

Abstract
Astellatol and nitidasin belong to a subset of sesterterpenoids that share a sterically encumbered trans-hydrindane motif with an isopropyl substituent. In addition, these natural products feature intriguing polycyclic ring systems posing significant challenges for chemical synthesis. Herein, we detail the evolution of our stereoselective strategy for isopropyl trans-hydrindane sesterterpenoids. Our endeavors included the synthesis of several building blocks, enabling studies toward all molecules of this terpenoid subclass, and of advanced intermediates of our initial route toward a biomimetic synthesis of astellatol. These findings provided the basis for a second-generation and a third-generation approach toward astellatol that eventually culminated in the enantioselective total synthesis of (−)-nitidasin. In particular, we orchestrated a series of substrate-controlled transformations to install the ten stereogenic centers of the target molecule and forged the carbocyclic backbone in a convergent fashion. Furthermore, we disclose our progress toward the synthesis of astellatol and provide insights into some observed yet unexpected diastereoselectivities by detailed quantum-mechanical calculations.
Keywords: Total Synthesis, Stereoselective Synthesis, Terpenoids, Natural Products, Computational Chemistry
Introduction
Among the large family of terpenoid natural products, sesterterpenoids form a small subclass with ca. 1000 isolated members.[1] During biosynthesis, the variety of cyclization modes, hydride- and methyl-shifts and oxidation events leads to a plethora of intricate C25-architectures ranging from mono- to pentacarbocyclic compounds. One member of the latter rare subset is the fungal metabolite astellatol isolated by Simpson and Sadler in 1989 (Figure 1).[2] Based on extensive NMR studies, the authors elucidated the relative configuration of the unique carbon framework with a total of ten stereogenic centers, an exo-methylene group attached to a four-membered ring and a single oxygenated site.[3] In 1997, the similarly complex sesterterpenoid nitidasin[4] was isolated from the Peruvian folk medicine “Hercampuri”, which has found an application as a remedy against hepatitis, hypertension and diabetes.[5] The rare 5-8-6-5 carbon framework is also decorated with ten stereogenic centers, together with its relative stereochemistry being unambiguously proven by single crystal X-ray diffraction. During their isolation program, Kawahara and co-workers found two structurally related metabolites in “Hercampuri”. While alborosin[6] seems to arise by degradation of nitidasin, the biosynthesis of nitiol[7] did only lead to partial cyclization, not forming the central eight-membered ring.
Figure 1.

Selected isopropyl trans-hydrindane sesterterpenoids.
From a structural perspective, astellatol, nitidasin and alborosin share a sterically encumbered trans-hydrindane motif that is substituted with an isopropyl residue cis to the angular methyl group. In addition, the five-membered ring has been oxidized to the carbonyl or the alcohol level. This portion has also been found in two inhibitors of mammalian GPI-anchor biosynthesis, YW-3699[8] and YW-3548.[9] Retigeranic acid B features the same moiety, although lacking the oxygenation at the five membered ring.[10] Interestingly, chemists isolated two additional sesterterpenoids, retigeranic acid A[10] and aleurodiscal,[11] the isopropyl residue of which adopts the epimeric trans relationship to the angular methyl group.
Despite their intriguing architectures and interesting biological profiles, only a few research groups have investigated these natural products.[12] This might be particularly due to the challenges associated with the synthesis of the polycarbocyclic ring-systems. Only the laboratories of Corey,[13] Paquette,[14] Hudlicky[15] and Wender[16] succeeded in the total synthesis of retigeranic acid A. Apart from these impressive achievements, Paquette[17] and Dake[18] reported the construction of the carbon framework of aleurodiscal and nitiol, respectively, and Tori and co-workers approached the YW-3699 skeleton, without addressing the hydrindane portion.[19]
A few years ago, we launched a program to develop a general approach to isopropyl trans-hydrindane sesterterpenoids.[20] Herein, we provide the full details of our stereoselective strategy that allowed access to advanced intermediates for a biomimetic synthesis of astellatol and resulted in the first total synthesis of (−)-nitidasin.[21]
Results and Discussion
First-generation retrosynthesis
As outlined in Scheme 1, our program initially focused on the total synthesis of astellatol. We envisioned accessing this natural product via a biomimetic cationic cascade from tricycle 1 (vide infra). The key intermediate 1, featuring a strained eleven-membered ring with two trisubstituted double bonds, could be elaborated in a stereospecific manner by different retrosynthetic disconnections that will be detailed in the relevant subsections. However, all approaches should be based on hydrindane 2 comprising a total of six stereogenic centers and bearing an alkene and a masked ketone as synthetic handles. We envisaged orchestrating a series of diastereoselective reactions to set the stereogenic centers of this key intermediate 2, giving ketone 3 as the logical precursor. While we initially considered employing a Pauson-Khand reaction to construct the five-membered ring of ketone 3, we ultimately decided to use diketone 4 as a starting point for our synthesis, due to the difficulties associated with the installation of the required trans-ring junction.[22] It should be stated that we also aimed at accessing the deoxygenated analogue 5 by diversifying one of the intermediates en route to building block 3. Therefore, our route would not only pave the way for the synthesis of astellatol, nitidasin and the YW compounds but it would also provide a key intermediate for studies toward retigeranic acid B and nitiol.
Scheme 1.

Retrosynthesis of astellatol based on a biomimetic cascade.
Scheme 2 illustrates the envisaged biomimetic keystep in line with the biosynthesis proposed by Simpson.[23] We envisioned treating tertiary allylic alcohol 1 with a Lewis or a Brønsted acid to form the stabilized tertiary carbocation 6 (with concomitant deprotection), which could then trigger a ring closure to form tetracycle 7. Next, a stereospecific 1,2-hydride shift would set the relative configuration at C17 and generate homoallylic carbocation 8 that could engage in a homoallyl-cyclopropylcarbinyl-cyclobutyl rearrangement[24] via intermediate 9 to furnish astellatol.
Scheme 2.

Proposed biomimetic cationic cascade toward astellatol.
Domino transformations of this kind are highly challenging to realize in the laboratory in the absence of the biosynthetic machinery, given the number of possible reaction pathways and side products. However, we assumed that the conformational bias associated with the already installed trans-hydrindane portion might influence the outcome of the cascade favoring the formation of astellatol. It should be stressed that this biomimetic cascade would rapidly build up structural complexity and provide insight into the biogenesis of astellatol. In addition, potential side reactions could open avenues toward structurally related natural products, e.g. nitidasin.
Synthesis of trans-hydrindenone 15
According to our retrosynthetic analysis, our synthetic plan started with the synthesis of diketone 4, which was readily prepared in both enantiomeric series in 100 g batches (Scheme 3).[25] Additionally, we elucidated the solid-state structure of this building block that is frequently used for terpenoid synthesis.[26] We originally intended to conduct a direct trans-selective reduction utilizing a bulky CuH species generated in situ.[27] Due to the sensitivity of this reaction and aiming to prepare decagram amounts of material, we decided instead to follow a known, more robust five-step protocol for the installation of the trans-ring junction.[28] Accordingly, chemo- and diastereoselective reduction of the more electrophilic ketone was followed by protection of the resulting alcohol as its tert-butyl ether to yield ketone 10.
Scheme 3.

Synthesis of trans-hydrindanone 14.
Next, reaction with Stiles’ reagent 11[29] introduced the carboxylic acid moiety as a traceless directing group to allow for a trans-selective hydrogenation. The reaction produced intermediate 12, the structure of which was confirmed by X-ray crystallography. Subsequently, hydrogenation in the presence of Rosenmund catalyst followed by heat and vacuum-promoted decarboxylation provided trans-hydrindanone 13 in 59% yield as a 19:1 mixture of diastereomers. An ensuing three-step protocol consisting of ether cleavage, dioxolane formation and Cr-mediated oxidation delivered tricycle 14. Purification by recrystallization not only removed any traces of the remaining cis isomer at this stage, but also provided crystals suitable for single crystal X-ray diffraction. Overall, this route was straightforward, robust and scalable. Thus, 19 g of ketone 14 was prepared in a single batch in 29% yield over eight steps.
With this material in hand, installing the required unsaturation in the five-membered ring was center stage. Initial attempts to achieve this oxidation by exposure of ketone 14 to IBX[30] or via synthesis of an α-bromo ketone were not successful. The same remained true for a protocol involving the treatment of a cyclic TIPS enol ether with Pearlman’s catalyst in the presence of tBuOOH under an O2 atmosphere.[31] Contrarily, α-selenylation was straightforward (Scheme 4). Upon oxidation of the resulting selenium species, the desired elimination occurred, but was accompanied by a Bayer-Villiger-oxidation to yield bicycle 15.[32] We next evaluated a Saegusa-Ito oxidation,[33] but attempts to render the reaction catalytic in Pd only gave low conversion to the desired product. Thus, we resorted to the classical conditions treating the intermediate trimethylsilyl enol ether with stoichiometric amounts of Pd(OAc)2. This protocol provided the desired enone 16 in 82% yield after recycling recovered ketone 14. The structure of enone 16 was also proven by single crystal X-ray diffraction.
Scheme 4.

Synthesis of enone 16 and lactone 15.
Diastereoselective Michael addition
We next investigated the addition of a suitable three-carbon nucleophile via a cuprate addition. To this end, we treated enone 16 with the higher order cuprate derived from isopropylene magnesium bromide and CuCN at −78 °C, obtaining a single diastereomer 17 (Scheme 5).[34] The selectivity of the addition was initially elucidated by NOESY NMR experiments indicating proximity between one methylene proton and the bridgehead methyl group. This stereochemical outcome was then unequivocally established by single crystal X-ray diffraction. Since we initially assumed that the addition of the cuprate from the β-face of the molecule would be disfavored due to sterical interactions with the bridgehead methyl group and because the selectivity is not obvious from inspecting the crystal structure of enone 16 alone, we investigated this transformation computationally.
Scheme 5.

Synthesis of ketone 17 by a diastereoselective 1,4-addition.
We were aware of the difficulties of determining the active species of higher order cuprates in our reaction mixture.[35] In order to develop a chemically reasonable model for the observed selectivity, we decided to use the less complicated low order cyanocuprate for the calculations. The most stable and reactive [Cu(isopropylene)CN·MgBr]–enone species were calculated to have the (µ2-cyano-C,N) ligand connecting the Mg2+ and Cu(I) centers (Figure 2). This coordination mode has been observed in X-ray structures of lithium cyanoorganocuprates.[36] In our model system, magnesium acts as a Lewis acid activating the enone and stabilizing the forming enolate upon isopropylene addition from the cuprate. Other possibilities including the two metal centers being linked by a (µ2-Br) ligand were considered. These species were significantly higher in energy and will not be discussed (see the Supporting Information). Michael additions of organocuprates have been proposed to proceed through an initial conjugate addition of the Cu(I) center to the α,β-unsaturated carbonyl system at low temperature leading to putative Cu(III) intermediates that subsequently transfer the organic nucleophile through reductive elimination upon warming of the reaction mixture.[37] Similarly to this proposed mechanism, we located diastereomeric reacting complexes (II) and transition states (III‡) for the coordination of the cuprate (R1) to bicyclic cyclopentenone (I). Approximation from the allegedly less sterically hindered Re face, in which the copper atom and the bridgehead methyl group are mutually anti (III‡anti), was calculated to be fast (ΔG‡ = +9.3 kcal mol−1 from II) and only 0.1 kcal mol−1 more favored than the approach by the opposite face (III‡syn). This was the first indication of steric factors not governing the stereochemical outcome of the reaction.
Figure 2.

