

Abstract
Papillary tumor of the pineal region (PTPR) is a recently recognized entity. We present the pathologic findings of two cases of PTPR as examples, and discuss the presence of cellular pleomorphism in these tumors. Patient 1 is a 48-year-old man with a pineal region mass. The tumor had unique biphasic patterns, papillary/pseudopapillary areas, and increased mitotic activity. Juxtaposed areas had marked pleomorphism, including nuclear enlargement, smudgy chromatin, nuclear pseudoinclusions, and cytoplasmic vacuolation. Mitoses were absent in these areas. Immunohistochemical staining revealed strong S100 expression. CAM 5.2 and CK18 were strongly positive in a patchy fashion. MIB1 labeling indices were high in classic PTPR regions but very low in pleomorphic areas. Patient 2 was a 35-year-old male with a pineal region tumor characterized by papillary architecture and overall cellular monotony, rare mitoses, and pleomorphism as a more isolated finding, with associated nuclear enlargement and crowding. S100 and CAM 5.2 labeling were present, and MIB1 labeling index was very low throughout the tumor. We discuss the pathologic and phenotypic features of PTPR. Variable pleomorphism may be present, reflected in size variation and nuclear hyperchromasia, but was not accompanied by increased proliferative activity in these cases, suggesting a degenerative phenomenon.
Keywords: Papillary tumor, Pineal, Pleomorphism, Brain, Immunohistochemistry
Introduction
Papillary tumor of the pineal region (PTPR) is a rare tumor described in 2003 by Jouvet et al. and a recently recognized entity in the 2007 World Health Organization classification [1]. Since its first description, approximately 60 cases have been reported [2]. PTPR are rare but usually have a distinct morphology, characteristically epithelioid cells, often with sheets of large cells arranged in papillary or pseudopapillary structures in which vessels are covered by layers of columnar to cuboidal cells [3, 4]. These cells frequently have a well defined cytoplasmic membrane, and occasionally the cytoplasm may be vacuolated and resemble signet ring cells [3]. Necrosis is a frequent finding [3]. There are currently no identifiable histologic features that predict prognosis in PTPR. Clinically, these tumors are characterized by frequent local recurrence, but craniospinal dissemination seems rare [5, 6].
We report unusual morphologic variations in two PTPR cases, including one presenting with a biphasic pattern including areas with prominent pleomorphism with a paradoxical decrease in proliferative indices. Implications for differential diagnosis in surgical neuropathology are discussed.
Materials
Tumor sections were evaluated using standard methods. All hematoxylin and eosin slides were reviewed. Immunohistochemical staining were performed using antibodies directed against GFAP (rabbit monoclonal, Ventana, prediluted), S100 (polyclonal, Venatana, prediluted), synaptophysin (clone 27G12, Novacastra, 1:400), chromogranin (clone LK2H10, Ventana, prediluted), cytokeratin 18 (clone DC 10, Dako, 1:100), cytokeratin CAM 5.2 (clone B22.1&B23, Cellmarque, prediluted), epithelial membrane antigen (EMA) (clone E29, Ventana, 1:1000), neuron-specific enolase (NSE) (BBS/NS/VIH14, Ventana, prediluted), NeuN (clone A60, Millipore, 1:250), vimentin (clone V9, Ventana, prediluted), and Ki67 (MIB1, Dako, 1:100) (Case 1), and cytokeratin AE1/3 (cocktail of monoclonal mouse antibodies AE1 and AE3, Millipore, 1:2000), cytokeratin CAM 5.2 (clone CAM 5.2, Becton– Dickinson, 1:5000), EMA (clone E29, Dako, 1:400), GFAP (polyclonal rabbit antibody, Dako, 1:800), S100 (rabbit polyclonal, Dako, 1:6000), and Ki67 (MIB1, Dako, 1:100) (case 2). Studies were approved by the Institutional Review Board at Johns Hopkins.
