

Abstract
Background
We have previously shown that palmitoylation is essential for NRAS leukemogenesis, suggesting that targeting RAS palmitoylation may be an effective therapy for NRAS-related cancers. For KRAS-driven cancer, although much research has been focused on the KRAS4B splice variant, which does not undergo palmitoylation, KRAS4A has recently been shown to play an essential role in the development of carcinogen-induced lung cancer in mice and to be widely expressed in human cancers. However, the role of palmitoylation in KRAS4A tumorigenesis is not clear.
Methods
The expression of KRAS4A in KRAS-mutated leukemia cell lines and acute myeloid leukemia (AML) cells were checked using western blotting and reverse transcriptions-quantitative polymerase chain reaction (RT-qPCR) analysis, respectively. The leukemogenic potentials of oncogenic KRAS4A and its palmitoylation-defective mutants were examined by a mouse bone marrow transduction and transplantation model and the in vitro transformation assays. The activation of the RAS downstream signaling pathways and the membrane localizations of the KRAS4A and its mutants were analyzed via western blot analysis and confocal microscopy, respectively.
Results
We show here that KRAS4A is expressed in human leukemia cell lines and in AML cells harboring KRAS mutations and that mutation at the palmitoylation site of oncogenic KRAS4A significantly abrogates its leukemogenic potential. However, unlike NRAS, palmitoylation-defective KRAS4A still induces leukemia in mice, albeit with a much longer latency. Using NRAS/KRAS4A chimeric constructs, we found that the KIKK motif of KRAS4A contributes to the transforming activity of KRAS4A. Mutations at both palmitoylation site and the KIKK motif abolish the ability of oncogenic KRAS4A to induce leukemia in mice.
Conclusions
Our studies suggest that therapies targeting RAS palmitoylation may also be effective in treating KRAS4A associated malignancies and that interfering the KIKK membrane-targeting motif would enhance the therapeutic effectiveness.
Keywords: RAS, Leukemogenesis, Drug target, Plasma membrane translocation, Signal transduction
Background
RAS small GTPases act as molecular binary switches in signal transduction regulating cell proliferation, survival, and differentiation [1]. When bound with GTP, RAS proteins can mediate diverse cellular processes by engaging many effector pathways like RAF-MEK-ERK and PI3K-AKT [2, 3]. Mammalian RAS family includes three RAS genes, which encode four highly homologous proteins: HRAS, NRAS, KRAS4A, and KRAS4B. The latter two are alternative splicing isoforms differing only at the carboxyl terminus. These isoforms possess over 90 % identity in the first 166 amino acid residues (G domain, including switch loops and the binding surfaces for downstream effectors) and are mainly diverse in the carboxyl terminal hypervariable region (HVR). Aberrant activation of the RAS signaling pathway is common in cancer, including 20–30 % cancers with RAS mutations [4]. Among RAS genes, KRAS mutations occur most frequently, accounting for 85 % of RAS mutations, followed by NRAS (12 %) [4]. HRAS mutation is relatively rare (3 %) [4].
Despite of intensive research over three decades, cancers harboring RAS mutations remain the most difficult to treat and are refractory to current targeted therapies [5]. Though strategies to target oncogenic RAS proteins are emerging, identification of alternative targets that block RAS signaling is critical to develop therapies for RAS-driven cancer [6]. The biological activities of RAS rely on post-translation modifications (PTMs) that target RAS proteins to cell membranes, particularly the plasma membrane [7]. One potential approach to block the RAS oncogenic signaling is, therefore, to inhibit RAS translocation to the plasma membrane. RAS are synthesized as cytosolic proteins. To translocate to membranes, they need first to be modified by prenylation at the cysteine of the carboxyl terminal CAAX motif by farnesyltransferases (FTase) or geranylgeranyltransferase (GGTase), followed by -AAX proteolysis by RAS converting enzyme (RCE) and methylation of the exposed, farnesylated cysteine residue by isoprenylcysteine carboxyl methyltransferase (Icmt) [8]. CAAX motif is the C-terminal tetrapeptide sequence of RAS proteins (C for cysteine, A for aliphatic amino acid, and X for serine or methionine). Since prenylation of RAS by FTase is the obligate step in RAS PTMs, much emphasis had been placed on developing therapies targeting RAS farnesylation, but successes are modest to date due to a redundancy of the FTase and GGTase [9]. Inhibitors targeting both FTase and GGTase in combination have been proved too toxic to be clinically useful [10, 11].
The prenylation of RAS proteins provides the minimal signal for their membrane association. NRAS, HRAS, and KRAS4A are further palmitoylated by palmitoylacyltransferases (PAT) at the cysteine residue(s) upstream of the CAAX motif [12–14]. On the other hand, KRAS4B, which lacks of cysteine residues at its C terminus to accept palmitoylation modification, traffics directly to the plasma membrane (PM) by associating its positively charged polylysine residues in HVR with the negatively charged component of the inner membrane through electrostatic interaction [15, 16].
We have previously shown that palmitoylation is essential for NRAS leukemogenesis, suggesting that targeting RAS palmitoylation may be an effective therapy for NRAS-related cancers [17]. For cancers with KRAS mutations, much research has been focused on KRAS4B, since KRAS4B transcript was shown to be more abundant [18]. However, since most oncogenic mutations occur in the G domain of RAS, which is identical for KRAS4A and KRAS4B, KRAS4A should be activated in cancers harboring KRAS mutations. Although KRAS4A is dispensable for mouse development [19], accumulating evidences indicate that the altered KRAS4A/4B ratios may correlate with progression of lung and colorectal adenocarcinoma [20, 21] and that KRAS4A plays an important role in lung carcinogenesis [22]. Furthermore, a recent study by improved quantitative RT-PCR revealed that the KRAS4A splice variant is widely expressed in human cancers [23]. Both KRAS isoforms, therefore, should be taken into account in developing effective cancer therapies.
The role of palmitoylation in KRAS4A tumorigenesis in vivo is not known. In this study, we compared the effect of palmitoylation on signaling and leukemogenic potential of oncogenic NRAS and KRAS4A. We found that palmitoylation also plays a critical role in KRAS4A leukemogenesis, but KRAS4A contains an additional membrane association motif that contributes to the oncogenic activity of KRAS4A in vivo.
Results
KRAS4A is expressed in human hematologic malignant cells with KRAS mutations
KRAS mutations have been found in hematologic malignancies [24]. To determine whether KRAS4A is expressed in blood cancer cells harboring KRAS mutations, we checked the KRAS4A protein levels with a KRAS4A specific antibody in acute myeloid leukemia cell lines SHI-1 and NB4, T cell acute lymphoblastic leukemia cell line CCRF-CEM, as well as diffuse large B cell lymphoma cell line Toledo. All of these four cell lines derived from human hematologic malignancies with KRAS mutations. We observed that KRAS4A is expressed at various levels in these cell lines, with a particularly high level in SHI-1 and Toledo cells (Fig. 1a).
Fig. 1.