Mechanism of the conjugate addition of organocuprate [Cu(isopropylene)CN·MgBr] to bicyclic cyclopentenone 16 calculated at the SMDTHF/ωB97X-D/6-311+G(2d,p)+SDD(Cu,Mg,Br)//ωB97X-D/6-31G(d)+SDD(Cu,Mg,Br) level. Free energies are given in kcal mol−1 and distances in angstroms. Different structures were considered for reactant (R1). Explicit solvation-desolvation of the metal atoms was not considered. Therefore, both the geometies of R1 and the initial complexation energies should be considered only qualitatively.
The structure of the resulting intermediates (IV) resemble Cu(I)-π-complexes more than Cu(III)-α-complexes. However, considering the short Cu–Cβ distance (2.0 Å), significant pyramidalization of Cβ carbon (28°) and closeness of the angle between the isopropylene and enone ligands to that of a square planar geometry (92°), as observed in the most stable isomer, the Cu(III) character of these species cannot be ruled out. These intermediates are unstable[38] and undergo migratory insertion of the isopropylene group into the enone (V‡) to give the conjugate addition intermediate (VI). This nucleophilic addition of the alkenyl group to the olefin is also fast, but entails the rate-limiting (ΔG‡ = +15.3 kcal mol−1 from II) and stereodetermining step of the reaction. At this stage, the alkenyl group transfer from the Si face of the enone (V‡syn) is significantly favored over the approximation from the Re face (ΔΔG‡syn-anti = –1.4 and –2.0 kcal mol−1 at the ωB97X-D and M06-2X levels, respectively, for the lowest energy diastereomeric transition states, see Figure 3), leading to a calculated kinetic ratio of the syn and anti adducts of >97:3 at −78 °C assuming Curtin-Hammett conditions. This calculated stereoselectivity matches the experimental observation, and proves that a larger driving force − favoring the approach of the nucleophile from the more hindered β-face of the enone − overcomes the steric repulsions. The origin of this trend is the different torsional strain arising at the transition states of the syn and anti additions. This well-established effect, proposed initially by Felkin[39] and applied to a multitude of nucleophilic, radical and electrophilic asymmetric processes, has been recently reviewed for reactions on unsaturated cyclic and bicyclic alkenes.[40]
Figure 3.

Lowest energy transition state structures for the rate-limiting and stereodetermining addition step for the addition of isopropylene cyanocuprate to bicyclic cyclopentenone 16, calculated at the SMDTHF/DFT/6-311+G(2d,p)+SDD(Cu,Mg,Br)//ωB97X-D/6-31G(d)+SDD(Cu,Mg,Br) [DFT= ωB97X-D and M06-2X] levels. Free energies are given in kcal mol−1 and distances in angstroms.
As illustrated in Figure 3, the Cβ–Cγ bond in transition state V‡anti shows a more eclipsed conformation (Hβ–Cβ–Cγ–Cδ dihedral angle = –12°) than in V‡syn (Hβ–Cβ–Cγ–Cδ dihedral angle = +62°) due to the pyramidalization of Cβ upon nucleophilic attack and the conformational restrictions imposed by the bicyclic backbone. The Cα–Cβ bond is also less eclipsed in both approximations. Regarding the steric repulsions, the H···H contacts between the incoming alkenyl and the bridgehead methyl groups (≥2.37 Å) are all larger than twice the van der Waals radius of hydrogen (2.20 Å) in V‡syn, and only contact between the alkenyl group and the axial Cδ–Hδ bond in the substrate is apparent (2.16 Å). A similar contact between the alkenyl group and the axial Cγ–Hγ bond in the substrate (2.18 Å) is present in V‡anti. In summary, steric repulsions are a minor contributor to the stereoselectivity of this reaction, which is largely controlled by torsional factors.
The Mg-enolates VI, in which copper forms a π-complex with the transferred isopropylene group, show the opposite and initially expected preference of a syn arrangement between the isopropylene and the bridgehead methyl group – both in axial positions of the five-membered ring – being highly repulsive (ΔΔGsyn-anti = +4.7 kcal mol−1). In the final products obtained after enolate protonation (VII), the relative stability of the anti diastereomer is even higher (ΔΔGsyn-anti = +6.7 kcal mol−1). This fact will be very relevant in the following step in our synthetic route (vide infra). Irrespective of their thermodynamic stability, syn conjugate addition products were obtained from 16 under kinetic control, since the alkenyl group transfer is highly exergonic and irreversible (ΔG = –25 kcal mol−1 from II).
A catalyst-controlled diversification
Since experiments on the use of isopropyl cuprate in the Michael addition gave unsatisfactory results in our hands, we next turned our attention to the seemingly straightforward hydrogenation of the isopropylene moiety and submitted bicycle 17 to standard hydrogenation conditions (Pd/C, H2, MeOH). Interestingly, we noticed a small impurity (less then 5%) by NMR spectroscopy that could be conveniently removed by recrystallization giving pure compound 18 in 91% yield (Scheme 6). Since the structure determination of compound 18 was thwarted by overlaying signals in NOESY NMR experiments, no unambiguous assignment could be established by these means. However, we obtained crystals of ketone 18 suitable for single crystal X-ray diffraction. To our surprise, the structure established that under the hydrogenation conditions epimerization of the former allylic stereogenic center at C4 had occurred. This was attributed to fact that transition metal-catalyzed hydrogenations can lead to double bond isomerization prior to reduction.[41] This process is often unnoticed in substrates where the isomerized product is subsequently reduced without any consequences, but holds the potential to result in epimerization or racemization. Notably, exposure of alkene 17 to Pd/C in the absence of hydrogen gas resulted in no isomerization to the tetrasubstituted alkene or into conjugation with the ketone functionality.
Scheme 6.

An unexpected epimerization to form ketone 18.
In order to shed light on this unexpected epimerization, we investigated possible mechanisms by DFT calculations. Although the experimental conditions for hydrogenation are heterogeneous, and the participation of solid Pd/C, or Pd nanoparticles in the epimerization process cannot be ruled out, we only considered homogeneous reactions. Thus, the formation of catalytically relevant Pd(II) species resulting from the oxidative addition of Pd(0) into H–H[42] (reactant) and MeO–H (solvent) bonds was hypothesized. The most plausible calculated mechanism involves two consecutive Pd(II)-catalyzed transformations (Figures 4 and S2, see the Supporting Information): (1) isomerization of the terminal alkene in the isopropylene group of VIIsyn into a more stable internal, tetrasubstituted alkene (XIV) through an initial barrierless migratory insertion of palladium hydride PdH(MeO)(MeOH)2 (IX‡syn), followed by a very fast β-elimination of MeOH (XII‡syn, ΔG‡ = +4.6 kcal mol−1 from XIsyn); (2) hydrogenation of XIV either with H2 on Pd/C (not considered), or initial stereoselective migratory insertion of homogenous PdH2(MeOH)2 into the terminal alkene (XVI‡anti-ipso), followed by reductive elimination of Pd(0) (XIX‡) to give ketone XXanti, which is 6.1 kcal mol−1 more stable than its epimer XXsyn. The first steps involving alkene isomerization are all calculated to be extremely fast and energetically downhill, except the last β-elimination step that is slightly endergonic. Nevertheless, the overall isomerization process to form the internal olefin is exergonic (ΔG = –7.0 kcal mol−1 from XIsyn). Migratory hydride insertion of the PdH2 π-complex XV was calculated to occur regio- and stereoselectively at the ipso carbon and from the Re face of the terminal olefin (XVI‡anti-ipso). Although this process involves a negligible activation barrier, the diastereomeric π-complex and transition state from the Si face are both disfavored by 1.6 kcal mol−1. This is due to the incipient repulsive 1,3-diaxial interactions with the bridgehead methyl group. These steric repulsions are even more apparent in the final and rate-limiting reductive elimination step (XIX‡anti-ipso, ΔG‡ = +17.4 kcal mol−1 from XVIIanti-ipso) (see the Supporting Information), in which the energy difference between the two epimeric transition states is at a maximum (ΔΔG‡syn-anti = +9.1 kcal mol−1). Overall, this mechanism explains the complete epimerization observed experimentally, and demonstrates the potential for Pd(II) hydrides generated in situ from heterogeneous H2 and Pd/C to promote fast alkene isomerizations.
Figure 4.

Minimum energy pathway for the epimerizing hydrogenation of ketone 17 involving an initial alkene isomerization, calculated at the SMDMeOH/ωB97X-D/6-311+G(2d,p)+SDD(Pd)//ωB97X-D/6-31G(d)+SDD(Pd) level. Free energies are given in kcal mol−1 and distances in angstroms.
As a consequence, the choice of the hydrogenation catalyst was revisited and Pd/C was replaced with Adam’s catalyst (PtO2). When conducting the reaction in MeOH, partial reduction of the carbonyl functionality in ketone 19 was observed. This was bypassed when submitting ketone 17 to PtO2 in EtOH under an atmosphere of hydrogen furnishing ketone 19 in 93% yield (Scheme 7). The obtained NMR spectroscopic data matched the ones of the impurity observed in the hydrogenation with Pd/C and we established the identity of ketone 19 by single crystal X-ray diffraction, indicating that under these reaction conditions the alkene isomerization process is suppressed.
Scheme 7.

Synthesis of ketone 19.
We then turned our attention to the transposition of the oxygen functionality via an alkene. Due to the unexpected epimerization, these studies were initially carried out with the α-isopropyl ketone 18. It should be emphasized that the transformations examined on this system provided helpful information for the further synthesis with the desired stereochemistry at C4. Our efforts commenced with the transformation of ketone 18 to tosylhydrazone 22 as a precursor for a Shapiro olefination.[43] However, the examined reaction conditions (TsNHNH2, TsOH, NaOAc, TsNHNH2, TsOH, MgSO4) did not provide the desired material (Scheme 8). Alternatively, a diastereoselective reduction enabled the synthesis of alcohol 20, which was subjected to elimination conditions without success. In contrast, formation of the corresponding xanthate 23 followed by Chugaev elimination furnished alkene 21 in low yield.[44] In parallel, we explored an alternative two-step protocol consisting of enol triflate formation and an ensuing Pd-catalyzed reductive detriflation[45] leading to the desired alkene 21 in excellent yield.
Scheme 8.

Initial attempts to prepare alkene 21 (top) and synthesis of ketone 24 (bottom).
Such material was then elaborated to ketone 24 by hydrogenation and cleavage of the dioxolane protecting group. We established the structure of building block 24, which intersects with the routes toward retigeranic acid A developed by Corey[13] and Hudlicky,[15] by single crystal X-ray diffraction. The synthesis of ketone 24 was shortened by one step when preparing diene 25, which again underwent the previously observed epimerization under the hydrogenation conditions.
Next, we were poised to transfer the reductive detriflation protocol to epimeric ketone 19 with the correct C4 stereochemistry. This reaction proceeded in excellent yield providing multigram quantities of alkene 26 (Scheme 9). Subsequently, alkene 26 was subjected to hydrogenation conditions and, in analogy to the hydrogenation of ketone 17, the use of Pd/C as a catalyst resulted in epimerization giving rise to a 1:2 ratio of diastereomers 27 as indicated by 1H NMR spectroscopy.
Scheme 9.