Case histories and pathology
Case 1
A 48 year-old-man presented with a history of mechanical falls. He was admitted and underwent a CT scan, followed by MRI. The MRI showed a non-calcified heterogeneously enhancing cystic and solid lesion within the dorsal aspect of the third ventricle, with growth on subsequent imaging studies (3.4 × 2.8 × 3.0 cm) (Fig. 1a). A craniotomy was performed. Microscopic analysis showed a cohesive neoplasm composed of areas with epithelial appearance, a largely sheet-like pattern, with focal pseudopapillary architecture, and cellular dehiscence (Fig. 1b). In those areas, the cells had well defined cytoplasmic membranes, with round to oval nuclei, sometimes indented, and stippled chromatin. Mitotic activity was particularly marked in sheet-like epithelial areas (approximately 6 per 10 HPF) (Fig. 1c). These areas, typical of PTPR, were sharply juxtaposed to areas of cytomegaly creating a biphasic appearance (Fig. 1d). Cells in this second area contained glassy and poorly defined cytoplasm, some with large vacuoles (Fig. 1e), and lacked pseudopapillae formation. Cytologic pleomorphism was pronounced, with hyperchromasia and smudged chromatin (Fig. 1f). Mitoses were not present. Immunohistochemical staining revealed focal strong CAM 5.2, specifically in pseudopapillae (Fig. 2a), with positivity in a few neoplastic cells in pleomorphic areas (Fig. 2b). Cytokeratin 18 was also positive in a patchy fashion. Synaptophysin was weakly expressed in papillary and pleomorphic areas, but preserved in adjacent pineal gland parenchyma (Fig. 2c). S100 was strongly expressed in all tumor areas (Fig. 2d), as also were NSE and vimentin. Neu-N, EMA, chromogranin, and GFAP immunostaining was uniformly negative. Ki-67 labeling index was moderately increased in epithelial areas (Fig. 2e), but very low in areas of cellular pleomorphism (Fig. 2f).
Fig. 1.
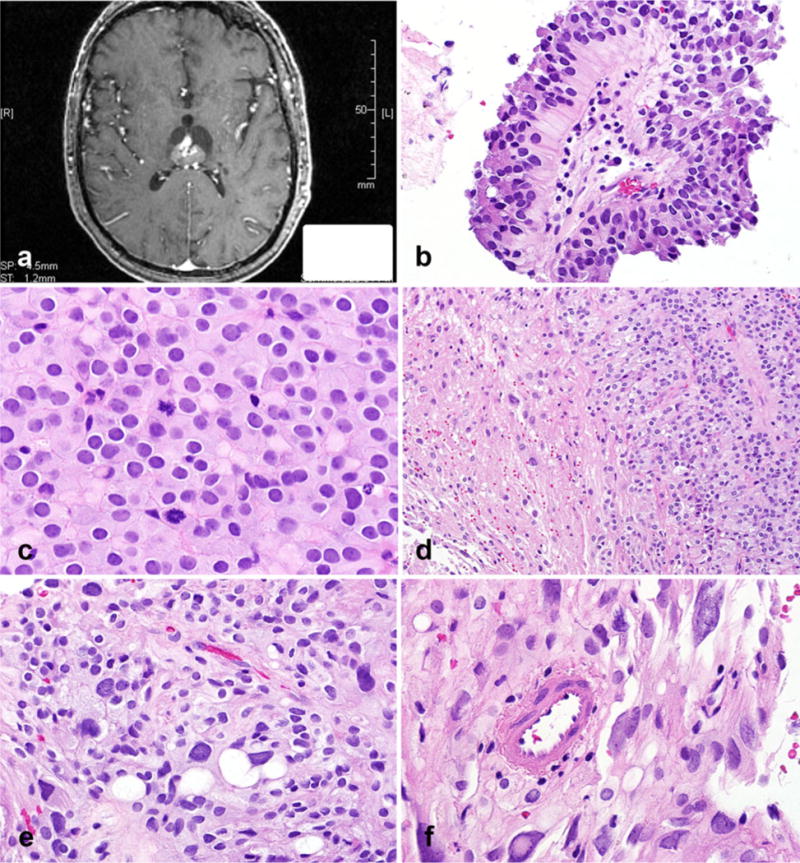
Papillary tumor of the pineal region with biphasic areas. Heterogeneously enhancing, partially cystic lesion within the dorsal aspect of the third ventricle (axial T1-weighted image, post-contrast) (a). Focal areas composed of pseudopapillae were a feature (b) as also were sheets of tumor cells with a vague perivascular arrangement, cellular monotony, and increased mitotic activity (c). Sharp interface between classic papillary areas and distinct pleomorphic cells (d). Vacuoles of different sizes in pleomorphic areas (e). Pleomorphic areas with cells containing glassy, poorly defined cytoplasm, nuclear pleomorphism, intranuclear inclusions with hyperchromasia, and smudged chromatin (f)
Fig. 2.