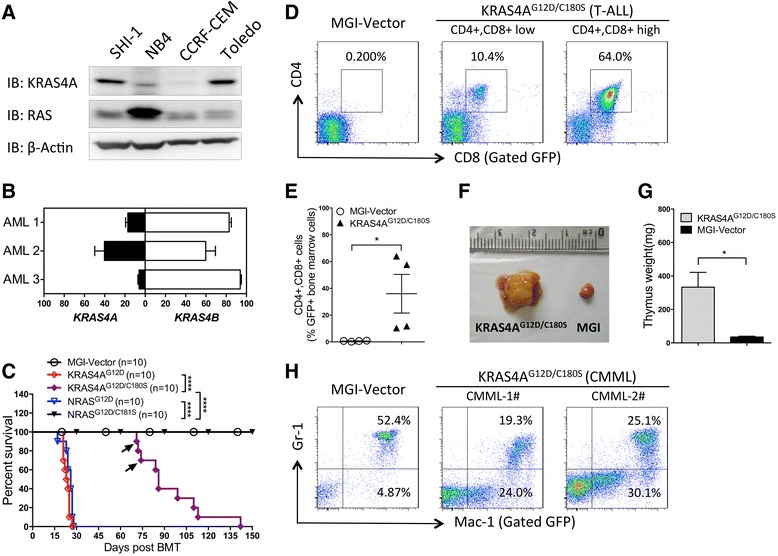
Effects of palmitoylation on leukemogenesis of oncogenic NRAS vs. KRAS4A. a The expression levels of KRAS4A in KRAS-mutated leukemia cell lines indicated were detected by western blot analysis using an anti-KRAS4A specific antibody. Total RAS and actin were detected as controls. b Quantification of the relative abundance of KRAS4A and KRAS4B slice variants in bone marrow cells from AML patients with oncogenic KRAS mutations. The results are presented as means ± SD. c Kaplan-Meier plot of cumulative survival of recipient mice transplanted with bone marrow cells infected by KRAS4AG12D, KRAS4AG12D/C180S, NRASG12D, or NRASG12D/C181S containing retroviruses. The observation period was over 1 year after BMT. The median survival is 23, 26, and 86 days for KRAS4AG12D, NRASG12D, and KRAS4AG12D/C180S, respectively. Disease incidences: KRAS4AG12D (9/10 CMML; 1/10 AML), KRAS4AG12D/C180S (2/10 CMML; 8/10 T-ALL), NRASG12D (5/10 CMML; 5/10 AML). Arrows represent the two moribund KRAS4AG12D/C180S mice diagnosed as CMML-like disease. P values were determined by the log-rank test. ****P < 0.0001; n = 10 in each group. d Representative analyses of leukemic bone marrow cells from the moribund KRAS4AG12D/C180S mice with T-ALL like disease (8 out of 10) by flow cytometry are shown. The MGI vector mice were used as control. e Comparison of CD4+, CD8+ cell percentages in GFP+ BM cells from the moribund KRAS4AG12D/C180S mice with T-ALL like disease (n = 4) and the MGI vector mice (n = 4). Student t test was used for the statistical analysis. *P < 0.05. f Representative thymus from moribund KRAS4AG12D/C180S mice with T-ALL like disease and that from the MGI vector mice. g Comparison of weight of thymus from moribund KRAS4AG12D/C180S mice with T-ALL like disease vs. that from MGI vector mice. The data were presented as mean ± SEM; *P < 0.05; n = 3 in each group. h Representative analyses of leukemic bone marrow cells from moribund KRAS4AG12D/C180S mice with CMML-like disease (2 out of 10). The MGI vector mice were used as control
To confirm our observation in the hematologic malignant cells from patients, we examined the relative abundance of KRAS4A and KRAS4B mRNA in bone marrow samples from patients suffering acute myeloid leukemia (AML) with oncogenic KRAS mutations by reverse transcriptions-quantitative polymerase chain reaction (RT-qPCR) as previously described [23]. We found that KRAS4A accounts for 6.08–40.21 % of total KRAS transcripts in these samples (Fig. 1b). The results demonstrate that KRAS4A is expressed in hematologic malignant cells with KRAS mutations.
Palmitoylation is critical but not essential for the leukemogenesis by oncogenic KRAS4A
We have previously shown that both oncogenic NRAS and KRAS4B can induce myeloid leukemia in a mouse bone marrow transduction/transplantation (BMTT) model [25, 26]. Here, we checked whether KRAS4A with the oncogenic mutation G12D (KRAS4AG12D) could also induce leukemia in mice and what is the role of palmitoylation if KRAS4AG12D does have leukemogenic potential. We constructed retroviral vectors expressing Myc-tagged KRAS4AG12D and its palmitoylation-defective mutant KRAS4AG12D/C180S. We infected BM cells isolated from 5-fluorouracil-treated mice with titer-matched retroviruses containing KRAS4AG12D or KRAS4AG12D/C180S and then transplanted these cells into lethally irradiated syngeneic recipient mice, as previously described [26]. As controls, mice receiving NRASG12D, NRASG12D/C181S, or the MGI vector-transduced bone marrow cells were generated. We found that KRAS4AG12D mice succumbed to disease in 20 to 40 days post bone marrow transplantation (BMT), with similar disease latency as NRASG12D (Fig. 1c). Mice receiving KRAS4AG12D/C180S-transduced bone marrow cells also died in succession but with a much longer disease latency compared with KRAS4AG12D (Fig. 1c). In contrast, all mice receiving MGI vector or NRASG12D/C181S-transduced bone marrow cells remained healthy for over one year (Fig. 1c and data not shown).
We sacrificed the moribund mice receiving KRAS4AG12D, KRAS4AG12D/C180S, and NRASG12D-transduced bone marrow cells to examine their disease phenotype. Analysis of peripheral blood, bone marrow, livers, and spleens revealed that KRAS4AG12D mice developed chronic myelomonocytic leukemia (CMML)- or AML-like diseases similar to NRASG12D mice as previously shown [25, 26] (data not shown). All KRAS4AG12D/C180S mice developed leukocytosis, anemia, and enlarged spleens and livers, in spite of the normal phenotype in the first analysis at 3 weeks post BMT (data not shown). Fluorescence-activated cell sorting (FACS) analyses revealed that 8 out of 10 KRAS4AG12D/C180S mice developed T cell acute lymphoblastic leukemia (T-ALL) like disease (Fig. 1d, e). Thymus is enlarged in these mice (Fig. 1f, g). The rest two KRAS4AG12D/C180S mice developed a CMML-like disease (Fig. 1h and data not shown).
These results demonstrate that like NRAS, HRAS, and KRAS4B, oncogenic KRAS4A also possesses leukemogenic activity, though it induces mostly T-ALL like disease rather than myeloid leukemia when palmitoylation is blocked. The difference in cellular localization between different RAS isoforms may account for the distinguished leukemia phenotypes. The data also show that though palmitoylation plays an important role in KRAS4A leukemogenesis, unlike NRAS, it is not essential.
Differential roles of palmitoylation on transformation and signal transduction by oncogenic NRAS and KRAS4A
To further compare the effect of palmitoylation defectiveness on the oncogenic potential of KRAS4A and NRAS, we infected the Ba/F3 cell line, an IL-3 dependent mouse hematopoietic progenitor cell line, with retroviruses containing green fluorescent protein (GFP) alone (MGI vector control), KRAS4AG12D, KRAS4AG12D/C180S, NRASG12D, or NRASG12D/C181S, and sorted for GFP-positive cells to generate corresponding cell lines. After removal of IL-3 from the culture medium, each group of cells was cultured and counted every three days in triplicate. Ba/F3 cells stably expressing GFP alone (MGI vector) stopped growing and died within first 3 days due to the lack of stimulation by IL-3, whereas cells expressing NRASG12D and KRAS4AG12D continued to proliferation after removal of IL-3 (Fig. 2a). Cells expressing KRAS4AG12D/C180S displayed a significantly slower proliferation rate compared with KRAS4A. NRASG12D/C181S, on the other hand, is incapable of rendering Ba/F3 cells IL-3 independent growth (Fig. 2a). Overall, the in vitro transforming activities of the oncogenic NRAS, KRAS4A, and their palmitoylation mutants correlate with their leukemogenic potentials in vivo.
Fig. 2.