Synthesis of ketone 5.
Interestingly, the hydrogenation in the presence of PtO2 also occurred with partial epimerization in the same ratio. We thus resorted to a non-metal mediated reduction protocol utilizing diimide,[46] and subsequent acetal cleavage furnished building block 5, suitable for a synthesis of retigeranic acid B. In addition, regioselective silyl enolether formation followed by ozonolysis could provide an entry for the synthesis of nitiol.[20]
Installation of the oxygen functionality
We next turned our attention to the introduction of the C5 alcohol. Initially, alkene 26 was converted to epoxide 30 with single crystal X-ray diffraction confirming once more the correct stereochemistry at C4 (Scheme 10). The structure indicates that the trajectory for an epoxide opening via a SN2 displacement is blocked due to geometric constraints. Consequently, treating epoxide 30 with LiEt3BH up to 140 °C did not result in any conversion.
Scheme 10.

Synthesis of ketone 3 and epoxide 30.
Next, a screening of alkene hydroboration conditions was carried out. We were aware that the outcome of this reaction would not be favored by electronical factors, but rather by steric effects. We initially employed a slight excess of BH3·SMe2 that delivered, after oxidative work up, a 2.7:1 mixture of regioisomeric alcohols 28 and 29 in favor of the desired product 29 (Table 1, Entry 1). Variation of temperature (Entry 2) and equivalents of hydroboration reagent (Entries 3,4) did not result in a significant improvement of the regioselectivity. Since the more bulky boron reagents ThxBH2, (sia)2BH and 9-BBN either led to no conversion of the starting material (Entry 5) or acetal cleavage (Entries 6,7), we revisited our findings and noticed that the amount of hydroboration reagent affected the regioselectivity.
Table 1.
Hydroboration of alkene 26: variation of the reaction conditions.[a]
| Entry | borane (equiv.) | T [°C] | t [h] | 29:28[b] |
|---|---|---|---|---|
| 1 | BH3·SMe2 (1.3) | 0 to rt | 0.5 | 2.7:1 |
| 2 | BH3·SMe2 (1.3) | rt | 0.5 | 2.6:1 |
| 3 | BH3·SMe2 (1.6) | 0 to rt | 0.5 | 2.1:1 |
| 4 | BH3·SMe2 (0.7) | 0 to rt | 0.5 | 2.9:1 |
| 5 | 9-BBN (5.0) | 0 to 66 | 15 | −[c] |
| 6[d] | ThxBH2 (1.6) | 0 to rt | 1 | −[e] |
| 7[d] | (sia)2BH (2.5) | 0 to rt | 1 | −[e] |
| 8[d] | (+)−IpcBH2 (1.5) | 0 to rt | 0.5 | 3.7:1[f] |
| 9[d] | (−)-IpcBH2 (1.5) | 0 to rt | 0.5 | 1:1 |
All reactions were carried out on 10 mg scale in THF (c 0.03M) unless otherwise stated. All reactions were followed by oxidative, basic work-up (NaOH, H2O2).
As determined by 1H NMR spectroscopy of the crude reaction mixture.
No conversion was observed.
The hydroboration agent was prepared immediately prior to use.
The reaction condition resulted in acetal cleavage
The ratio was enhanced to 5.5:1 (84% yield) to 12:1 (67% yield) by flash column chromatography. rt = room temperature.
Whereas the best result was obtained by quick addition of 0.7 equivalents of BH3·SMe2, the ratio dropped to 2.1:1 by employing 1.6 equivalents. This observation was tentatively explained by the in situ formation of a chiral hydroboration agent RBH2. Consequently, alkene 26 was exposed to (+)-IpcBH2 (31), prepared in situ from (S)-Alpine Boramine™ and BF3·OEt2,[47] which improved the regioisomeric ratio to 3.7:1 (Entry 8). Alcohols 28 and 29 were difficult to separate, but column chromatography enhanced the diastereomeric ratio ranging from 5.5:1 (84% yield) to 12:1 (67% yield), providing 5.20 g of alcohol 29 on the largest scale. Interestingly, the use of (−)-IpcBH2 (ent-31) gave an approximately 1:1 mixture of regioisomers 28 and 29 (Entry 9). Therefore, the reaction of alkene 26 with borane 31 constituted a matched case of double diastereoselection. The carbonyl group was then unmasked to yield bicycle 32 and column chromatography allowed for an improvement of the regioisomeric ratio (12:1 to ≥19:1). Subsequently, alcohol 32 was converted to TBS ether 3, the structure of which was unambiguously proven by single crystal X-ray diffraction.
Installation of the C9 and C10 stereogenic centers
In the following experiments, we focused on the installation of the C9 and C10 stereogenic centers (astellatol numbering). Based on literature precedence, we were aware that such a setting would be difficult to access. A Tsuji crotylation via a mixed carbonate was ruled out due to the difficulties associated with diastereoselectivity and mixtures of branched and linear products.[48] In order to evaluate a viable synthetic pathway, these studies were initially carried out with model ketone 13. Firstly, we aimed to establish the required regioselectivity in reactions involving the carbonyl functionality in ketone 13. To this end, we treated ketone 13 with PhMe3NBr3 (PTAB) in the presence of HOAc to generate diastereomeric α-bromo carbonyl compounds 33 that were further elaborated, with enone 34 being isolated as the sole product (Scheme 11). On the other hand, we conducted a kinetic deprotonation with LiHMDS and trapped the enolate with Manders’ reagent 36 to give β-keto-ester 37.[49] Furthermore, we investigated the conversion of compound 37 to vinyl allyl ether 38 under Mitsunobu conditions.[50] However, a brief screen of reaction conditions (e.g. ADDP, PBu3) did not result in any conversion. This was unfortunate as we were particularly interested in this transformation since the latter intermediate 38 could serve as a precursor for a Claisen rearrangement to install the C10 stereogenic center.
Scheme 11.

Proof of regioselective reactivity of ketone 13: synthesis of enone 34 and attempts toward Claisen rearrangement precursor 38.
Since proof of the desired regioselectivity in transformations of ketone 13 had been obtained, we next focused on alkylation protocols utilizing different electrophiles such as allyl iodide or α-halo acetic acid derivatives. Unfortunately, ketone 13 was largely resistant to the attempts to form product 35 or an O-allylated analogue suitable for a Claisen rearrangement.[49]
Thus, we resorted to a Pd-mediated allylation protocol pioneered by Negishi[51] and recently applied by Covey[52] that provided hydrindane 39 on multigram scale (Scheme 12). Subsequently, we examined the reduction of the ketone moiety and found a reagent-controlled reversal of diastereoselectivity. Whereas exposure of the ketone 39 to NaBH4 delivered alcohol 41 in 79% yield, treatment of intermediate 39 with K-Selectride® exclusively led to α-diastereomer 40. The structure of alcohol 41 was elucidated based on NOESY experiments and analysis of coupling constants. In order to incorporate a γ-lactone moiety for the diastereoselective installation of the C10 methyl group, we sought to convert alkene 40 into the corresponding aldehyde by ozonolysis. Since the spontaneously formed lactol was quantitatively converted to the acetals by reaction with the protic co-solvents EtOH or MeOH, we submitted alkene 40 to a Lemieux-Johnson oxidation instead.[53] The desired reaction proceeded smoothly and the obtained crude lactol was oxidized with PCC to yield tricycle 42 in an excellent yield of 90%.
Scheme 12.

Preparation of lactone 42.
The stage was now set for the installation of the C10 methyl group taking advantage of the conformational bias of lactone 42. To this end, lactone 42 was sequentially treated with LiHMDS and MeI at −78 °C yielding, as expected, lactone 43 as a single diastereomer (Scheme 13). The stereochemical outcome of this transformation was initially confirmed by analysis of 1H NMR coupling constants. As shown in Scheme 13, the C10 methine proton was observed as a quartet, indicating a dihedral angle H10−C10−C9−H9 of around 90° according to the Karplus curve. A comparison with the DFT-calculated dihedral angle of 87° for this structure supported the identity of lactone 43. In addition, we unambiguously assigned the structure of tricycle 43 by single crystal X-ray diffraction.
Scheme 13.

Diastereoselective synthesis of lactone 43.
The focus was then turned to the installation of a terminal alkene. We initially reduced lactone 43 to the corresponding lactol (DIBAL-H, −78 °C) and exposed the intermediate to a Wittig methylenation. In our hands, this reaction required elevated temperatures (THF, 66 °C) to proceed and provided the desired product only in low yield as a mixture of diastereomers. In contrast, the reaction with ylene 44 at 81 °C yielded ester 45 as a single diastereomer, albeit in moderate yield (Scheme 14).
Scheme 14.

Synthesis of ester 45 and of alkene 47.
In view of these results, we decided to pursue another strategy and synthesized the corresponding diol by reaction of lactone 43 with LiAlH4. In order to avoid an orthogonal protecting group strategy, we exploited the potential of Swern oxidation conditions to convert primary triethylsilyl ethers directly to aldehydes and prepared intermediate 46.[54] Careful control of the external temperature not exceeding −60 °C and prolonged reaction times (7−8 h) were crucial for a success of the ensuing oxidation since otherwise lower yields and/or the bisoxidized carbonyl compound were observed. Furthermore, the aldehyde was isolated as a single diastereomer, indicating that no epimerization occured under the reaction conditions. Ultimately, a Wittig methylenation provided access to intermediate 47, which served as the branching point for several routes toward the target sesterterpenoids (vide infra).We then set out to adapt the reaction conditions to trans-hydrindane 3 with the correct substitution pattern on the five-membered ring in place. Initially, the TBS protected analogue 3 was converted to ketone 49, which in turn was elaborated to tricycle 51 (Scheme 15). Compared to the model system, the diastereoselective methylation required a slightly higher reaction temperature of −40 °C to achieve full conversion. In analogy to the preparation of the model studies, lactone 51 was carried forward in four steps to alkene 2. Since we found in initial experiments that a chemoselective TES deprotection in bicycle 2 (e.g. CSA, PPTS or HOAc in CH2Cl2/MeOH) was accompanied by cleavage of the TBS ether, we decided to employ the more robust, but fluoride labile SEM protecting group in upscale experiments. Thus, protection of alcohol 32 as its SEM ether 48 followed by allylation gave rise to ketone 50, which was converted to alkene 53 via tricycle 52. Overall, the established protocol proved to be excellent, affording building block 53 in 47% yield over nine steps on a reasonable scale.
Scheme 15.

Synthesis of alkenes 2 and 53.
Toward construction of the macrocycle − the metathesis approach
Having successfully installed the C9 and C10 stereogenic centers, we next turned our attention to the preparation of the precursor for the biomimetic key step. One major challenge concerning this envisaged route was the construction of the highly strained eleven-membered ring in tricycle 1 comprising two (E)-configured trisubstituted alkenes (cf. Scheme 1).[55] In order to conserve our precious building block 53, these studies were conducted using model compound 47. In our first retrosynthetic proposal, we aimed at closing the macrocycle 54 by a ring-closing metathesis (RCM)[56] from allylic alcohol 55, which in turn could arise from alkene 47 by a sequential B-alkyl Suzuki coupling[57] and an alkenyl lithium addition (Scheme 16).
Scheme 16.

