

Abstract
Age-related depletion of GSH levels and perturbations in its redox state may be especially deleterious to metabolically active tissues, such as the heart and brain. We examined the extent and the mechanisms underlying the potential age-related changes in cerebral and myocardial GSH status in young and old F344 rats and whether administration of (R)-α-lipoic acid (LA) can reverse these changes. Our results show that GSH/GSSG ratios in the aging heart and the brain declined by 58 and 66% relative to young controls, respectively (p < 0.001). Despite a consistent loss in GSH redox status in both tissues, only cerebral GSH levels declined with age (p < 0.01). To discern the potential mechanisms underlying this differential loss, the levels and the activities of γ-glutamylcysteine ligase (GCL) and cysteine availability were determined. There were no significant age-related changes in substrate or enzyme levels, or GCL activity when saturating amounts of substrates were provided. However, kinetic analysis of GCL in brains of old rats displayed a significant increase (p < 0.05) in the apparent Km for cysteine (Kmcys) vs. young rats (84.3± 25.4 vs. 179.0± 49.0; young and old, respectively), resulting in a 40% loss in apparent catalytic turnover of the enzyme. Thus, the age-related decline in total GSH appears to be mediated, in part, by a general decrement in GCL catalytic efficiency. Treating old rats with LA (40 mg/kg body wt; by i.p.) markedly increased tissue cysteine levels by 54% 12 h following treatment and subsequently restored the cerebral GSH levels. Moreover, LA improved the age-related changes in the tissue GSH/GSSG ratios in both heart and the brain. These results demonstrate that LA is an effective agent to restore both the age-associated decline in thiol redox ratio as well as increase cerebral GSH levels that otherwise decline with age.
Keywords: Aging, Oxidative stress, Antioxidants, Glutathione, Lipoic acid, γ-Glutamylcysteine ligase, Km, Redox status, Brain, Heart
Oxidative stress is a major factor of the aging process and in the initiation of many chronic diseases associated with aging [1,2]. A heightened pro-oxidant cellular environment is particularly evident in metabolically active tissues, such as the heart and brain; in turn, these organs are prone to develop life-threatening pathophysiologies, such as congestive heart failure and Alzheimer’s disease, respectively [3–6]. The susceptibility of these organs to oxidative stress stems partly from their post-mitotic nature, which limits tissue turnover and allows oxidative damage to accumulate, leading to impaired function.
The brain and heart are especially vulnerable to oxidative insult owing to their relatively limited glutathione (GSH)1-dependent antioxidant capacity [7]. For example, the heart has markedly less GSH synthetic capacity relative to the liver and also cannot effectively efflux GSSG, which limits this organ’s ability to maintain a high GSH/GSSG ratio during acute oxidative insult [8,9]. Similarly, neurons lack high levels of GSH-dependent antioxidant enzymes (e.g., GSH peroxidase and GSSG reductase) [10,11]. Thus, both the heart and brain are generally more susceptible to redox changes associated with the enhanced oxidant-rich cellular environment associated with aging.
Based on these observations, maintenance of GSH is critical to preserving normal thiol redox state in the aging heart and brain. GSH is synthesized by the concerted action of two cystosolic, ATP-requiring enzymes, γ-glutamylcysteine ligase (GCL) and GSH synthetase (GS) [12]. GCL appears to be the rate-controlling enzyme for the pathway. It exists as a heterodimer composed of a catalytic (GCLC; 73 kDa) and a modulatory (GCLM; 30 kDa) subunit [12–14]. While GCLC retains all of the catalytic activity, GCLM improves overall catalytic rate by lowering the Km for glutamate and suppressing negative feedback inhibition by GSH itself [12].
GSH levels can be increased following oxidative insult by heightened expression of GCL [15]. Additionally, GSH concentrations can also be elevated by increasing cytosolic cysteine availability; cysteine is the limiting substrate for GSH synthesis and is normally found in concentrations below the Kmcys for GCL [16]. Thus, GSH synthesis can be markedly upregulated to meet the heightened demands for the antioxidant in times of acute oxidative stress.
Given that the aging heart and brain are in a more pro-oxidant state and that GSH synthesis is highly inducible, it is surprising that these organs do not contain increased levels of GSH relative to those found in young animals. Indeed, we and others have shown that aging results in depletion of GSH in the brain while the heart exhibits no significant age-related loss [17–19]. The mechanism(s) for this differential decline and the lack of induced GSH levels in these organs are presently unclear.
Because oxidative stress appears to be a fundamental aspect of the aging process, supplementation regimens that directly augment the endogenous cellular antioxidant pool may be beneficial in restoring the normal cellular pro- vs. antioxidant ratio. For myocardial and neuronal cells, a significant effort has focused on increasing or maintaining GSH status, as it is the most abundant low molecular weight antioxidant in these tissues [13]. Unfortunately, dietary strategies aimed at increasing GSH levels in these cell types have had limited success, owing to the lack of uptake of intact GSH into these tissues and/or the inability to efficiently target substrates, such as N-acetylcysteine, to these organs [20,21]. Thus, different and innovative strategies will be necessary to ameliorate chronic oxidative stress associated with neuronal and myocardial aging.
Recently, we showed that giving old rats (R)-α-lipoic acid (LA) restored vitamin C and GSH levels in the aging rat heart and liver, respectively [6,17,22]. Moreover, this dithiol compound effectively improved hepatocellular redox balance, as indicated by a higher GSH/GSSG ratio, and reversed the age-associated increased susceptibility to toxicological insult [17]. The mechanism(s) how LA reverses the age-associated decline in endogenous antioxidants and their redox states are presently not well understood. However, in vitro studies show that LA induces cysteine uptake, thereby removing the constraints of this limiting substrate on GSH synthesis [23]. Independent of increasing critical substrate availability, LA may improve cellular redox state by increasing NADPH availability, which would supply reducing equivalents to GSSG reductase and glutaredoxin and thus maintain thiol status in a normal redox state [24,25].
Thus, the major aims of the present study were to define the levels and redox nature of GSH in the aging rat heart and brain as well as how aging affects GSH synthetic capacity in these tissues. Moreover, we sought to determine whether LA treatment to old rats improves thiol redox status and GSH homeostasis and if so, the mechanisms involved in this improvement. Herein, we show that while both the heart and brain display an age-related loss in GSH/GSSG redox ratio, only the brain has lower overall GSH levels relative to young controls. However, treating old rats with LA reverses both the age-related GSH/GSSG redox decline as well as restores cerebral GSH. This improved cerebral GSH status appears to be through an LA-mediated increased cysteine accumulation.