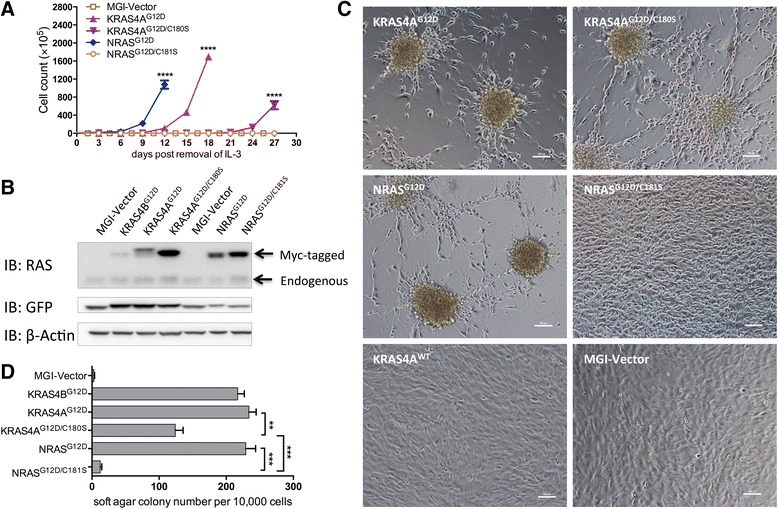
Effects of palmitoylation on transformation potential of oncogenic NRAS vs. KRAS4A in vitro. a The growth curves of Ba/F3 cells transduced with the MGI vector control, KRAS4AG12D, KRAS4AG12D/C180S, NRASG12D, or NRASG12D/C181S after removal of IL-3. Student t test was used for the statistical analysis of the cell numbers in triplicate at each time point. ****P < 0.0001. b Immunoblotting of lysates of NIH3T3 cells stably expressing the GFP control, KRAS4AG12D, KRAS4AG12D/C180S, NRASG12D, or NRASG12D/C181S using a pan anti-RAS antibody. The top band represents Myc-tagged-RAS; the bottom band represents endogenous RAS. c Morphology of cultured NIH3T3 cells stably expressing the indicated proteins was observed under light microscope (original magnification, ×200). d Quantification of anchorage-independent growth of NIH3T3 cells expressing the indicated proteins. Colonies were counted on day 14 after plating in triplicate. Student t test was used for the statistical analysis. **P < 0.01, ***P < 0.001
We previously showed that palmitoylation-deficient NRASG12D confers morphologic changes, abrogates cell density-dependent inhibition of growth but does not confer anchorage-independent growth to NIH3T3 cells [17]. To determine the effect of palmitoylation on transformation of NIH3T3 cells by oncogenic KRAS4A, we infected NIH3T3 cells with KRAS4AG12D and KRAS4AG12D/C180S retroviruses. NRASG12D, NRASG12D/C181S, KRAS4BG12D, and the MGI vector were included as controls. To rule out the possibility whether the myc tag could modify tumorigenic activity of oncogenic RAS, we also infected NIH3T3 cells with myc-tagged wild type KRAS4A retrovirus and found that the myc tag does not alter the biological activity of wild type KRAS4A in this assay (Fig. 2c). RAS protein expression levels in NIH3T3 were detected by western blot analysis (Fig. 2b). It is known that high levels of RAS can induce cellular senescence [27]. The low expression level of oncogenic KRAS4B, hence, may be due to the negative selection of cells with high expression of KRAS4BG12D.
We found that similar to the morphology of NIH3T3 cells expressing NRASG12D, cells expressing KRAS4AG12D and KRAS4AG12D/C180S both appeared smaller and more spindle-shaped than the NRASG12D/C181S and MGI control cells and began to grow into spheres from the nodules in a lattice (Fig. 2c), which resemble the cancer stem cells (CSCs)-like properties ([28] and unpublished data by Frank McCormick and colleagues). We further assessed the effect of palmitoylation deficiency on the transforming potential of KRAS4AG12D by soft agar colony assay. NIH3T3 cells expressing KRAS4AG12D/C180S generated fewer colonies than KRAS4AG12D but significantly more colonies than NRASG12D/C181S (Fig. 2d). These results also correlate in general with the leukemogenic potentials of NRAS, KRAS4A, and their palmitoylation mutants in vivo.
As the in vitro transforming activities of NRAS, KRAS4A, and their palmitoylation mutants in NIH3T3 cells correlate well with their transforming activities in Ba/F3 cells as well as their in vivo leukemogenic potentials, we went on to check the effect of palmitoylation on NRAS and KRAS4A signaling using the NIH3T3 cell lines expressing RAS variants described above. It is well known that the transforming potential of oncogenic RAS relies on multiple downstream signaling pathways, including the RAS/RAF/MEK/ERK and PI3K/AKT pathways [1, 2, 29]. We examined known activating phosphorylation sites of well-identified signaling proteins in the RAS/RAF/MEK/ERK and PI3K/AKT pathways in KRAS4AG12D-, KRAS4AG12D/C180S-, NRASG12D-, or NRASG12D/C181S-expressing NIH3T3 cells by Western blotting analysis using specific phospho-protein antibodies. NIH3T3 cells expressing GFP alone or KRAS4BG12D were included as controls. Cells were serum-starved for 22 h before lysis to minimize RAS signaling background activated by serum growth factors.
As expected, the RAF/MEK/ERK pathway is activated by oncogenic KRAS4B, KRAS4A, and NRAS (Fig. 3a). Interestingly, palmitoylation deficiency greatly abrogates the ability of NRASG12D, but not KRAS4AG12D, on activating c-RAF (Fig. 3a). On the other hand, palmitoylation deficiency markedly reduces the ability of both NRASG12D and KRAS4AG12D in activating MEK1/2 and ERK1/2 (Fig. 3a). The results confirm that activation of MEK/ERK requires conditions other than c-RAF activation [30, 31]. It is also known that c-RAF possesses biological activities other than that through MEK/ERK activation [32]. The above results suggest that in addition to inhibition of the MEK/ERK signaling, completely blocking RAS transformation may involve downstream effectors of c-RAF other than MEK and ERK.
Fig. 3.
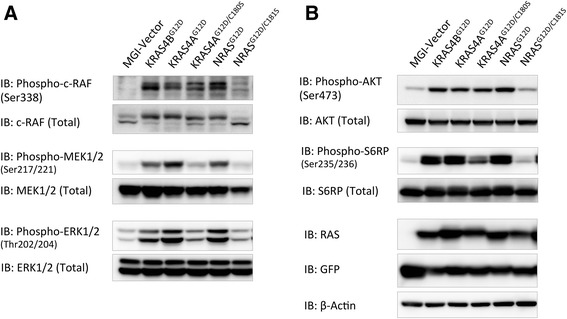
Effects of palmitoylation on signaling transduction of oncogenic NRAS vs. KRAS4A. Serum-starved lysates of NIH3T3 cells expressing GFP, KRAS4AG12D, KRAS4AG12D/C180S, NRASG12D, or NRASG12D/C181S were analyzed by western blotting. a Phospho-c-RAF, MEK, and ERK1/2 antibodies were used to detect the effects of palmitoylation deficiency on the MEK-ERK signaling pathway. b Phospho-AKT and S6RP antibodies were used to detect the effects of palmitoylation deficiency on the PI3K-AKT signaling pathway. Expression levels of RAS, GFP, and β-Actin were detected as loading controls
Also as expected, AKT and S6RP are activated by oncogenic KRAS4B, KRAS4A, and NRAS (Fig. 3b). Similar to its effect on c-RAF activation, palmitoylation deficiency greatly abrogates the ability of NRASG12D, but not KRAS4AG12D, on activating AKT (Fig. 3b). Palmitoylation deficiency also abrogates the ability of NRASG12D and, to a lesser extent, of KRAS4AG12D, to activate S6RP (Fig. 3b).
The results presented here suggest that palmitoylation deficiency differentially impinges upon signaling pathways of NRASG12D and KRAS4AG12D, which correlates well with their in vitro and in vivo transforming activities and that abolishing RAS oncogenic activity may involve blocking its ability to activate c-RAF, MEK, ERK, AKT, and S6RP.
The KIKK polybasic motif in HVR of KRAS4A contributes to the oncogenic activity of KRAS4A
To determine which amino acids contribute to the differential role of palmitoylation in the oncogenesis of KRAS4A and NRAS, the HVR sequence of KRAS4AG12D/C180S and NRASG12D/C181S (Fig. 4a) was swapped to generate KRAS4A/NRAS chimeric genes named NK and KN (Fig. 4b). We then ectopically expressed these two chimeric genes in Ba/F3 and NIH3T3 cell lines. As controls, cells expressing KRAS4AG12D/C180S, NRASG12D/C181S, or GFP alone were generated. After removal of IL-3, Ba/F3 cells expressing NK continued to proliferate, whereas Ba/F3 expressing KN stopped growing and died soon (Fig. 4c). Similarly, NIH3T3 cells expressing NK grew into spheres in a lattice as previously described, whereas NIH3T3 cells expressing KN had a slight morphology change but did not form any sphere (Fig. 4d). Soft agar colony assay also shows that cells expressing NK generate significantly greater number of colonies than that of KN (Fig. 4e). These results indicate that the HVR domain of KRAS4A render KRAS4AG12D/C180S a higher transforming activity than NRASG12D/C181S.
Fig. 4.
