

Epidemiology of uric acid nephrolithiasis and its association to the metabolic syndrome
There are global diversities in uric acid stone prevalence. Prevalence is highest in the Middle East and a few countries in Europe. In fact, uric acid stone occurrence was reported to be 28 % in Pakistan and 22 % in Israel [1-3], while comprising only 8-10 % of all kidney stones in the United States [4]. The incidence of uric acid stones in Japanese and Chinese descendants in San Francisco was reported to be 15-16 %. Moreover, the prevalence of uric acid nephrolithiasis and gout among the Hmong immigrant population in the U.S. is significantly higher [5, 6].
The exact cause for global diversity in uric acid (UA) nephrolithiasis prevalence has not yet been fully elucidated. However, a high prevalence of obesity, diabetes and hypertension is commonly associated with uric acid nephrolithiasis in Western societies and has been shown to be present in Hmong populations born in the United States [7-9]. This is novel as there is generally a low incidence (approximately 20 %) of UA stones in Northeastern Thailand, a geographic region adjacent to the Hmong homeland of Laos with an ethnically distinct population [10]. Therefore, it appears that both genetic factors and the adoption of Western dietary behaviors play an important role in the high incidence of uric acid stone prevalence.
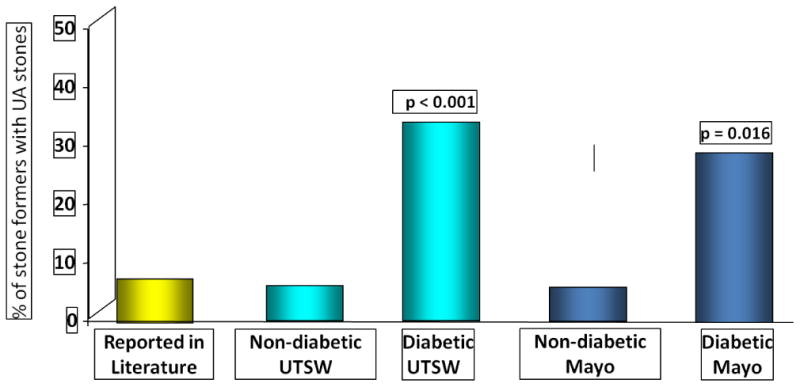
It has been established that stone formers with type 2 diabetes mellitus (T2DM) have UA stones as the main stone constituent more frequently than non-diabetic stone formers [11] (Fig. 1). This greater prevalence among T2DM stone formers has since been confirmed by other investigators [8, 9, 12]. A higher prevalence of UA stones has also been demonstrated among obese stone formers [13, 14]. Moreover, it was shown that greater body mass index (BMI) and T2DM are independent risk factors for UA nephrolithiasis [15].
Figure 1. Prevalence of Uric Acid Stones in Diabetic versus Non-diabetic Stone Formers.
Etiologic causes of uric acid stone formation
The etiologic cause(s) of the formation of uric acid nephrolithiasis are complex; they may include genetic and/or acquired factors [16]. However, features of metabolic syndrome (MS) have emerged as the most common cause of uric acid stone formation [7, 9]. In fact, chronic diarrhea has a major impact on stone formation. In inflammatory bowel diseases, including ulcerative colitis and Chron's disease, the majority of stones contain calcium. However, 1/3 of stones are composed of uric acid, an incidence which is higher than the 8-10% incidence reported for general populations with uric acid nephrolithiasis [17-19]. Moreover, in patients following ileostomy, uric acid stones are common and comprise 2/3 of all stones [17]. In these subjects unduly acidic urine pH (< 5.5) is commonly encountered compared to control subjects [20-22]. However, urinary uric acid excretion has been reported to be normal in this population [23]. The underlying pathophysiologic mechanism for undue urine acidity in this population has not yet been fully investigated. However, loss of bicarbonate in the stool and systemic metabolic acidosis may play an important role in lowering urinary pH. The major therapeutic modality in these subjects should involve administration of alkali treatment to combat abnormally acidic urine [23] in conjunction with increased intake of fluids as urine volume is significantly low due to persistent diarrheal fluid loss.
