

Abstract
Background
The current extensive use of the domestic goat (Capra hircus) is the result of its medium size and high adaptability as multiple breeds. The extent to which its genetic variability was influenced by early domestication practices is largely unknown. A common standard by which to analyze maternally-inherited variability of livestock species is through complete sequencing of the entire mitogenome (mitochondrial DNA, mtDNA).
Results
We present the first extensive survey of goat mitogenomic variability based on 84 complete sequences selected from an initial collection of 758 samples that represent 60 different breeds of C. hircus, as well as its wild sister species, bezoar (Capra aegagrus) from Iran. Our phylogenetic analyses dated the most recent common ancestor of C. hircus to ~460,000 years (ka) ago and identified five distinctive domestic haplogroups (A, B1, C1a, D1 and G). More than 90 % of goats examined were in haplogroup A. These domestic lineages are predominantly nested within C. aegagrus branches, diverged concomitantly at the interface between the Epipaleolithic and early Neolithic periods, and underwent a dramatic expansion starting from ~12–10 ka ago.
Conclusions
Domestic goat mitogenomes descended from a small number of founding haplotypes that underwent domestication after surviving the last glacial maximum in the Near Eastern refuges. All modern haplotypes A probably descended from a single (or at most a few closely related) female C. aegagrus. Zooarchaelogical data indicate that domestication first occurred in Southeastern Anatolia. Goats accompanying the first Neolithic migration waves into the Mediterranean were already characterized by two ancestral A and C variants. The ancient separation of the C branch (~130 ka ago) suggests a genetically distinct population that could have been involved in a second event of domestication. The novel diagnostic mutational motifs defined here, which distinguish wild and domestic haplogroups, could be used to understand phylogenetic relationships among modern breeds and ancient remains and to evaluate whether selection differentially affected mitochondrial genome variants during the development of economically important breeds.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-015-2342-2) contains supplementary material, which is available to authorized users.
Keywords: Goat mitochondrial genome, mtDNA haplogroups, Domestication, Origin of Capra hircus, Capra aegagrus
Background
The domestic goat (Capra hircus), which counts a worldwide population of more than 800,000,000 specimens and about 1200 breeds described (http://dad.fao.org), is among the “big five” livestock species defined by the Food and Agriculture Organization (FAO) [1] and is an invaluable source of milk, meat, skin and fiber for poor small holders and shepherds in developing countries and marginal areas [2].
On the basis of bone morphological changes associated with progressive taming [3], zooarchaeology suggests that goat domestication began about 11,000 years (ka) ago in an area stretching from the high Euphrates valleys in Southeastern Anatolia (Turkey) to the Zagros Mountains in Central Iran [4, 5] and located within the natural distribution of the wild ancestor species, the bezoar Capra aegagrus [6, 7]. This event is now considered as the final outcome of a gradual change from hunting to management of wild captive animals [3, 5, 8]. Due to their high rusticity and adaptability to harsh environments, goats represented a key resource during the Neolithic agricultural revolution and for the human migration waves that spread Neolithic culture out of the Fertile Crescent [9].
Previous studies of mitochondrial control-region haplotypes described six highly divergent haplogroups (Hg.s) in domestic goats: A, B, C [10], D [11], F [12] and G [13]. An additional haplogroup, named E, has been described by Joshi et al. [9] on the basis of two highly divergent control-region haplotypes. This was recognized as a sub-clade of haplogroup A when compared to a larger dataset [13]. The reported weak geographical structuring of goat mitochondrial variability was often interpreted as a consequence of the frequent transportation of goats along terrestrial and maritime routes of migration and commerce, probably during the early domestication phases [7, 10, 14, 15]. A subsequent comparison with wild stocks confirmed the presence of all “domestic” haplogroups in the current C. aegagrus populations, which could result from early translocations of animals and/or feralization before the worldwide spread of goats [7]. The haplogroup A is largely predominant (>90 %) among domestic goats, but rare (6 %) in the bezoar and never observed in the Iranian Zagros Mountains. The probable origin of haplogroup A occurred in Eastern Anatolia, where it is still present among wild populations, and its presence in Eastern Iran probably is the result of a subsequent feralization of domestic goats. The most frequent haplogroup in wild populations is C (39 %) detected in most of the bezoar distribution area and more common in Southern Zagros/Central Iranian Plateau. The evidence that C control-region haplotypes from Pakistan are the farthest from the domestic-related ones [7] disproved the Indus Valley domestication hypothesis suggested by archaeological remains from Mehrgarh (Baluchistan, Pakistan) [5]. Haplogroup F is still found in wild populations (from Northern Caucasus to lower Indus Valley), but it is very rare in domestic goats (<0.2 %), as it was identified only in three Sicilian samples [12]. The other haplogroups were found only in Iranian (D and G) or in both Northern Iranian and Eastern Anatolian bezoars (B). It has been proposed that these haplotypes might have entered the domestic goat gene pool either during the early spread of domestic goats, or due to small-scale domestication events. These findings indicate that the process of goat domestication occurred not only in Eastern Anatolia, as marked by haplogroup A and supported by zooarchaeological data [5, 8], but possibly also in Central Iran (Zagros Mountains and Iranian Plateau). This additional easternmost domestication event has been marked by haplogroup C, although it led only to a small contribution detectable in the mitochondrial gene pool of current domestic goats (1.5 %) and no archaeological substantiations [7].
MtDNA haplotypes belonging to haplogroups A and C have been found in ancient goat samples retrieved from an early Neolithic site in Southern France [16]. The two haplogroups occurred with almost the same frequency among the analyzed bones (i.e. 8 samples carrying A haplotypes and 11 samples carrying C haplotypes), suggesting that domestic goat populations were already characterized by the mtDNA variants A and C [16] during the first colonization waves that brought Neolithic farmers into the Mediterranean area about 7.5 ka ago [3].
Haplogroup divergence times calculated on different control-region datasets, usually by employing different calibration points, span from 100 to 940 ka [9–11, 17], thus largely predating the domestication events (~11 ka). These data suggest that many sub-haplogroups were already present among the bezoar populations and, therefore, that many lineages were included in the domesticated stocks. Previous studies on livestock species showed that phylogenies based on short mitochondrial sequences can be heavily affected by the confounding effects of homoplasies [18–21] and mitochondrial pseudogenes [22–24], which blur the real extent of lineage divergences/similarities. The available goat sequencing data are usually restricted to a few hundreds of control-region base pairs (bps), spanning from np 15431 to np 16643. Moreover, several complete goat mitochondrial genomes deposited in GenBank are probably chimeric or affected by NuMtS (i.e. nuclear sequences of mitochondrial origin) [22], including the previously adopted mtDNA goat reference sequence NC_005044 [22, 25]. In order to overcome these drawbacks, two recent papers have already extended the analysis to the mtDNA coding genes, but either failed to explore the complete mitochondrial molecule [17] or focused only on a new mtDNA haplotype [26].
We present the first extensive survey of the entire mitogenome variability based on 81 novel complete sequences from domestic goats (n = 76) and wild relatives (n = 5). This analysis allowed us to accurately define those (sub-) haplogroups involved in the domestication process(es), and to provide haplogroup coalescence estimates falling at the interface between Epipaleolithic and early Neolithic periods and expansion times falling into the Neolithic.
