous plant growing in the Mediterranean area (Fig. 1). Advantage of the skin irritating effects of the plant has been taken in traditional Arabian medicine for millennia [1], and the resin of the root was last included in the 1937 edition of the French Pharmacopoeia. Also the toxic effects of parts of the plant in fodder have been known for centuries [1]. In spite of the ancient knowledge of the effects of the plant the chemistry and pharmacology was not understood until the early 1980's.
Fig. 1.

Thapsia garganica photographed ultimo June when the fruits are ripened and dry.
2. Phytochemical investigation of the genus Thapsia
A bioguided isolation afforded a fraction with potent skin irritating properties [2]. Spectroscopic studies of the active principle revealed that the compound was a hexaoxygenated guaianolide, which was named thapsigargin (1, Fig. 2) [3]. The relative configuration was established by solving the crystal structure of the crystalline epoxide (2) formed by treating 1 with thionyl chloride (Scheme 1) [4]. This conversion of a vicinal diol into an epoxide is a very unusual reaction and only very few analogous reaction are described [5]. Finally the absolute configuration was solved taken advantage of exciton coupling [6]. Phytochemical investigation of the genus Thapsia revealed a number of other hexaoxygenated guaianolides (thapsigargicin (3), thapsitranstagin (4), thapsivillosin A–E (5–9), thapsivillosin G–K (10–14) and 2-acet-oxytrilobolide (15) Fig. 2) [7,8] and in addition some pentaoxygenated guaianolides (trilobolide (16), nortrilobolide (17) and thapsivillosin F (18) (Fig. 3) [7]. Except for Laser trilobum L. (Borkh) (Apiaceae) hexaoxygenated and pentaoxygenated guaianolides have only been found within species belonging to the genus Thapsia. In addition to the presence of these unique specialized metabolites other unusual metabolites like thapsanes (Fig. 4) [7], tethered lipids (Fig. 4) [9] and C19 terpenoids (Fig. 4) [10] have been found in plants belonging to the genus. Inspired by poor correlation between the species assigned by morphological characteristics and the specialized metabolites a reinvestigation of the taxonomy of the genus has been initiated [11].
Fig. 2.
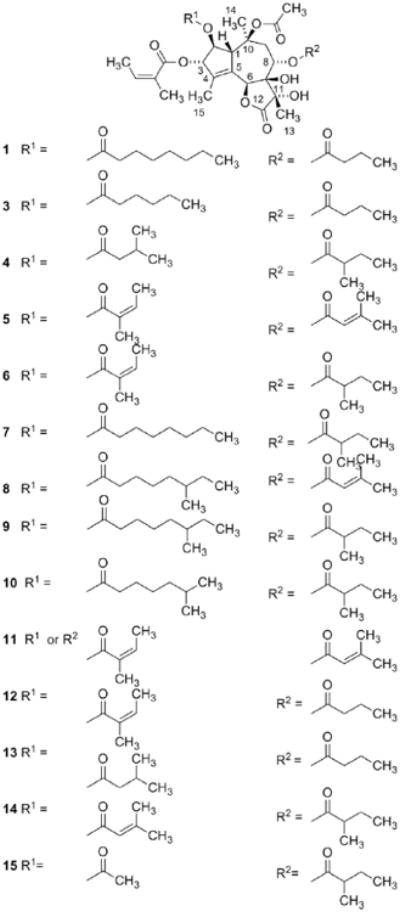
Structure of thapsigargin (1), thapsigargicin (3), thapsitranstagin (4), thapsivillosin A–E (5–9), thapsivillosin G–K (10–14) and 2-acetoxytrilobolide (15).
Scheme 1.
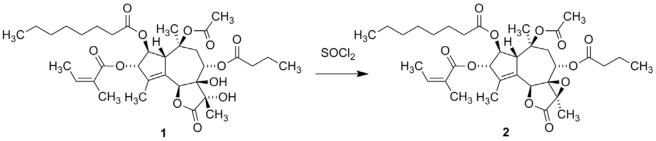
Conversion of thapsigargin (1) into thapsigargin epoxide (2).