Materials and methods
Materials
(R)-α-lipoic acid (LA) was a gift from Asta Medica (Frankfurt, Germany). Rabbit polyclonal antibodies to GCLC (catalytic subunit) were obtained as a reference [18]. All high performance liquid chromatography (HPLC) solvents were HPLC grade reagents from Fisher Scientific (Houston, TX). All other chemicals were of reagent grade or the highest quality available from Sigma (St. Louis, MO).
Animals
Rats (Fischer 344 [F344], virgin male, outbred albino), both young (2–5 mo, N = 15) and old (24–28 mo, N = 15; National Institute of Aging animal colonies), were acclimatized in the Oregon State University animal facilities for at least one week prior to experimentation. Animals were maintained on standard chow diet. Both food and water were given ad libitum.
LA (40 mg/ml) was dissolved in 2M NaOH containing 154 mM NaCl and the pH was adjusted to 7.4 with concentrated HCl. LA solutions were sterile-filtered and were freshly prepared just prior to use. LA (40 mg/kg body wt.) or saline vehicle (control) was administered by i.p. injection. This dose was used as our previous studies showed that old rats given this regimen have significantly higher tissue GSH values without any untoward side-effects [6,17,22].
To reduce diurnal variations, animals were sacrificed between 10 and 11 am each morning. Rats were anesthetized with diethyl ether and a midline incision was made in the abdomen. Heparin (0.4 mg/ml) was injected via the femoral vein and blood was removed by retrograde perfusion with Hanks’ balanced salt buffer (pH 7.4) for 5 min via the thoracic aorta followed by cutting the posterior vena cava above the renal vein. Brains and hearts were removed and rinsed in ice-cold Chelex-treated phosphate-buffered saline (PBS; pH 7.4).
Total tissue glutathione and cysteine analysis
Tissue glutathione (GSH) and GSSG concentrations were determined according to the methods of Fariss and Reed [26]. This HPLC method has been used extensively to monitor tissue GSH levels and GSH/GSSG ratio by numerous investigators and proven to be highly reproducible in discerning potential age-related changes in GSH redox state in tissues. Brain and heart tissue (200 mg) were minced and homogenized in 10% (w/v) perchloric acid (PCA) containing 5 mM EDTA. After centrifugation, 170 μl of the supernatant containing internal standard (γ-glutamyl glutamate; 100 μM final) was mixed with 50 μl iodoacetic acid (100 mM dissolved in 0.2 mM m-cresol purple) and the pH was adjusted to 10 by using KOH–KHCO3 buffer (2M KOH:2.4M KHCO3). Samples were placed in the dark at room temperature for 1 h. Following completion of S-carboxymethyl formation, an equal volume of 1-fluoro-2,4-dinitrobenzene (1% v/v in absolute ethanol) was added and incubated overnight in the dark at room temperature. Samples (50 μl) were separated by HPLC [26] and detected using a Bioanalytical Systems (BAS; West Lafayette, IN) UV-116A spectrophotometer with the absorbance set at 365 nm. Quantitation was obtained by integration relative to internal standard. GSH/GSSG redox ratios were determined by dividing GSH values obtained by GSSG levels.
Measurement of GSSG reductase (GR) and glutaredoxin (Grx) activities
Tissues were homogenized in 0.1M potassium phosphate buffer (pH 7.4) containing 1 mM EDTA and 0.1% (v/v) protease inhibitor cocktail P8340 (Sigma Life sciences). The homogenates were centrifuged at 14,000 g for 15 min. The soluble fractions were collected for determining GR and Grx acitivity.
Tissue GR activities was monitored as described previously [27]. Briefly, tissue homogenates (40–50 μg of protein) were added to a reaction buffer consisting of 0.1 mM dithionitrobenzoic acid (DTNB), 0.5 mM NADPH, and 2 mM GSSG and the rates of DTNB reduction at 412 nm were monitored for 5 min at 25 °C using a 96-well plate reader (Spectromax 340; Molecular Devices, Sunnyvale, CA). As a control, blanks without the addition of GSSG were run simultaneously. The final reaction rates were obtained by subtracting the background rates of DTNB reduction and activity was expressed relative to protein.
Tissue Grx activities were estimated spectrophometrically, using 2-hydrozyethyldisulfide (HEDS) as substrate [28]. The reaction mixture consisting of 0.1M potassium phosphate (pH 7.5), 0.5 mM GSH, 2 U/ml glutathione reductase (Sigma life sciences), and 0.35 mM NADPH. HEDS (2 mM) was then added and the decrease in absorbance at 340 nm was measured for 3 min to obtain the background rate. The reaction was initiated by the addition of sample (50–100 μg of protein) and the rate was monitored for 3 min at 25 °C. The differences between the two slopes were used to obtain the enzymatic activity.
Measurement of γ-glutamylcysteine ligase activity and kinetic analysis
Tissue GCL activity was monitored as described previously [18]. Briefly, brain and heart tissues (200 mg) were homogenized in 0.25M sucrose containing 1 mM EDTA, 20 mM Tris–HCl (pH 7.4), and 0.1% (v/v) protease inhibitor cocktail P8340 (Sigma Life sciences). The homogenates were centrifuged at 3000 g for 10 min at 4 °C. The supernatants were centrifuged at 20,000 g for 30 min. To remove endogenous substrates and inhibitors of GCL, these samples were filtered in Microcon-10 (Amicon) tubes for 45 min at 4 °C and washed twice with 0.2 ml lysis solution (0.1M Tris–HCl, pH 8.2, 150 mM KCl, 20 mM MgCl2, and 2 mM EDTA). Proteins were quantified using a standard Lowry assay. The enzyme reaction was initiated by adding protein (0.5 mg/ml) to the reaction buffer containing 20 mM L-glutamic acid, 5 mM cysteine, 5 mM dithiothreitol, 10 mM ATP, 0.1M Tris–HCl (pH 8.2), 150 mM KCl, 20 mM MgCl2, 2 mM EDTA, and 0.04 mg/ml acivicin. The samples were incubated in a water bath at 37 °C for 45 min and the reactions were stopped by mixing 150 μl of the sample with an equal volume of 10% PCA. The amount of GCL formed was detected using the same analytical method as described for GSH. Quantitation was obtained by integration relative to GCL external standard.
The kinetic analysis of GCL was performed by determining the reaction rates over a range of cysteine concentrations (0.1–5 mM) in the presence of fixed concentrations of 20 mM L-glutamic acid. The results were plotted via double reciprocal plot format of the rate vs. substrate concentration. The apparent Km for cysteine was obtained by linear regression. Results were also calculated by direct linear plot using the equation of Vmax = v + (v/S)Km. The apparent Kcat for GCL was obtained by dividing Vmax by Km.