Physiochemical properties of uric acid stone formation
Mammals produce UA as an end product of purine metabolism [4]. UA is then metabolized by the hepatic enzyme uricase to a more soluble allantoin, which is then excreted into the urine. However, humans and higher primates lack uricase and, due to their inability to metabolize uric acid, disclose serum and urinary acid concentrations many folds higher than other mammals [24]. Since urinary UA excretion in humans generally exceeds 600 mg/day, the limited urinary UA solubility of 96 mg/l poses a great risk for UA precipitation [25]. Urine pH plays the most dominant role in UA solubility, as UA is a weak organic acid with an ionization constant (pKa 5.35 at 37° C) [26]. Unduly acidic urine (urine pH ≤ 5.5) leads to precipitation of the sparingly soluble UA, increasing the predisposition to UA nephrolithiasis [27].
Normal urine is often metastably supersaturated with respect to UA; thus, the presence of a urinary inhibitor of crystallization also plays some role in UA precipitation. Therefore, acidic urine may be necessary but is not sufficient to cause UA nephrolithiasis. It is plausible that the lack of an inhibitor or the presence of a promoter, may account for the difference in the propensity for UA stone formation. In this situation unlike that of calcium oxalate, the roles of inhibitors have not been clearly identified. However, an in vitro experiment has identified that the presence of molecules inhibit the adhesion of UA renal epithelial cells which supports the inhibitory role of this micromolecule against UA precipitation [28-30].
Pathophysiology of acidic urine pH in uric acid nephrolithiasis
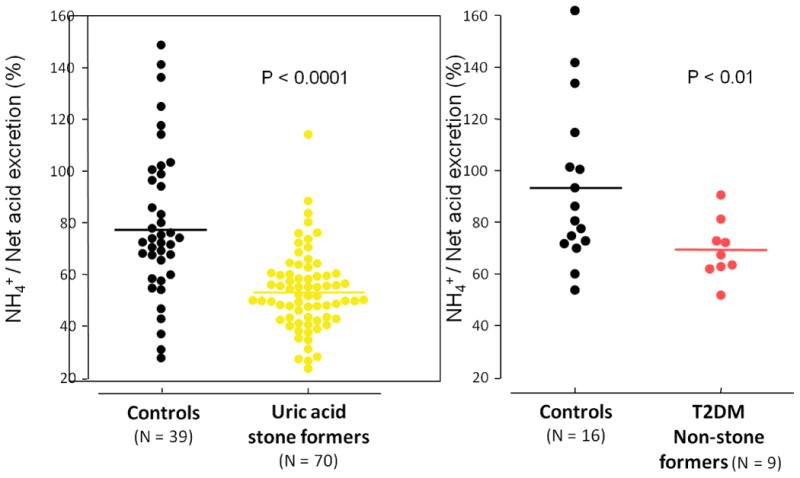
Three significant urinary abnormalities have been described in patients with uric acid nephrolithiasis: low urinary pH, hyperuricosuria, and low urinary volume [16]. In uric acid stone forming patients, the most important and invariant features are an overly acidic urine which increases the urinary content of disassociated uric acid, and the propensity for uric acid precipitation [7, 31]. We have previously shown that two major etiologic factors result in unduly low urinary pH of uric acid stone formers: reduced renal ammonium (NH4+) excretion and increased net acid excretion (NAE), with the combination resulting in overly acidic urine [7, 9]. These abnormalities have been encountered under both fixed metabolic diets and ad-lib diets. In addition, overly acidic urine, defective NH4+ excretion and increased NAE were also demonstrated in diabetic non-stone formers who share some phenotypic features with uric acid stone formers under a fixed metabolic diet (Fig. 2) [32]. Given that the net acid excretion matches net acid production at a steady state, these findings of significant net acid excretion in uric acid stone formers and T2DM patients without kidney stones suggest that net acid production is significantly higher in this population.
Figure 2. Inpatient Controlled Diet: NH4+ Excretion in UA Stone Formers and T2DM Non-stone Formers.