Results
The phylogeny of goat mitochondrial genomes
An initial collection of 758 mtDNA samples from C. aegagrus (n = 19) and C. hircus (n = 739; mostly from Western Eurasian breeds) was preliminarily characterized through control-region sequencing (Additional file 1: Table S1). This dataset, including 70 previously published samples [7], was used to build a haplotype network (data not shown). Overall, the network evidenced a high number of homoplasies and crosslinks, but it was useful to select 81 samples (from 76 domestic goats and five bezoars) for complete sequencing (Additional file 1: Table S2) using the criterion of including the widest possible range of mtDNA variation. The selected mitogenomes belonged to all known haplogroups, with few notable exceptions: i) the very rare haplogroup F, identified so far only in three domestic goats from Sicily [12], was not represented within our initial dataset of domestic goats; ii) the single control-region haplotype A in our C. aegagrus samples was re-classified as C by preliminary sequencing of some informative coding-region segments, which encompass diagnostic single nucleotide polymorphism (SNP) markers of A (at nps 3194/7839) and C (at nps 2885/3002/3131/3293/7657); iii) lastly, the few G control-region haplotypes were obtained from degraded DNA molecules, which allowed only partial coding-region sequencing that most likely confirmed the G affiliation (Additional file 1: Table S3). The 81 novel mitogenomes were compared with the revised goat reference sequence (GRS; NC_005044.2 – Additional file 1: Table S4). Various measures of molecular diversity were evaluated on the final dataset of 84 mitogenomes, which included 83 different haplotypes. After excluding ambiguous sites and indels (insertions/deletions), we identified a total number of 1003 variant sites (Table 1): 774 in the coding region (15414 nucleotides) and 229 in the D-loop (1213 nucleotides). Overall we observed an average number of 60.470 ± 16.628 nucleotide differences between two randomly chosen sequences. Figure S1 illustrates the distributions of nucleotide diversity (π) and total number of substitutions (continuous and dotted lines, respectively) along the mitogenome (Additional file 1: Figure S1). As expected, the highest diversity was observed around the HVS-I segment (hypervariable segment-I, from np 15,707 to np 16,187), with a peak of π = 0.059. The latter value is higher than those previously reported in horses [21]. Within the coding region, the highest number of variant sites was found in protein-coding genes (n = 655), mostly synonymous mutations. These data are consistent with previous studies on human and horse mitochondrial genomes [21, 27–29].
Table 1.
Distribution and recurrence of mutations in the 84 goat mtDNA sequences
| CONTROL | CODING | |||
|---|---|---|---|---|
| D-loop | rRNA | tRNA | mRNA | |
| Length in base pairsa | 1213 | 2529 | 1513 | 11370 |
| Invariable sites | 984 | 2456 | 1467 | 10715 |
| No. of variable sites | 229 | 73 | 46 | 655 |
| Proportion of variable sites | 0.233 | 0.030 | 0.031 | 0.061 |
| Sites with a single hit | 96 | 50 | 36 | 470 |
| Sites with two hits | 25 | 10 | 4 | 51 |
| Sites with three or more hits | 108 | 13 | 6 | 134 |
| Transitions | 221 | 70 | 45 | 627 |
| Transversions | 12 | 3 | 1 | 28 |
| Transition/Transversion ratio | 18.4 | 23.3 | 45.0 | 22.4 |
aLengths of protein-coding genes were readjusted and extended by considering the overlapping segments. Regarding the tRNA loci, the overlapping portions were counted only once
A parsimony approach was applied to infer evolutionary relationships from the final dataset of 84 complete mitogenomes. Eventually, only coding-region substitutions were included in the tree (Additional file 1: Figure S2) because of the extraordinary control-region variability (mainly around HVS-I, see Additional file 1: Figure S1) and high indels’ instability. The obtained topology confirmed all previously known control-region branches (A-G), but also revealed many different sub-branches, particularly within the major branch A. Seven novel sub-branches, named A1 to A7, were identified (at least three different haplotypes were required here to nominate a new clade, with the only notable exception of D1), all marked by coding-region transitions. In order to convert mutational distances into time over the entire mitogenome, the goat mtDNA sequences were compared with a published complete mitogenome (KF302445) from a Comisana sheep (Ovis aries) [30], used as an outgroup. Diverse maximum likelihood (ML) and Bayesian analyses were employed, as described in Materials and Methods. Initially, an ML tree based on synonymous mutations alone was estimated (Fig. 1). In fact, contrary to the number of potential factors causing time-dependent rates [31], synonymous mutations are virtually neutral and not subject to the effect of purifying selection, even though saturation might still be an issue with long time frames. Using CODEML and the mammalian mtDNA genetic code, we calibrated the synonymous molecular clock at 7.77 × 10−8 substitutions per year (at 3790 codons) or 1 substitution every 3397 years, after verifying the clock model hypothesis (p-value = 0.214). This mutation rate was also used to convert into time the rho estimates based on synonymous mutations (Table 2). The deepest node corresponds to the single Ancestral Goat Mitogenome (AGM), from which all modern goat mtDNA sequences derive, and was dated ~460 ka. The F bezoar mitogenome radiates first in the tree, then a major split (~130 ka ago) separates haplogroup C (dated ~80 ka) from the remaining mtDNA haplotypes. A subsequent branching separates two sister clades (B and A’D’G) both dated about 50 ka. Haplogroup B encompasses four samples, including GRS and one bezoar. The remaining wild samples belong to haplogroup D together with two domestic goats from Kyrgyzstan (with the same haplotype); affiliation to haplogroup D was also confirmed by considering recently published partial coding sequences [17]. The other bezoar mitogenomes, within haplogroups B and C, are ancestral to their most closely related domestic clusters B1 and C1a, dated 14.2 and 9.2 ka, respectively. These estimates are very similar to those of the A (12.8 ka) and G (9.0 ka) clusters, which include only C. hircus mitogenomes. In summary, all domestic clusters (A, B1, C1a and G) originated between 14 and 9 ka ago, as confirmed by both ML and rho statistics estimates (Table 2).
Fig. 1.
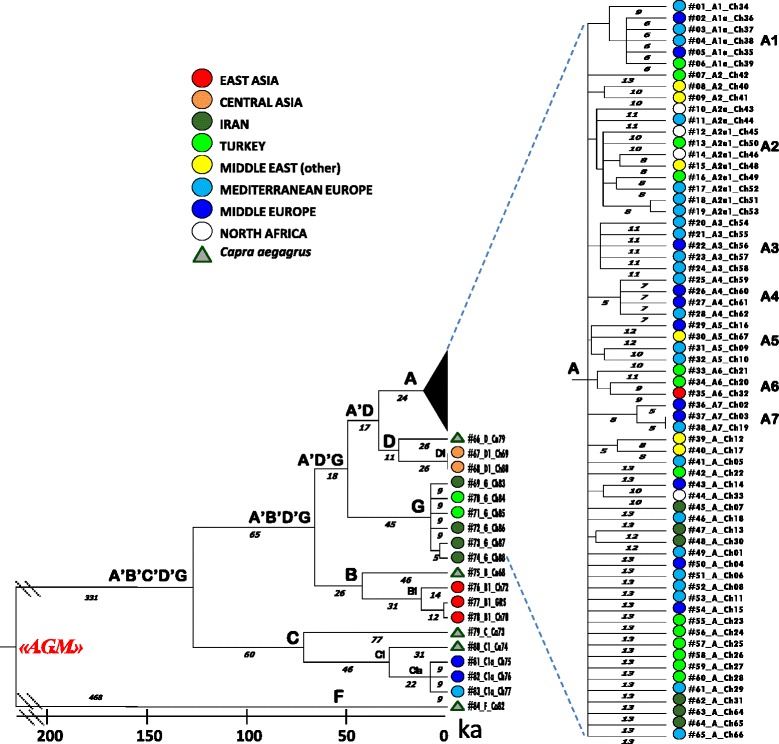
Schematic phylogeny of complete mtDNAs from modern domestic and wild goats. The tree encompasses 84 sequences and was rooted by using a published sheep (O. aries) sequence (not displayed). “A.G.M.” indicates the reconstructed Ancestral Goat Mitogenome. The topology was inferred by a maximum parsimony approach, while maximum likelihood (ML) time divergences based on synonymous substitutions are shown below the branches. The right inset shows the complete A branch rooted in D. Further details are given in Additional file 1: Table S4 and Figure S2
Table 2.