Western blot analysis
Tissues were homogenized and processed as described for the analysis of GCL activity. An aliquot of tissue homogenate (100 μl) was used for determining GCL protein content by Western blotting as described before [18]. GCLC was identified according to molecular weight markers. Relative densities of the bands were digitally quantified using NIH image analysis software.
Statistical analysis
The statistical significance of differences between means of two independent groups was determined by Student’s t test, assuming equal variances. For the comparison of treatment effects of LA in young and old rats, two-way analysis of variance test with Bonferroni’s post hoc test was used. For all of the analysis, the results were considered significant if the p value was less than 0.05. Statistical analysis was performed using GraphPad Prism version 4.0 software (GraphPad Software, San Diego, CA).
Results
Age-related changes in cerebral and myocardial thiol redox state
To gauge the extent of age-related changes in thiol redox status, the GSH/GSSG ratios in brains and hearts of young and old rats were examined. The cerebral GSH/GSSG ratios declined from 28.3± 3.8 to 13.8± 0.3 with age, a significant (p < 0.01) 50% loss in this important redox parameter (Table 1). Paralleling the patterns observed in the brain, myocardial GSH/GSSG ratios were also markedly lower on an age basis (18.2± 0.3 vs. 7.7± 0.8 in young compared to old rats, respectively; p < 0.01; Table 1), indicating heightened levels of oxidative stress in both the aging brain and the heart.
Table 1.
Age-related changes in cerebral and myocardial GSH status
| Young (N = 6)
|
Old (N = 6)
|
p< | |||
|---|---|---|---|---|---|
| Brain | Heart | Brain | Heart | ||
| GSH (nmol/mg protein) | 1.7 ± 0.2 | 2.0 ± 0.1 | 1.1 ± 0.1† | 2.0 ± 0.1 | 0.05 |
| GSSG (nmol/mg protein) | 0.06 ± 0.01 | 0.11 ± 0.02 | 0.08 ± 0.01 | 0.26 ± 0.02† | 0.05 |
| GSH/GSSG | 28.3 ± 3.8 | 18.2 ± 0.3 | 13.8 ± 0.7‡ | 7.7 ± 0.8‡ | 0.01 |
| Total GSH (nmol/mg protein) | 1.8 ± 0.1 | 2.2 ± 0.2 | 1.3 ± 0.1† | 2.5 ± 0.3 | 0.05 |
Values are expressed as means± SEM. † and ‡ denote statistically significant differences between young and old animals.
p < 0.05;
p < 0.01; N = 4.
Because GSH is normally maintained in a highly reduced state via the NADPH-dependent enzymes, GSSG reductase (GR) and glutaredoxin (Grx), one possible mechanism contributing to the decline in GSH redox state could be due to a loss in activities of one or both of these enzymes. To examine this possibility, GR and Grx activities were determined. Results showed no apparent age-associated changes in GR or Grx activity in the brain. The cerebral, GR activities in old animals (1.3± 0.1 nmol/min/mg protein) were similar to typical values seen in young animals (1.0± 0.1 nmol/min/mg protein). Grx activity in the senescent brain (2.7± 0.1 μmol/min/mg protein) was also indistinguishable from average activity seen in brains from young rats (2.3± 0.24 μmol/min/mg protein). Likewise, no apparent age-related deficits in GR or Grx activity were seen in the heart (3.5± 0.9 nmol/min/mg protein vs. 3.48± 0.6 nmol/min/mg protein; young vs. old, respectively). Grx activity in the aging heart was higher in old (2.6± 0.25 μmol/min/mg protein) vs. young animals (1.91± 0.5 μmol/min/mg protein); however, this difference was not statistically significant. These results suggest that age-related alterations in GR and Grx activities do not contribute to the observed decline in GSH redox status.
Age-related changes in cerebral and myocardial GSH and cysteine levels
To discern the extent of age-related changes in tissue GSH, brains and hearts obtained from young and old rats were isolated and GSH content was determined. Results show a significant (p < 0.05) 32% decline in cerebral reduced GSH levels in old compared to young rats (1.7± 0.2 vs. 1.1± 0.1 μmol/g tissue wet wt.; young and old, respectively (Table 1). In contrast, myocardial reduced GSH levels were unaffected by age (Table 1). Moreover, cerebral total GSH levels were on average 28% lower in old (1.3± 0.1 nmol/mg protein) vs. young (1.8± 0.1 nmol/mg protein; p < 0.05). Again, no differences in total GSH were observed in the heart. These results confirm previous reports showing differential age-related declines in GSH in these two organs.
Because cellular GSH concentrations are largely governed by the rate of its de novo synthesis, we sought to determine whether this differential age-related loss of GSH levels were in part, due to changes in tissue levels of cysteine, the rate-controlling substrate for GSH synthesis. Results show a small but statistically insignificant loss of cerebral cysteine levels in old when compared to young rats (young; 239.5 ± 17.7 nmol/g tissue and old; 207.8 ± 13.0 nmol/g tissue). Similarly, there were no age-associated differences in tissue cysteine levels in the heart (data not shown). Thus, cysteine availability in both the heart and brain appears largely unaffected in senescent rats.
Age-related changes in cerebral and myocardial γ-glutamylcysteine ligase activity and expression
Because GCLC levels correlate with overall GCL activity, we monitored GCLC content in tissues of young and old rats by Western analysis to gauge whether the loss of cerebral GSH in the aging rat brain was due to lower synthetic capacity. Despite a significant decrease in cerebral steady-state levels of GSH, no apparent age-associated differences in GCLC expression were observed (Table 2). Similarly, there were no discernable differences in GCLC levels in hearts from young and old rats (Table 2). These results thus show that GCLC levels, the main protein subunit governing overall GSH synthetic capacity, remain unchanged in the aging rat brain and heart.
Table 2.
Age-related changes in GCL levels and activities
| Young
|
Old
|
|||
|---|---|---|---|---|
| GCLC levels (% change from young) | GCL activity (nmol/min/mg protein) | GCLC levels (% change from young) | GCL activity (nmol/min/mg protein) | |
| Brain | — | 1.11 ± 0.10 | 10 ± 5.78 | 1.00 ± 0.18 |
| Heart | — | 0.72 ± 0.06 | 9 ± 18.78 | 0.84 ± 0.06 |
Values are expressed as means ± SEM.
To determine whether catalytic activity of GCL was affected by age, we monitored activity in tissue extracts from young and old rats where substrates for the enzyme were provided at saturation levels (5 mM cysteine, 20 mM glutamate). In young rat brain, the basal GCL activity was 1.1 ± 0.1 nmol/min/mg protein, which remained unaffected with age (1.0 ± 0.2 nmol/min/mg protein; Table 2). Like the brain, there were no significant age-dependent changes in GCL activity for the heart (Table 2). Thus, GCL activity under substrate saturating conditions does not appear to be affected in the heart and the brain, and general GSH synthetic capacity in both these aging tissues remains intact.