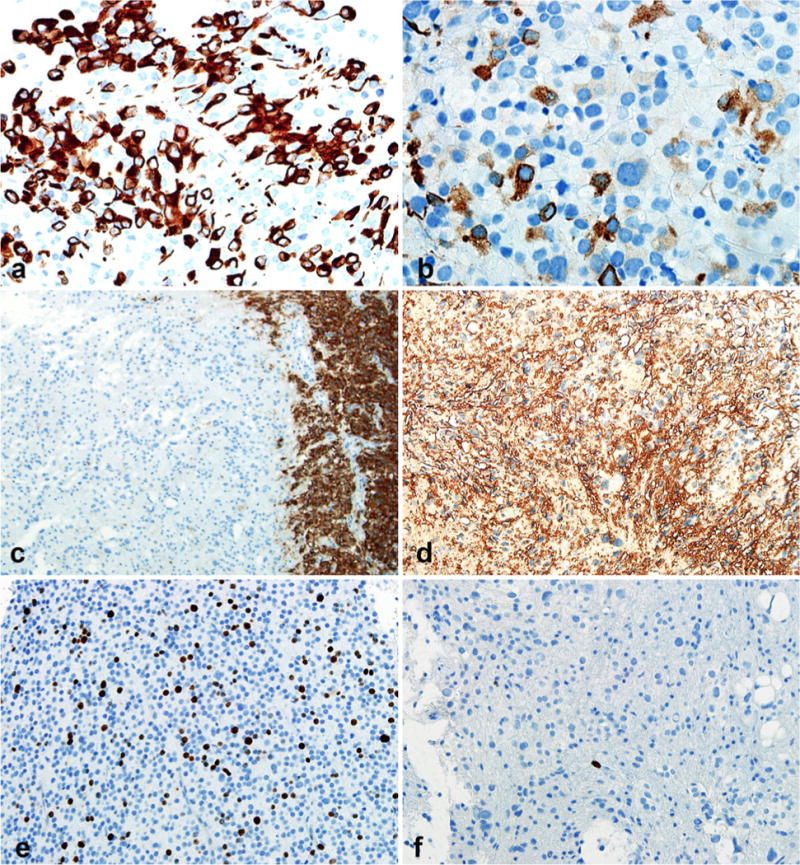
Contrasting immunohistochemical features in biphasic areas. Papillary areas with strong cytokeratin (CAM 5.2) expression (a). The strength of CAM 5.2 labeling was weaker in pleomorphic areas (b). Synaptophysin was not expressed in pleomorphic areas (left) in contrast with adjacent pineal gland (right) (c), whereas strong S-100 expression was present in all areas (d). Paradoxical MIB1 labeling indices were present, being elevated in cellular monotonous areas (e) and extremely low in pleomorphic areas (f)
Case 2
A 35-year-old male presented with worsening blurry vision. An MRI revealed a solid and cystic mass in the posterior third ventricle/pineal region (Fig. 3a). The patient had an endoscopic biopsy of the mass at the time of ventriculostomy performed to treat hydrocephalus. The tumor had a papillary architecture and bland cytologic features in most regions (Fig. 3b). The papillary architecture was more conspicuous and well developed than for case 1. Well formed flat epithelial surfaces were evident. A rare mitosis was present. Focally, in the center of the biopsy an increase in cell crowding, nuclear hyperchromasia, and variation were present (Fig. 3c). These findings differed from those in case 1 by merging with the more conventional papillary areas rather than making a sharply circumscribed region within the tumor. The chromatin quality in this area was finer than for case 1, and nuclear inclusions were not present. Cytoplasmic vacuolation was also absent. S100 and cytokeratin (CAM 5.2) were uniformly expressed throughout the tumor including cells lining the papillae and the focally pleomorphic area (Fig. 3d), whereas cytokeratin AE1/AE3 was patchy, and synaptophysin very weak. GFAP and EMA were negative. MIB1 labeling index was very low, and similar in papillary and pleomorphic areas (<1 %).