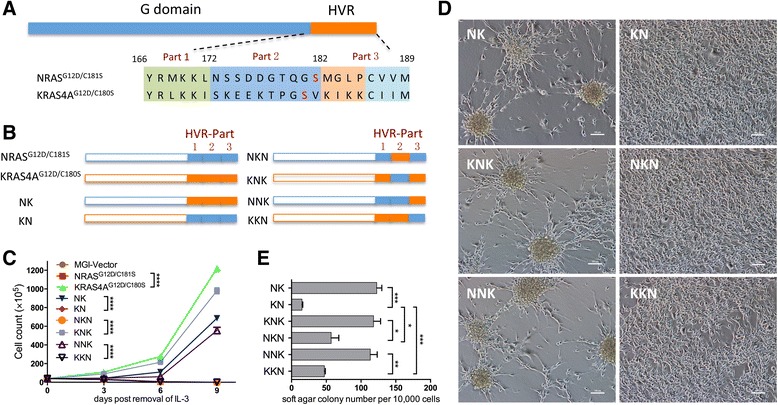
Identification of sequences rendering palmitoylation defective oncogenic KRAS4A transformation active. a The sequence alignment of the hypervariable regions (HVRs) of NRAS vs. KRAS4A. The shaded areas in green, blue, or orange indicate the sequences separated according to the lysine enrichment. Part 1: amino acids 166–172; part 2: amino acids 173–182; part 3: amino acids 183–189. b Diagram of NRASG12D/C181S and KRAS4AG12D/C180S C-terminal domain chimeras. The total C-terminal domain, part 2 or part 3 was swapped respectively to generate NK, KN, NKN, KNK, NNK, and KKN. c Growth curves of Ba/F3 cells overexpressing the indicated proteins after removal of IL-3. Student t test was used for the statistical analysis of the cell numbers in each timepoint. ****P < 0.001. d Morphology of cultured NIH3T3 cells stably expressing the indicated proteins was observed under light microscope (original magnification, ×200). e Quantification of anchorage-independent growth of NIH3T3 cells expressing the indicated proteins. Colonies were counted on day 14 after plating in triplicate. Student t test was used for the statistical analysis. *P < 0.05, **P < 0.01, ***P < 0.001
It was reported that HVR of KRAS4A contains a bipartite polybasic region, designated as PB1 (RLKK, aa167-170) and PB2 (KIKK, aa182-185), which can independently deliver KRAS4A to the plasma membrane [23]. Sequence comparison shows that PB1 is also contained in NRAS HVR sequence (Fig. 4a). To further clarify which amino acids in the HVR sequence contribute to the differential effect of palmitoylation defectiveness on oncogenesis of KRAS4A and NRAS, we divided HVR of KRAS4A into three parts: aa166-171, aa172-181, and aa182-189 (part 1, 2, 3 respectively; Fig. 4a, b). We swapped the sequence of aa172-181 and aa182-189 of KRAS4AG12D/C180S and NRASG12D/C181S, respectively, and constructed the corresponding retroviral vectors expressing these chimeric RAS genes named NKN, KNK, NNK, or KKN (Fig. 4b). We then expressed these genes in Ba/F3 and NIH3T3 cell lines. We found that Ba/F3 cells expressing KNK or NNK continue to proliferate after removal of IL-3, whereas Ba/F3 cells expressing NKN or KKN stop growing and die in the absence of IL-3 (Fig. 4c). Consistently, NIH3T3 cells expressing KNK or NNK formed spheres as previously described, whereas cells expressing KNN or NKN did not (Fig. 4d). Soft agar colony assay shows that NIH3T3 cells expressing KNK or NNK generate significant greater numbers of colonies compared with those expressing KNN or NKN, indicating that chimeras containing aa182-189 of KRAS4A possesses higher transforming activity (Fig. 4e). Interestingly, we found that the colony number of KKN or NKN expressing cells was significantly more than KNN or NRASG12D/C181S, suggesting that sequence aa172-181 contributes to the transforming potential of KRAS4A as well (Fig. 4e). These results indicate that the sequence aa182-189 mainly accounts for the differential role of palmitoylation on the oncogenesis of KRAS4A and NRAS.
Amino acids between position 182 and 189 consist of two parts: the KIKK polybasic motif (PB2) and the CAAX box, the latter of which is conserved in all RAS proteins and undergoes prenylation, -AAX proteolysis, and methylation. Hence, the data indicate that the KIKK polybasic motif in HVR of KRAS4A contributes to the differential role of palmitoylation in the oncogenesis of KRAS4A and NRAS.
The role of the KIKK motif on cellular localization of KRAS4A
To reveal the function of KIKK polybasic motif on KRAS4A plasma membrane association, we expressed N-terminal GFP-fusion KRAS4AG12D, KRAS4AG12D/C180S, KRAS4AG12D/QIQQ (substitution of lysine residuals in KIKK with glutamines), and KRAS4AG12D/C180S/QIQQ in NIH3T3 cells and checked the protein membrane localization by confocal microscope. A clear and intensive localization of KRAS4AG12D on the plasma membrane of NIH3T3 was observed, whereas nonpalmitoylated KRAS4AG12D/C180S localized mainly on the endomembrane, with only a small fraction of KRAS4AG12D/C180S proteins localize on the plasma membrane, particular at some focal areas (Fig. 5a, b, e). KRAS4AG12D/QIQQ, on the other hand, mainly localizes on the plasma membrane, with a slight increase of localization on endomembranes (Fig. 5c, e). The plasma membrane localization of KRAS4A is completely abolished by mutating both the palmitoylation site and KIKK motif (Fig. 5d, e). These results indicate that palmitoylation plays a major role in translocating KRAS4A proteins from endomembranes to the plasma membrane. The KIKK polybasic motif only plays a minor role in translocating KRAS4A from endomembranes to the plasma membrane; it may help the protein to localize on special domains of the plasma membrane, allowing palmitoylation-deficient KRAS4AG12D signals distinctively from palmitoylation-deficient NRASG12D (Fig. 3).
Fig. 5.
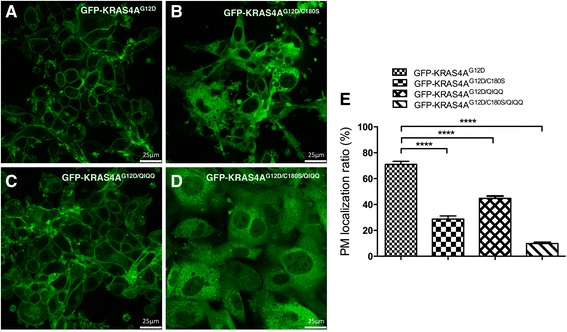
KIKK of KRAS4A functions as a second plasma membrane association motif. NIH3T3 cells expressing GFP-fused KRAS4AG12D a, KRAS4AG12D/C180S b, KRAS4AG12D/QIQQ c, or KRAS4AG12D/C180S/QIQQ d were visualized on a Leica TCS 7300 Spectral Confocal Microscope (original magnification, ×630). e The comparison of plasma membrane localization ratios of each KRAS4A mutant. Ten representative cells of each group were photographed and analyzed by LAS AF Lite Software. PM plasma membrane; Student t test was used for the statistical analysis. ****P < 0.0001
The plasma membrane-targeting function of KIKK motif depends on its positively charged residuals
To determine whether the KIKK motif of KRAS4A functions by purely providing electrostatic interaction with membrane through its basic amino acids or by unknown interactions, we substituted the lysine residuals in the KIKK motif of KRAS4AG12D/C180S with one or two uncharged glutamine residuals (named QIKK, KIQK, KIKQ, QIQK, QIKQ, and KIQQ, respectively) and overexpressed the mutated genes in NIH3T3 cells. We observed that NIH3T3 cells expressing QIKK, KIQK, or KIKQ formed spheres in a lattice and had other morphology close to KRAS4AG12D/C180S, whereas cells expressing QIQK, QIKQ, or KIQQ had spindle-shaped morphology but did not form any sphere (Fig. 6a). Soft agar colony assay also showed a gradual decline in the colony numbers with the decrease of lysine residual numbers in the KIKK motif (Fig. 6b). Meanwhile, no obvious changes in the sphere morphology and the transforming potential were observed after substitution of the lysine residuals in the KIKK motif with arginine residuals (named KRAS4AG12D/RIRR and KRAS4AG12D/C180S/RIRR, Fig. 6c, d). These results indicate that the KIKK motif functions by providing electrostatic interaction with the plasma membrane through its positively charged amino acids.
Fig. 6.