Age estimates based on different datasets
| ML (synonymous sub.s) | Rho (synonymous sub.s) | ML (all substitutions)a | ML (only coding region) | Rho (only coding region) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Node | T(ka) | ΔT(ka) | T(ka) | ΔT(ka) | T(ka) | ΔT(ka) | T(ka) | ΔT(ka) | T(ka) | ΔT(ka) |
| A.G.M. b | 467.7 | 59.2 | 457.9 | 37.3 | 652.0 | 34.0 | 734.9 | 61.1 | 715.5 | 51.2 |
| A’B’C’D’G | 136.7 | 21.3 | 125.7 | 17.4 | 186.6 | 12.2 | 214.3 | 24.0 | 187.4 | 23.0 |
| A’B’D’G | 71.2 | 12.2 | 57.3 | 10.6 | 115.9 | 8.3 | 124.8 | 15.6 | 95.6 | 14.9 |
| A’D’G | 53.5 | 9.8 | 42.5 | 8.8 | 89.3 | 6.8 | 93.1 | 12.8 | 68.5 | 12.0 |
| A’D | 36.9 | 8.1 | 25.4 | 5.8 | 66.0 | 5.9 | 63.6 | 11.3 | 42.0 | 8.1 |
| A | 12.8 | 1.9 | 14.1 | 1.4 | 23.4 | 1.4 | 23.0 | 2.2 | 23.7 | 1.9 |
| A1 | 9.2 | 2.8 | 10.2 | 3.5 | 20.9 | 1.9 | 18.6 | 3.5 | 19.2 | 4.9 |
| A2 | 12.8 | 2.9 | 16.2 | 3.9 | 19.7 | 1.4 | 21.0 | 2.5 | 26.3 | 4.9 |
| A3 | 10.7 | 1.9 | 15.6 | 3.3 | 19.7 | 1.5 | 19.5 | 2.7 | 24.7 | 4.5 |
| A4 | 7.4 | 2.5 | 6.8 | 2.4 | 16.8 | 1.9 | 16.1 | 3.9 | 14.4 | 3.9 |
| A5 | 12.8 | 3.7 | 12.7 | 3.5 | 21.5 | 1.8 | 19.9 | 3.4 | 19.6 | 4.7 |
| A6 | 11.2 | 2.1 | 15.9 | 4.5 | 17.1 | 1.9 | 20.4 | 3.0 | 24.7 | 6.1 |
| A7 | 4.8 | 4.2 | 3.4 | 4.4 | 11.1 | 1.9 | 9.3 | 4.0 | 6.9 | 3.6 |
| B | 45.7 | 9.4 | 47.6 | 9.1 | 70.9 | 6.2 | 76.7 | 11.9 | 78.3 | 12.7 |
| B1 | 14.2 | 5.1 | 12.5 | 4.4 | 23.9 | 3.4 | 24.9 | 7.1 | 23.4 | 6.9 |
| C | 77.2 | 14.1 | 78.1 | 12.4 | 120.1 | 9.1 | 122.3 | 16.9 | 114.5 | 16.2 |
| C1 | 31.4 | 7.6 | 31.4 | 7.3 | 54.3 | 5.2 | 51.6 | 10.1 | 49.4 | 9.8 |
| C1a | 9.2 | 3.4 | 9.1 | 3.2 | 15.9 | 2.2 | 17.4 | 5.0 | 16.5 | 4.7 |
| D | 26.3 | 7.2 | 32.8 | 7.8 | 45.2 | 5.6 | 49.4 | 10.9 | 60.4 | 11.8 |
| D1 | 0.0 | 11.0 | 0.0 | 5.1 | 0.0 | 7.9 | 0.0 | 16.5 | 0.0 | 6.2c |
| G | 9.0 | 2.7 | 9.1 | 2.5 | 23.1 | 2.2 | 22.1 | 4.6 | 22.7 | 4.3 |
aThe entire genome was partitioned into six datasets: 1st, 2nd and 3rd positions of the codons, RNAs, HVS-I and the remainder of the control region
bAncestral Goat Mitogenome
cA 95 % C.I. for the age of D1 is 0 to ln(20)/n in units of 4120 years
A Bayesian Skyline Plot (BSP) analysis was carried out to assess population expansions. The overall BSP points to a steep increase of the female effective population size about 12–10 ka ago (Fig. 2). This analysis was performed on the complete molecule after establishing six different partitions (1st, 2nd and 3rd codon positions, RNA genes, HVS-I and other control-region segments). The same partitions were also considered to perform ML analyses on the entire mitogenome. The final outcomes were an overall mutational rate of 3.95 × 10−8 substitutions per nucleotide per year (1 mutation every 1522 years) on the entire molecule and 1.57 × 10−8 substitutions per nucleotide per year (or 1 mutation every 4120 years) on the coding region alone. Both analyses revealed that the molecular clock could not be rejected (p-values > 0.05) when employing the complex GTR/REV model. A clear sign of purifying selection is apparent when calculating the non-synonymous/synonymous ratio, i.e. (ɷ) = 0.190 (Table 3). As expected, this ratio is significantly lower (p-value < < 0.001; Fisher’s exact test) in the deep portion of the tree and the comparison among domestic branches reveals slightly significant differences (χ2 p-value = 0.026) (Table 3); in particular the ɷ ratio of A is much higher than previously reported (ɷ = 0.049) [17]. Similarly, age values of younger clades are much higher when considering the entire panel of mutations rather than those based on synonymous changes only (Fig. 3), probably because the effect of purifying selection is incomplete and all the negatively selectable characters are still included [29].
Fig. 2.
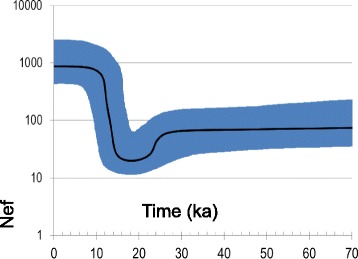
Bayesian Skyline Plot showing the goat population size trend with a generation time of 4.5 years [60]. The Y axis indicates the effective number of females. The thick solid line is the median estimate and the grey shading shows the 95 % highest posterior density limits. The time axis is limited to 100 ka, beyond that time the curve remains linear
Table 3.
Rates of non-synonymous/synonymous differences (dN/dS) on the goat phylogeny
| dN | dS | dN/dS | |
|---|---|---|---|
| Entire Phylogeny | 114 | 600 | 0.190 |
| Pre-domestic branches | 46 | 361 | 0.127 |
| Domestic branches | 68 | 239 | 0.284 |
| Comparisons within domestic haplogroups | |||
| A | 46 | 199 | 0.231 |
| B1 | 3 | 9 | 0.333 |
| C1a | 3 | 8 | 0.375 |
| D1 | 4 | 9 | 0.444 |
| G | 12 | 14 | 0.857 |
Fig. 3.