Fig. 3.
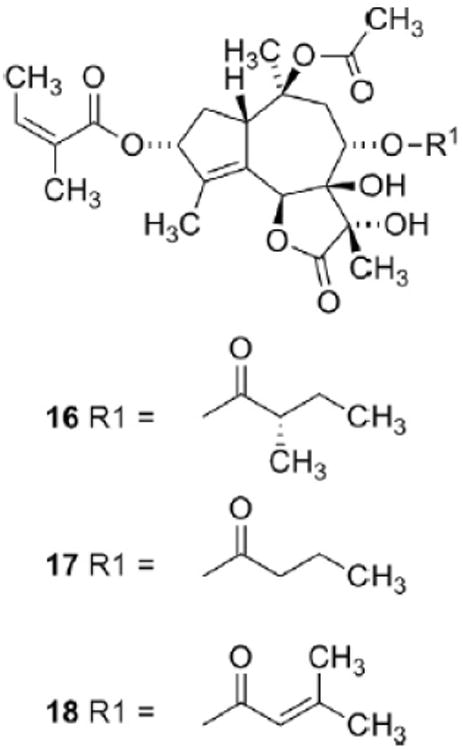
Structure of trilobolide (16), nortrilobolide (17) and thapsivillosin F (18).
Fig. 4.
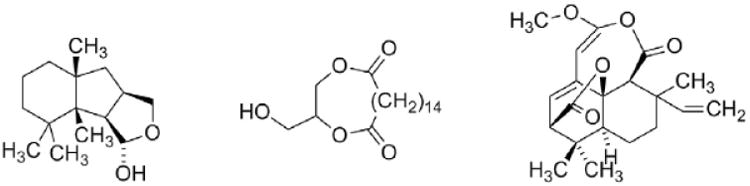
A representative example of a thapsanes, tethered lipid and a C19 diterpenoid isolated from T. garganica.
3. Pharmacological effects of the thapsigargins
The potent skin irritating effect of the isolated compound 1 provoked an investigation of the mechanism of action. Incubation of peritoneal mast cells in the presence of calcium ions with 1 even in low concentrations provoked a release of histamine [12]. This mediator release probably contributes to the skin irritating effects. Expansion of the studies revealed that 1 provoked a release of his-tamine and other mediators from a broad spectrum of cells involved in the immunologic response [13,14] and even had an effect on muscle cells [15]. The skin irritating effects made Fujiki suggest that 1 like the phorbols was a tumor promoter [16]. Systemic administration of 1 revealed a LD100 value of 0.8 mg/kg in mice [17]. A positive correlation between the lipophilicity of the thapsigargins and their effects on rat mast cells was demonstrated [18].
4. The SERCA pump, the biologic target of thapsigargin
The observation that the biological effects of 1 always are related to an increase in the cytosolic Ca2+ concentration indicates an effect on the Ca2+ homeostasis. A final proof of this hypothesis was found when inhibition of the Sarco-EndoPlasmic Reticulum Ca2+ ATPase (SERCA) in the subnanomolar range [19] was observed [20]. The SERCA pump is bound to the membranes of the endo- or sarcoplasmic reticulum. The pump is a P-type ATPase, which pumps Ca2+ ions from the cytosol into the plasmic reticulum. The mechanism of action has been intensively explored and five of the intermediate conformations of the pump are now known [21]. In depth understanding of the interactions of 1 to SERCA became possible when an X-ray structure of 1 bound to SERCA was published [22]. Based on a grid analysis of the binding pocket a model of the pharmacophore of 1 was suggested [23]. According to this model lipophilic interactions from the acetyl group, the C15-methyl group, the butanoate moiety and the angeloate moiety to the SERCA pump are of major importance for the binding (Fig. 5). A better resolved X-ray structure of 1 bound to the pump revealed that water mediated hydrogen bonds between the carbonyl group of the butanoate moiety and the C7-hydroxy group might also be of importance for the binding [24].
Fig. 5.