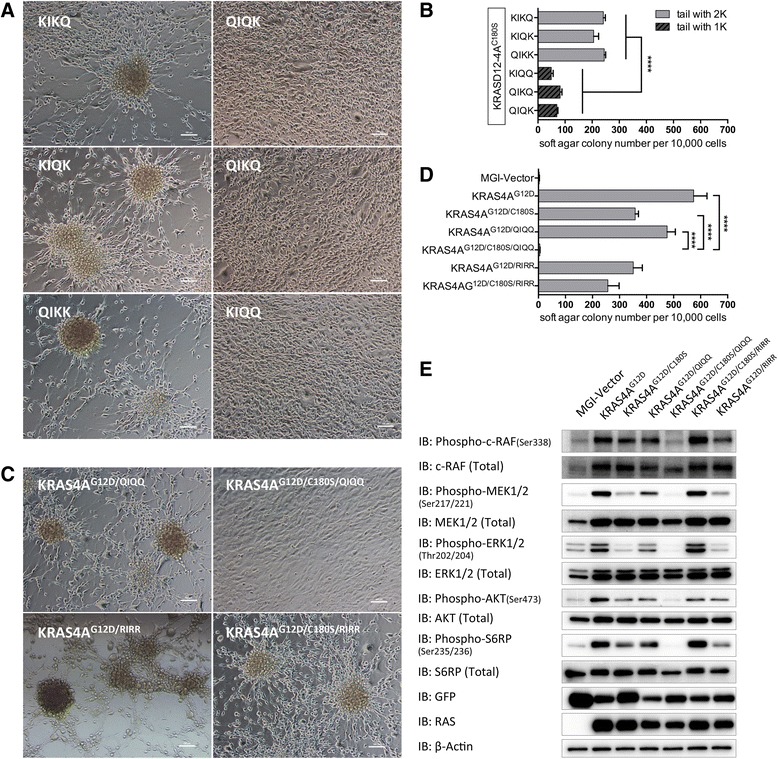
KIKK motif of KRAS4A functions by providing a positive charged region. a Morphology of cultured NIH3T3 cells stably expressing KRAS4AG12D/C180S with KIKK motif mutated to QIKK, KIQK, KIKQ or QIQK, QIKQ, KIQQ was observed under light microscope (original magnification, ×200). b, d Quantification of anchorage-independent growth of NIH3T3 cells expressing the indicated proteins. Colonies were counted on day 14 after plating in triplicate. Student t test was used for the statistical analysis. ****P < 0.0001. c Morphology of cultured NIH3T3 cells stably expressing KRAS4AG12D, KRAS4AG12D/C180S, KRAS4AG12D/QIQQ, or KRAS4AG12D/C180S/QIQQ was observed under light microscope (original magnification, ×200). e Serum-starved lysates of NIH3T3 cells expressing the indicated proteins were analyzed by western blotting to detect the phosphorylation levels of c-RAF, MEK, ERK1/2, AKT, and S6RP. Expressions of RAS, GFP, and β-actin were used as loading controls
To check the effect of KIKK mutations on signaling of KRAS4A, we infected NIH3T3 cells with retroviral constructs expressing KRAS4AG12D, KRAS4AG12D/C180S, KRAS4AG12D/QIQQ, or KRAS4AG12D/C180S/QIQQ. Cells were starved for 22 h and then lysed for detection of the activation of signaling proteins c-RAF, MEK1/2, ERK1/2, AKT, and S6RP. We observed that in KRAS4AG12D/QIQQ expressing cells, the phosphorylation levels of MEK1/2, ERK1/2, AKT, and S6RP were lower than KRAS4AG12D but a slightly higher than KRAS4AG12D/C180S (Fig. 6e). No obvious change in the phosphorylation level of c-RAF Ser338 in KRAS4AG12D/QIQQ cells was observed compared with that of KRAS4AG12D and KRAS4AG12D/C180S. Interestingly, the ability of KRAS4AG12D to active c-RAF, MEK1/2, ERK1/2, AKT, and S6RP is abolished by mutating both the palmitoylation site and the KIKK motif (KRAS4AG12D/C180S/QIQQ). In contrast, substitution of the lysine residuals with arginine residuals in the KIKK motif does not significantly affect KRAS4AG12D signaling activity. These results further support the conclusion that the KIKK polybasic motif in HVR of KRAS4A contributes to the differential role of palmitoylation in the oncogenic signaling of KRAS4A and NRAS.
The relative importance of palmitoylation and the KIKK motif on leukemogenic potential of KRAS4A
To further examine the role of KIKK motif in KRAS4A leukemogenesis, we used the BMTT model with retroviruses containing KRAS4AG12D/QIQQ or KRAS4AG12D/C180S/QIQQ. Mice receiving KRAS4AG12D, KRAS4AG12D/C180S, or MGI vector-transduced bone marrow cells were used as controls. As previously shown, KRAS4AG12D rapidly and efficiently induces a CMML- or AML-like disease in mice. Under the same experimental conditions, the disease latency of KRAS4AG12D/QIQQ was only slightly prolonged compared to KRAS4AG12D, while KRAS4AG12D/C180S mice survived a much longer time (Fig. 7a). In contrast, all KRAS4AG12D/C180S/QIQQ mice remained healthy during the course of experiment (Fig. 7a). Immunophenotype analysis showed that mice transfected KRAS4AG12D/QIQQ also developed CMML- or AML-like disease (Fig. 7b), while the majority of KRAS4AG12D/C180S mice developed T-ALL like disease as previously described (Fig. 1d and data not shown). All groups of mice in moribund or over 1 year were sacrificed for pathological analysis. The spleens of KRAS4AG12D/C180S/QIQQ mice are normal compared with KRAS4AG12D, KRAS4AG12D/QIQQ, or KRAS4AG12D/C180S (Fig. 7c). These results indicate that palmitoylation plays a more important role than the KIKK plasma membrane-targeting motif in KRAS4AG12D leukemogenesis, yet blocking both could completely abolish the ability of oncogenic KRAS4A to induce leukemia.
Fig. 7.
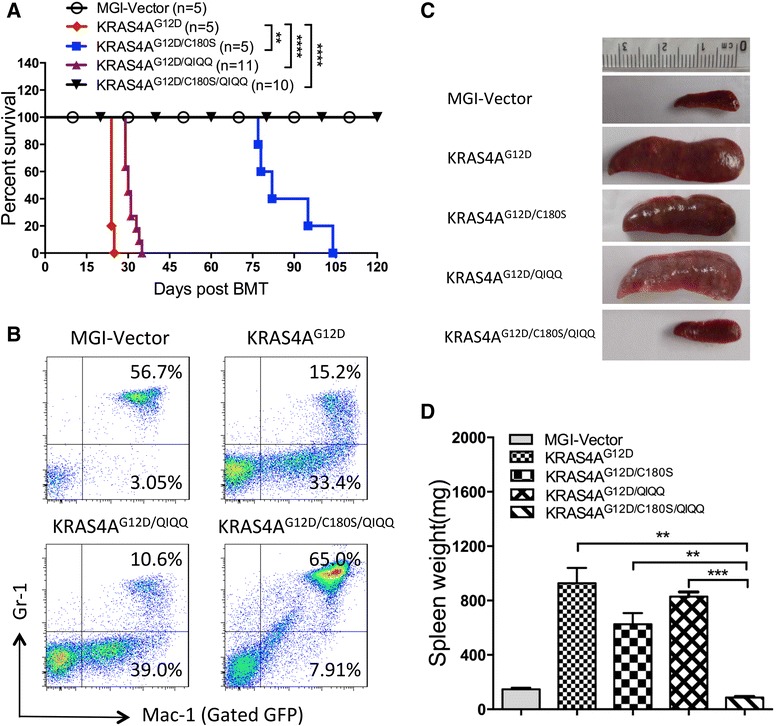
Contributions of palmitoylation and KIKK motif on leukemogenic potential of oncogenic KRAS4A. a Kaplan-Meier plot of cumulative survival of recipient mice transplanted with bone marrow cells infected by KRAS4AG12D (n = 5), KRAS4AG12D/C180S (n = 5), KRAS4AG12D/QIQQ (n = 11), KRAS4AG12D/C180S/QIQQ (n = 10) containing retroviruses. The MGI vector mice (n = 5) were observed as control. P values were determined by the log-rank test. **P < 0.01, ****P < 0.0001. b Immunophenotyping of the leukemic bone marrow cells isolated from the KRAS4AG12D, KRAS4AG12D/QIQQ, and KRAS4AG12D/C180S/QIQQ moribund mice by flow cytometry. The myeloid lineage markers include Gr-1 and Mac-1. All the cells are GFP positive gated. c Spleens from the mice of each group were shown as indicated. d Comparison of the spleen weights of each group mice. The data were presented as mean ± SEM. Student t test was used for the statistical analysis. **P < 0.01, ***P < 0.001; n = 3 in each group
Discussion
Accumulating evidence has indicated that the KRAS4A splice variant plays an important role in tumorigenesis [20–23]. Understanding the oncogenic potential of KRAS4A in vivo and the ways to block its oncogenic signaling is important for developing effective cancer therapies. Here, we show that KRAS4A has a similar leukemogenic potential as NRAS (Fig. 1), as well as KRAS4B (data not shown), when expressed under the same promoter. Palmitoylation is very important for KRAS4A leukemogenesis, but it is not essential as that in case of NRAS. The KIKK motif in KRAS4A functions as an alternative membrane-targeting motif and makes a contribution to leukemogenesis by oncogenic KRAS4A.