Retrosynthesis of macrocycle 54 involving a RCM.
This approach commenced with a coupling of the in situ generated alkyl-boron species of alkene 47 (9-BBN, 40 °C) with alkenyl iodide 56[58] and provided intermediate bis-silylether 57 that was submitted to the previously established deprotective Swern oxidation conditions (Scheme 17). Upon Wittig olefination, diene 58 was obtained in a good yield of 66% over the three steps. In order to complete the synthesis of the model RCM precursor 55, the silyl ether within trans-hydrindane 58 was cleaved and the resulting secondary alcohol was oxidized with Dess-Martin periodinane (DMP).[59] The subsequent addition of isopropenyl lithium at −78 °C was surprisingly facile given the steric encumbrance at the carbonyl functionality, and furnished tertiary alcohol 55 as a single diastereomer. The relative configuration at the newly formed stereogenic center could not be assigned unambiguously, but we assumed an equatorial attack due to the shielding effect of the adjacent substituent, blocking an axial attack from the Si face.
Scheme 17.

Synthesis of triene 55 and observed dimer 59.
Next, we embarked on the investigation of the key RCM step.[60] We initially tested the use of Grubbs second-generation catalyst in refluxing CH2Cl2. These conditions led to the formation of a new product, which was identified as the homocoupled dimer 59 as an undetermined mixture of (E)- and (Z)-isomers by 1H NMR and HSQC spectroscopy and ESI mass spectrometry. We assumed that this outcome might be attributed to a high activation barrier for the formation of the strained macrocycle and thus used higher boiling solvents (benzene, DCE, toluene). However, this did not provide any new reaction products. Since the use of the Stewart-Grubbs catalyst,[61] particularly developed for the synthesis of sterically hindered alkenes by RCM, also resulted only in the formation of dimer 59, we decided to turn our attention to other ring-forming reactions.
Toward construction of the macrocycle − the B-alkyl-Suzuki approach
In addition to the RCM approach, further strategies were pursued toward closing the eleven-membered macrocycle in tricycle 54 for the envisioned bioinspired cationic cascade. As outlined in Scheme 18, one strategy was based on an intramolecular B-alkyl Suzuki-coupling.[62] The retrosynthetic precursor 60 could arise via a Negishi carbalumination and addition of an alkenyl lithium species to a trans-hydrindanone prepared from silyl ether 47.
Scheme 18.

Retrosynthesis of macrocycle 54 with a B-alkyl Suzuki coupling.
Our route commenced with a two-step deprotection/oxidation sequence providing ketone 61 in 90% yield (Scheme 19). Next, the alkenyl lithium species derived from alkenyl iodide 62[63] was reacted with ketone 61 at −78 °C to afford allylic alcohol 63 as a single diastereomer. The stereochemical outcome of this reaction was verified by NOESY NMR experiments and analysis of the spectroscopic data revealed the preferred conformation of the stereogenic center at C10. Based on the lacking NOE correlation between the C10 methyl group and 9−H, both groups could potentially point into opposite directions. This assumption was further supported by the scalar coupling of the C9 methine (ddd at δ = 1.86 ppm with J = 12.7, 3.7 and 2.3 Hz). While the first two values could be identified to an axial-axial coupling and an axial-equatorial coupling within the six-membered ring (to 8-H), the remaining coupling constant of J = 2.3 Hz accounts for the coupling of 10-H. Thus, this value indicates a dihedral angle of around 67° according to the Karplus curve. Consequently, this observation suggested that a rotation around the C9−C10 bond would be required for an efficient ring closure, constituting an important factor for the activation barrier.
Scheme 19.

Synthesis of alkenyl silane 67 and observed iodoetherification.
Next, a three-step sequence consisting of oxidative PMB ether cleavage, Swern oxidation and homologation with the Ohira-Bestmann reagent (64)[64] cleanly provided access to alkyne 65. Notably, the oxidation of the intermediate primary alcohol with DMP only proved successful under elevated temperature and a large excess of the reagent. Since a direct Negishi carbalumination of alkyne 65 resulted in an intractable product mixture, we resorted to a Pd-catalyzed silylstannylation.[65]
It has been shown that alcohol functionalities usually thwart this catalytic process. Thus, we aimed at protecting the tertiary alcohol.[65b] However, silylation attempts (TMSOTf, 2,6-lutidine, CH2Cl2, 40 °C; Et3SiCl, pyridine, 90 °C; BSTFA, DMF) failed. Consequentially, we assumed that the alcohol might be too burdened in its steric environment to affect the envisaged transformation. Indeed, exposing alkyne 65 to Bu3SnSiMe3 under Pd-catalysis gave rise to a single regioisomer 66. Subsequent iododestannylation (I2, 2,6-di-tert-butyl-4-methylpyridine, −40 °C)[66] and reaction with Me2CuLi delivered alkenyl silane 67. In order to access macrocyclization precursor 60, we treated silane 67 with excess NIS (≈ 2.5 equivalents) in MeCN to affect an iododesilylation. Upon isolation of a new product, 1H NMR spectroscopy revealed that the iododesilylation took place as indicated by a downfield shift of the alkenyl proton from δ = 5.20 ppm to δ = 5.89 ppm. However, the characteristic protons signals for the terminal alkene group disappeared. In addition, proton signals at δ = 4.53 and 3.61 ppm indicated that heteroatoms were attached to the respective carbon atoms. Thus, we tentatively assigned the new compound as a diastereomeric mixture of tricycle 68. While employing excess NIS in MeCN is usually required for the iododesilylation to secure retention of the double bond geometry,[67] the use of hexafluoroisopropanol as solvent allows for stoichiometric quantities of NIS.[68] This protocol resulted in decomposition of alkenyl silane 66 in our hands. We rationalized that a protection of the alcohol functionality might avoid the cyclization, but the examined conditions (KHMDS, Et3SiCl; Ac2O, pyridine, 90 °C) were not met with success. Due to material constraints, no further investigations were undertaken at this stage.[69]
Second-generation approach
In light of our previous endeavors, we sought to establish a more convergent strategy for a potential biomimetic total synthesis of astellatol. Particularly, we opted at a route involving a 5-8-6-5 ring system having the correct relative configuration at C14 already in place and reducing the number of events in the key reaction cascade.
As outlined in Scheme 20, we envisaged diene 69 as key precursor for a biomimetic synthesis of astellatol. Upon treatment with acid, protonation could trigger the proposed homoallyl-cyclopropylcarbinyl-cyclobutyl rearrangement. Closure of the central eight-membered carbocycle should be achieved by a McMurry coupling and it was envisaged that the required diketone could arise from trans-hydrindane 53 and triflate 71 using B-alkyl-Suzuki coupling conditions. The latter fragment could in turn be prepared from 3-pentyn-1-ol (70) via an intramolecular 1,3-dipolar nitrile oxide cycloaddition and subsequent reductive ring opening. In addition, an auxiliary controlled alkylation could render our approach asymmetric.
Scheme 20.

Second generation retrosynthetic analysis of astellatol: diene 69 as precursor for the biomimetic cationic cascade.
In the forward direction, Myers alkylation[70] of pseudo-ephedrinepropionamide (72) with iodide 73 resulted in clean formation of alkyne 74 (Scheme 21).[71] Subsequently, hydrogenation of alkyne 74 in the presence of Pd/BaSO4 in pyridine yielded the corresponding cis alkene.[72] Other attempts mostly resulted in over reduction to the respective alkane. Cleavage of the chiral auxiliary with lithium amidotrihydroborate[70] provided the respective alcohol and characterization of the corresponding Mosher ester[73] revealed an enantiomeric excess of 98%. Oxidation to the corresponding volatile aldehyde under Swern conditions and transformation to oxime 75 proceeded smoothly and gave an inconsequential 5:2 E/Z mixture of isomers in good yield over the four steps.[74] Among the various methods for the in situ generation of nitrile oxides and ensuing 1,3-dipolar cycloadditions,[75] treatment of oxime 75 with aqueous NaOCl proved best and gave clean conversion to the 4,5-dihydroisoxazole as an inseparable 2:3 mixture of syn/anti isomers 76 and 77.[76] Chemoselective hydrogenation to the corresponding β-keto alcohol by reaction with Raney® Ni in the presence of a boron-based buffer[77] was followed by Swern oxidation to provide ketoenol 78. Notably, exposure of the intermediate alcohol to DMP or TPAP/NMO led to decomposition. Unfortunately, the ensuing conversion of intermediate 78 to triflate 71 by sequential treatment with NaH and PhNTf2 suffered from a very poor yield.
Scheme 21.

Enantioselective synthesis of enol triflate 71.
The low yield of the last step was very surprising to us, since the conversion of commercially available diketone 79 to enol triflate 80[78] was high yielding under identical reaction conditions (Scheme 22).
Scheme 22.

Synthesis of enol triflate 80 (top) and formation of ether 83 and hemiacetal 84 under reductive carbonyl coupling conditions (bottom).
Consequently, we first explored the key step of our proposed strategy with building block 80 and trans-hydrindane ent-47[79] as model systems. Again, fragment coupling under B-alkyl-Suzuki conditions worked smoothly, delivering product 81 in quantitative yield. Subsequently, desilylation and oxidation yielded ketone 82. With this material in hand, we were poised to investigate the envisaged McMurry coupling for the synthesis of key intermediate 69.[80,81] To this end, substrate 81 was slowly added to a suspension of low-valent titanium, prepared in situ from TiCl3(THF)3 and Zn−Cu couple in DME.[82] Interestingly, these reaction conditions did not result in the formation of the desired eight-membered ring in tetracycle 69, but pentacyclic product 83 was isolated as a single diastereomer. Despite extensive NMR spectroscopic studies, however, we could not unambiguously assign the configuration of the newly formed stereocenters. To the best of our knowledge, this cascade to form the complex 5-5-6-6-5-membered ring system is unprecedented in the literature.
Since reductive carbonyl-carbonyl coupling can also be achieved by means of diol formation, we turned our attention towards SmI2-mediated reactions.[83,84] In agreement with the previous result, the formation of an eight-membered ring was not observed. Instead, the same cyclization mode occurred, affording hemiacetal 84 as a single diastereomer.
Mechanistically, we propose the following reaction pathway for this transformation (Scheme 23). Upon formation of the ketyl radical at the more electrophilic carbonyl in diketone 82, the resulting intermediate 85 undergoes a Michael addition in a 6-exo-trig fashion.[85] Subsequent one-electron reduction of the resulting acyl radical (not shown) with a second equivalent of SmI2 forms dimetallic species 87. Protonation and ketoenol tautomerization − presumably during aqueous work-up − gives ketone 86 that immediately cyclizes to form hemiacetal 84. Under the McMurry-coupling conditions, the latter process seems to occur in the reaction mixture and is followed by deoxygenation of the hemiacetal to form an oxonium ion. These steps might be assisted by the Lewis acidic nature and the oxophilicity of the generated titanium species. A final two-electron reduction step would then lead to the corresponding ether 83. Presumably, the less Lewis acidic properties of SmI2 combined with the milder reaction conditions account for the fact that in this case no final reductive deoxygenation was observed. A further screening of reaction conditions for a reductive carbonyl-carbonyl coupling proved less fruitful. In light of these findings and the fact that the low yielding enol triflate formation remained a bottleneck in our synthetic roadmap for building block 71, we abandoned this strategy at this point.[86]
Scheme 23.