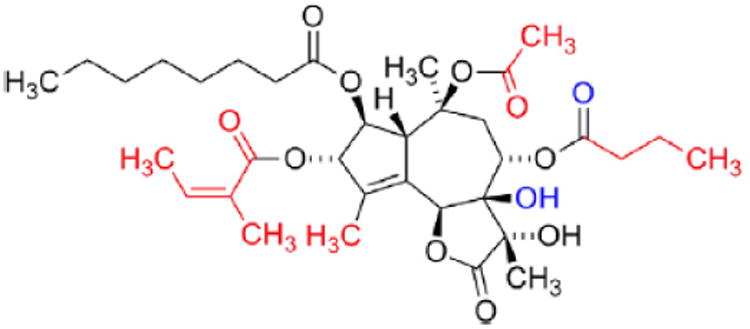
The pharmacophore of thapsigargin, the carbon atoms marked with red are in a group forming hydrophobic interactions with SERCA, the oxygen atoms marked with blue form water mediated hydrogen bonds to the backbone of SERCA.
5. Medicinal chemistry of thapsigargin
The chemistry of 1 has been intensively investigated and methods for selective transformations of the molecule have been performed [7]. Based on the activities as pump inhibitors of derivatives prepared by these methods the above mentioned pharmacophore has been verified. Reversing the absolute configuration at C3 or C8 strongly decrease the affinity demonstrating the importance of correct stereochemistry at these positions [25], similar removal of the hydrophobic acyl groups at O3, O8 or O10 also reduces the affinity [25,26]. To confirm a similar binding of different analogues X-ray structures of the complex of the analogue with high affinity and SERCA have been solved [27]. A very interesting result of these studies is that substitution with long flexible acyl groups at O8 still give analogues with high affinity for the pump. Advantage was taken of this finding when constructing prodrug for treatment of cancer diseases [28].
6. Apoptosis
Binding of 1 to the pump locks the transmembrane segments of the pumps and prevents the changes of conformation needed for a proper function of the pump. As a consequence the pump is prevented from removing Ca2+ ions from the cytosol and bringing them into the reticulum. Blockage of the pump in addition affords an efflux of Ca2+ ions from the reticulum into the cytosol. The increased cytosolic Ca2+ concentration also mediates an opening of Ca2+ channels in the cell membrane affording an additional influx of Ca2+. As a consequence the SERCA pump is prevented from maintaining a low concentration of Ca2+ in the cytosol and a high Ca2+ concentration in the reticulum. If this situation continues for some hours the cells undergo apoptosis by a cascade reaction, which at the present is not fully understood [29,30].
7. Targeting thapsigargin: the prodrug approach
A prodrug is defined as a drug which per se has no pharmacological effect but in the body is transformed into the active therapeutic. Approaches, which increasingly are being used, are to conjugate drugs to peptides or proteins, which can target the drug towards the malign tissue. One approach is to conjugate a drug to an antibody that selectively binds to the surface of a cancer cell. After internalization the toxin is released and kills the cancer cell. Some drugs based on this principle are currently in clinical trials [31]. In the case of 1 a prodrug concept also had to be chosen since proper function of SERCA is essential for almost all cells in the organism. Consequently systemic administration of 1 will induce general toxicity as is evidenced from the low LD100 value towards mice. In this case, however, advantage was taken of the proteolytic activities of enzymes found in malign tissues. Prostate specific antigen (PSA) is exclusively expressed in the prostate [32]. PSA diffuses into the blood but is inactivated by protein binding. Consequently the proteolytic activity is only found in the vicinity of the prostate cells or prostate cancer cells. In addition PSA has a substrate specificity, which makes it possible to construct a pep-tide that only will be cleaved by this protease [17,32]. Fig. 6 depicts the prodrug (G115) designed for treatment of prostate cancer. The peptide part of the molecule prevents the prodrug from penetrating the cell membrane and thereby the drug from inhibiting SERCA, which is only found in the intracellular membrane of the reticu-lum. Systemic administration of the drug into mice inoculated with prostate cancer cells proved that the prodrug only was cleaved to give 20 in tumors expressing PSA. The enzyme present in the tumors cleaved the prodrug sufficiently efficient to give a concentration of 20 in the tumor, which significantly decreased their sizes [17]. At the present no drug exists for treatment of androgen-inde-pendent metastatic prostate cancer.