Consistent with the results in our previous work, NRASG12D palmitoylation mutant completely loses the capacity to activate MEK-ERK and PI3K-AKT pathways in NIH3T3 cells [17]. Unlike NRASG12D, although defective in activating MEK, ERK, and S6RP, the KRAS4AG12D palmitoylation mutant retains most of its ability to activate c-RAF and AKT, coincident with the sphere morphology and the more colony numbers of KRAS4AG12D/C180S-expressing NIH3T3 cells compared with NRASG12D/C181S-expressing cells. These data suggest that though the plasma membrane localization of KRAS4AG12D is greatly reduced by the palmitoylation mutation (Fig. 5), the KIKK motif may target KRAS4A to a special domain in the plasma membrane, allowing it to activate c-RAF and AKT. These results indicates that the KIKK motif, assisting in the KRAS4A plasma membrane translocation, may mainly contribute to the differential effect of palmitoylation in the leukemogenesis of oncogenic KRAS4A and NRAS. These results also suggest that palmitoylation would be an effective target for both oncogenic KRAS4A and NRAS associated cancer therapy and that the KIKK motif should also be taken into account in the case of oncogenic KRAS4A associated cancer.
The KIKK motif has been recognized as a membrane-targeting motif [12, 23], but the mechanism was unknown. Electrostatical interaction of polybasic motifs with negatively charged lipids promotes protein binding with plasma membrane [33, 34]. But, the polybasic region of KRAS4B consists of a stretch of six lysine residuals. To test whether the KIKK motif target KRAS4A to membrane through electrostatical interaction or specific protein-protein interaction, we substituted all lysine residuals in the KIKK motif with arginine residuals and found no obvious functional change of oncogenic KRAS4A. In addition, substitution of lysine residuals in the KIKK motif with one, two, or three glutamine residuals results in its gradual decrease of transforming potential, suggesting that the KIKK motif of KRAS4A functions by providing electrostatic interaction with the plasma membrane through its positively charged amino acids.
Sequence analysis shows that both KRAS4A and NRAS harbor an RxKK (x: methionine or leucine) motif, which is designated as PB1 (polybasic domain 1) and was shown to promote the membrane localization [12, 23]. Our data show that mutating both the palmitoylation site and the KIKK motif like the effect of mutating the palmitoylation site in NRASG12D, completely abolishes KRAS4AG12D’s leukemogenic potential. These data suggest that the RxKK motif is a much weaker membrane-targeting motif comparing to palmitoylation and KIKK. In addition to the KIKK motif and the RxKK motif, amino acids between 172 and 181 from KRAS4A increases the transforming activity of NRASG12D in NIH3T3, but not Ba/F3, cells (Fig. 4c, e), indicating that this region in KRAS4A may also help its plasma membrane localization.
The fraction of KRAS4AG12D, even the QIQQ mutant KRAS4AG12D, localized on the plasma membrane is significantly more than that of NRASG12D (Fig. 5 and data not shown). It is possible that palmitoylation of KRAS4A is more efficient than that of NRAS. It is noticeable that the amino acid sequence around the palmitoylation site is different between NRAS and KRAS4A, raising the possibility that palmitoylation of NRAS and KRAS4A may be catalyzed by different palmitoylacyltransferases. Given its implication in developing targeted cancer therapies, this possibility should be tested in the future.
Recurrent mutations of slicing factor genes have been found in hematological malignances, as well as some solid tumors, indicating aberrant splicing is involved in the pathogenesis of cancer [35–38]. Our data show that KRAS4A is expressed in all hematological malignant cell lines tested and that some cell lines harbor a particularly high level of KRAS4A (Fig. 1a). It is possible that deregulated splicing facilitates the expression of the KRAS4A splice variant in certain hematological malignant cells. Understanding the mechanism by which the KRAS4A/4B splicing is regulated might help to reveal alternative strategies for cancer therapy.
Conclusions
Palmitoylation palys a critical role in KRAS4A leukemogenesis, but it is not essential as that in case of NRAS. The KIKK motif in KRAS4A functions as an additional membrane-targeting motif and makes a contribution to leukemogenesis by oncogenic KRAS4A. The studies suggest that therapies targeting KRAS4A palmitoylation might be effective in treating related malignancies and that interfering the KIKK membrane-targeting motif would enhance the therapeutic effectiveness.
Methods
DNA constructs
NRASG12D- and NRASG12D/C181S-containing MSCV-GFP-IRES (MGI) retroviral constructs were generated as previously described [17]. Wild-type KRAS4A human cDNA (NCBI accession number: NM_033360.3) was cloned into the MGI vector with N-terminal Myc-tag. KRAS4AG12D (replacing the glycine residual at codon 12 with aspartic acid), KRAS4AG12D/C180S, KRAS4AG12D/QIQQ, KRAS4AG12D/C180S/QIQQ, KRAS4AG12D/RIRR, KRAS4AG12D/C180S/RIRR, KRAS4AG12D/C180S/QIKK, KRAS4AG12D/C180S/KIQK, KRAS4AG12D/C180S/KIKQ, KRAS4AG12D/C180S/QIQK, KRAS4AG12D/C180S/QIKQ, and KRAS4AG12D/C180S/KIQQ mutants were generated using Quickchange Site-Directed Mutagenesis Kit (Stratagene, #200518). Genes containing fragment-swapped mutants NK, KN, NKN, KNK, NNK, and KKN were synthesized by GENEWIZ, followed by subcloning them into the MGI vector. Retroviral vectors expressing KRAS4AG12D, KRAS4AG12D/C180S, KRAS4AG12D/QIQQ, and KRAS4AG12D/C180S/QIQQ mutants as N-terminal GFP-fusion proteins were created by cloning these mutational genes into EGFP-C1 plasmids, followed by subcloning them into the MSCV-IRES (without GFP) vector to generate MSCV-GFP/RAS mutants-IRES plasmids. Genes containing fragment-switching mutants NK, KN, NKN, KNK, NNK, and KKN were synthesized by GENEWIZ, followed by subcloning them into the MGI vector. All constructs were confirmed by DNA sequencing before use.
RT-qPCR
Bone marrow total RNA of three AML patient samples with oncogenic KRAS mutations were collected from the centers of Shanghai Institute of Hematology (SIH). The primers for KRAS4A and KRAS4B were designed as previously described [23]. Primer sequences are as follows: for KRAS4A, forward: 5′-agacacaaaacaggctcagga-3′, reverse: 5′-ttcacacagccaggagtcttt-3′; for KRAS4B, forward: 5′-gactggggagggctttcttt- 3′, reverse: 5′-gcatcatcaacaccctgtct-3′. Results were normalized to a standard curve built by a dilution of MGI-KRAS4A and MGI-KRAS4B plasmid DNA.
The study was approved by the ethics committees of all participating centers. All patients were given informed consent for cryopreservation of bone marrow (BM) according to the Declaration of Helsinki.
Retrovirus production and tittering
Retroviruses were produced with the constructs described above by individually cotransfecting them with the Pcl-Eco retrovirus packaging vector into the Bosc23 retroviral packaging cell line using a calcium phosphate precipitation method as previously described [25, 26]. Retroviral supernatant was harvested 48 h after transfection and tittered as previously described [25, 26].