Age-associated changes in kinetic parameters of γ-glutamylcysteine ligase
Because cerebral GSH declines without any apparent differences in cysteine levels, GCLC expression, or activity, we sought to examine whether a potential age-related change in substrate binding affinity for GCL may, in part, explain this conundrum. Because cysteine is the rate-controlling substrate for GCL activity and hence GSH synthesis, we elected to monitor apparent enzyme kinetic properties with respect to cysteine utilization (see Materials and methods). Brain GCL kinetic parameters vs. cysteine concentrations are shown in Figs. 1A and B. Old rats exhibited a significant (p < 0.05) increase in the apparent Km for cysteine (Kmcys) (Figs. 1A and B). The calculated Kmcys for young rats was 84.3 ± 25.4 vs. 179.0 ± 49.0 μM seen in old animals, a 112% increase with age. These results show a compromised substrate-binding affinity of GCL.
Fig. 1.

The Kmcys for γ-GCL is increased in the aging brain. The potential age-related changes in apparent substrate binding affinity for γ-GCL were examined in brains from young (N = 3) and old (N = 5) rats. Kinetic analysis of γ-GCL was determined by the reaction rates over a range of cysteine concentrations (0.1–5 mM) in the presence of L-glutamic acid (20 mM). The Eadie–Hofstee plot (A) and the corresponding double reciprocal plots (B) are shown. Results show a significant 112% increase in Kmcys with age (p < 0.05). Units are as follows: Vmax (nmol/min/mg protein) and Kmcys (mmol).
The apparent Vmax of GCL was not significantly changed with age (young rats=0.64 ± 0.04 μmol/min/mg protein; old rats=0.80 ± 0.06 μmol/min/mg protein; Fig. 1B). This small increase in Vmax may be a potential adaptation for the significant loss in substrate affinity.
The age-related changes in Kmcys significantly (p < 0.05) lowered overall efficiency of GCL as exemplified by an altered catalytic turnover (Kcat) of GCL. The apparent Kcat for the enzyme, as approximated by the ratio of Vmax/Km, was 68% higher in young (0.0075 s−1) vs. old (0.0045 s−1) rats.
LA-mediated changes in tissue cysteine availability and improvement in GSH levels
Given the age-related decline in cysteine binding affinity of GCL, treatment strategies that would improve cysteine uptake may be beneficial in attenuating the loss of GSH seen in senescent rat brains. Previous studies indicate that LA effectively crosses the blood brain barrier and also enhances the rate of cysteine uptake in vitro [23]. Thus, a rationale exists suggesting that LA may induce cysteine uptake in vivo, which could compensate for the age-related increase in Kmcys for GCL. We examined this hypothesis by monitoring cysteine levels following LA treatment in brains and hearts of young and old rats.
As shown in Fig. 2A, LA-treated old rats exhibited a significant 54% increase in cerebral cysteine levels over controls 12 h following treatment (p < 0.01). This rapid accumulation in tissue cysteine quickly returned to baseline, 48 h following LA treatment. GSH concentrations increased in parallel along with heightened cysteine levels (Fig. 3A). LA induced a 50 and 57% increase in cerebral GSH, 24 and 48 h following LA administration, respectively (p < 0.01; Fig. 3A). Heightened cysteine levels preceded any increases in tissue GSH, suggesting that LA elicited its effects by increasing availability of cysteine for GSH synthesis. These results thus suggest that the age-dependent increase in Kmcys can be compensated by LA treatment.
Fig. 2.
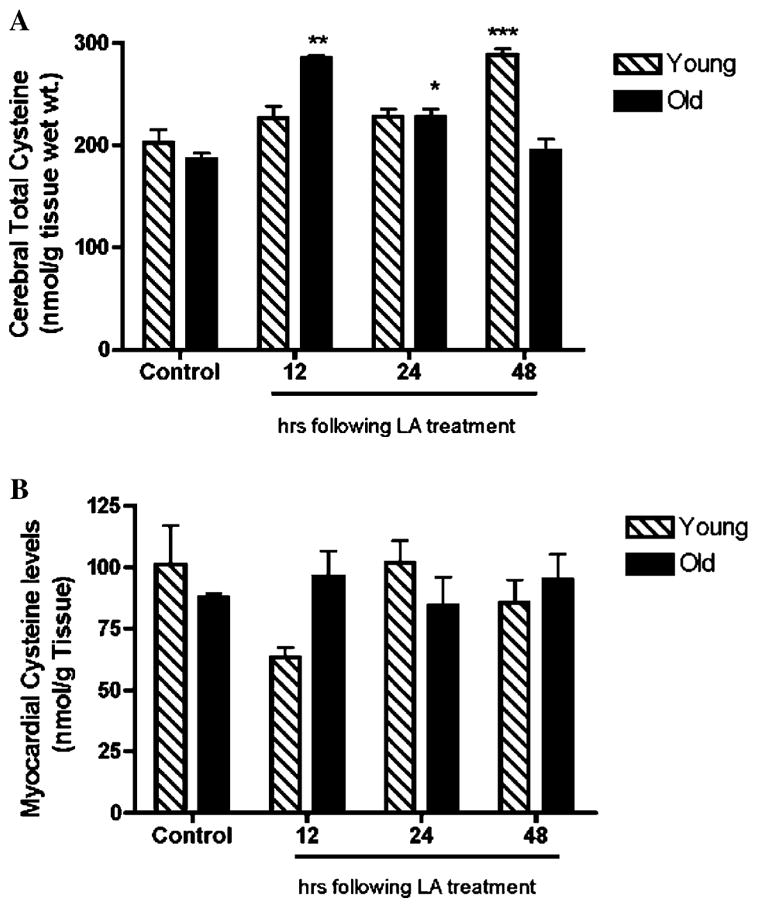
Total cysteine increases in response to LA treatment in the brain. Changes in brain (A) and heart (B) cysteine levels following LA treatment were examined in young (N = 3) and old (N = 3) rats following LA treatment (40 mg/kg body wt.). Results show a transient but significant increase in total cysteine content within 12 h following LA administration in the aging brain (A). In contrast, myocardial cysteine levels were unaffected by LA treatment in both young and old animals. Statistical significance was determined by two-way ANOVA using Bonferroni’s post hoc tests. (* denotes statistically significant differences between controls and treatment; *p < 0.05; **p < 0.01; and ***p < 0.001.)
Fig. 3.