Fig. 6.
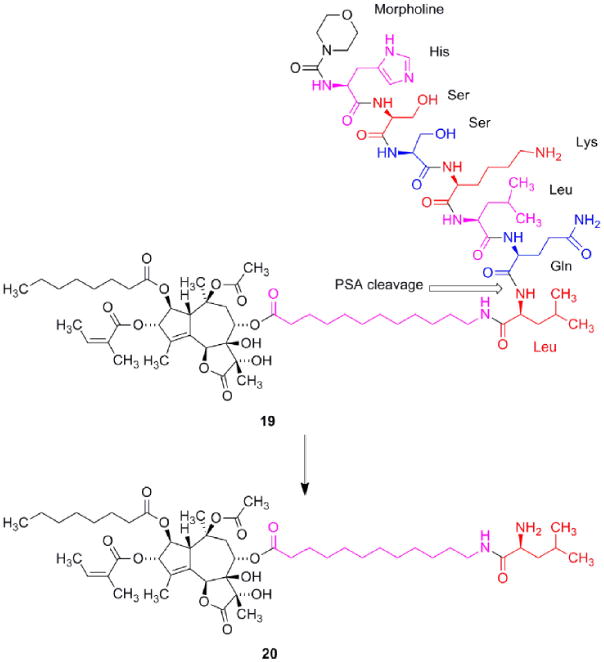
Structure of G115 (19) and the PSA cleavage product 20. In spite of the ionic structure of 20 this molecule is able to penetrate the cell membrane and induce apoptosis. The linker is marked with magenta.
The proteolytic enzyme prostate specific membrane antigen (PSMA) is not specific for prostate as the name indicates, but is also found in neovascular tissue of a broad number of tumors including hepatocellular-, bladder-, renal-, cell-, breast- and in glioblastoma multiforme cancer [33]. The enzyme is a glutamate carboxypeptidase indicating that the enzyme preferentially cleaves poly-γ-glut-amyl peptides [34]. Advantage was taken of this substrate specificity to design G202 (21, Fig. 7). A further advantage of the tetra- γ -glutamyl peptide is the water solubility, which facilitates administration of the prodrug. Inspection of the conformation of PSMA reveals the presence of an approximately 20 Å long funnel leading to the active site explaining the need for a linker between the guaianolide moiety and the peptide [34].
Fig. 7.
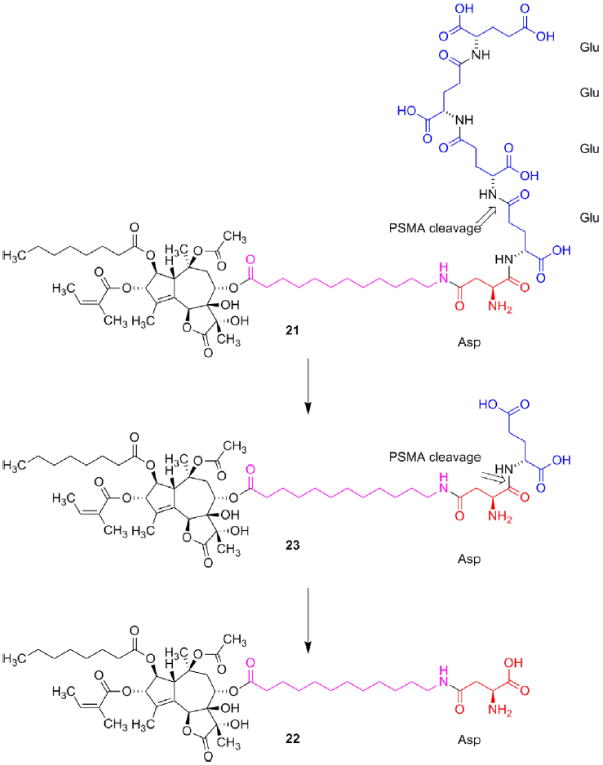
Structure of G202 (21) and the major PMSA cleavage product 22. In spite of the zwitterionic structure of 22 this molecule is able to penetrate the cell membrane and induce apoptosis. The linker is marked with magenta.