Fig. 3.
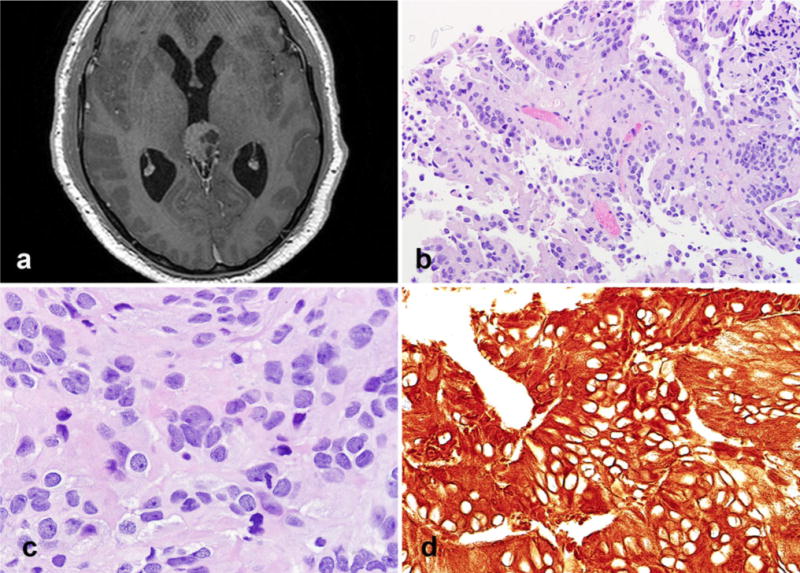
Radiologic and pathologic features of case 2. Axial MR image reveals a well circumscribed solid and cystic mass with its epicenter in the pineal gland (a). The tumor had more overt epithelial features, including well delimited surfaces (b). Cellular pleomorphism was limited to small isolated tumor cell clusters (c). Strong cytokeratin expression (CAM 5.2) was a feature (d)
Discussion
The pineal region is known for the diversity of tumors that may occur on this unique anatomic site. Neoplasms centered in this region include pineal parenchymal tumors, meningiomas, ependymomas, gliomas, germ cell tumors, non-neoplastic cysts, and metastases. Differential diagnosis is generally difficult, given the scant material obtained during pineal biopsies, and requires several immunohistochemical markers. Biopsy specimens from pineal region tumors might not provide any useful material for accurate diagnosis, because it is a region of difficult access [7].
The most unique tumors of the pineal gland are the pineal parenchymal tumors and PTPR. The current 2007 WHO classification of pineal parenchymal tumors divides them largely on the basis of features seen in routine hematoxylin–eosin-stained sections. A range of biological aggressiveness is observed for these tumors [1]. PTPR is a recently described entity thought to originate from the specialized cells of the subcommissural organ. On the basis of a series of six tumors with identical histological features, PTPR was first described as a distinct entity in 2003 [3]. A precise grade has not been assigned but may correspond to WHO grade II or III. It typically afflicts adults, but may also occur in children [7, 8].
The histologic features of most PTPR are typical, but as the cases we present illustrate well, they may be variable in individual cases. Case 1 had a curious biphasic pattern that required detailed histologic evaluation and immunophenotyping. Classic epithelial areas with pseudopapillae were present, but alternated with areas of pronounced nuclear variation, hyperchromasia, pseudoinclusions, and cytoplasmic vacuolation. Given the low proliferative indices, these findings were interpreted as degenerative nuclear atypia. The second case had more monotonous cytologic features, with pleomorphism as a focal finding. Cellular pleomorphism has been recognized as occurring in PTPR [8–12], although this usually takes the form of scattered enlarged cells with hyperchromatic nuclei. The biphasic pattern with a solid area containing degenerative atypia present in case 1 is distinctly unusual in our experience.