Bone marrow transduction/transplantation
Retroviral transduction and BM transplantation experiments were performed as previously described [26]. Briefly, BM cells were isolated from the 6–8-week-old donor mice pretreated with 5-fluorouracil (250 mg/kg). BM cells were infected with retroviruses each day for 2 days in transplant medium, followed by injecting 4 × 105 cells into the tail vein of each lethally irradiated (2 × 3.8 Gy, 4 h between each dose) female recipient BALB/c mice. Retroviral titers were matched to 2 × 106 TFUs before BM infection. Recipient mice were monitored weekly for signs of disease beginning on day 14 after transplantation. All the animal experiments were approved by The Animal Care and Welfare Committee of Shanghai Jiao Tong University School of Medicine.
Hematological and immunophenotypic analysis
Peripheral blood (PB) was obtained from mice by tail bleeding and analyzed by using a pocH-100iV Diff hematology analyzer (Sysmex Corporation). After lysing the RBC, PB, bone marrow, spleen, or thymus cells were stained with conjugated antibodies to specific surface antigens. Mac-1, Gr-1, CD19, B220, CD3, CD4, and CD8 were used separately or in combination for immunophenotypic analysis of hematopoietic or leukemic cells (BD Pharmingen). All flow cytometric analyses were performed on an LSR II system (BD Biosciences). Data were analyzed with FlowJo software (Tree Star Inc.).
Cell culture
Bosc23 cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM), 10 % Fetal bovine serum (GIBCO, Catalog 10099-141), and 1× penicillin/streptomycin. NIH3T3 cells were cultured in DMEM, 10 % bovine serum (GIBCO, catalog #16170078), and 1× penicillin/streptomycin. Ba/F3 cells were maintained in 1640 medium supplemented with 10 % fetal bovine serum (FBS), 1× penicillin/streptomycin, and 10 % IL-3 containing WEHI-3B conditional medium. NIH3T3 and Ba/F3 cell lines stably expressing KRAS4AG12D and mutant KRAS4AG12D genes were generated by retroviral transduction as described [17, 26]. All cell lines were sorted by FACS to >95 % GFP positive homogeneity.
Cell proliferation assay and microscopic analysis
After sorting by FACS, Ba/F3 cells transduced with KRAS4AG12D or mutant KRAS4AG12D genes expressing retroviruses were washed with 1640 twice to remove IL-3 and resuspended with 1640 containing 10 % FBS and 1× penicillin/streptomycin. Cells were plated in triplicate into 6-cm dishes in the absence of IL-3 and were stained with trypan blue to exclude nonviable cells and counted manually under a light microscope every 3 days until the cell lines fully grew or died. Student t test was used for the comparison of the cell numbers in each time point. NIH3T3 cells expressing KRAS4AG12D or mutant KRAS4AG12D genes formed tight spheres after culturing for 6 days and were photographed using an Olympus digital camera.
Soft agar colony forming assay
To generate 0.6 % soft agar, 1.5 ml 4 % agarose with 8.5-ml cell culture media (Dulbecco’s modified Eagle’s medium + 20 % bovine serum + 1.2 times Pen-Strep) was mixed. A total of 2 mL of 0.6 % bottom agar (incubated in 42 °C) was then platted into each well of a 6-well tissue culture plate. FACS-purified NIH3T3 cell lines expressing MGI vector or RAS mutants were diluted to 2 × 105 cells/ml, 2 × 104 cells/ml, or 2 × 103 cells/ml in. Triplicate 3-mL cells suspended in 0.3 % soft agar (cell dilution + 0.6 % soft agar with 1:1 ratio) were added on top of the jellified bottom agar for each type of transduced cells and incubated in CO2 incubator at 37 °C. Colonies were counted under a light microscope at day 14 after plating.
Western blot analysis
Western blot analyses were performed as previously described [17, 26, 39]. Briefly, 90 % confluent NIH3T3 cells were serum-starved for 22 h. Cells were counted and lysed in 1× SDS running buffer, heated at 100 °C for 10 min, and centrifuged to remove debris. Lysates were resolved on 10 % polyacrylamide gels, transferred to nitrocellulose membranes, and blotted with the following primary antibodies: anti-KRAS4A specific antibody (1:500; Santa Cruz Biotechnology, sc-522), anti-RAS antibody (1:2000; Cell Signaling Technology, #3965), anti-actin (1:5000; Sigma-Aldrich, A1978), anti-GFP (1:2000; Cell Signaling Technology, #2555), and phospho-c-RAF, c-RAF, phoshpo-MEK1/2, MEK1/2, phospho-ERK1/2, ERK1/2, phospho-AKT, AKT, phospho-S6RP, and S6RP (All 1:2000; Cell Signaling Technology). HRP-labeled goat anti-mouse IgG or goat anti-rabbit IgG was used as a secondary antibody.
Cellular localization analysis of RAS proteins
NIH3T3 cells expressing KRAS4AG12D with GFP fused to its N terminus or expressing GFP-fusion versions of KRAS4AG12D PTM or KIKK motif mutant proteins were grown overnight. Fluorescence was visualized and photographed on a Leica TCS 7300 Spectral Confocal Microscope. The images were analyzed by using LAS AF Lite Software (Leica). The plasma membrane localization ratios of KRAS4A mutants were counted by (pixel(plasma membrane)/pixel(sum)). Ten representative cells of each group were counted and analyzed. Student t test was used for statistical data analysis.
Statistical analysis
Graphpad Prism 6 software was used for statistical data analysis. Student t test was used for mean comparison, whereas the Kaplan-Meier survival curve and log-rank test were used for survival analysis.
Acknowledgements
We thank Wanbin Fu, Zheng Ruan, Xiangqin Weng, Yan Sheng, Wu Zhang, Jing Lu and Yi Jin for technical assistance. This work was supported by the National Natural Science Foundation of China (81230055 to R.R.), Shanghai Outstanding Academic Leader Program (12XD1403500 to R.R.) and the Samuel Waxman Cancer Research Foudation Co-PI Program.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
HZ performed research, analyzed data, and drafted the manuscript; PL, RZ, DL, XZ, and CZ assisted in performing in vivo experiments and analyzing data; MW and BJ assisted in performing experiments in vitro; BC assisted in performing experiments with patient samples; ZC provided suggestions, analyzed the data, and wrote the paper; RR designed the study, analyzed data, and wrote the paper. All authors read and approved the final manuscript.
Contributor Information
Huanbin Zhao, Email: hbzhao@sibs.ac.cn.
Ping Liu, Email: liupingjize@126.com.
Ruihong Zhang, Email: zhangruihong007@163.com.
Min Wu, Email: wumin806@163.com.
Donghe Li, Email: lidonghes@163.com.
Xuemei Zhao, Email: xuemeiyxx@163.com.
Chun Zhang, Email: zhangchun84135@126.com.
Bo Jiao, Email: bjiao@sibs.ac.cn.
Bing Chen, Email: chenbing.rjsih@yahoo.com.
Zhu Chen, Email: zchen@stn.sh.cn.
Ruibao Ren, Email: rbren@brandeis.edu.