Mechanisms of impaired ammonium excretion
NH4+ is a high capacity urinary buffer (pKa of 9.2) which efficiently buffers most of the hydrogen secreted by the kidney. In patients with UA stones, the defective NH4+ excretion in most of the secreted hydrogen molecules is buffered by titratable acid (TA) to maintain acid-base homeostasis [33]. Therefore, the trade-off will be unduly acidic urine which poses a high risk for UA precipitation. Defective NH4+ has been demonstrated at a steady state and also following a single dose of acid load with ammonium chloride [7]. Renal proximal tubular cells are the main segment for the synthesis and secretion of NH4+ [34]. Ammonium made in proximal renal tubular cells is transported across the apical membrane either directly as NH4+ or as non-ionic diffusion of free ammonia (NH3). The sodium-hydrogen exchanger NHE3 plays a key role in both of these processes [34, 35]. The underlying cellular mechanism(s) associating MS to the development of UA stones has been extensively studied. In obesity, diabetes and MS, disequilibrium occurs between caloric intake and caloric utilization. Hence, the name of “lipotoxicity” has been adopted for this process [36]. This process involves fat distribution in non adipocyte tissue including cardiac myocytes, pancreatic β cells, skeletal muscle cells and parenchymal liver cells [36-38]. Renal proximal tubal cells are specifically vulnerable to lipotoxicity due to increased filter load of free fatty acids [39]. The supporting role of renal steatosis in the pathogenesis of urinary acidification defects was found in opossum proximal tubular cell cultures and in Zucher Diabetic Fatty (ZDF) rats, and established an animal model of obesity and MS [40]. The comparison of ZDF rats to lean litter mates demonstrated a higher renal triglyceride content, associated with low urinary NH4+ and pH, as well as lower levels of brush border membranes NHE3 activity and proteins. Furthermore, treatment of ZDF rats with thiazoladinediones, which is known to reduce non-adipocyte tissue steatosis, was shown to restore urinary profiles to that of the controlled litter mates as well as significantly reduce renal triglyceride accumulation [40]. However, an established link between renal steatosis and defective ammonium excretion in humans with uric acid stone formations has not yet been fully elucidated.
Tissue injury is principally due to the accumulation of non-esterified fatty acid and their toxic metabolites including acyl-CoA, diacylglycerol, and ceramide [33, 41].
Mechanisms of increased acid production
Under a fixed metabolic diet and a steady state when the urine specimens were collected under mineral oil, compared to control subjects patients with UA nephrolithiasis and T2DM patients without kidney stones exhibited significantly higher NAE, approximately 1.5 fold, suggestive of increased acid production in these two populations.
To date, no study has explored the source and nature of these putative acid anions. A plausible mechanism is increased organic production by intestinal and aerobic metabolism [42]. This may occur in diabetics, and potentially in UA stone formers, due to differences in the gut microflora [43, 44]. Consequently, this process will result in greater acid production and/or net gastrointestinal (GI) alkali loss by increased excretion of the base precursors in the stool [7, 9, 32].
Conclusion
With the worldwide problem of obesity of epidemic proportions, it has emerged that uric acid prevalence increases with metabolic syndrome (obesity and T2DM). Fundamental progress has been made in the elucidation of the pathophysiologic mechanisms of uric acid stone formation and its link to metabolic syndrome. The two principal defects examined by careful metabolic studies include defective ammonium excretion and increased net acid production. Previous studies using ZDF and opossum kidney cell culture have shown that renal steatosis may be responsible for reduced ammonium excretion. Future studies are needed to elucidate whether renal fat accumulation will play a similar role in human subjects with uric acid stones and whether metabolic syndrome and diabetes play an important pathogenetic role in increased acid production in this population.
Acknowledgments
The author would like to acknowledge Ashlei L. Johnson for her primary role in the preparation and editorial review of this manuscript.