Cellular pleomorphism is often encountered in pineal parenchymal tumors [13, 14], and was the main differential diagnosis in case 1, specifically a “collision tumor” between a PTPR and a pineal parenchymal tumor. However, the almost complete absence of synaptophysin staining, in particular in the pleomorphic areas, which contrasted with adjacent non-neoplastic pineal gland, argues against this possibility. An unusual glial neoplasm was another possibility but GFAP staining was negative whereas cytokeratin CAM 5.2 was positive. Other diagnostic considerations included ependymoma and choroid plexus tumor, for both of which cellular pleomorphism may be observed [15]. Cytokeratin expression and lack of EMA and GFAP immunoreactivity argue against ependymoma, while the anatomic location (i.e. pineal gland proper rather than intraventricular) and less well developed papillae makes a choroid plexus tumor unlikely. The observed immunophenotype is typical for PTPR, including cytokeratin expression, and lack of GFAP and EMA. GFAP and synaptophysin immunoreactivity in PTPR, in particular, is usually focal or absent [5, 16, 17]. It must be noted that choroid plexus tumors frequently stain for membranous Kir7.1 or cytoplasm stanniocalcin-1, which are absent from most PTPR [17]. Finally, papillary meningioma and metastatic carcinoma are two additional entities that enter the differential diagnosis. The frequent lack of EMA expression argues against meningioma, and co-expression of S100 and cytokeratin, and the earlier age of onset, suggests PTPR rather than metastatic carcinoma. However, we cannot emphasize enough the importance of integrating histologic, radiologic, and immunophenotypic findings in evaluation of these tumors.
In conclusion, we have discussed the presence of cellular pleomorphism in PTPR, including the presentation of a unique case with a biphasic morphologic pattern with an area of pronounced degenerative atypia. It is unclear what the prognostic relevance of this finding is, but given the reduced proliferation present in these areas, a negative effect on clinical outcome is unlikely. The histologic features presented emphasize the histologic variation observed in PTPR from case to case, and the importance of an integrative diagnostic approach when encountering these tumors in surgical neuropathology.
Contributor Information
Juliana Magalhães, Division of Neuropathology, Department of Pathology, Johns Hopkins University, Sheikh Zayed Tower, Room M2101, 1800 Orleans Street, Baltimore, MD 21231, USA.
Steven Rostad, Department of Pathology, Swedish Medical Center, Seattle, WA, USA.
Greg Foltz, Department of Neurosurgery, Swedish Medical Center, Seattle, WA, USA.
Peter Pytel, Department of Pathology, University of Chicago, Chicago, IL, USA.
Fausto J. Rodriguez, Email: frodrig4@jhmi.edu, Division of Neuropathology, Department of Pathology, Johns Hopkins University, Sheikh Zayed Tower, Room M2101, 1800 Orleans Street, Baltimore, MD 21231, USA.
References
- 1.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, editors. WHO classification of tumours of the central nervous system. International Agency for Research on Cancer; Lyon: 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rickard KA, Parker JR, Vitaz TW, Plaga AR, Wagner S, Parker JC., Jr Papillary tumor of the pineal region: two case studies and a review of the literature. Ann Clin Lab Sci. 2011;41:174–181. pii: 41/2/174. [PubMed] [Google Scholar]
- 3.Jouvet A, Fauchon F, Liberski P, Saint-Pierre G, Didier-Bazes M, Heitzmann A, Delisle MB, Biassette HA, Vincent S, Mikol J, Streichenberger N, Ahboucha S, Brisson C, Belin MF, Fevre-Montange M. Papillary tumor of the pineal region. Am J Surg Pathol. 2003;27:505–512. doi: 10.1097/00000478-200304000-00011. [DOI] [PubMed] [Google Scholar]