Reference
- 1.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11(11):761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akinleye A, Avvaru P, Furqan M, Song Y, Liu D. Phosphatidylinositol 3-kinase (PI3K) inhibitors as cancer therapeutics. J Hematol Oncol. 2013;6(1):88. doi: 10.1186/1756-8722-6-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang T, Karsy M, Zhuge J, Zhong M, Liu D. B-Raf and the inhibitors: from bench to bedside. J Hematol Oncol. 2013;6:30. doi: 10.1186/1756-8722-6-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov. 2014;13(11):828–851. doi: 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gysin S, Salt M, Young A, McCormick F. Therapeutic strategies for targeting ras proteins. Genes Cancer. 2011;2(3):359–372. doi: 10.1177/1947601911412376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith AD, Roda D, Yap TA. Strategies for modern biomarker and drug development in oncology. J Hematol Oncol. 2014;7(1):70. doi: 10.1186/s13045-014-0070-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prior IA, Hancock JF. Compartmentalization of Ras proteins. J Cell Sci. 2001;114(Pt 9):1603–1608. doi: 10.1242/jcs.114.9.1603. [DOI] [PubMed] [Google Scholar]
- 8.Hancock JF. Ras proteins: different signals from different locations. Nat Rev Mol Cell Biol. 2003;4(5):373–384. doi: 10.1038/nrm1105. [DOI] [PubMed] [Google Scholar]
- 9.Harousseau JL. Farnesyltransferase inhibitors in hematologic malignancies. Blood Rev. 2007;21(4):173–182. doi: 10.1016/j.blre.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 10.Whyte DB, Kirschmeier P, Hockenberry TN, Nunez-Oliva I, James L, Catino JJ, et al. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J Biol Chem. 1997;272(22):14459–14464. doi: 10.1074/jbc.272.22.14459. [DOI] [PubMed] [Google Scholar]
- 11.Lobell RB, Omer CA, Abrams MT, Bhimnathwala HG, Brucker MJ, Buser CA, et al. Evaluation of farnesyl:protein transferase and geranylgeranyl:protein transferase inhibitor combinations in preclinical models. Cancer Res. 2001;61(24):8758–8768. [PubMed] [Google Scholar]
- 12.Laude AJ, Prior IA. Palmitoylation and localisation of RAS isoforms are modulated by the hypervariable linker domain. J Cell Sci. 2008;121(Pt 4):421–427. doi: 10.1242/jcs.020107. [DOI] [PubMed] [Google Scholar]
- 13.Apolloni A, Prior IA, Lindsay M, Parton RG, Hancock JF. H-ras but not K-ras traffics to the plasma membrane through the exocytic pathway. Mol Cell Biol. 2000;20(7):2475–2487. doi: 10.1128/MCB.20.7.2475-2487.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cox AD, Der CJ, Philips MR. Targeting RAS membrane association: back to the future for anti-RAS drug discovery? Clin Cancer Res. 2015;21(8):1819–1827. doi: 10.1158/1078-0432.CCR-14-3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jang H, Abraham SJ, Chavan TS, Hitchinson B, Khavrutskii L, Tarasova NI, et al. Mechanisms of membrane binding of small GTPase K-Ras4B farnesylated hypervariable region. J Biol Chem. 2015;290(15):9465–9477. doi: 10.1074/jbc.M114.620724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bivona TG, Quatela SE, Bodemann BO, Ahearn IM, Soskis MJ, Mor A, et al. PKC regulates a farnesyl-electrostatic switch on K-Ras that promotes its association with Bcl-XL on mitochondria and induces apoptosis. Mol Cell. 2006;21(4):481–493. doi: 10.1016/j.molcel.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 17.Cuiffo B, Ren R. Palmitoylation of oncogenic NRAS is essential for leukemogenesis. Blood. 2010;115(17):3598–3605. doi: 10.1182/blood-2009-03-213876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pells S, Divjak M, Romanowski P, Impey H, Hawkins NJ, Clarke AR, et al. Developmentally-regulated expression of murine K-ras isoforms. Oncogene. 1997;15(15):1781–1786. doi: 10.1038/sj.onc.1201354. [DOI] [PubMed] [Google Scholar]
- 19.Plowman SJ, Williamson DJ, O'Sullivan MJ, Doig J, Ritchie AM, Harrison DJ, et al. While K-ras is essential for mouse development, expression of the K-ras 4A splice variant is dispensable. Mol Cell Biol. 2003;23(24):9245–9250. doi: 10.1128/MCB.23.24.9245-9250.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Plowman SJ, Berry RL, Bader SA, Luo F, Arends MJ, Harrison DJ, et al. K-ras 4A and 4B are co-expressed widely in human tissues, and their ratio is altered in sporadic colorectal cancer. J Exp Clin Canc Res. 2006;25(2):259–267. [PubMed] [Google Scholar]
- 21.Patek CE, Arends MJ, Wallace WAH, Luo FJ, Hagan S, Brownstein DG, et al. Mutationally activated K-ras 4A and 4B both mediate lung carcinogenesis. Exp Cell Res. 2008;314(5):1105–1114. doi: 10.1016/j.yexcr.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 22.To MD, Wong CE, Karnezis AN, Del Rosario R, Di Lauro R, Balmain A. Kras regulatory elements and exon 4A determine mutation specificity in lung cancer. Nat Genet. 2008;40(10):1240–1244. doi: 10.1038/ng.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsai FD, Lopes MS, Zhou M, Court H, Ponce O, Fiordalisi JJ, et al. K-Ras4A splice variant is widely expressed in cancer and uses a hybrid membrane-targeting motif. Proc Natl Acad Sci U S A. 2015;112(3):779–784. doi: 10.1073/pnas.1412811112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kranenburg O. The KRAS oncogene: past, present, and future. Bba-Rev Cancer. 2005;1756(2):81–82. doi: 10.1016/j.bbcan.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 25.Parikh C, Subrahmanyam R, Ren R. Oncogenic NRAS rapidly and efficiently induces CMML- and AML-like diseases in mice. Blood. 2006;108(7):2349–2357. doi: 10.1182/blood-2004-08-009498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parikh C, Subrahmanyam R, Oncogenic RR, NRAS KRAS, and HRAS exhibit different leukemogenic potentials in mice. Cancer Res. 2007;67(15):7139–7146. doi: 10.1158/0008-5472.CAN-07-0778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88(5):593–602. doi: 10.1016/S0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- 28.McCormick F. KRAS as a therapeutic target. Clin Cancer Res. 2015;21(8):1797–1801. doi: 10.1158/1078-0432.CCR-14-2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Akinleye A, Furqan M, Mukhi N, Ravella P, Liu D. MEK and the inhibitors: from bench to bedside. J Hematol Oncol. 2013;6:27. doi: 10.1186/1756-8722-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Marusiak AA, Edwards ZC, Hugo W, Trotter EW, Girotti MR, Stephenson NL, et al. Mixed lineage kinases activate MEK independently of RAF to mediate resistance to RAF inhibitors. Nat Commun. 2014;5:3901. doi: 10.1038/ncomms4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bae D, Ceryak S. Raf-independent, PP2A-dependent MEK activation in response to ERK silencing. Biochem Biophys Res Commun. 2009;385(4):523–527. doi: 10.1016/j.bbrc.2009.05.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hindley A, Kolch W. Extracellular signal regulated kinase (ERK)/mitogen activated protein kinase (MAPK)-independent functions of Raf kinases. J Cell Sci. 2002;115(Pt 8):1575–1581. doi: 10.1242/jcs.115.8.1575. [DOI] [PubMed] [Google Scholar]
- 33.Okeley NM, Gelb MH. A designed probe for acidic phospholipids reveals the unique enriched anionic character of the cytosolic face of the mammalian plasma membrane. J Biol Chem. 2004;279(21):21833–21840. doi: 10.1074/jbc.M313469200. [DOI] [PubMed] [Google Scholar]
- 34.Yeung T, Terebiznik M, Yu L, Silvius J, Abidi WM, Philips M, et al. Receptor activation alters inner surface potential during phagocytosis. Science. 2006;313(5785):347–351. doi: 10.1126/science.1129551. [DOI] [PubMed] [Google Scholar]
- 35.Venables JP. Aberrant and alternative splicing in cancer. Cancer Res. 2004;64(21):7647–7654. doi: 10.1158/0008-5472.CAN-04-1910. [DOI] [PubMed] [Google Scholar]
- 36.Adamia S, Pilarski PM, Bar-Natan M, Stone RM, Griffin JD. Alternative splicing in chronic myeloid leukemia (CML): a novel therapeutic target? Curr Cancer Drug Targets. 2013;13(7):735–748. doi: 10.2174/15680096113139990083. [DOI] [PubMed] [Google Scholar]
- 37.Adamia S, Haibe-Kains B, Pilarski PM, Bar-Natan M, Pevzner S, Avet-Loiseau H, et al. A genome-wide aberrant RNA splicing in patients with acute myeloid leukemia identifies novel potential disease markers and therapeutic targets. Clin Cancer Res. 2014;20(5):1135–1145. doi: 10.1158/1078-0432.CCR-13-0956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim M, Slack FJ. MicroRNA-mediated regulation of KRAS in cancer. J Hematol Oncol. 2014;7(1):84. doi: 10.1186/s13045-014-0084-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gross AW, Zhang X, Ren R. Bcr-Abl with an SH3 deletion retains the ability to induce a myeloproliferative disease in mice, yet c-Abl activated by an SH3 deletion induces only lymphoid malignancy. Mol Cell Biol. 1999;19(10):6918–6928. doi: 10.1128/MCB.19.10.6918. [DOI] [PMC free article] [PubMed] [Google Scholar]