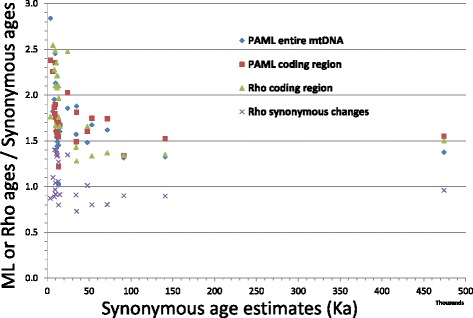
Comparison of ML and rho-statistics ages relative to the ML synonymous estimates. See Table 2 for further details
Geographic distributions of goat mtDNA haplogroups
An analysis of the geographic distribution of goat haplogroups in Eurasia and Africa (Fig. 4), based on the control-region data currently available in literature or deposited in GenBank, confirms an overwhelming predominance of haplogroup A (~90.5 %) among domestic goats all over the world (including the Americas, Additional file 2: Table S5). The second most frequent haplogroup is B (~6.5 %), more common in Asia and Southern Africa, but also present in Europe. Haplogroup G (0.9 %) is restricted to North-central Africa and Asia, while the presence of haplogroups C and D is limited to Eurasia with an average frequency of 1.4 % and 0.6 %, respectively. Finally, haplogroup F was identified only in three Sicilian domestic samples. The bezoar samples analyzed so far are mainly from the Middle East (Iran, Turkey and Jordan) and South Asia (India, Pakistan and Bangladesh), where they are mostly characterized by the occurrence of haplogroups C (37.4 and 42.4 %) and F (28.9 and 18.2 %). C. aegagrus is also represented by haplogroups D (12.9 %), A (6.4 %), B (3.7 %) and G (2.2 %) in the Middle East. Wild goats are also found on some Mediterranean islands (C. aegagrus cretica), even though the widely accepted opinion is that they derive from the feralization of very early domestic animals [32]. Finally, the few ancient mtDNA haplotypes (Additional file 2: Table S5) were found in goat remains excavated in Central/East Asia (A, B and D), the Near East (only A) and Europe (B in mainland; A and C in the Mediterranean area). The presence of A and C in the Mediterranean area since ancient times could be due to the Neolithic spread attested by the first appearance of Cardial pottery in the Eastern Adriatic since 8.5 ka ago [33–35].
Fig. 4.
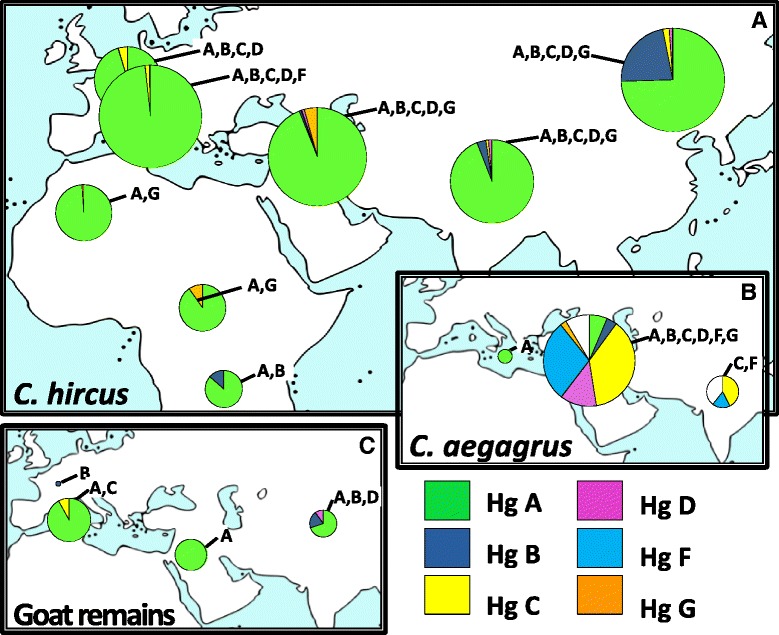
Spatial frequency distributions of goat mtDNA haplogroups in different geographic areas based on different datasets: modern breeds (C. hircus) a ; wild goats (C. aegagrus) b; and ancient goat remains c. See Additional file 2 (Table S5) for more information
Discussion and conclusions
Domestication of C. aegagrus
Wild goats were widely distributed throughout Southwestern Eurasia during the Middle Paleolithic (300 to 45 ka ago). In an attempt to reconstruct past demographic histories based on phylogenetic inferences, the tree in Fig. 1 suggests that a drastic bottleneck occurred during the Late Eurasian Saalian glacial (~130–160 ka ago) [36–39], which is compatible with the age of the major macro-haplogroup A’B’C’D’G. During the following Riss-Würm interglacial relapse (~115–130 ka ago), the A’B’D’G lineages evolved and expanded independently from the C-derived populations. Many lineages did not survive the following Würm glacial period (~12–71 ka ago) and the drastic climatic changes during the Last Glacial Maximum (LGM, ~19.5–25 ka ago), but some of them, corresponding to the principal goat haplogroups, were preserved in the Near East refuge areas [40], survived the severe drop in temperature known as the Younger Dryas (~11.5–12.7 ka ago) [41, 42], and provided the necessary substrate of variability for later goat domestication and management in an area from the Zagros Mountains to Southeastern Anatolia, as testified by the abundance of goat remains in Neolithic sites from that regions [8, 43].
The main goat haplogroups were identified through control-region analyses following an approach similar to that adopted for other domesticated species, such as sheep [3, 30, 44–46]. However, in the case of both horses and bovines, the mtDNA haplogroup classification received a major makeover from the exploration of the entire mitogenome variability [21, 47–49]. A common feature of all livestock phylogenies is that the control-region molecular clocks turned out to be very inadequate for an accurate dating of mitochondrial lineage divergences. When mtDNA protein-coding genes were considered [17], goat haplogroups were dated to the Middle Paleolithic, thus suggesting that multiple related lineages were domesticated ~11 ka ago in an area spanning from Southeastern Anatolia in Turkey to Zagros Mountains in Iran, by incorporating pre-existing variation [7–9, 17]. This domestication center, further confirmed by zooarchaeological data [43], left a clear signature in the mtDNA gene pool of current domestic breeds, as attested by the large predominance of A haplotypes. Our analyses allowed us to assess the coding-region variability within this clade (Additional file 2: Table S6) and to date for the first time this major goat haplogroup to the Epipaleolithic period; a few A sub-haplogroups were also phylogenetically defined and dated to the early Neolithic. The implication is that more than 90 % of current domestic goats might descend from a single foundress, represented by the internal node A in our phylogeny. Obviously, the absence of A bezoars in our dataset is an issue, as well as the long-term calibration point, which might represent an underestimate of the true sheep/goat divergence. Thus, by also considering the alternative hypothesis of many founder lineages within haplogroup A independently proposed by Naderi et al. [7] and Nomura et al. [17], we might suggest that at least seven of them have been identified in our dataset as represented by the ancestral mitogenomes of sub-haplogroups A1–A7.