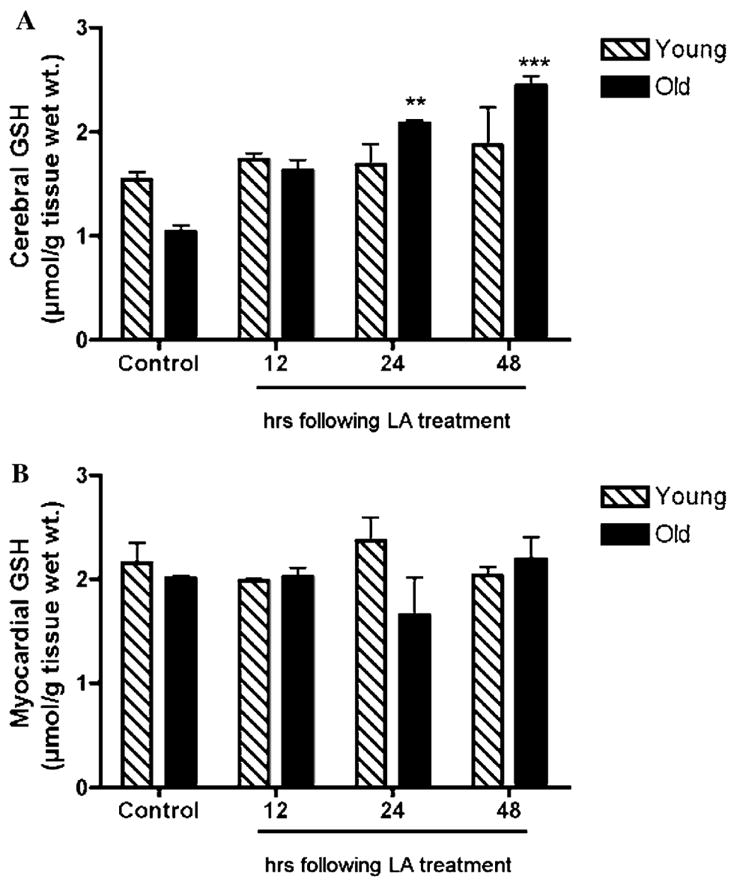
LA effectively reverses the age-related loss in cerebral GSH. Both cerebral (A) and myocardial (B) GSH levels following LA treatment (40 mg/kg body wt.) were determined at indicated times. LA restored the age-related loss in cerebral GSH within 24 h following LA injection (A). In contrast, cerebral GSH levels were unaffected in young animals (A). In heart tissue, no apparent age-related loss of GSH was seen and LA did not further affect total GSH levels in either young or old animals (B). Statistical significance was determined by two-way ANOVA using Bonferroni’s post hoc tests. (* denotes statistically significant differences between controls and treatment; *p < 0.05; **p < 0.01; and ***p < 0.001.) N = 4 for both young and old animals.
Similarly, young rats treated with LA also displayed a time-dependent increase in cysteine levels. However, in comparison to old rats, there was a marked lag before any appreciable cysteine accumulation over the norm occurred; cerebral cysteine levels did not accumulate maximally until 48 h following LA treatment (Fig. 3A). Moreover, even this delayed cysteine accumulation did not cause any changes in cerebral GSH levels in young animals (Fig. 3A). Thus, the LA-mediated improvement in tissue GSH appears to be age-specific in that it effectively reverses losses seen in old rats but does not enhance GSH levels in young rats.
In contrast to the brain, both young and old rats did not show any significant changes in myocardial cysteine levels following LA treatment (Fig. 2B). Moreover, myocardial GSH levels remained unaffected by LA administered to old rats (Fig. 3B). These results suggest a differential regulation in cysteine uptake in the heart relative to the brain.
LA-dependent changes in GCLC expression and activity
To discern whether the LA-induced increase in cerebral GSH levels is also mediated by a potential increase in GSH synthetic capacity, the cerebral and myocardial levels of GCLC and also GCL activities were assessed following LA treatment. As shown in Table 3, LA did not affect either cerebral or myocardial GCLC protein content or activities in young or old rats. These results suggest that LA does not induce expression of GCL in these tissues; rather, it augments cerebral cysteine availability for GSH synthesis.
Table 3.
LA does not affect cerebral or myocardial GCLC levels or activities in old rats
| Young
|
Old
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Brain
|
Heart
|
Brain
|
Heart
|
|||||
| GCLC levels (% change) | GCL activity (nmol/mg/protein) | GCLC levels (% change) | GCL activity (nmol/mg/protein) | GCLC levels (% change) | GCL activity (nmol/mg/protein) | GCLC levels (% change) | GCL activity (nmol/mg/protein) | |
| Control | — | 1.1± 0.1 | — | 0.7± 0.1 | 10± 6 | 1.0± 0.2 | 9± 19 | 0.8± 0.1 |
| LA 12 h | −17± 14 | 1.2± 0.2 | 12± 13 | 0.7± 0.1 | 86± 17 | 1.1± 0.1 | 16± 7 | 0.9± 0.1 |
| LA 24 h | −10± 11 | 1.1± 0.1 | −2± 5 | 0.8± 0.1 | 82± 16 | 1.0± 0.04 | −6± 17 | 0.8± 0.1 |
| LA 48 h | −20± 17 | 1.1± 0.1 | −16± 32 | 0.7± 0.02 | 90± 19 | 1.3± 0.1 | −26± 12 | 0.8± 0.01 |
Values are expressed as means ± SEM. GCLC levels are expressed as % of young control.
LA-mediated improvement in thiol redox status with age
We determined whether LA may also improve the decline in the GSH/GSSG ratio seen in the aging rat heart and brain. Results showed that treating old rats with LA reversed the profound age-related loss in GSH/GSSG ratios in both these organs. For the brain, the GSH/GSSG ratio was normalized to that of young vehicle-treated rats 12 h following LA administration and this ratio was maintained at this heightened level throughout the remainder of the time course (Fig. 4A).
Fig. 4.

LA improves the age-dependent declines in cerebral and myocardial GSH redox states. The GSH/GSSG ratios in brain (A) and heart (B) obtained from young (N = 3) and old (N = 3) rats were determined at indicated times following LA treatment (40 mg/kg body wt.). LA in old rats reversed the profound age-related declines in GSH redox state in both brain (A) and heart (B) within 12 and 24 h, respectively, following LA injection. Statistical significance was determined by two-way ANOVA using Bonferroni’s post hoc tests. (* denotes statistically significant differences between controls and treatment; *p < 0.05; **p < 0.01; and ***p < 0.001.)
To further discern whether the improvements in cerebral GSH/GSSG ratios may partly be explained by potential increases in activities of GR and Grx following LA treatment, tissue GR and Grx in controls, and LA-treated rat brains were monitored. Results show no apparent differences in GR activities between the controls and LA-treated old rats (2.7 ± 0.1 vs. 2.1 ± 0.53 mmol/min/mg protein; old control vs. old LA treated, respectively). Similarly, LA treatment did not modulate tissue Grx activities in old animals (1.25 ± 0.11 vs. 1.21 ± 0.23 μmol/min/mg protein; old controls vs. old LA treated, respectively). These results indicate that the LA-mediated reversal of GSH redox state is not due to improvements in Grx or GR activities.