References
- 1.Rafique M, Bhutta RA, Rauf A, Chaudhry IA. Chemical composition of upper renal tract calculi in Multan. J Pak Med Assoc. 2000 May;50(5):145–8. [PubMed] [Google Scholar]
- 2.Herbstein FH, Kleeberg J, Shalitin Y, Wartski E, Wielinski S. Chemical and x-ray diffraction analysis of urinary stones in Israel. Isr J Med Sci. 1974 Dec;10(12):1493–9. [PubMed] [Google Scholar]
- 3.Hossain RZ, Ogawa Y, Hokama S, Morozumi M, Hatano T. Urolithiasis in Okinawa, Japan: a relatively high prevalence of uric acid stones. Int J Urol. 2003 Aug;10(8):411–5. doi: 10.1046/j.1442-2042.2003.00656.x. [DOI] [PubMed] [Google Scholar]
- 4.Sakhaee K. Uric Acid Metabolism and Uric Acid Stones. In: Rao PP J, Kavanagh J, editors. Urinary Tract Stone Disease. Manchester, UK: Springer; 2011. pp. 185–93. [Google Scholar]
- 5.Portis AJ, Hermans K, Culhane-Pera KA, Curhan GC. Stone disease in the Hmong of Minnesota: initial description of a high-risk population. J Endourol. 2004 Nov;18(9):853–7. doi: 10.1089/end.2004.18.853. [DOI] [PubMed] [Google Scholar]
- 6.Portis AJ, Laliberte M, Tatman P, Moua M, Culhane-Pera K, Maalouf NM, Sakhaee K. High prevalence of gouty arthritis among the Hmong population in Minnesota. Arthritis Care Res (Hoboken) 2010 Oct;62(10):1386–91. doi: 10.1002/acr.20232. [DOI] [PubMed] [Google Scholar]
- 7.Sakhaee K, Adams-Huet B, Moe OW, Pak CY. Pathophysiologic basis for normouricosuric uric acid nephrolithiasis. Kidney Int. 2002 Sep;62(3):971–9. doi: 10.1046/j.1523-1755.2002.00508.x. [DOI] [PubMed] [Google Scholar]
- 8.Daudon M, Lacour B, Jungers P. High prevalence of uric acid calculi in diabetic stone formers. Nephrol Dial Transplant. 2005 Feb;20(2):468–9. doi: 10.1093/ndt/gfh594. [DOI] [PubMed] [Google Scholar]
- 9.Cameron MA, Maalouf NM, Adams-Huet B, Moe OW, Sakhaee K. Urine composition in type 2 diabetes: predisposition to uric acid nephrolithiasis. J Am Soc Nephrol. 2006 May;17(5):1422–8. doi: 10.1681/ASN.2005121246. [DOI] [PubMed] [Google Scholar]
- 10.Prasongwatana V, Sriboonlue P, Suntarapa S. Urinary stone composition in North-East Thailand. Br J Urol. 1983 Aug;55(4):353–5. doi: 10.1111/j.1464-410x.1983.tb03320.x. [DOI] [PubMed] [Google Scholar]
- 11.Pak CY, Sakhaee K, Moe O, Preminger GM, Poindexter JR, Peterson RD, Pietrow P, Ekeruo W. Biochemical profile of stone-forming patients with diabetes mellitus. Urology. 2003 Mar;61(3):523–7. doi: 10.1016/s0090-4295(02)02421-4. [DOI] [PubMed] [Google Scholar]
- 12.Lieske JC, de la Vega LS, Gettman MT, Slezak JM, Bergstralh EJ, Melton LJ, 3rd, Leibson CL. Diabetes mellitus and the risk of urinary tract stones: a population-based case-control study. Am J Kidney Dis. 2006 Dec;48(6):897–904. doi: 10.1053/j.ajkd.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 13.Ekeruo WO, Tan YH, Young MD, Dahm P, Maloney ME, Mathias BJ, Albala DM, Preminger GM. Metabolic risk factors and the impact of medical therapy on the management of nephrolithiasis in obese patients. J Urol. 2004 Jul;172(1):159–63. doi: 10.1097/01.ju.0000128574.50588.97. [DOI] [PubMed] [Google Scholar]
- 14.Daudon M, Lacour B, Jungers P. Influence of body size on urinary stone composition in men and women. Urol Res. 2006 Jun;34(3):193–9. doi: 10.1007/s00240-006-0042-8. [DOI] [PubMed] [Google Scholar]
- 15.Daudon M, Traxer O, Conort P, Lacour B, Jungers P. Type 2 diabetes increases the risk for uric acid stones. J Am Soc Nephrol. 2006 Jul;17(7):2026–33. doi: 10.1681/ASN.2006030262. [DOI] [PubMed] [Google Scholar]