Moreover, in some instances, our phylogenetic reconstruction clearly shows that other domestic clusters are nested within wild branches and each of them descends from a unique wild haplotype. Therefore, excluding the rare but possible occurrence of stochastic backcrossing between wild and domestic animals, the first domestication process probably involved only four additional female goat populations corresponding to one founder haplotype for each of the other domestic lineages, i.e. B1, D1, C1a and G,. We cannot exclude that, when analyzing further C. aegagrus samples at the maximum level of resolution, several additional sub-groups could be identified within each haplogroup, as previously discussed for the A lineages. Likewise, the possibility that some of the mitochondrial haplotypes belonging to lineages B1, D1 and C1a and presently found in domestic animals might derive from introgression of wild lineages cannot be completely ruled out. Yet, events of introgression from wild animals into domestic herds are incidental and usually male-mediated [50], and they were not identified so far even when analyzing a larger dataset of wild and domestic control-region sequences [7, 13]. We have also verified that all available control-region haplotypes (including those from the present study) belonging to haplogroups B, C and D could be specifically assigned either to wild or domestic branches as expected from individual phenotypes (i.e. wild or domestic), without finding any notable exceptions.
The Bayesian analysis of our goat sequences also shows that, after domestication, the early domestic populations soon experienced a drastic demographic expansion about 12–10 ka ago (Fig. 2). However, since our tree includes samples from a very wide geographical area, we cannot exclude that additional and perhaps more recent signals of local demographic expansions could be detected by analyzing larger and more geographically structured datasets, particularly along the domestication routes.
A more fascinating scenario could also be envisioned for haplogroup C since this haplogroup might represent a marker of a concomitant secondary domestication, as already suggested by control-region analyses [7]. To support this scenario, our age estimate for C1a is compatible with the first domestication wave and, more importantly, the C clade represents an independent branch in our tree, which is separated from the A’B’D’G cluster by over one hundred thousand years, thus suggesting a genetically distinct population origin.
The prevalence of haplogroup A: a close similarity between cattle and goat domestication
Phylogenetic analyses based on entire mitogenomes revealed that, similarly to horses and taurine cattle, the ancient goat populations might have passed through a severe bottleneck during the Late Saalian glacial maximum with a single sequence (A’B’C’D’G) from that period as the common ancestor of most goat mitogenomes. However, the recent goat evolutionary history suggests more similarities with Bos primigenius domestication (Additional file 2: Table S7). Present-day cattle and goat mitogenomes belong to very few haplogroups, with most of them (>85 % in Eurasia) falling within a single branch (T3 and A, respectively), whose sequence coalescence time corresponds to a very similar time estimate of about 11–13 ka. As in the case of the cattle T3 branch, this study and other published data support a Neolithic origin for all goat A mitogenomes (possibly differentiated into some sub-haplogroups, e.g. A1–A7) from a Middle Eastern bezoar population. At least four other minor lineages (B1, C1a, D1 and G) directly related to bezoar ancestors (i.e. B, C1 and D) were domesticated. However, unlike the bovine minor clades P and Q that suggest a possible European domestication or introgression, the geographical distribution of the low-frequency goat clusters indicates that secondary domestication foci were located in the same area between Anatolian Turkey and Iran. This leaves open the possibility of a more eastward Iranian center involving the furthest phylogenetically-related haplotype C1. Moreover, the new coding-region diagnostic mutational motifs defined in the present study (Additional file 2: Table S6) could be employed to revise phylogenetic relationships between modern breeds and ancient remains with the overall objective of testing the proposed scenario of secondary early domestication processes that took place before the occurrence of morphological modifications, and evaluating whether selection differentially affected mitogenome variants during the development of economically important breeds.
Methods
Sequencing of goat mitochondrial genomes
All experimental procedures were reviewed and approved by the Animal Research Ethics Committee of the University of Pavia, Prot. 2/2010 (October 15th, 2010), in accordance with the European Union Directive 86/609. Prior to the sequencing of entire mtDNA molecules, a preliminary sequence analysis of the control regions was performed. This allowed for the selection of 81 samples with good quality DNA and encompassing the widest range of mutational motifs. During the first phases of the experimental work we used the established Sanger sequencing approach rather than new sequencing technologies since, at that time, the latter did not yet guarantee complete coverage and could be still prone to artificial ambiguities [51]. The sequencing protocol was similar to those previously and successfully used for human and livestock mitogenomes [21, 30, 47, 52]. The oligonucleotides used to amplify and sequence the goat mitochondrial genome are reported in Additional file 2: Table S8. They were checked through GenBank BLAST to avoid amplification of nuclear insertions of mitochondrial sequences (NuMtS) [53]. In the last phases of the experimental work, thanks to the acquired affability and accuracy of the next generation techniques, additional 28 samples were amplified with the same oligonucleotide pairs, pooled together (5 μg of DNA in total) and sequenced on the Illumina Genome Analyzer IIx, platform at the IGA Technology Services, Udine, Italy.
Several parameters of the mtDNA sequence variation were estimated by using DnaSP 5.1. The variation of nucleotide diversity (π) along the entire mtDNA was estimated by assessing windows of 100 bps with step size of 50 bps centered at the midpoint. For an estimation of the synonymous/non-synonymous sites we created an alignment containing only the protein-coding genes, with the ND6 gene adjusted to present the same reading direction as the other genes. The “stop codons” were excluded from the analysis. Overlapping loci were counted twice leading to a final alignment of protein-coding genes equal to 11370 bps.
Phylogeny construction and molecular divergence
The phylogeny construction was performed as described elsewhere [21, 30, 47, 52] and confirmed using an adapted version of mtPhyl 3.0 for a maximum parsimony (MP) analysis [54]. The modified .txt files are available upon request.
We constructed an MP tree including 84 goat mitogenomes (without D-loop) rooted on sheep, O. aries, mitogenome (KF302445). A first maximum likelihood (ML) analysis was performed using PAML X [55] by considering only the protein coding genes (and synonymous mutations). As already mentioned, the ND6 gene was reverse-complemented to present the same reading direction as the other genes and, under the vertebrate mitochondrial genetic code, the non-synonymous substitutions were excluded from the alignment and replaced with the ancestral base pairs. The “stop codons” were excluded from the analysis. Lengths of protein-coding genes were readjusted and extended by considering the overlapping segments. The final alignment (11370 bps long) was analyzed with CODEML, which calculates a synonymous mutation rate taking into account the mitochondrial genetic code. The second survey was carried out by considering six partitions in the molecule: one corresponds to the RNA genes (tDNA and 12S/16S rDNA), one to each codon position of the protein-coding genes (CDS), one to HVS-I, and one to the remaining D-loop sequences. In the final alignment, the rDNA and tDNA, CDS, HVS-I and D-loop segments were 4042 (overlapping sites between rDNA and tDNA were counted once), 11370 (see above), 481 and 732 bps long respectively. The non-coding region between np 5160 and np 5191 was not considered, but we checked that it was invariable. The best model able to describe the phylogenetic relationships among taxa was selected by using jModelTest [56]. Eventually, separate analyses were performed on the coding region and on the entire mitogenome by assuming a GTR/REV mutation model, a molecular clock and gamma-distributed rates, approximated by a discrete distribution with 32 categories. In order to check the clock hypothesis, likelihood ratio tests were applied with and without molecular clocks. The ML estimates were also compared with those directly obtained on the MP trees by using mtPhyl as averaged distances (ρ) of the haplotypes of a clade to the respective root haplotype, also known as rho-statistics [57], accompanied by a heuristic estimate of the standard error (σ).
We also obtained a Bayesian skyline plot (BSP) [58] from the goat phylogeny using BEAST 2.2.1 software [59]. We run 10,000,000 iterations with samples drawn every 5000 steps and used a generation time of 4.5 years [60]. BSPs provided a good visualization of the increase in diversity in the tree by estimating effective population sizes through time.