While writing this manuscript we were informed that the USAN council has named G202 Mipsagargin.
8. Preclinical development of G202
8.1. In vitro studies
Incubation of purified SERCA with the cleavage products of G202 (22) or 1 revealed that the two compounds were almost equi-potent as inhibitors of the enzyme. Compound 23 an intermediate during the cleavage of G202 was found to be only half as potent. Incubation of prostate cancer cell cultures (a strain producing PSMA (LNCaP) and a strain not producing PSMA (TSU) revealed that the IC50 value of 22 was approximately twice the value of 1 meaning that the zwitterionic structure of the aspartyl group only to a limited extend prevents the compound from penetrating the cell membrane. In contrast, however, 23 was approximately 250 time less potent than 1 towards TSU cells probably because the hydrophilic dipeptide affords a poor ability to diffuse through the cell membrane. This was confirmed by incubating PSMA producing cells (LNCaP) with 23. In this case 23 only was ten times less potent. A possible explanation is cleavage of the dipeptide moiety affording 22. The poorer potency might be explained by a slow cleavage of the dipeptide [33]. A recent study, however, has found 22 to be less potent [30] towards LNCaP cells. In the present approach, however, the cancer cells are not the target for the drug. The mechanism of action relies on a cleavage of the prodrug in the neovascular tissue of the tumor releasing the active therapeutic, which destroys the vessels and thereby deprives the tumor from nutrients (Fig. 8).
Fig. 8.
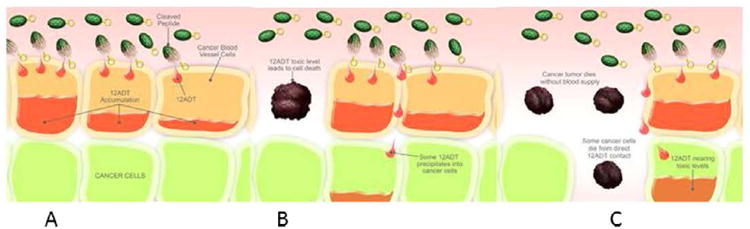
Cartoon displaying the mechanism of action of the prodrug (depicted as a grenade). When the prodrug (the grenade) appears in the neovascular tissue the peptide is cleaved (the pin is removed) (A), the active drug diffuses into the cell (B), and kills the cell (C).
The rate of hydrolysis of G202 was determined by treatment with PSMA. The three terminal glutamic acid residues were found to be cleaved very fast to give 23, whereas the last residue was found to be hydrolyzed somewhat slower [33]. Very satisfactory G202 was found to be inert towards a number of proteases found in the human body.
8.2. In vivo studies
Since injection of 56 mg/kg/day in three consecutive days only produced less than 10% lethality, this dose was chosen for observing the effect on prostate cancer xenografts growing on mice. The treatment afforded approximately 50% regression of the tumors whereas the size of the tumors in the control group expanded more than four times. Treatment with docetaxel only stabilized the size of the tumors [33]. Pharmacokinetic studies showed that only vanishing amounts of 22 and 23 were found in the normal tissue whereas significant accumulation was found in PSMA forming tumors [33]. Toxicological studies in monkeys enabled identification of a starting dose of 1.5 mg/m2 in clinical trial I studies [33].
9. Clinical trials
A human phase I clinical study has demonstrated the safety and tolerability of G202 in advanced cancer patients [35]. In addition the study revealed that two patients suffering from hepatocellular cancer (HCC) experienced a prolonged period of stable disease. This result has encouraged to a phase II trial in HCC patients, which is running at the present.
It might appear curious that a compound suspected to have tumor promoting properties was selected for clinical trials. Only one report, however, mentions this effect of 1 [16], and no preclinical investigations confirmed the risk. Consequently, FDA allowed clinical tests to be performed.