- 16.Maalouf NM, Cameron MA, Moe OW, Sakhaee K. Novel insights into the pathogenesis of uric acid nephrolithiasis. Curr Opin Nephrol Hypertens. 2004 Mar;13(2):181–9. doi: 10.1097/00041552-200403000-00006. [DOI] [PubMed] [Google Scholar]
- 17.Deren JJ, Porush JG, Levitt MF, Khilnani MT. Nephrolithiasis as a complication of ulcerative colitis and regional enteritis. Ann Intern Med. 1962 Jun;56:843–53. doi: 10.7326/0003-4819-56-6-843. [DOI] [PubMed] [Google Scholar]
- 18.Gelzayd EA, Breuer RI, Kirsner JB. Nephrolithiasis in inflammatory bowel disease. Am J Dig Dis. 1968 Dec;13(12):1027–34. doi: 10.1007/BF02233547. [DOI] [PubMed] [Google Scholar]
- 19.Knudsen L, Marcussen H, Fleckenstein P, Pedersen EB, Jarnum S. Urolithiasis in chronic inflammatory bowel disease. Scand J Gastroenterol. 1978;13(4):433–6. doi: 10.3109/00365527809181917. [DOI] [PubMed] [Google Scholar]
- 20.Bambach CP, Robertson WG, Peacock M, Hill GL. Effect of intestinal surgery on the risk of urinary stone formation. Gut. 1981 Apr;22(4):257–63. doi: 10.1136/gut.22.4.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fukushima T, Yamazaki Y, Sugita A, Tsuchiya S. Prophylaxis of uric acid stone in patients with inflammatory bowel disease following extensive colonic resection. Gastroenterol Jpn. 1991 Aug;26(4):430–4. doi: 10.1007/BF02782810. [DOI] [PubMed] [Google Scholar]
- 22.Clarke AM, McKenzie RG. Ileostomy and the risk of urinary uric acid stones. Lancet. 1969 Aug 23;2(7617):395–7. doi: 10.1016/s0140-6736(69)90108-1. [DOI] [PubMed] [Google Scholar]
- 23.Obialo CI, Clayman RV, Matts JP, Fitch LL, Buchwald H, Gillis M, Hruska KA. Pathogenesis of nephrolithiasis post-partial ileal bypass surgery: case-control study. The POSCH Group. Kidney Int. 1991 Jun;39(6):1249–54. doi: 10.1038/ki.1991.158. [DOI] [PubMed] [Google Scholar]
- 24.Rafey MA, Lipkowitz MS, Leal-Pinto E, Abramson RG. Uric acid transport. Curr Opin Nephrol Hypertens. 2003 Sep;12(5):511–6. doi: 10.1097/00041552-200309000-00005. [DOI] [PubMed] [Google Scholar]
- 25.Asplin JR. Uric acid stones. Semin Nephrol. 1996 Sep;16(5):412–24. [PubMed] [Google Scholar]
- 26.Finlayson B, Smith A. Stability of first dissociable proton of uric acid. J Chemical and Engineering Data1974 January. 1974;19(1):94–7. [Google Scholar]
- 27.Riese RJ, Sakhaee K. Uric acid nephrolithiasis: pathogenesis and treatment. J Urol. 1992 Sep;148(3):765–71. doi: 10.1016/s0022-5347(17)36715-0. [DOI] [PubMed] [Google Scholar]
- 28.Sakhaee K, Nigam S, Snell P, Hsu MC, Pak CY. Assessment of the pathogenetic role of physical exercise in renal stone formation. J Clin Endocrinol Metab. 1987 Nov;65(5):974–9. doi: 10.1210/jcem-65-5-974. [DOI] [PubMed] [Google Scholar]
- 29.Reddy ST, Wang CY, Sakhaee K, Brinkley L, Pak CY. Effect of low-carbohydrate high-protein diets on acid-base balance, stone-forming propensity, and calcium metabolism. Am J Kidney Dis. 2002 Aug;40(2):265–74. doi: 10.1053/ajkd.2002.34504. [DOI] [PubMed] [Google Scholar]
- 30.Koka RM, Huang E, Lieske JC. Adhesion of uric acid crystals to the surface of renal epithelial cells. Am J Physiol Renal Physiol. 2000 Jun;278(6):F989–98. doi: 10.1152/ajprenal.2000.278.6.F989. [DOI] [PubMed] [Google Scholar]