Proposed mechanism for the formation of pentacycle 84 under reductive carbonyl coupling conditions.
Third-generation retrosynthesis
Although we had established the B-alkyl Suzuki coupling as a robust and viable C−C-bond formation reaction for our model hydrindane hydrindane 47, we ultimately decided to pursue other retrosynthetic disconnections. As depicted in Scheme 24, we assumed that we could utilize allylic alcohol 88 to trigger a biomimetic cascade for the synthesis of astellatol. Upon ionization, a syn specific 1,2-hydride shift could initiate the homoallyl-cyclopropylcarbinyl-cyclobutyl rearrangement. Notably, the same intermediate 88 could also serve as a synthetic precursor for a total synthesis of nitidasin by sequential oxidations. It was envisaged that the central eight-membered ring of our advanced key intermediate 88 would be constructed by a RCM[87] and a subsequent chemoselective hydrogenation of the less substituted double bond. Further retrosynthetic simplification would give two building blocks, alkenyl lithium species 89 and ketone 90, that should be joined by a nucleophilic addition. As we had learned from our previous studies (vide supra), such an addition was most likely to be feasible and should result in the correct relative configuration at the tertiary alcohol required for a stereoselective total synthesis of nitidasin.
Scheme 24.

Third-generation retrosynthesis: convergent access to tetracycle 88 as precursor for astellatol and nitidasin.
Synthesis of alkenyl iodide coupling partners
Our studies commenced with the synthesis of the precursor for fragment 89 and related alkenyl iodides. We were aware that the installation of the cis relationship of the two adjacent ring substituents would be challenging, therefore we aimed at exploiting substrate control once more and resorted to the chiral pool to implement an asymmetric route (Scheme 25). Accordingly, (−)-citronellene (91) was converted in a two-step oxidative protocol to a literature-known aldehyde.[88] This was followed by a Corey-Fuchs homologation[89] with quenching of the intermediate lithium species with MeI to furnish enyne 92. The latter compound was very volatile complicating its isolation and accounted for the poor yield of the four-step sequence. Next, we employed a cyclometallation protocol developed by Negishi[90] to establish the correct alkene geometry of alkenyl iodide 93. In this reaction, exposure of enyne 92 to stoichiometric amounts of Cp2Zr generated in situ gave a zirconacycle, which was quenched with excess NIS.[91,92] The resulting inconsequential mixture of diastereomers 93 was briefly purified before eliminating the primary iodide by treatment with DBU. The so formed diene proved to be highly unstable and was immediately used in a diastereoselective hydroboration (9-BBN, 45 °C) furnishing alcohol 95 as the major isomer in good yield over the three steps. At this stage, we prepared the corresponding Mosher ester and determined the enantiomeric excess of alcohol 95 to be 60%. This was due to the low optical purity of the commercially available (−)-citronellene (91).
Scheme 25.

Diastereoselective synthesis of alkenyl iodides 94, 95 and 95.
In order to explore the envisioned fragment coupling, we next synthesized two building blocks. On the one hand, silylation of alcohol 95 afforded the corresponding silyl ether 94. On the other hand, sequential Swern oxidation and Wittig olefination delivered diene 96 as a single isomer, indicating that no epimerization occurred at the aldehyde oxidation state. This was assumed to be due to the high 1,3-allylic strain an intermediate enolate would encounter. Overall, the route was straightforward and diene 96 was accessed in 16% yield over nine steps.
Model studies − fragment combination and RCM
Having prepared sufficient amounts of the two alkenyl iodide building blocks 94 and 96, we were poised to investigate their coupling to the trans-hydrindanone 90. In analogy to our earlier studies and in order to conserve our precious isopropyl substituted fragment 90, these studies were first carried out with ketone 61.
Since we assumed that the alkenyl lithium species of iodide 96 might be prone to undergo a 1,3-metallotropic rearrangement, we initially utilized silyl ether 94 in these explorations. To our delight, preparation of the alkenyl lithium compound and subsequent slow addition of ketone 61 resulted in a facile reaction and, after purification, we obtained adduct 97 as a single diastereomer in 48% yield (Scheme 26). This reactivity is particularly worth noting since tetrasubstituted alkenyl lithium species are rarely used in organic synthesis and addition to sterically encumbered ketones such as 61 are unprecedented in the literature.[93] In order to elaborate the metathesis precursor, we next cleaved the silyl ether and oxidized the primary alcohol under Swern conditions. Interestingly, the formed aldehyde underwent a spontaneous intramolecular cyclization forming lactol 99 as a single diastereomer, the relative configuration of which was not further investigated. While direct olefination of this intermediate 99 proved unviable, this outcome was an indirect proof that the double bond geometry of the alkenyl iodide was retained upon lithiation and under the addition reaction conditions.
Scheme 26.

Model fragment coupling: synthesis of triene 98 and lactol 99.
Encouraged by this result, we next prepared the alkenyl lithium species derived from iodide 96 at −78 °C. Upon addition of ketone 61, the fragment coupling was again efficient and provided adduct 98 in 75% yield. It is worth noting that the obtained 10:1 mixture of diastereomers reflects a kinetic resolution of scalemic alkenyl lithium species 89 under the reaction conditions. In addition, we verified the relative configuration of all stereogenic centers and the alkene geometry by single crystal X-ray diffraction. The solid-state structure also revealed a close proximity between the two terminal alkenes, solely requiring a 120° rotation around the C9−C10 bond (astellatol numbering) for the envisaged metathesis. With this in mind, we were excited to explore the key RCM (Scheme 27).
Scheme 27.

RCM of triene 98: unexpected synthesis of tricycle 101.
Our efforts commenced by using the Grubbs first-generation catalyst in CH2Cl2, resulting in slow consumption of the major diastereomer 98, but no formation of the desired product 100. Interestingly, we observed traces of a cyclized product by 1H NMR spectroscopy and EI mass spectrometry, which we tentatively assigned as tetracycle 102.[94] Similar results were obtained when employing the Grubbs second-generation catalyst in CH2Cl2 or DCE. When exchanging the solvent for benzene, quick consumption of the starting material 98 occurred and a new product was selectively formed. However, NMR spectroscopic analysis revealed that the tetrasubstituted double bond unexpectedly participated in the metathesis reaction yielding tricycle 101 in 60% yield. Chromatography employing 1% Et3N in the eluent mixture was required, since otherwise an allylic transposition of the alcohol functionality to a diastereomeric mixture was observed. In analogy to our earlier studies, attempts to increase the sterical demand at the tetrasubstituted alkene in triene 98 by protecting the adjacent alcohol (e.g. Et3SiCl; Et3SiOTf; TMSOTf; TMSCl; MeI; Me2SO4; FSO3Me) remained unfruitful.
Although our intial attempts to construct cyclooctadiene 100 via RCM had been hampered by the formation of tricycle 101, we were intrigued by the seemingly smooth formation of the minor diastereomer 102. We rationalized that a relay metathesis strategy (Scheme 28) could prevent initiation of the catalyst at the terminal alkene adjacent to C10 (astellatol numbering), thus possibly giving rise to the desired eight-membered ring.[95,96]
Scheme 28.

Relay RCM of tetraene 104.
Modifying the already developed Swern oxidation and Wittig olefination sequence, we were able to access triene 103 from alcohol 95. Subsequent lithiation and addition to model system 61 furnished the desired metathesis precursor 104 in excellent yield. Again, a kinetic resolution of scalemic alkenyl iodide 103 took place, although in somewhat less of an extent than in the previous explorations. Despite the flexible alkene handle in intermediate 104, single crystals suitable for X-ray diffraction were obtained. Next, we screened alkene metathesis reaction conditions for the reaction of tetraene 104. When using the Grubbs first- or second-generation catalyst in benzene, tricycle 101 was formed accompanied with varying amounts of homo-dimer 105 and the previously observed diastereomer 102. In contrast, the use of DCE as solvent only resulted in observation of the dimer 105. In order to enhance the catalyst preference to initiate at the least substituted double bond, we next employed catalyst 106[97] in benzene, resulting in significantly increased amounts of dimer 105 (62% yield). However, only traces of dimer 105 were observed with this catalyst in DCE.
In view of these results, we decided to block the tetrasubstituted alkene for metathesis by epoxidation, additionally addressing the synthesis of nitidasin. Gratifyingly, exposure of triene 98 to mCPBA resulted in a diastereoselective reaction of the tetrasubstituted alkene, furnishing RCM substrate 107 with the correct epoxide geometry for nitidasin (Scheme 29). The structure of this intermediate 107 was again confirmed by single crystal X-ray diffraction and indicated a close proximity of the terminal alkenes necessary for the metathesis. Since we had observed unique reactivity of triene 98 using the Grubbs second-generation catalyst in benzene, we submitted diene 107 to the same reaction conditions. To our delight, the envisaged ring closure occurred smoothly and delivered tetracarbocycle 108 in 98% yield.[98] Next, we aimed at completing the synthesis of the carbon framework of nitidasin. To this end, alkene 108 was hydrogenated in the presence of Pd/C and the resulting saturated analogue 109 was fully characterized by NMR spectroscopy in benzene-d6. In order to compare our model compound with the isolation data of nitidasin, we next employed CDCl3 as the NMR solvent. Interestingly, this resulted in a rapid conversion of epoxide 109 to exo-methylene compound 110 that was attributed to traces of acid present in CDCl3.
Scheme 29.

Model studies: synthesis of the nitidasin skeleton 109.
Total synthesis of nitidasin
After successfully establishing a fragment union and the subsequent RCM with model ketone 61, we were excited to transfer these conditions to a system with the correct substitution pattern at the five-membered ring. To this end, chemoselective cleavage of the triethylsilyl ether in bicycle 53 was followed by oxidation of the resulting secondary alcohol to furnish ketone 90 (Scheme 30). This intermediate was then slowly added to the alkenyl lithium species derived from iodide 96 and tBuLi at −78 °C. In analogy to the model system, the reaction proceeded in a facile manner and with complete stereocontrol with respect to the newly formed stereogenic center. The isomers arising from scalemic reagent 89 were separable by column chromatography yielding 7% of the undesired adduct 112. Interestingly, the desired isomer 111 decomposed upon complete evaporation of the solvents and was thus immediately subjected to the epoxidation conditions. The intermediate was isolated in 71% yield, indicating that again a kinetic resolution of scalemic iodide 96 took place. The ensuing RCM proceeded smoothly to deliver tetracarbocycle 113 in 66% yield for the three steps. At this stage, single crystal X-ray diffraction verified the relative configuration of all eleven stereogenic centers.
Scheme 30.

Construction of the nitidasin backbone 114.
In order to complete the total synthesis of nitidasin, we identified the hydrogenation of the cyclooctene and the SEM deprotection as the major tasks. We initially investigated the hydrogenation of alkene 113 under the conditions employed for the model system (0.2 eq. Pd/C, H2 (1 bar), MeOH). This resulted in comparably slow conversion and the prolonged reaction times caused a mixture of compounds to be formed that we attributed to degradation of product 114. Thus, we screened further conditions (0.2 eq. Pd/C, H2 (8, 20 or 30 bar), MeOH or EtOAc; PtO2, H2 (1 bar), EtOH; 5 mol-% Rh/Al2O3, H2 (1 bar), EtOH; 10 mol-% Crabtree’s catalyst, H2 (1 bar), CH2Cl2), however, without achieving any satisfying result.
Finally, we decided to revisit our initial reaction conditions, but using superstoichiometric amounts of Pd/C in MeOH. This resulted in a fast and relatively clean conversion and delivered the saturated compound 114 in good yield. The structure of this intermediate was again confirmed by single crystal X-ray diffraction. However, the observed sensitivity of tetracycle 114 under the hydrogenation conditions combined with little amounts of material available for testing purposes prompted us to investigate the SEM deprotection at an earlier stage of our synthetic route.
Thus, we screened a variety of conditions on ketone 90 to identify a suitable protocol for this transformation. Among the reagents examined (e.g. HF/py; TFA; TASF; MgBr2/MeNO2; TBAF; HCl), HF/py proved to be the best (Scheme 31). Interestingly, the reaction did not lead to full conversion and was accompanied by the partial epimerization at C3 (nitidasin numbering). The isomers were separable and the undesired ketone could be equilibrated by subjection to the reaction conditions together with the recovered starting material, increasing the overall yield. With intermediate 115 in hand, we reasoned that the previously established reaction sequence might also be feasible with the free alcohol functionality. To this end, the fragment combination was carried out with a sacrificial equivalent of the alkenyl lithium species 89 giving a 25:1 mixture of diastereomers 116 originating from scalemic alkenyl iodide 96.
Scheme 31.