Calibrating the goat mtDNA molecular clock
For the calibration point in the maximum likelihood analyses, we assumed an estimated bifurcation time between sheep and goat of 6,000,000 years (assuming a 95 % interval of 5–7,000,000 years in the BEAST analysis) based on fossil evidence as already used by Sultana et al. [11]. Internal calibration points were not available. Considering the time-dependency of molecular rate estimates [31], the use of a paleontological calibration point means that we are prone to possible biases mainly generated by non-synonymous substitutions and mutations affecting tRNA/rRNA genes (purifying selection) and the control region (tendency to saturation due to the high evolution rate). This long-term calibration point probably represents an underestimate of the true sheep/goat divergence and might have had a considerable impact on the date estimates.
Availability of supporting data
Sequences of the novel goat mitogenomes have been deposited in GenBank under accession numbers KR059146 - KR059226 (81 complete mtDNAs) and KR059227 - KR059851 (625 mtDNA control regions). Phylogenetic data have been deposited in TreeBase (http://purl.org/phylo/treebase/phylows/study/TB2:S18595).
Acknowledgments
We thank Dr. Ugo A. Perego and Dr. Ivan Fowler for proofreading the manuscript. This research received support from the Italian Ministry of Education, University and Research: Progetti FIRB-Futuro in Ricerca 2008 and 2012 (to AA and AO), and PRIN-Progetti Ricerca di Interesse Nazionale 2007 (to PAM), 2009 and 2012 (to AA); and from the EU FP7 project NextGen (Grant KBBE-2009-1-1-03).
Abbreviations
- AGM
Ancestral Goat Mitogenome
- BEAST
Bayesian Evolutionary Analysis Sampling Trees
- BLAST
Basic Local Alignment Search Tool
- Bps
Base pairs
- BSP
Bayesian Skyline Plot
- CDS
Coding DNA Sequence
- D-loop
Displacement Loop
- dN/dS
Non-synonymous/synonymous ratio
- DnaSP
DNA Sequence Polymorphism
- FAO
Food and Agriculture Organization
- GRS
Goat Reference Sequence
- GTR/REV
General Time Reversible
- Hg
Haplogroup
- HVS-I
Hypervariable segment-I
- Indels
Insertions/deletions
- Ka
Kilo annos
- LGM
Last Glacial Maximum
- ML
Maximum Likelihood
- mtDNA
Mitochondrial DNA
- ND6
NADH, ubiquinone oxidoreductase core subunit 6
- NuMt
Nuclear Mitochondrial DNA
- PAML
Phylogenetic Analysis Using Maximum Likelihood
- rDNA
Ribosomal DNA
- rRNA
Ribosomal RNA
- SNP
Single Nucleotide Polymorphism
- tDNA
Transfer DNA
- tRNA
Transfer RNA
Additional files
Table S1. Sources for the 758 goat control-region sequences. Table S2. Control-region haplotypes and haplogroup classification of the 758 mtDNA sequences from Capra aegagrus (n = 19) and Capra hircus (n = 739). Table S3. Partial coding-region haplotypes and haplogroup classification of two bezoar mtDNAs. Table S4. Source and haplogroup affiliation of the goat complete mtDNA sequences. Figure S1. Nucleotide diversity and total number of substitutions along the entire mtDNA. Figure S2. A putative most parsimonious tree of 84 complete mtDNA sequences from goats. (XLSX 1268 kb)
Table S5. Goat haplogroup frequencies based on modern and ancient control-region mtDNA data from this study and downloaded from GenBanka. Table S6. Diagnostic mutational motifs of goat mtDNA haplogroups and sub-haplogroups. Table S7. A comparison of the phylogeographic features of goat, taurine and horse mtDNA haplogroups identified by analyzing domestic breeds from Eurasia. Table S8. Oligonucleotides used to amplify and to sequence (Sanger method) the goat mitochondrial genome. (PDF 652 kb)
Footnotes
Licia Colli and Hovirag Lancioni contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
This study was conceived by LC, HL, FP, PT, PAM and AA. LC, HL, IC and AA designed and performed molecular experiments. LC, HL, IC, AO, MRC, MP, MR, FG, VB, PAM and AA conducted the genetic analysis. ER, MTS, BP, FB, EC, MP, MR, FG, PAM and AA contributed reagents/materials/analysis tools. WZ, SN, SMFV, SA, HRR and PL performed the collection of biological samples. LC, HL, PAM and AA wrote the paper. All authors read and approved the final manuscript.
Contributor Information
Licia Colli, Email: licia.colli@unicatt.it.
Hovirag Lancioni, Email: hovirag.lancioni@unipg.it.
Irene Cardinali, Email: cardinali_irene@libero.it.
Anna Olivieri, Email: anna.olivieri@unipv.it.
Marco Rosario Capodiferro, Email: marcocapo87@yahoo.it.
Marco Pellecchia, Email: marcopellecchia@koineambiente.com.
Marcin Rzepus, Email: Marcin.Rzepus@unicatt.it.
Wahid Zamani, Email: zamani_wahid@yahoo.com.
Saeid Naderi, Email: naderi@guilan.ac.ir.
Francesca Gandini, Email: francesca.gandini01@universitadipavia.it.
Seyed Mohammad Farhad Vahidi, Email: smf.vahidi@gmail.com.
Saif Agha, Email: safsoof23@hotmail.com.
Ettore Randi, Email: ettore.randi@isprambiente.it.
Vincenza Battaglia, Email: vincenza.battaglia@unipv.it.
Maria Teresa Sardina, Email: mariateresa.sardina@unipa.it.
Baldassare Portolano, Email: baldassare.portolano@unipa.it.
Hamid Reza Rezaei, Email: hamid.r.rezaei@gmail.com.
Petros Lymberakis, Email: lyberis@nhmc.uoc.gr.
Frédéric Boyer, Email: frederic.boyer@ujf-grenoble.fr.
Eric Coissac, Email: eric.coissac@inria.fr.
François Pompanon, Email: francois.pompanon@ujf-grenoble.fr.
Pierre Taberlet, Email: pierre.taberlet@ujf-grenoble.fr.
Paolo Ajmone Marsan, Email: paolo.ajmone@unicatt.it.
Alessandro Achilli, Email: alessandro.achilli@unipv.it.
References
- 1.FAO. The State of the World’s Animal Genetic Resources for Food and Agriculture. In: Pilling D, Rischkowsky B, editors. Rome; 2007.