10. Conclusion
Forty years of work with an active principle from a Mediterranean plant has led to a drug candidate. Originally the compound was isolated to satisfy academic curiosity of knowing the skin irritating principle in T. garganica. The potent properties of the compound, however, inspired to intensive studies to understand the chemistry and the pharmacology of this natural product. As a consequence an international network consisting of highly qualified experts within different fields in as well the academic society as in the field of private enterprise developed. This collaboration eventually led to development of the intellectual and financial platform needed for bringing a compound into clinical trial II (G202). In addition the project has led to development of a new and heavily used biological tool (thapsigargin) for exploring the Ca2+ homeostasis [36] and for understanding the SERCA pump.
Acknowledgments
This work has been supported by The Danish Cancer Society, The Danish Research Council for Strategic Research, Carlsbergfondet.
References
- 1.Perrot E. Matière Première usuelle du Règne Vègètale. Paris: Masson et cie; 1943. pp. 1630–2. [Google Scholar]
- 2.Rasmussen U, Christensen SB, Sandberg F. Thapsigargin and Thapsigargicin, two new histamine liberators from Thapsia garganica L. Acta Pharm Suec. 1978;15:133–40. [PubMed] [Google Scholar]
- 3.Christensen SB, Rasmussen U, Christophersen C. Thapsigargin, constitution of a sesquiterpene lactone histamine liberator from Thapsia garganica. Tetrahedron Lett. 1980;21:3829–30. [Google Scholar]
- 4.Christensen SB, Larsen IK, Rasmussen U, Christophersen C. Thapsigargin and thapsigargicin, two histamine liberating sesquiterpene lactones from Thapsia garganica. X-ray analysis of the 7,11-epoxide of thapsigargin. J Org Chem. 1982;47:649–52. [Google Scholar]
- 5.Holub M, Samek Z, deGroote R, Herout V, Sorm F. On terpenes. CCXXVII. The structure of the sesquiterpene triester lactone trilobolide. Collect Czech Chem Commun. 1973;38:1551–62. [Google Scholar]
- 6.Christensen SB, Norup E. Absolute configuration of the histamine liberating sesquiterpene lactones thapsigargin and trilobolide. Tetrahedron Lett. 1985;26:107–11. [Google Scholar]
- 7.Christensen SB, Andersen A, Smitt UW. Sesquiterpenoids from thapsia species and medicinal chemistry of the thapsigargins. Program Chem Natl Prod. 1997;71:131–67. doi: 10.1007/978-3-7091-6529-4_2. [DOI] [PubMed] [Google Scholar]
- 8.Harmatha J, Budesinsky M, Vokac K, Kostecka P, Kmonickova E, Zidek Z. Trilobolide and related sesquiterpene lactones from Laser trilobum possessing immunobiological properties. Fitoterapia. 2013;89C:157–66. doi: 10.1016/j.fitote.2013.05.025. [DOI] [PubMed] [Google Scholar]
- 9.Liu H, Olsen CE, Christensen SB. Tethered lipids from Thapsia garganica. J Nat Prod. 2004;67:1339–40. doi: 10.1021/np049781y. [DOI] [PubMed] [Google Scholar]
- 10.Appendino G, Prosperini S, Valdivia C, Ballero M, Colombano G, Billington RA, Genazzani AA, Sterner O. SERCA-inhibiting activity of C-19 terpenolides from Thapsia garganica and their possible biogenesis. J Nat Prod. 2005;68:1213–7. doi: 10.1021/np050115m. [DOI] [PubMed] [Google Scholar]
- 11.Weitzel C, Rønsted N, Spalik K, Simonsen HT. Resurrecting deadly carrots: towards a revision of Thapsia (Apiaceae) based on phylogenic analysis of nrITS sequences and chemical profiles. Bot J Linn Soc. 2014;174:620–36. [Google Scholar]