Interestingly, despite the fact that no apparent increases in total GSH levels were observed in the heart, old rats treated with LA displayed a significant improvement in GSH/GSSG ratio in a time-dependent manner (Fig. 4B). The cerebral GSH levels in young rats remained unaffected following LA treatment, demonstrating that the GSH/GSSG ratios are normally held within a tightly regulated range. Thus, these results suggest that in addition to its effect in improving cysteine uptake, LA also augments tissue reductive capacity, which otherwise appears to decline with age.
Discussion
Maintaining the ability to respond to oxidant challenges is critical for overall survival of organisms and for ameliorating increased risks for morbidity and mortality associated with aging. In particular, a robust and responsive GSH synthesizing capacity is needed in aging cells to protect against endogenous and exogenous oxidative insults [15,29]. Thus, it is not surprising that the expression and activities of enzymes critical for GSH synthesis are sensitively controlled to ensure timely compensation for oxidative challenge and when GSH levels otherwise become limiting [30–32]. Despite these regulatory mechanisms, we observed a significant loss of total cerebral GSH levels in the aging rat brain. This overall loss was reflected primarily by the decline in reduced GSH levels. Similar changes were not observed in the heart. However, both the heart and the brain exhibited a significant age-associated change in GSH redox state.
The diminished capacity to maintain normal GSH redox state in these tissues indicates potential lesions in electron flux via NADPH with age. Theoretically, this may be caused by a number of factors, including lower NADPH generation, GSSG export, and/or activity of enzymes critical for GSSG reduction. To initially characterize potential factors involved, the age-dependent changes in activities of GR and Grx were examined and reveal no significant alterations in their activities, indicating that aging may compromise cellular capacity to maintain pyridine nucleotide redox states and/or to export GSSG. NADPH production in tissues can arise from multiple pathways, including the pentose phosphate pathway (PPP), NADP+-dependent isocitrate dehydrogenase (ICDH), and transhydrogenation reactions [33–35]. Among these pathways, ICDH has been previously identified as the principal generator of NADPH in the heart, while the primary modes of NADPH production in the brain have not been fully defined [33,34]. Due to the multiplicity of pathways involved in NADPH maintenance, it is beyond the scope of this current study to fully examine potential lesions in electron flux upstream of GR and Grx.
In previous studies, (R)-α-lipoic acid (LA) supplementation to old rats restored GSH and vitamin C levels in the liver and heart [6,17]. Moreover, this dithiol compound effectively improved cellular redox balance (as indicated by a higher GSH/GSSG ratio) and reversed the age-associated increased susceptibility to toxicological insult [17]. Results from the present study add to these previous findings and show the effectiveness of LA in improving thiol redox state in the aging heart and the brain.
The beneficial effects of LA do not appear to be completely dependent upon its potent antioxidant capacity. Because the liver is the initial and the primary site of LA uptake and reduction, it is likely that both oxidized and reduced forms of LA are quickly available in the plasma for uptake by the brain and heart [36,37]. Although the exact ratio between LA and DHLA in the brain and the heart is not known, it is unlikely that direct antioxidant effects of DHLA are fully responsible for the observed improvements in GSH redox state. This is because free non-protein bound LA does not accumulate to any appreciable degree in tissues [37,38]. It is worth noting that in certain cell types, LA increases the rate of NADPH generation and may also improve potential deficits in NADPH production capacity in these tissues [24].
Given that the rate of GSH synthesis is largely influenced by cysteine availability [39], we assessed whether the level of this rate-limiting substrate may decline with age. However, examination of tissue cysteine levels in young and old rats showed no significant alterations in its levels in the heart, whereas in the brain, there was a small insignificant loss of cysteine. Considering that the normal intracellular concentration of cysteine is kept well below the Km for cysteine (0.1–0.3 mM), it suggests that small changes in the cysteine pool may adversely or positively impact the velocity of GCL catalytic activity. Decrements in the apparent Kmcys evident in aging rat brains, therefore, would markedly lower GCL activity far below the half maximal velocity of the enzyme. Based on cerebral cysteine availability and the lack of differences in enzyme levels, we estimate that this adverse Km effect should lower cerebral GSH levels by nearly 50% in aging rats, assuming equal rates of utilization and/or efflux. This estimate is in good agreement with the 32% decline in GSH levels shown in the present study.
Because purified GCL was not used in this study, it is not possible to conclude whether the observed loss of Kmcys stems from direct modification of GCL itself or is due to bias introduced from using a crude homogenate. However, it is noteworthy that GCL activities were determined in the absence of any small endogenous molecules less than 30 kDa, suggesting that any potential source of interference is not likely due to small molecules capable of interacting with the cysteine binding site.
Other factors may also further exacerbate the catalytic efficiency of GCL in the aging brain. Although not examined in this study, GCLM levels were previously shown to decline significantly without a concomitant loss of GCLC [19]. Loss of GCLM from the holoenzyme radically affects allosteric regulation of GSH on enzyme activity where the apparent Ki for GSH feedback inhibition declines from 8.2 to 1.8 mM, a level below the normal cerebral GSH concentration (typically 2.0 mM). Furthermore, the absence of GCLM also increases the Km for glutamate from 1.4 to 18.2 mM. The age-related loss of GCLM previously reported translated to a roughly 63% loss of overall GCL activity in the presence of physiological levels of GSH (10 mM) and glutamate (1.5 mM) [19]. This, in conjunction with our current study, provides strong evidence that GCL activity under physiological substrate conditions may be drastically lower in the aging rat brain.
The age-associated changes in Km do not appear to be unique to GCL. A number of enzymes display changes in either Km or Vmax as a function of age. For example, δ-6-desaturase exhibits a similar age-dependent increase in the Km for linoleic acid, resulting in lower enzymatic efficiency [40]. Cytosolic proteins, such as tryptophan hydroxylase from old rat brains, also display age-associated increases in Km, which may result in lower serotonin levels [41]. Mitochondrial proteins, such as carnitine acyl-CoA transferase [42], which are particularly prone to oxidative modification, also show decreased affinity for substrates with age, suggesting that the pro-oxidant milieu of aging tissues may enhance damage to enzymes.