Total synthesis of (−)-nitidasin.
Subsequent epoxidation and RCM gave tetracarbocycle 117 in an excellent yield of 72% over three steps. The hydrogenation of the double bond under the previously established reaction conditions proceeded smoothly to deliver ‘nitidasol’, presumably the biosynthetic precursor of nitidasin. Finally, oxidation with TPAP/NMO[99] furnished the natural product. These conditions had been established with alcohol 117, in which case DMP had resulted in decomposition. The spectroscopic data of our product matched the isolation data.[4] However, the use of rigorously acid-free CDCl3 was required since otherwise, in accordance with our model studies, a conversion to exo-methylene compound 118 was observed. This transformation occurred in time frames ranging from minutes to days, depending on the batch of solvent employed. In addition to the correlating spectroscopic data, we verified the structure of our synthetic material by single crystal X-ray diffraction. Furthermore, comparison of the optical rotation of our sample with the authentic value – both being levorotatory – established that we synthesized natural nitidasin.
While this first synthetic route proved viable for a total synthesis of nitidasin, it used a sacrificing equivalent of the alkenyl iodide 96 in the fragment coupling compared to the route with the SEM ether in place. This prompted us to investigate the deprotection in tetracarbocycle 114. First attempts, using TBAF in DMPU only led to slow decomposition. Interestingly, when using TBAF in THF in the presence of molecular sieves, loss of the TMS moiety was observed rather then complete cleavage of the SEM group. In contrast, the use of excess TASF in HMPA at 75 °C[100] resulted in clean deprotection along with recovered starting material (Scheme 32). The use of molecular sieves proved crucial for the success of this transformation. The so obtained alcohol 117 was then elaborated to nitidasin as described previously. Overall, 16 mg of the natural product have been prepared via the two routes.
Scheme 32.

Late stage SEM ether cleavage using substrate 113.
Progress toward a biomimetic synthesis of astellatol
As outlined in our third-generation retrosynthetic analysis (cf. Scheme 24), we also aimed at accessing allylic alcohol 88 as a precursor for the proposed biomimetic cationic cascade toward the construction of the astellatol ring system. Since our efforts to obtain alkene 88 via a cyclooctadiene capitalizing from a RCM proved unsuccessful in model studies (vide supra), we changed our strategy and were drawn to the transformation of epoxides into the respective alkenes by the use of a low valent tungsten species generated in situ from WCl6 and nBuLi.[101] In the event, epoxide 114 was cleanly converted to allylic alcohol 88 in excellent yield (Scheme 33).[102] At room temperature, signal broadening originating from the rotameric nature of cyclooctene 88 thwarted the structure analysis by 13C NMR spectroscopy, but performing the studies at 40 °C allowed for clean characterization. In addition, we resorted to single crystal X-ray diffraction to prove the identity of compound 88.
Scheme 33.