- 31.Pak CY, Sakhaee K, Peterson RD, Poindexter JR, Frawley WH. Biochemical profile of idiopathic uric acid nephrolithiasis. Kidney Int. 2001 Aug;60(2):757–61. doi: 10.1046/j.1523-1755.2001.060002757.x. [DOI] [PubMed] [Google Scholar]
- 32.Maalouf NM, Cameron MA, Moe OW, Sakhaee K. Metabolic basis for low urine pH in type 2 diabetes. Clin J Am Soc Nephrol. 2010 Jul;5(7):1277–81. doi: 10.2215/CJN.08331109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sakhaee K. Recent advances in the pathophysiology of nephrolithiasis. Kidney Int. 2009 Mar;75(6):585–95. doi: 10.1038/ki.2008.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nagami GT. Ammonia production and secretion by the proximal tubule. Am J Kidney Dis. 1989 Oct;14(4):258–61. doi: 10.1016/s0272-6386(89)80198-2. [DOI] [PubMed] [Google Scholar]
- 35.Bobulescu IA, Di Sole F, Moe OW. Na+/H+ exchangers: physiology and link to hypertension and organ ischemia. Curr Opin Nephrol Hypertens. 2005 Sep;14(5):485–94. doi: 10.1097/01.mnh.0000174146.52915.5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee Y, Hirose H, Ohneda M, Johnson JH, McGarry JD, Unger RH. Beta-cell lipotoxicity in the pathogenesis of non-insulin-dependent diabetes mellitus of obese rats: impairment in adipocyte-beta-cell relationships. Proc Natl Acad Sci U S A. 1994 Nov 8;91(23):10878–82. doi: 10.1073/pnas.91.23.10878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Szczepaniak LS, Nurenberg P, Leonard D, Browning JD, Reingold JS, Grundy S, Hobbs HH, Dobbins RL. Magnetic resonance spectroscopy to measure hepatic triglyceride content: prevalence of hepatic steatosis in the general population. Am J Physiol Endocrinol Metab. 2005 Feb;288(2):E462–8. doi: 10.1152/ajpendo.00064.2004. [DOI] [PubMed] [Google Scholar]
- 38.Bachmann OP, Dahl DB, Brechtel K, Machann J, Haap M, Maier T, Loviscach M, Stumvoll M, Claussen CD, Schick F, Haring HU, Jacob S. Effects of intravenous and dietary lipid challenge on intramyocellular lipid content and the relation with insulin sensitivity in humans. Diabetes. 2001 Nov;50(11):2579–84. doi: 10.2337/diabetes.50.11.2579. [DOI] [PubMed] [Google Scholar]
- 39.Weinberg JM. Lipotoxicity. Kidney Int. 2006 Nov;70(9):1560–6. doi: 10.1038/sj.ki.5001834. [DOI] [PubMed] [Google Scholar]
- 40.Bobulescu IA, Dubree M, Zhang J, McLeroy P, Moe OW. Effect of renal lipid accumulation on proximal tubule Na+/H+ exchange and ammonium secretion. Am J Physiol Renal Physiol. 2008 Jun;294(6):F1315–22. doi: 10.1152/ajprenal.00550.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, Grundy SM, Hobbs HH. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004 Dec;40(6):1387–95. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- 42.Halperin ML, Kamel KS. D-lactic acidosis: turning sugar into acids in the gastrointestinal tract. Kidney Int. 1996 Jan;49(1):1–8. doi: 10.1038/ki.1996.1. [DOI] [PubMed] [Google Scholar]
- 43.Larsen N, Vogensen FK, van den Berg FW, Nielsen DS, Andreasen AS, Pedersen BK, Al-Soud WA, Sorensen SJ, Hansen LH, Jakobsen M. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One. 2010;5(2):e9085. doi: 10.1371/journal.pone.0009085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Iber FL, Parveen S, Vandrunen M, Sood KB, Reza F, Serlovsky R, Reddy S. Relation of symptoms to impaired stomach, small bowel, and colon motility in long-standing diabetes. Dig Dis Sci. 1993 Jan;38(1):45–50. doi: 10.1007/BF01296772. [DOI] [PubMed] [Google Scholar]