- 12.Patkar SA, Rasmussen U, Diamant B. On the mechanism of histamine release induced by thapsigargin from Thapsia garganica L. Agents Actions. 1979;9:53–7. doi: 10.1007/BF02024109. [DOI] [PubMed] [Google Scholar]
- 13.Ali H, Christensen SB, Foreman JC, Pearce FL, Piotrowski W, Thastrup O. The ability of thapsigargin and thapsigargicin to activate cells involved in the inflammatory response. Br J Pharmacol. 1985;85:705–12. doi: 10.1111/j.1476-5381.1985.tb10567.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kmonickova E, Melkusova P, Harmatha J, Vokac K, Farghali H, Zidek Z. Inhibitor of sarco-endoplasmic reticulum Ca2+-ATPase thapsigargin stimulates production of nitric oxide and secretion of interferon-gamma. Eur J Pharmacol. 2008;588:85–92. doi: 10.1016/j.ejphar.2008.03.037. [DOI] [PubMed] [Google Scholar]
- 15.Mikkelsen EO, Thastrup O, Christensen SB. Effects of thapsigargin in isolated rat thoracic aorta. Pharmacol Toxicol. 1988;62:7–11. doi: 10.1111/j.1600-0773.1988.tb01835.x. [DOI] [PubMed] [Google Scholar]
- 16.Hakii H, Fujiki H, Suganuma M, Nakayasu M, Tahira T, Sugimura T, Scheuer PJ, Christensen SB. Thapsigargin, a histamine secretagogue, is a non-12-O-tetradecanoylphorbol-13-acetate (TPA) type tumor promoter in two-stage mouse skin carcinogenesis. J Cancer Res Clin Oncol. 1986;111:177–81. doi: 10.1007/BF00389230. [DOI] [PubMed] [Google Scholar]
- 17.Denmeade SR, Jakobsen CM, Janssen S, Khan SR, Garrett ES, Lilja H, Christensen SB, Isaacs JT. Prostate-specific antigen-activated thapsigargin prodrug as targeted therapy for prostate cancer. J Natl Cancer Inst. 2003;95:990–1000. doi: 10.1093/jnci/95.13.990. [DOI] [PubMed] [Google Scholar]
- 18.Norup E, Smitt UW, Christensen SB. The potencies of thapsigargin and analogues as activators of rat peritoneal mast cells. Planta Med. 1986:251–5. doi: 10.1055/s-2007-969144. [DOI] [PubMed] [Google Scholar]
- 19.Thastrup O, Cullen PJ, Drøbak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc Natl Acad Sci USA. 1990;87:2466–70. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davidson GA, Varhol RJ. Kinetics of thapsigargin-Ca2+-ATPase (sarcoplasmic reticulum) interaction reveals a two-step binding mechanism and picomolar inhibition. J Biol Chem. 1995;270:11731–4. doi: 10.1074/jbc.270.20.11731. [DOI] [PubMed] [Google Scholar]
- 21.Møller JV, Olesen C, Winther AML, Nissen P. The sarcoplasmic Ca2+-ATPase: design of a perfect chemiosmotic pump. Q Rev Biophys. 2010;43:501–66. doi: 10.1017/S003358351000017X. [DOI] [PubMed] [Google Scholar]
- 22.Toyoshima C, Nakasako M, Nomura H, Ogawa H. Crystal structure of the calcium pump of the of sarcoplasmic reticulum at 2.6 Å resolution. Nature. 2000;405:647–55. doi: 10.1038/35015017. [DOI] [PubMed] [Google Scholar]
- 23.Sohoel H, Liljefors T, Ley SV, Oliver SF, Antonello A, Smith MD, Olsen CE, Isaacs JT, Christensen SB. Total synthesis of two novel subpicomolar sarco/endoplasmatic reticulum Ca2+-ATPase inhibitors designed by an analysis of the binding site of thapsigargin. J Med Chem. 2005;48:7005–11. doi: 10.1021/jm058036v. [DOI] [PubMed] [Google Scholar]
- 24.Paulsen ES, Villadsen J, Tenori E, Liu H, Bonde DF, Lie MA, Bublitz M, Olesen C, Autzen HE, Dach I, Sehgal R, Nissen P, Møller JV, Schiott B, Christensen SB. Water mediated interactions influence the binding of thapsigargin to sarco/endoplasmic reticulum calcium adenosinetriphosphatase. J Med Chem. 2013;56:3609–19. doi: 10.1021/jm4001083. [DOI] [PubMed] [Google Scholar]