- 2.MacHugh DE, Bradley DG. Livestock genetic origins: goats buck the trend. Proc Natl Acad Sci U S A. 2001;98(10):5382–5384. doi: 10.1073/pnas.111163198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zeder MA. Domestication and early agriculture in the Mediterranean Basin: Origins, diffusion, and impact. Proc Natl Acad Sci U S A. 2008;105(33):11597–11604. doi: 10.1073/pnas.0801317105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zeder MA, Emshwiller E, Smith BD, Bradley DG. Documenting domestication: the intersection of genetics and archaeology. Trends Genet. 2006;22(3):139–155. doi: 10.1016/j.tig.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 5.Vigne JD, Peters J, Helmer D. The first steps of animal domestication: new archaeozoological approaches. Oxford: Oxbow; 2005. [Google Scholar]
- 6.Taberlet P, Coissac E, Pansu J, Pompanon F. Conservation genetics of cattle, sheep, and goats. C R Biol. 2011;334(3):247–254. doi: 10.1016/j.crvi.2010.12.007. [DOI] [PubMed] [Google Scholar]
- 7.Naderi S, Rezaei HR, Pompanon F, Blum MG, Negrini R, Naghash HR, et al. The goat domestication process inferred from large-scale mitochondrial DNA analysis of wild and domestic individuals. Proc Natl Acad Sci U S A. 2008;105(46):17659–17664. doi: 10.1073/pnas.0804782105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zeder MA, Hesse B. The initial domestication of goats (Capra hircus) in the Zagros mountains 10,000 years ago. Science. 2000;287(5461):2254–2257. doi: 10.1126/science.287.5461.2254. [DOI] [PubMed] [Google Scholar]
- 9.Joshi MB, Rout PK, Mandal AK, Tyler-Smith C, Singh L, Thangaraj K. Phylogeography and origin of Indian domestic goats. Mol Biol Evol. 2004;21(3):454–462. doi: 10.1093/molbev/msh038. [DOI] [PubMed] [Google Scholar]
- 10.Luikart G, Gielly L, Excoffier L, Vigne JD, Bouvet J, Taberlet P. Multiple maternal origins and weak phylogeographic structure in domestic goats. Proc Natl Acad Sci U S A. 2001;98(10):5927–5932. doi: 10.1073/pnas.091591198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sultana S, Mannen H, Tsuji S. Mitochondrial DNA diversity of Pakistani goats. Anim Genet. 2003;34(6):417–421. doi: 10.1046/j.0268-9146.2003.01040.x. [DOI] [PubMed] [Google Scholar]
- 12.Sardina MT, Ballester M, Marmi J, Finocchiaro R, van Kaam JB, Portolano B, et al. Phylogenetic analysis of Sicilian goats reveals a new mtDNA lineage. Anim Genet. 2006;37(4):376–378. doi: 10.1111/j.1365-2052.2006.01451.x. [DOI] [PubMed] [Google Scholar]
- 13.Naderi S, Rezaei HR, Taberlet P, Zundel S, Rafat SA, Naghash HR, et al. Large-scale mitochondrial DNA analysis of the domestic goat reveals six haplogroups with high diversity. PLoS One. 2007;2(10):e1012. doi: 10.1371/journal.pone.0001012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pereira F, Queiros S, Gusmao L, Nijman IJ, Cuppen E, Lenstra JA, et al. Tracing the history of goat pastoralism: new clues from mitochondrial and Y chromosome DNA in North Africa. Mol Biol Evol. 2009;26(12):2765–2773. doi: 10.1093/molbev/msp200. [DOI] [PubMed] [Google Scholar]
- 15.Piras D, Doro MG, Casu G, Melis PM, Vaccargiu S, Piras I, et al. Haplotype affinities resolve a major component of goat (Capra hircus) MtDNA D-loop diversity and reveal specific features of the Sardinian stock. PLoS One. 2012;7(2):e30785. doi: 10.1371/journal.pone.0030785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fernández H, Hughes S, Vigne JD, Helmer D, Hodgins G, Miquel C, et al. Divergent mtDNA lineages of goats in an Early Neolithic site, far from the initial domestication areas. Proc Natl Acad Sci U S A. 2006;103(42):15375–15379. doi: 10.1073/pnas.0602753103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nomura K, Yonezawa T, Mano S, Kawakami S, Shedlock AM, Hasegawa M, et al. Domestication process of the goat revealed by an analysis of the nearly complete mitochondrial protein-encoding genes. PLoS One. 2013;8(8):e67775. doi: 10.1371/journal.pone.0067775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCracken K, Sorenson M. Is homoplasy or lineage sorting the source of incongruent mtDNA and nuclear gene trees in the stiff-tailed ducks (Nomonyx-Oxyura)? Syst Biol. 2005;54(1):35–55. doi: 10.1080/10635150590910249. [DOI] [PubMed] [Google Scholar]
- 19.Achilli A, Bonfiglio S, Olivieri A, Malusa A, Pala M, Kashani BH, et al. The multifaceted origin of taurine cattle reflected by the mitochondrial genome. PLoS One. 2009;4(6):e5753. doi: 10.1371/journal.pone.0005753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bonfiglio S, Ginja C, De Gaetano A, Achilli A, Olivieri A, Colli L, et al. Origin and spread of Bos taurus: new clues from mitochondrial genomes belonging to haplogroup T1. PLoS One. 2012;7(6):e38601. doi: 10.1371/journal.pone.0038601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Achilli A, Olivieri A, Soares P, Lancioni H, Kashani BH, Perego UA, et al. Mitochondrial genomes from modern horses reveal the major haplogroups that underwent domestication. Proc Natl Acad Sci U S A. 2012;109(7):2449–2454. doi: 10.1073/pnas.1111637109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hassanin A, Bonillo C, Nguyen BX, Cruaud C. Comparisons between mitochondrial genomes of domestic goat (Capra hircus) reveal the presence of numts and multiple sequencing errors. Mitochondrial DNA. 2010;21(3–4):68–76. doi: 10.3109/19401736.2010.490583. [DOI] [PubMed] [Google Scholar]
- 23.Kim H, Lee T, Park W, Lee JW, Kim J, Lee BY, et al. Peeling back the evolutionary layers of molecular mechanisms responsive to exercise-stress in the skeletal muscle of the racing horse. DNA Res. 2013;20(3):287–298. doi: 10.1093/dnares/dst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moyle RG, Jones RM, Andersen MJ. A reconsideration of Gallicolumba (Aves: Columbidae) relationships using fresh source material reveals pseudogenes, chimeras, and a novel phylogenetic hypothesis. Mol Phylogenet Evol. 2013;66(3):1060–1066. doi: 10.1016/j.ympev.2012.11.024. [DOI] [PubMed] [Google Scholar]
- 25.Parma P, Feligini M, Greeppi G, Enne G. The complete nucleotide sequence of goat (Capra hircus) mitochondrial genome. Goat mitochondrial genome. DNA Seq. 2003;14(3):199–203. doi: 10.1080/1042517031000089487. [DOI] [PubMed] [Google Scholar]
- 26.Doro MG, Piras D, Leoni GG, Casu G, Vaccargiu S, Parracciani D, et al. Phylogeny and patterns of diversity of goat mtDNA haplogroup A revealed by resequencing complete mitogenomes. PLoS One. 2014;9(4):e95969. doi: 10.1371/journal.pone.0095969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mishmar D, Ruiz-Pesini E, Golik P, Macaulay V, Clark AG, Hosseini S, et al. Natural selection shaped regional mtDNA variation in humans. Proc Natl Acad Sci U S A. 2003;100(1):171–176. doi: 10.1073/pnas.0136972100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kivisild T, Shen P, Wall DP, Do B, Sung R, Davis K, et al. The role of selection in the evolution of human mitochondrial genomes. Genetics. 2006;172(1):373–387. doi: 10.1534/genetics.105.043901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Soares P, Ermini L, Thomson N, Mormina M, Rito T, Rohl A, et al. Correcting for purifying selection: an improved human mitochondrial molecular clock. Am J Hum Genet. 2009;84(6):740–759. doi: 10.1016/j.ajhg.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lancioni H, Di Lorenzo P, Ceccobelli S, Perego UA, Miglio A, Landi V, et al. Phylogenetic relationships of three Italian merino-derived sheep breeds evaluated through a complete mitogenome analysis. PLoS One. 2013;8(9):e73712. doi: 10.1371/journal.pone.0073712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ho SY, Lanfear R, Bromham L, Phillips MJ, Soubrier J, Rodrigo AG, et al. Time-dependent rates of molecular evolution. Mol Ecol. 2011;20(15):3087–3101. doi: 10.1111/j.1365-294X.2011.05178.x. [DOI] [PubMed] [Google Scholar]