In spite of these changes, treating old rats with LA effectively attenuated the age-related loss in cerebral GSH. In addition to the potential improvements in NADPH redox state, LA enhanced tissue GSH status by increasing cellular cysteine availability. In vitro studies showed that LA augmented cellular cysteine levels via DHLA-mediated reduction of extracellular cystine to cysteine, which facilitated rapid uptake of cysteine [23]. A recent study also reported that LA acts as a potent inducer of Phase II detoxication proteins in astroglial cells, a principal cell type responsible for GSH synthesis in the brain [43]. Because the Xc− transporter is a Phase II detoxication enzyme [44], there is a strong possibility that LA may increase the expression of the Xc− transporter via transactivation of genes containing the antioxidant response element.
The potential use of LA in reversing the age-associated [45] alterations in GSH may be beneficial in many respects. First, the targeted delivery of exogenous GSH to tissues like the heart and brain is not feasible owing to its inability to be transported into these tissues. Second, conventional treatment with cysteine delivery agents (e.g., N-acetylcysteine) may not work as efficiently to increase cerebral GSH, mainly due to the low bioavailability of these compounds in the brain [21]. Because LA is readily taken up into neuronal tissues [46,47], LA represents a potentially safe, yet effective, means to beneficially modulate the age-related alterations in GSH levels. Thus, the results from our study serve as the basis for identifying LA as a novel therapeutic agent in improving the age-related loss of cerebral GSH, which could be important to mitigate oxidative stress-induced pathophysiologies, such as Alzheimer’s and Parkinson’s diseases.
Because of the multifaceted functions of GSH as an antioxidant, its role in maintaining critical protein thiols in a reduced state, and in regulating the cell cycle [15,29,32], any alterations in its levels or redox state are likely to have a profound impact on overall organ function. Studies show that human polymorphisms in GCL that lower GSH synthetic capacity are significantly associated with increased risks for myocardial infarction [48]. Moreover, depletion of cerebral GSH severely compromises the capacity of the central nervous system to protect against oxidant injury and is causally involved in the pathogenesis of degenerative disorders, such as Parkinson’s disease [4,49,50]. Thus, the ability of LA to improve GSH status in multiple organs including the brain may prove useful in maintaining cerebrovascular functions in aging.
Acknowledgments
This work was supported by NIH Grants RO1 AG17141A and P01 AT002034-01 to T.M.H. and RO1 ES11831-01 to RLU. This work was also supported by the Environmental Health Sciences Center, Oregon State University (ES00210). The authors thank Ms. Swapna Shenvi and Mrs. Du Heath for their expert technical assistance. We are also grateful to Stephen Lawson for careful review of the manuscript.
Footnotes
Abbreviations used: LA, R-α-lipoic acid; GCL, γ-glutamylcysteine; GCLC, catalytic GCL; GCLM, modulatory GCL; GSH, glutathione; HPLC, high performance liquid chromatography; PCA, perchloric acid; DTNB, dithionitrobenzoic acid; HEDS, 2-hydroxyethyldisulfide; GR, GSSG reductase; Grx, glutaredoxin; PPP, pentose phosphate pathway; ICDH, isocitrate dehydrogenase.
References
- 1.Harman D. Mutat Res. 1992;275:257–266. doi: 10.1016/0921-8734(92)90030-s. [DOI] [PubMed] [Google Scholar]
- 2.Ames BN, Shigenaga MK, Hagen TM. Proc Natl Acad Sci USA. 1993;90:7915–7922. doi: 10.1073/pnas.90.17.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sawada M, Carlson JC. Mech Ageing Dev. 1987;41:125–137. doi: 10.1016/0047-6374(87)90057-1. [DOI] [PubMed] [Google Scholar]
- 4.Head E, Liu J, Hagen TM, Muggenburg BA, Milgram NW, Ames BN, Cotman CW. J Neurochem. 2002;82:375–381. doi: 10.1046/j.1471-4159.2002.00969.x. [DOI] [PubMed] [Google Scholar]
- 5.Smith CD, Carney JM, Starke-Reed PE, Oliver CN, Stadtman ER, Floyd RA, Markesbery WR. Proc Natl Acad Sci USA. 1991;88:10540–10543. doi: 10.1073/pnas.88.23.10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Suh JH, Shigeno ET, Morrow JD, Cox B, Rocha AE, Frei B, Hagen TM. FASEB J. 2001;15:700–706. doi: 10.1096/fj.00-0176com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Farooqui MY, Day WW, Zamorano DM. Comp Biochem Physiol B. 1987;88:177–180. doi: 10.1016/0305-0491(87)90097-6. [DOI] [PubMed] [Google Scholar]
- 8.Ishikawa T, Zimmer M, Sies H. FEBS Lett. 1986;200:128–132. doi: 10.1016/0014-5793(86)80524-5. [DOI] [PubMed] [Google Scholar]
- 9.Ishikawa T, Sies H. J Biol Chem. 1984;259:3838–3843. [PubMed] [Google Scholar]
- 10.Benzi G, Marzatico F, Pastoris O, Villa RF. Exp Gerontol. 1989;24:137–148. doi: 10.1016/0531-5565(89)90024-7. [DOI] [PubMed] [Google Scholar]
- 11.Floyd RA. Proc Soc Exp Biol Med. 1999;222:236–245. doi: 10.1046/j.1525-1373.1999.d01-140.x. [DOI] [PubMed] [Google Scholar]
- 12.Huang CS, Chang LS, Anderson ME, Meister A. J Biol Chem. 1993;268:19675–19680. [PubMed] [Google Scholar]
- 13.Meister A. Pharmacol Ther. 1991;51:155–194. doi: 10.1016/0163-7258(91)90076-x. [DOI] [PubMed] [Google Scholar]