Deoxygenation of epoxide 114 and proposed pathway for the investigated biomimetic cationic cascade toward astellatol.
We were then in the position to examine the proposed cationic cascade for the synthesis of astellatol. In order to trigger the initial ionization step to transform allylic alcohol 88 into carbocation 119, we investigated the use of Brønsted acids (TsOH; CSA; HCl; HF; TFA; HClO4) in a variety of solvent systems of different polarities (CDCl3; CH2Cl2; DCE; toluene; CH3CN; THF/H2O; 1,4-dioxane; DMSO/H2O; CH3NO2; TFE; MeOH; acetone, pyridine) at temperatures ranging from room temperature up to 110 °C. Unfortunately, the only observation repeatedly made was the formation of several kinds of elimination products that were occasionally accompanied by SEM group removal. The characteristic signals of the exo-methylene group of astellatol were never observed by 1H NMR analysis of the reaction mixtures. Furthermore, the addition of perrhenate species such as Re2O7, which are known to catalyze the transposition of allylic alcohols,[103] could not avoid undesired elimination reactions. The same held true when Lewis acids (e.g. BF3·OEt2, Ti(OiPr)4, TiCl4, Cp2ZrCl2, AlCl3) were employed instead. Presumably, the required initial 1,2-hydride shift to form intermediate 8 exhibits an energetic barrier too high to compete with elimination processes. Accordingly, future work will be directed to the synthesis of the corresponding homoallylic alcohol of cation 8.
Conclusions
In summary, we have presented the evolution of our campaign for the total synthesis of isopropyl trans-hydrindane sesterterpenoids. Starting from diketone 4, we have developed a robust and scalable synthetic route to several versatile building blocks that was based on a series of substrate-controlled and diastereoselective transformations. Since some of the selectivities were surprising at first glance, we investigated their origin by DFT calculations and established reasonable reaction pathways. The initial efforts of our unified approach aimed at a biomimetic synthesis of astellatol. However, attempts to close a strained eleven-membered ring remained unsuccessful and prompted us to pursue alternative routes by preparing a 5-8-6-5 tetracarbocyclic backbone in a convergent fashion. In our first strategy to close the central eight-membered ring by a reductive coupling of two carbonyl functionalities, we identified the intriguing yet undesired pentacyclic products 83 and 84, which from a cascade that sets four stereogenic centers in a single step. Our second approach involved the addition of a complex alkenyl lithium species to a trans-hydrindanone, a chemo- and stereoselective epoxidation and an olefin-metathesis to construct a 5-6-8-5 tetracyclic carbon framework. While our attemots to access astellatol based on this intermediate remained unfruitful, we successfully synthesized (−)-nitidasin and established its absolute configuration. Our stereoselective route proceeded in a longest linear sequence of 34 steps and delivered the natural product in 1.9% overall yield. A second-generation synthesis of key ketone 90 and studies toward the synthesis of astellatol and the YW compounds are currently under investigation in our laboratories and will be reported in due course.
Experimental Section
Experimental, computational and crystallographic details as well as compound characterization data and copies of 1H and 13C NMR spectra are available in the Supporting Information for this article on the WWW under http://dx.doi.org/10.1002/chem.2015XXXXXX.
Supplementary Material
Acknowledgements
We are indebted to the Fonds der Chemischen Industrie (PhD scholarship, D.T.H.), the Deutsche Forschungsgemeinschaft (SFB 749, D.T.) and NIH-NIGMS (GM075962, K.N.H.) for financial support. This work used the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by the National Science Foundation (OCI-1053575) along with the UCLA Institute of Digital Research and Education (IDRE). The authors gratefully acknowledge M. Sc. Giulio Volpin for assistance in NMR measurements and M. Sc. Martin Maier, Dr. David Barber, Dr. Pavol Jakubec, Dr. Robert Webster, Dr. Dominik Hager, Dr. Pascal Ellerbrock, Dr. Martin Olbrich, Dr. Nicolas Guimond, Dr. Olga Schöne, Dr. Jesús García-López and Dr. Sharon R. Neufeldt for insightful discussions. We would like to thank the analytical facilities of LMU Munich for excellent support and undergraduate researchers Sebastian Rappenglück, Thomas Wildenhof, Martin Rossa and Florian Weinzierl for skillful experimental assistance.
Footnotes
Supporting information for this article is given via a link at the end of the document.
Contributor Information
Gonzalo Jiménez-Osés, Email: gjimenez@chem.ucla.edu.
Dirk Trauner, Email: Trauner@cup.uni-muenchen.de.
References
- 1.Wang L, Yang B, Lin Xr.-P, Zhou X-F, Liu Y. Nat. Prod. Rep. 2013;30:455–473. doi: 10.1039/c3np20089b. and earlier reviews in this series. [DOI] [PubMed] [Google Scholar]
- 2.Sadler IH, Simpson TJ. J. Chem. Soc. Chem. Commun. 1989:1602–1604. [Google Scholar]
- 3.Sadler IH, Simpson TJ. Magn. Res. Chem. 1992;30:S18–S30. [Google Scholar]
- 4.Kawahara N, Nozawa M, Flores D, Bonilla P, Sekita S, Satake M, Kawai K-i. Chem. Pharm. Bull. 1997;45:1717–1719. [Google Scholar]
- 5.Senatore F, Feo VD, Zhou ZL. Ann. Chim. (Rome) 1991;81:269–274. [Google Scholar]
- 6.Kawahara N, Nozawa M, Flores D, Bonilla P, Sekita H, Satake M. Phytochemistry. 2000;53:881–884. doi: 10.1016/s0031-9422(99)00518-x. [DOI] [PubMed] [Google Scholar]
- 7.Kawahara N, Nozawa M, Kurata A, Hakamatsuka T, Sekita S, Satake M. Chem. Pharm. Bull. 1999;47:1344–1345. doi: 10.1248/cpb.47.1344. [DOI] [PubMed] [Google Scholar]
- 8.Wang Y, Dreyfuss M, Ponelle M, Oberer L, Riezman H. Tetrahedron. 1998;54:6415–6426. [Google Scholar]
- 9.Wang Y, Oberer L, Dreyfuss M, Sütterlin C, Riezman H. Helv. Chim. Acta. 1998;81:2031–2042. [Google Scholar]
- 10.Kaneda M, Takahashi R, Iiataka Y, Shibata S. Tetrahedron Lett. 1972;13:4609–4611.Kaneda M, Takahashi R, Shibata S. Acta Crystallogr. 1974;B30:358–364.
- 11.Lauer U, Anke T, Sheldrick WS, Scherer A, Steglich W. J. Antibiotics. 1989;42:875–882. doi: 10.7164/antibiotics.42.875. [DOI] [PubMed] [Google Scholar]
- 12.For a review on synthetic approaches toward sesterterpenoids, see: Hog DT, Webster R, Trauner D. Nat. Prod. Rep. 2012;29:752–779. doi: 10.1039/c2np20005h.
- 13.Corey EJ, Desai MC, Engler TA. J. Am. Chem. Soc. 1985;107:4339–4341. [Google Scholar]
- 14.Paquette LA, Wright J, Drtina GJ, Roberts RA. J. Org. Chem. 1987;52:2960–2962.Wright J, Drtina GJ, Roberts RA, Paquette LA. J. Am. Chem. Soc. 1988;110:5806–5817.
- 15.Hudlicky T, Radesca-Kwart L, Li L-q, Bryant T. Tetrahedron Lett. 1988;29:3283–3286.Hudlicky T, Fleming A, Radesca L. J. Am. Chem. Soc. 1989;111:6691–6707.
- 16.Wender PA, Singh SK. Tetrahedron Lett. 1990;31:2517–2520. [Google Scholar]
- 17.Paquette LA, Heidelbaugh TM. Synthesis. 1998:495–507. [Google Scholar]
- 18.Wilson MS, Dake GR. Org. Lett. 2001;3:2041–2044. doi: 10.1021/ol016002l.Wilson MS, Woo JCS, Dake GR. J. Org. Chem. 2006;71:4237–4245. doi: 10.1021/jo0604585.
- 19.Mizutani R, Nakashima K, Saito Y, Sono M, Tori M. Tetrahedron Lett. 2009;50:2225–2227. [Google Scholar]
- 20.Hog DT. Dissertation, LMU Munich. 2013 [Google Scholar]
- 21.Parts of this work have been communicated previously, see: Hog DT, Mayer P, Trauner D. J. Org. Chem. 2012;77:5838–5843. doi: 10.1021/jo300726z.Hog DT, Huber FME, Mayer P, Trauner D. Angew. Chem. 2014;126:8653–8657. doi: 10.1002/anie.201403605.Angew. Chem. Int. Ed. 2014;53:8513–8517. doi: 10.1002/anie.201403605.
- 22.For a recent publication addressing this problem, see: Jeso V, Aquino C, Cheng X, Mizoguchi H, Nakashige M, Micilanzio GC. J. Am. Chem. Soc. 2014;136:8209–821. doi: 10.1021/ja504374j.
- 23.Simpson TJ. J. Chem. Soc. Perkin Trans. 1. 1994:3055–3056. [Google Scholar]
- 24.For an overview of this rearrangement as part of a review on the use of cyclopropanes and their derivatives in organic synthesis, see: Wong HNC, Hon M-Y, Tse C-W, Yip Y-C, Tanko J, Hudlicky T. Chem. Rev. 1989;89:165–198.
- 25.Hajos ZG, Parrish DR. Org. Synth. 1985;63:26–31. [Google Scholar]
- 26.CCDC 865613, 865614, 865615, 865616, 865617, 865618, 865619, 934938, 934939, 934940, 934941, 987776, 987777, 987778, 988636, 990612, 1015223, 1015224 and 1015225, contain the supplementary crystallographic data for this publication. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif
- 27.Daniewski AR, Kiegiel J. Synth. Commun. 1988;18:115–118.Daniewski AR, Kiegiel K, Piotrowska E, Warchol T, Wojchiechowska W. Liebigs Ann. Chem. 1988:593–594.
- 28.Micheli RA, Hajos ZG, Cohen N, Parrish DR, Portland LA, Sciamanna W, Scott MA, Wehrli PA. J. Org. Chem. 1975;40:675–681. doi: 10.1021/jo00894a003. For selected applications in total synthesis; see: Pearson RCA, Digrandi MJ, Danishefsky SJ. J. Org. Chem. 1993;58:3938–3941.Nicolaou KC, Sun YP, Peng XS, Polet D, Chen DY-K. Angew. Chem. Angew. Chem. Int. Ed. 2008;2008;12047:7420–7423. 7310–7313. doi: 10.1002/anie.200803550.Flyer AN, Si C, Myers AG. Nat. Chem. 2010;2:886–892. doi: 10.1038/nchem.794.
- 29.The chemical structure of Stiles’ reagent is believed to be as drawn, but additional CO2 might be incorporated dependent on temperature and solvent, see: L. N. Mander, ‘Methyl Magnesium Carbonate’ in e-EROS Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, 2001. DOI: 10.1002/047084289X.rm206m.
- 30.Nicoalou KC, Zhong Y-L, Baran PS. J. Am. Chem. Soc. 2000;122:7596–7597. [Google Scholar]
- 31.For a similar finding, see: Isaka M, Tamiya M, Hasegawa A, Ishiguro M. Eur. J. Org. Chem. 2012:665–668.
- 32.Yu J-Q, Wu H-C, Corey EJ. Org. Lett. 2005;7:1415–1417. doi: 10.1021/ol050284y. [DOI] [PubMed] [Google Scholar]
- 33.Ito Y, Hirao T, Saegusa T. J. Org. Chem. 1978;43:1011–1013. [Google Scholar]
- 34.Similar selectivity has been observed previously, see: Bull JR, Loedolff MC. J. Chem. Soc. Perkin Trans. 1. 1996:1269–1276.Molander GA, Quirmbach MS, Silva LF, Jr, Spencer KC, Balsells J. Org. Lett. 2001;3:2257–2260. doi: 10.1021/ol015763l.
- 35.For a report on the nature of organocuprates with DFT calculations, see: Stemmler TL, Barnhart TM, Penner-Hahn JE, Tucker CE, Knochel P, Böhme M, Frenking G. J. Am. Chem. Soc. 1995;117:12489–12497. For a recent overview on the mechanism of nucleophilic organocopper(I) reactions, see: Yoshikai N, Nakamura E. Chem. Rev. 2012;112:2339–2372. doi: 10.1021/cr200241f.
- 36.Hwang C-S, Power PP. Bull. Korean Chem. Soc. 2003;24:605–609. [Google Scholar]
- 37.Bertz SH, Cope S, Murphy M, Ogle CA, Taylor BJ. J. Am. Chem. Soc. 2007;129:7208–7209. doi: 10.1021/ja067533d. [DOI] [PubMed] [Google Scholar]
- 38.Intermediates III were located as true minima in the gas phase at the ω-B97X-D/6-31G(d)+SDD(Cu,Mg,Br) level, and were ∼1 kcal mol−1 lower in energy than the preceding transition states (II‡) at the ω-B97X-D/6-311+G(2d,p)+SDD(Cu,Mg,Br) level in THF solution. However, these structures were ∼2.5 kcal mol−1 higher in energy than II‡ at the M06-2X/6-311+G(2d,p)+SDD(Cu,Mg,Br) level, reflecting their high instability.
- 39.Chérest M, Felkin H, Prudent N. Tetrahedron Lett. 1968;9:2199–2204. [Google Scholar]
- 40.Wang H, Houk KN. Chem. Sci. 2014;5:462–470. doi: 10.1039/C3SC52538D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smit C, Fraaje MW, Minnaard AJ. J. Org. Chem. 2008;73:9482–9485. doi: 10.1021/jo801588d. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 42.Nag NK. J. Phys. Chem. B. 2001;105:5945–5949.Stakheev AY, Mashkovskii IC, Tkachenko OP, Klementiev KV, Grünert W, Baeva GN, Kustov LM. Russ. Chem. Bull. 2009;58:280–283.
- 43.Shapiro RH, Heath MJ. J. Am. Chem. Soc. 1967;89:5734–5735. [Google Scholar]
- 44.Trost BM, Fray MJ. Tetrahedron Lett. 1988;29:2163–2166. [Google Scholar]
- 45.Cacchi S, Morera E, Ortar G. Tetrahedron Lett. 1984;25:4821–4824. [Google Scholar]
- 46.Webster R, Boyer A, Fleming MJ, Lautens M. Org. Lett. 2010;12:5418–5421. doi: 10.1021/ol1022239. [DOI] [PubMed] [Google Scholar]
- 47.Brown HC, Jadhav PK, Mandal AK. J. Org. Chem. 1982;47:5074–5083. [Google Scholar]