- 25.Christensen SB, Andersen A, Poulsen JC, Treiman M. Derivatives of thapsigargin as probes of its binding site on endoplasmic reticulum Ca2+ ATPase. Stereoselectivity and important functional groups. FEBS Lett. 1993;335:345–8. doi: 10.1016/0014-5793(93)80416-r. [DOI] [PubMed] [Google Scholar]
- 26.Skytte DM, Moller JV, Liu HZ, Nielsen HO, Svenningsen LE, Jensen CM, Olsen CE, Christensen SB. Elucidation of the topography of the thapsigargin binding site in the sarco-endoplasmic calcium ATPase. Bioorg Med Chem. 2010;18:5634–46. doi: 10.1016/j.bmc.2010.06.032. [DOI] [PubMed] [Google Scholar]
- 27.Winther AML, Liu HZ, Sonntag Y, Olesen C, le Maire M, Soehoel H, Olsen CE, Christensen SB, Nissen P, Møller JV. Critical roles of hydrophobicity and orientation of side chains for inactivation of sarcoplasmic reticulum Ca2+-ATPase with thapsigargin and thapsigargin analogs. J Biol Chem. 2010;285:28883–92. doi: 10.1074/jbc.M110.136242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jakobsen CM, Denmeade SR, Isaacs JT, Gady A, Olsen CE, Christensen SB. Design, synthesis, and pharmacological evaluation of thapsigargin analogues for targeting apoptosis to prostatic cancer cells. J Med Chem. 2001;44:4696–703. doi: 10.1021/jm010985a. [DOI] [PubMed] [Google Scholar]
- 29.Christensen SB, Skytte DM, Denmeade SR, Dionne C, Møller JV, Nissen P, Isaacs JT. A trojan horse in drug development: targeting of thapsigargins towards prostate cancer cells. Anti-Cancer Agents Med Chem. 2009;9:276–94. doi: 10.2174/1871520610909030276. [DOI] [PubMed] [Google Scholar]
- 30.Dubois C, Abeele FV, Sehgal P, Olesen C, Junker S, Christensen SB, Prevarskaya N, Møller JV. Differential effects of thapsigargin analogues on apoptosis of prostate cancer cells. Febs J. 2013;280:5430–40. doi: 10.1111/febs.12475. [DOI] [PubMed] [Google Scholar]
- 31.Weidle UH, Tiefenthaler G, Georges G. Proteases as activators for cytotoxic prodrugs in antitumor therapy. Cancer Genomics Proteomics. 2014;11:67–79. [PubMed] [Google Scholar]
- 32.Denmeade SR, Isaacs JT. The SERCA pump as a therapeutic target: Making a “Smart bomb” for prostate cancer. Cancer Biol Ther. 2005;4:14–22. doi: 10.4161/cbt.4.1.1505. [DOI] [PubMed] [Google Scholar]
- 33.Denmeade SR, Mhaka AM, Rosen DM, Brennen WN, Dalrymple S, Dach I, Olesen C, Gurel B, Demarzo AM, Wilding G, Carducci MA, Dionne CA, Møller JV, Nissen P, Christensen SB, Isaacs JT. Engineering a prostate-specific membrane antigen-activated tumor endothelial cell prodrug for cancer therapy. Sci Transl Med. 2012;4:140ra86. doi: 10.1126/scitranslmed.3003886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Davis MI, Bennett MJ, Thomas LM, Bjorkman PJ. Crystal structure of prostate-specific membrane antigen, a tumor marker and peptidase. Proc Natl Acad Sci USA. 2005;102:5981–6. doi: 10.1073/pnas.0502101102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mahalingam D, Dionne C. Final phase I data on GenSpera's G202 presented at symposium on molecular targets and cancer therapeutics. 2013 [Google Scholar]
- 36.Treiman M, Caspersen C, Christensen SB. A tool coming of age – Thapsigargin as an inhibitor of sarco-endoplasmic reticulum Ca2+-ATPases. Trends Pharmacol Sci. 1998;19:131–5. doi: 10.1016/s0165-6147(98)01184-5. [DOI] [PubMed] [Google Scholar]