- 14.Gipp JJ, Bailey HH, Mulcahy RT. Biochem Biophys Res Commun. 1995;206:584–589. doi: 10.1006/bbrc.1995.1083. [DOI] [PubMed] [Google Scholar]
- 15.Dickinson DA, Forman HJ. Biochem Pharmacol. 2002;64:1019–1026. doi: 10.1016/s0006-2952(02)01172-3. [DOI] [PubMed] [Google Scholar]
- 16.Richman PG, Meister A. J Biol Chem. 1975;250:1422–1426. [PubMed] [Google Scholar]
- 17.Hagen TM, Vinarsky V, Wehr CM, Ames BN. Antioxid Redox Signal. 2000;2:473–483. doi: 10.1089/15230860050192251. [DOI] [PubMed] [Google Scholar]
- 18.Liu R, Choi J. Free Radic Biol Med. 2000;28:566–574. doi: 10.1016/s0891-5849(99)00269-5. [DOI] [PubMed] [Google Scholar]
- 19.Liu RM. J Neurosci Res. 2002;68:344–351. doi: 10.1002/jnr.10217. [DOI] [PubMed] [Google Scholar]
- 20.Witschi A, Reddy S, Stofer B, Lauterburg BH. Eur J Clin Pharmacol. 1992;43:667–669. doi: 10.1007/BF02284971. [DOI] [PubMed] [Google Scholar]
- 21.McLellan LI, Lewis AD, Hall DJ, Ansell JD, Wolf CR. Carcinogenesis. 1995;16:2099–2106. doi: 10.1093/carcin/16.9.2099. [DOI] [PubMed] [Google Scholar]
- 22.Hagen TM, Moreau R, Suh JH, Visioli F. Ann N Y Acad Sci. 2002;959:491–507. doi: 10.1111/j.1749-6632.2002.tb02119.x. [DOI] [PubMed] [Google Scholar]
- 23.Han D, Handelman G, Marcocci L, Sen CK, Roy S, Kobuchi H, Tritschler HJ, Flohe L, Packer L. Biofactors. 1997;6:321–338. doi: 10.1002/biof.5520060303. [DOI] [PubMed] [Google Scholar]
- 24.Jones W, Li X, Qu ZC, Perriott L, Whitesell RR, May JM. Free Radic Biol Med. 2002;33:83–93. doi: 10.1016/s0891-5849(02)00862-6. [DOI] [PubMed] [Google Scholar]
- 25.Roy S, Sen CK, Tritschler HJ, Packer L. Biochem Pharmacol. 1997;53:393–399. doi: 10.1016/s0006-2952(96)00764-2. [DOI] [PubMed] [Google Scholar]
- 26.Fariss MW, Reed DJ. Methods Enzymol. 1987;143:101–109. doi: 10.1016/0076-6879(87)43018-8. [DOI] [PubMed] [Google Scholar]
- 27.Cribb AE, Leeder JS, Spielberg SP. Anal Biochem. 1989;183:195–196. doi: 10.1016/0003-2697(89)90188-7. [DOI] [PubMed] [Google Scholar]
- 28.Ehrhart J, Gluck M, Mieyal J, Zeevalk GD. Parkinsonism Relat Disord. 2002;8:395–400. doi: 10.1016/s1353-8020(02)00020-2. [DOI] [PubMed] [Google Scholar]
- 29.Dickinson DA, Forman HJ. Ann N Y Acad Sci. 2002;973:488–504. doi: 10.1111/j.1749-6632.2002.tb04690.x. [DOI] [PubMed] [Google Scholar]
- 30.Wild AC, Moinova HR, Mulcahy RT. J Biol Chem. 1999;274:33627–33636. doi: 10.1074/jbc.274.47.33627. [DOI] [PubMed] [Google Scholar]
- 31.Liu RM, Borok Z, Forman HJ. Am J Respir Cell Mol Biol. 2001;24:499–505. doi: 10.1165/ajrcmb.24.4.4307. [DOI] [PubMed] [Google Scholar]
- 32.Lu SC. FASEB J. 1999;13:1169–1183. [PubMed] [Google Scholar]
- 33.Andres A, Satrustegui J, Machado A. Biochem J. 1980;186:799–803. doi: 10.1042/bj1860799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goodwin GW, Cohen DM, Taegtmeyer H. Am J Physiol Endocrinol Metab. 2001;280:E502–E508. doi: 10.1152/ajpendo.2001.280.3.E502. [DOI] [PubMed] [Google Scholar]
- 35.Andres A, Satrustegui J, Machado A. Biol Neonate. 1983;43:198–204. doi: 10.1159/000241629. [DOI] [PubMed] [Google Scholar]
- 36.Teichert J, Hermann R, Ruus P, Preiss R. J Clin Pharmacol. 2003;43:1257–1267. doi: 10.1177/0091270003258654. [DOI] [PubMed] [Google Scholar]
- 37.Schupke H, Hempel R, Peter G, Hermann R, Wessel K, Engel J, Kronbach T. Drug Metab Dispos. 2001;29:855–862. [PubMed] [Google Scholar]
- 38.Spence JT, McCormick DB. Arch Biochem Biophys. 1976;174:13–19. doi: 10.1016/0003-9861(76)90318-0. [DOI] [PubMed] [Google Scholar]
- 39.Bannai S. Biochim Biophys Acta. 1984;779:289–306. doi: 10.1016/0304-4157(84)90014-5. [DOI] [PubMed] [Google Scholar]
- 40.Hrelia S, Celadon M, Rossi CA, Biagi PL, Bordoni A. Biochem Int. 1990;22:659–667. [PubMed] [Google Scholar]
- 41.Hussain AM, Mitra AK. Drug Metab Dispos. 2000;28:1038– 1042. [PubMed] [Google Scholar]
- 42.Liu J, Killilea DW, Ames BN. Proc Natl Acad Sci USA. 2002;99:1876–1881. doi: 10.1073/pnas.261709098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Flier J, Van Muiswinkel FL, Jongenelen CA, Drukarch B. Free Radic Res. 2002;36:695–699. doi: 10.1080/10715760290029155. [DOI] [PubMed] [Google Scholar]
- 44.Sasaki H, Sato H, Kuriyama-Matsumura K, Sato K, Maebara K, Wang H, Tamba M, Itoh K, Yamamoto M, Bannai S. J Biol Chem. 2002;277:44765–44771. doi: 10.1074/jbc.M208704200. [DOI] [PubMed] [Google Scholar]
- 45.Peter G, Borbe HO. Arzneimittelforschung. 1995;45:293– 299. [PubMed] [Google Scholar]
- 46.Packer L, Tritschler HJ, Wessel K. Free Radic Biol Med. 1997;22:359–378. doi: 10.1016/s0891-5849(96)00269-9. [DOI] [PubMed] [Google Scholar]
- 47.McGahon BM, Martin DS, Horrobin DF, Lynch MA. Neurobiol Aging. 1999;20:655–664. doi: 10.1016/s0197-4580(99)00050-0. [DOI] [PubMed] [Google Scholar]
- 48.Nakamura S, Kugiyama K, Sugiyama S, Miyamoto S, Koide S, Fukushima H, Honda O, Yoshimura M, Ogawa H. Circulation. 2002;105:2968–2973. doi: 10.1161/01.cir.0000019739.66514.1e. [DOI] [PubMed] [Google Scholar]
- 49.Gu M, Owen AD, Toffa SE, Cooper JM, Dexter DT, Jenner P, Marsden CD, Schapira AH. J Neurol Sci. 1998;158:24–29. doi: 10.1016/s0022-510x(98)00095-1. [DOI] [PubMed] [Google Scholar]
- 50.Spencer JP, Jenner P, Daniel SE, Lees AJ, Marsden DC, Halliwell B. J Neurochem. 1998;71:2112–2122. doi: 10.1046/j.1471-4159.1998.71052112.x. [DOI] [PubMed] [Google Scholar]