- 48.Nicolaou KC, Vassilikogiannakis G, Mägerlein W, Kranich R. Angew. Chem. 2001;2001;113:2543–2547. doi: 10.1002/1521-3773(20010702)40:13<2482::AID-ANIE2482>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 40:2482–2486. [Google Scholar]
- 49.Scaglione JB, Rath NP, Covey DF. J. Org. Chem. 2005;70:1089–1092. doi: 10.1021/jo048561m. [DOI] [PubMed] [Google Scholar]
- 50.Uyeda C, Rötheli AR, Jacobsen EN. Angew. Chem. 2010;2010;122:9947–9950. doi: 10.1002/anie.201005183. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 49:9753–9756. [Google Scholar]
- 51.Negishi E-i, Matsushita H, Chatterjee S, A John R. J. Org. Chem. 1982;47:3190–3192.Negishi E-i, Luo F-T. J. Org. Chem. 1983;48:2427–2430.
- 52.Jastrzebska I, Scaglione JB, DeKoster GT, Rath NP, Covey DF. J. Org. Chem. 2007;72:4837–4843. doi: 10.1021/jo070530e. [DOI] [PubMed] [Google Scholar]
- 53.Pappo R, Allen DS, Jr, Lemieux RU, Johnson WS. J. Org. Chem. 1956;21:478–479.Xu J, Trzoss L, Chang WK, Theodorakis EA. Angew. Chem. Int. Ed. 2011;50:3672–3676. doi: 10.1002/anie.201100313.
- 54.Mancuso AJ, Swern D. Synthesis. 1981:165–181. For an example of a deprotective Swern oxidation, see: Becker J, Bergander K, Fröhlich R, Hoppe D. Angew. Chem. 2008;2008;120:1678–1681. doi: 10.1002/anie.200704678.Angew. Chem. Int. Ed. 47:1654–1657.
- 55.Eleven-membered rings with two trisubstituted (E)-configured alkenes are present in dolabellane diterpenoids and have been addressed by total synthesis. For a recent example, see: Snyder SA, Corey EJ. J. Am. Chem. Soc. 2006;128:740–742. doi: 10.1021/ja0576379. For a review, see: Hiersemann M, Helmboldt H. Top. Curr. Chem. 2005;243:73–136.
- 56.For selcted reviws on the application of RCM in natural product synthesis, see: Fürstner A. Chem. Commun. 2011;47:6505–6511. doi: 10.1039/c1cc10464k.Gradillas A, Pérez-Castells J. Angew. Chem. 2006;2006;118:6232–6247. doi: 10.1002/anie.200600641.Angew. Chem. Int. Ed. 45:6086–6101.
- 57.For selected reviews on B-alkyl Suzuki cross couplings, see: Seidel G, Fürstner A. Chem. Commun. 2012;48:2055–2070. doi: 10.1039/c2cc17070a.Chemler SR, Trauner D, Danishefsky SJ. Angew. Chem. 2001;2001;113:4676–4701. doi: 10.1002/1521-3773(20011217)40:24<4544::aid-anie4544>3.0.co;2-n.Angew. Chem. Int. Ed. 40:4544–4568.
- 58.Clausen DJ, Wan S, Floreancig PE. Angew. Chem. 2011;2011;123:5284–5287. doi: 10.1002/anie.201007757. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 50:5178–5181. [Google Scholar]
- 59.Dess DB, Martin JC. J. Org. Chem. 1983;48:4155–4156. [Google Scholar]
- 60.For examples on the synthesis of eleven-membered rings using RCM, see: Cai Z, Yongpruksa N, Harmata M. Org. Lett. 2012;14:1661–1663. doi: 10.1021/ol300400x. and references cited therein
- 61.Stewart IC, Ung T, Pletnev AP, Berlin JM, Grubbs RH, Schrodi Y. Org. Lett. 2007;9:1589–1592. doi: 10.1021/ol0705144. [DOI] [PubMed] [Google Scholar]
- 62.For selected examples of closing medium-sized rings using a B-alkyl Suzuki coupling, see: Chemler SR, Danishefsky SJ. Org. Lett. 2000;2:2949–2951. doi: 10.1021/ol0161357.Mohr PJ, Halcomb RL. J. Am. Chem. Soc. 2003;125:1712–1713. doi: 10.1021/ja0296531.
- 63.Stivala CE, Gu Z, Smith LL, Zakarian A. Org. Lett. 2012;14:804–807. doi: 10.1021/ol203342e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ohira S. Synth. Commun. 1989;19:561–564.Müller S, Liepold B, Roth GJ, Bestmann H-J. Synlett. 1996:521–522.
- 65.Chenard BL, Laganis ED, Davidson F, RajanBabu TV. J. Org. Chem. 1985;50:3666–3667.Apte S, Radetich B, Shin S, RajanBabu TV. Org. Lett. 2004;6:4053–4056. doi: 10.1021/ol048265w.
- 66.Williams DR, Walsh MJ, Miller NA. J. Am. Chem. Soc. 2009;131:9038–9045. doi: 10.1021/ja902677t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stamos DP, Taylor AG, Kishi Y. Tetrahedron Lett. 1996;37:8647–8650. [Google Scholar]
- 68.Ilardi EA, Stivala CE, Zakarian A. Org. Lett. 2008;10:1727–1730. doi: 10.1021/ol800341z. [DOI] [PubMed] [Google Scholar]
- 69.An alternative solution for the problem of the undesired etherification could be a late-stage installation of the terminal alkene in cyclization precursor 60 via a Grieco alkene synthesis. First steps toward such a strategy have been conducted (see the Supporting Information). However, we abandoned this strategy realizing that our route became increasingly lengthy and resource consuming.
- 70.Myers AG, Yang BH, Chen H, McKinstry L, Kopecky DJ, Gleason JL. J. Am. Chem. Soc. 1997;119:6496–6511. [Google Scholar]
-
71.We initially envisaged synthesizing enol triflate 71 starting from Evans alkylation of oxazolidinone 120 with triflate 121 yielding alkyne 122 as a single diastereomer. However, the semi-hydrogenation of alkyne 122 to alkene 123 remained unsuccessful under a variety of reaction conditions.
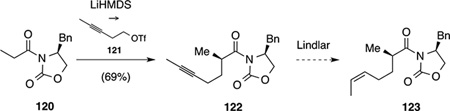
- 72.Juan JJW, Smith AB., III J. Org. Chem. 1993;58:3073–3711. [Google Scholar]
- 73.Dale JA, Dull DL, Mosher HS. J. Org.Chem. 1969;34:2543–2549.Hoye TR, Jeffery CS, Shao F. Nat. Protoc. 2007;2:2451–2458. doi: 10.1038/nprot.2007.354.
- 74.The major oxime isomer was tentatively assigned to be (E)-configured based on the general stability of oximes.
- 75.For selected examples on the in situ generation of nitrile oxides, see: Kozikowski AP, Stein PD. J. Am. Chem. Soc. 1982;104:4023–4024.Jung ME, Vu BT. J. Org. Chem. 1996;61:4427–4433. doi: 10.1021/jo952254m.Irie O, Fujiwara Y, Nemoto H, Shishido K. Tetrahedron Lett. 1996;37:9929–9232.Bonne D, Salat L, Duclère J-P, Rodriguez J. Org. Lett. 2008;10:5409–5412. doi: 10.1021/ol8023133.
- 76.The relative configuration of the major isomer was assigned based on 2D NOESY experiments, see the Supporting Information.
- 77.Curran DP. J. Am. Chem. Soc. 1982;104:4024–4026. [Google Scholar]
- 78.A synthesis of enol triflate 80 has been published previously, see: Nakatani K, Tanabe K, Saito I. Tetrahedron. 1997;38:1207–1210. Modification of the reported protocol was required, since these conditions mainly led to decomposition of substance 80 in our hands
- 79.These studies were carried out with ketone ent-47 that we had prepared for studies toward a synthesis of natural retigeranic acid B, see: F. M. E. Huber, Dissertation, LMU Munich, 2014
- 80.For reviews on carbonyl-couplings using low valent titanium see: Fürstner A, Bogdanović B. Angew. Chem. 1996;108:2582–2609.Angew. Chem. Int. Ed. 1996;35:2442–2469.McMurry JE. Chem. Rev. 1989;89:1513–1524.
- 81.For an application of McMurry conditions constructing an eight-membered ring, see: Kende AS, Johnson S, Sanfilippo P, Hodges JC, Jungheim LN. J. Am. Chem. Soc. 1988;108:3513–3515.
- 82.McMurry JE, Fleming MP, Kees KL, Kepski LR. J. Org. Chem. 1978;43:3255–3266.McMurry JE, Lectka T, Rico JG. J. Org. Chem. 1989;54:3748–3749.
- 83.For selected reviews on carbonyl-couplings using SmI2, see: Edmonds DJ, Johnston D, Procter DJ. Chem. Rev. 2004;104:3371–3403. doi: 10.1021/cr030017a.Krief A, Laval A-M. Chem. Rev. 1999;99:745–777. doi: 10.1021/cr980326e.Molander GA, Harris CR. Chem. Rev. 1996;96:307–338. doi: 10.1021/cr950019y.
- 84.For the utilization of SmI2 in taxane synthesis, see: Arseniyadis S, Yashunsky D, Pereira de Freitas R, Toromanoff E, Portier P. Tetrahedron Lett. 1993;34:1137–1140.Swindell CS, Fan W. J. Org. Chem. 1996;61:1109–1118.
- 85.A similar mechanism has been proposed for vinylogous esters, see: Kito M, Sakai T, Haruta N, Shirahama H, Matsuda F. Synlett. 1996;11:1057–1060.
-
86.Both fragments enol triflate 71 and trans-hydrindane 53 were combined in good yield to furnish compound 124. We observed a second diastereomer in this coupling indicating that partial epimerization of the selectively installed stereogenic center took place in the pursued route toward enol triflate 71.
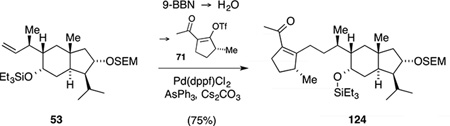
- 87.Tori M, Mizutani R. Molecules. 2010;15:4242–4260. doi: 10.3390/molecules15064242.Michaut A, Rodriguez J. Angew. Chem. 2006;2006;118:5870–5881.Angew. Chem. Int. Ed. 45:5740–5750.
- 88.Tu YQ, Hübener A, Zhang H, Moore CJ, Fletcher MT, Hayes P, Dettner K, Francke W, McErlean CSP, Kitching W. Synthesis. 2000:1956–1978.Magauer T, Martin HJ, Mulzer J. Angew. Chem. 2009;2009;121:6148–6152.Angew. Chem. Int. Ed. 48:6032–6036.
- 89.Corey EJ, Fuchs PL. Tetrahedron Lett. 1972;36:3769–3772. [Google Scholar]
- 90.Agnel G, Owczarczyk Z, Negishi E-i. Tetrahedron Lett. 1992;33:1543–1546. [Google Scholar]
- 91.García AM, Mascareñas JL, Castedo L, Mouriño A. J. Org. Chem. 1997;62:6353–6358. [Google Scholar]
- 92.This reaction could also be carried out with Cp2TiCl2 with similar yield.
- 93.For selected examples on the addition of tetrasubstituted alkenyl lithium species to aldehydes, see: Melekhov A, Forgione P, Legoupy S, Fallis AG. Org. Lett. 2000;2:2793–2796. doi: 10.1021/ol0062042.Chen J, Song Q, Li P, Guan H, Jin X, Xi Z. Org. Lett. 2002;4:2269–2271. doi: 10.1021/ol0261478.Nakajima R, Delas C, Takayama Y, Sato F. Angew. Chem. 2002;2002;114:3149–3151. doi: 10.1002/1521-3773(20020816)41:16<3023::AID-ANIE3023>3.0.CO;2-N.Angew. Chem. Int. Ed. 41:3023–3025.
- 94.Although the diastereomeric mixture 98 was inseparable by column chromatography, a slight Rf by TLC analysis. The minor component was consumed fast under all reaction conditions examined. Due to the small amounts of isolated material and the conversion of the major isomer to the 5-6-5 ring system in benzene, we assumed that the minor isomer underwent the desired eight-membered ring formation to tetracycle 102. The identity of 102 was supported by 1H, COSY and HSQC NMR experiments and mass spectrometry.
- 95.Hoye TR, Jeffrey CS, Tennakoon MA, Wang J, Zhao H. J. Am. Chem. Soc. 2004;126:10210–10211. doi: 10.1021/ja046385t. [DOI] [PubMed] [Google Scholar]
- 96.For selected recent examples of applying relay metathesis in natural product synthesis, see: Roethle PA, Chen IT, Trauner D. J. Am. Chem. Soc. 2007;129:8960–8961. doi: 10.1021/ja0733033.McGrath NA, Lee CA, Araki H, Brichacek M, Njardarson JT. Angew. Chem. 2008;2008;120:9592–9595. doi: 10.1002/anie.200804237.Angew. Chem. Int. Ed. 47:9450–9453.Wei H, Qiao C, Liu G, Yang Z, Li C-c. Angew. Chem. 2013;2013;125:648–652.Angew. Chem. Int. Ed. 52:620–624. For selected examples of unsuccessful relay RCM approaches, see: Trost BM, Yang H, Thiel OR, Frontier AJ, Brindle CS. J. Am. Chem. Soc. 2007;129:2206–2207. doi: 10.1021/ja067305j.Fürstner A, Fasching B, O’Neil GW, Fenster MDB, Godbout C, Ceccon J. Chem. Commun. 2007:3045–3047. doi: 10.1039/b707835h.
- 97.Stewart IC, Douglas CJ, Grubbs RH. Org. Lett. 2008;10:441–444. doi: 10.1021/ol702624n. [DOI] [PubMed] [Google Scholar]
- 98.The success of this transformation is also owed to the mild reaction conditions of the Grubbs catalyst, tolerating the tetrasubstituted epoxide. The number of natural product syntheses by metathesis with an epoxide in place is not overall high. For leading examples, see: Larrosa I, Da Silva MI, Gómez PM, Hannen P, Ko E, Lenger SR, Linke SR, White APJ, Wilton D, Barrett AGM. J. Am. Chem. Soc. 2006;128:14042–14043. doi: 10.1021/ja0662671.Kanada RM, Itoh D, Nagai M, Niijima J, Asai N, Mizui Y, Abe S, Kotake Y. Angew. Chem. 2007;2007;119:4428–4433. doi: 10.1002/anie.200604997.Angew. Chem. Int. Ed. 46:4350–4355.Fürstner A, Bouchez LC, Funel J-A, Liepins V, Porée F-H, Gilmour R, Beaufils F, Laurich D, Tamiya M. Angew. Chem. 2007;2007;119:9425–9430. doi: 10.1002/anie.200704024.Angew. Chem. Int. Ed. 46:9265–9270.
- 99.Griffith WP, Ley SV, Whitcombe AD, White GP. J. Chem. Soc., Chem. Commun. 1987:1625–1627. [Google Scholar]
- 100.Gladding JA, Bacci JP, Shaw SA, Smith AB., III Tetrahedron. 2011;67:6697–6706. doi: 10.1016/j.tet.2011.04.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Umbreit MA, Sharpless KB. Org. Synth. 1981;60:29–32.Molawi K, Delpont N, Echavarren AM. Angew. Chem. 2010;2010;122:3595–359. doi: 10.1002/anie.201000890.Angew. Chem. Int. Ed. 49:3517–3519.
- 102.Initially, we explored these conditions on our model system 109, see the Supporting Information.
- 103.For selected examples of perrhenate based transposition of alcohols, see: Nakasaka K, Kusama H, Hayashi Y. Chem. Lett. 1991:1413–1416.Bellemin-Laponnaz S. ChemCatChem. 2009;1:357–362.Xie Y, Floreancig PE. Angew. Chem. 2013;125:653–656.Angew. Chem. Int. Ed. 2013;52:625–628. doi: 10.1002/anie.201208132.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.