- 4.Epari S, Bashyal R, Malick S, Gupta T, Moyadi A, Kane SV, Bal M, Jalali R. Papillary tumor of pineal region: report of three cases and review of literature. Neurol India. 2011;59:455–460. doi: 10.4103/0028-3886.82773. [DOI] [PubMed] [Google Scholar]
- 5.Fevre-Montange M, Hasselblatt M, Figarella-Branger D, Chauveinc L, Champier J, Saint-Pierre G, Taillandier L, Coulon A, Paulus W, Fauchon F, Jouvet A. Prognosis and histopathologic features in papillary tumors of the pineal region: a retrospective multicenter study of 31 cases. J Neuropathol Exp Neurol. 2006;65:1004–1011. doi: 10.1097/01.jnen.0000240462.80263.13. [DOI] [PubMed] [Google Scholar]
- 6.Yano H, Ohe N, Nakayama N, Shinoda J, Iwama T. Clinicopathological features from long-term observation of a papillary tumor of the pineal region (PTPR): a case report. Brain Tumor Pathol. 2009;26:83–88. doi: 10.1007/s10014-009-0246-z. [DOI] [PubMed] [Google Scholar]
- 7.Buffenoir K, Rigoard P, Wager M, Ferrand S, Coulon A, Blanc JL, Bataille B, Listrat A. Papillary tumor of the pineal region in a child: case report and review of the literature. Childs Nerv Syst. 2008;24:379–384. doi: 10.1007/s00381-007-0500-9. [DOI] [PubMed] [Google Scholar]
- 8.Li J, Recinos PF, Orr BA, Burger PC, Jallo GI, Recinos VR. Papillary tumor of the pineal region in a 15-month-old boy. J Neurosurg Pediatr. 2011;7:534–538. doi: 10.3171/2011.2.PEDS10434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fevre-Montange M, Vasiljevic A, Champier J, Jouvet A. Histopathology of tumors of the pineal region. Future Oncol. 2010;6:791–809. doi: 10.2217/fon.10.28. [DOI] [PubMed] [Google Scholar]
- 10.Matyja E, Grajkowska W, Nauman P, Bonicki W. Histopathological patterns of papillary tumour of the pineal region. Folia Neuropathol. 2011;49:181–190. pii: 17401. [PubMed] [Google Scholar]
- 11.Dhillon J, Krishnamurthy S, Fuller GN. Papillary tumour of the pineal region: diagnosis by cytology smear. Cytopathology. 2010;21:267–269. doi: 10.1111/j.1365-2303.2009.00696.x. [DOI] [PubMed] [Google Scholar]
- 12.Kuchelmeister K, Hugens-Penzel M, Jodicke A, Schachenmayr W. Papillary tumour of the pineal region: histodiagnostic considerations. Neuropathol Appl Neurobiol. 2006;32:203–208. doi: 10.1111/j.1365-2990.2006.00741.x. [DOI] [PubMed] [Google Scholar]
- 13.Sasaki A, Horiguchi K, Nakazato Y. Pineal parenchymal tumor of intermediate differentiation with cytologic pleomorphism. Neuropathology. 2006;26:212–217. doi: 10.1111/j.1440-1789.2006.00676.x. [DOI] [PubMed] [Google Scholar]
- 14.Fevre-Montange M, Szathmari A, Champier J, Mokhtari K, Chretien F, Coulon A, Figarella-Branger D, Polivka M, Varlet P, Uro-Coste E, Fauchon F, Jouvet A. Pineocytoma and pineal parenchymal tumors of intermediate differentiation presenting cytologic pleomorphism: a multicenter study. Brain Pathol. 2008;18:354–359. doi: 10.1111/j.1750-3639.2008.00128.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zec N, De Girolami U, Schofield DE, Scott RM, Anthony DC. Giant cell ependymoma of the filum terminale. A report of two cases. Am J Surg Pathol. 1996;20:1091–1101. doi: 10.1097/00000478-199609000-00007. [DOI] [PubMed] [Google Scholar]
- 16.Kawahara I, Tokunaga Y, Yagi N, Iseki M, Abe K, Hayashi T. Papillary tumor of the pineal region. Neurol Med Chir (Tokyo) 2007;47:568–571. doi: 10.2176/nmc.47.568. [DOI] [PubMed] [Google Scholar]
- 17.Hasselblatt M, Blumcke I, Jeibmann A, Rickert CH, Jouvet A, van de Nes JA, Kuchelmeister K, Brunn A, Fevre-Montange M, Paulus W. Immunohistochemical profile and chromosomal imbalances in papillary tumours of the pineal region. Neuropathol Appl Neurobiol. 2006;32:278–283. doi: 10.1111/j.1365-2990.2006.00723.x. [DOI] [PubMed] [Google Scholar]