- 32.Kahila Bar-Gal G, Smith P, Tchernov E, Greenblatt C, Ducos P, Gardeisen A, et al. Genetic evidence for the origin of the agrimi goat (Capra aegagrus cretica) J Zool. 2002;256(3):369–377. doi: 10.1017/S0952836902000407. [DOI] [Google Scholar]
- 33.Battaglia V, Fornarino S, Al-Zahery N, Olivieri A, Pala M, Myres NM, et al. Y-chromosomal evidence of the cultural diffusion of agriculture in Southeast Europe. Eur J Hum Genet. 2009;17(6):820–830. doi: 10.1038/ejhg.2008.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paschou P, Drineas P, Yannaki E, Razou A, Kanaki K, Tsetsos F, et al. Maritime route of colonization of Europe. Proc Natl Acad Sci U S A. 2014;111(25):9211–9216. doi: 10.1073/pnas.1320811111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fernández E, Pérez-Pérez A, Gamba C, Prats E, Cuesta P, Anfruns J, et al. Ancient DNA analysis of 8000 B.C. near eastern farmers supports an early neolithic pioneer maritime colonization of Mainland Europe through Cyprus and the Aegean Islands. PLoS Genet. 2014;10(6):e1004401. doi: 10.1371/journal.pgen.1004401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Richter J. When Did the Middle Paleolithic Begin? In: Conard NJ, Richter J, editors. Neanderthal lifeways, subsistence and technology: one hundred fifty years of Neanderthal study Vertebrate Paleobiology and Paleoanthropology. Berlin: Springer; 2011. [Google Scholar]
- 37.Ehlers J, Gibbard PL, Hughes PD. Quaternary glaciations - extent and chronology: a closer look. Amsterdam: Elsevier; 2011. [Google Scholar]
- 38.Colleoni F. On the Late Saalian glaciation (160–140 ka) – a climate modeling study. Stockholm: Stockholm University; 2009. [Google Scholar]
- 39.Ferrigno JG. Glaciers of the Middle East and Africa - Glaciers of Iran. In: Williams RS, Ferrigno JG, editors. Satellite Image Atlas of Glaciers of the World. Washington, DC: Dept. of the Interior, U.S. Geological Survey; 1991. [Google Scholar]
- 40.Pala M, Olivieri A, Achilli A, Accetturo M, Metspalu E, Reidla M, et al. Mitochondrial DNA signals of late glacial recolonization of Europe from Near Eastern refugia. Am J Hum Genet. 2012;90(5):915–924. doi: 10.1016/j.ajhg.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clutton-Brock J. A natural history of domesticated mammals, 2nd Edition edn. Cambridge: Cambridge University Press; 1999. [Google Scholar]
- 42.MacFadden BJ. Fossil horses: systematics, paleobiology, and evolution of the family Equidae. Cambridge: Cambridge University Press; 1992. [Google Scholar]
- 43.Groves C, Leslie D, Huffman B, Valdez R, Habibi K, Weinberg P, et al. Bovidae (Hollow-Horned Ruminants) In: Wilson DE, Mittermeier RA, et al., editors. Handbook of the Mammals of the World Volume 2 Hoofed Mammals. Barcelona: Lynx Edicions; 2011. [Google Scholar]
- 44.Driscoll CA, Macdonald DW, O’Brien SJ. From wild animals to domestic pets, an evolutionary view of domestication. Proc Natl Acad Sci U S A. 2009;106(1):9971–9978. doi: 10.1073/pnas.0901586106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Groeneveld LF, Lenstra JA, Eding H, Toro MA, Scherf B, Pilling D, et al. Genetic diversity in farm animals--a review. Anim Genet. 2010;41(1):6–31. doi: 10.1111/j.1365-2052.2010.02038.x. [DOI] [PubMed] [Google Scholar]
- 46.Taberlet P, Valentini A, Rezaei HR, Naderi S, Pompanon F, Negrini R, et al. Are cattle, sheep, and goats endangered species? Mol Ecol. 2008;17(1):275–284. doi: 10.1111/j.1365-294X.2007.03475.x. [DOI] [PubMed] [Google Scholar]
- 47.Achilli A, Olivieri A, Pellecchia M, Uboldi C, Colli L, Al-Zahery N, et al. Mitochondrial genomes of extinct aurochs survive in domestic cattle. Curr Biol. 2008;18(4):R157–R158. doi: 10.1016/j.cub.2008.01.019. [DOI] [PubMed] [Google Scholar]
- 48.Lippold S, Matzke NJ, Reissmann M, Hofreiter M. Whole mitochondrial genome sequencing of domestic horses reveals incorporation of extensive wild horse diversity during domestication. BMC Evol Biol. 2011;11:328. doi: 10.1186/1471-2148-11-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bonfiglio S, Achilli A, Olivieri A, Negrini R, Colli L, Liotta L, et al. The enigmatic origin of bovine mtDNA haplogroup R: sporadic interbreeding or an independent event of Bos primigenius domestication in Italy? PLoS One. 2010;5(12):e15760. doi: 10.1371/journal.pone.0015760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kikkawa Y, Takada T, Sutopo, Nomura K, Namikawa T, Yonekawa H, et al. Phylogenies using mtDNA and SRY provide evidence for male-mediated introgression in Asian domestic cattle. Anim Genet. 2003;34(2):96–101. doi: 10.1046/j.1365-2052.2003.00956.x. [DOI] [PubMed] [Google Scholar]
- 51.Bandelt HJ, Salas A. Current next generation sequencing technology may not meet forensic standards. Forensic Sci Int Genet. 2012;6(1):143–145. doi: 10.1016/j.fsigen.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 52.Olivieri A, Achilli A, Pala M, Battaglia V, Fornarino S, Al-Zahery N, et al. The mtDNA legacy of the Levantine early Upper Palaeolithic in Africa. Science. 2006;314(5806):1767–1770. doi: 10.1126/science.1135566. [DOI] [PubMed] [Google Scholar]
- 53.Nergadze SG, Lupotto M, Pellanda P, Santagostino M, Vitelli V, Giulotto E. Mitochondrial DNA insertions in the nuclear horse genome. Anim Genet. 2010;41(2):176–185. doi: 10.1111/j.1365-2052.2010.02130.x. [DOI] [PubMed] [Google Scholar]
- 54.Eltsov NP, Volodko NV. mtPhyl program. 2011. [Google Scholar]
- 55.Yang Z. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997;13(5):555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- 56.Posada D, Buckley TR. Model selection and model averaging in phylogenetics: advantages of akaike information criterion and bayesian approaches over likelihood ratio tests. Syst Biol. 2004;53(5):793–808. doi: 10.1080/10635150490522304. [DOI] [PubMed] [Google Scholar]
- 57.Forster P, Harding R, Torroni A, Bandelt HJ. Origin and evolution of Native American mtDNA variation: a reappraisal. Am J Hum Genet. 1996;59(4):935–945. [PMC free article] [PubMed] [Google Scholar]
- 58.Drummond AJ, Rambaut A, Shapiro B, Pybus OG. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol. 2005;22(5):1185–1192. doi: 10.1093/molbev/msi103. [DOI] [PubMed] [Google Scholar]
- 59.Drummond AJ, Rambaut A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007;7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thirstrup JP, Bach LA, Loeschcke V, Pertoldi C. Population viability analysis on domestic horse breeds (Equus caballus) J Anim Sci. 2009;87(11):3525–3535. doi: 10.2527/jas.2008-1760. [DOI] [PubMed] [Google Scholar]