

Abstract
Translocator protein 18 kDa (TSPO) is a mitochondrial outer membrane protein and is abundantly expressed in a variety of organ and tissues. To date, the functional role of TSPO on vascular endothelial cell activation has yet to be fully elucidated. In the present study, the phorbol 12-myristate 13-acetate (PMA, 250 nM), an activator of protein kinase C (PKC), was used to induce vascular endothelial activation. Adenoviral TSPO overexpression (10–100 MOI) inhibited PMA-induced vascular cell adhesion molecule-1 (VCAM-1) and intracellular cell adhesion molecule-1 (ICAM-1) expression in a dose dependent manner. PMA-induced VCAM-1 expressions were inhibited by Mito-TEMPO (0.1–0.5 μM), a specific mitochondrial antioxidants, and cyclosporin A (1–5 μM), a mitochondrial permeability transition pore inhibitor, implying on an important role of mitochondrial reactive oxygen species (ROS) on the endothelial activation. Moreover, adenoviral TSPO overexpression inhibited mitochondrial ROS production and manganese superoxide dismutase expression. On contrasts, gene silencing of TSPO with siRNA increased PMA-induced VCAM-1 expression and mitochondrial ROS production. Midazolam (1–50 μM), TSPO ligands, inhibited PMA-induced VCAM-1 and mitochondrial ROS production in endothelial cells. These results suggest that mitochondrial TSPO can inhibit PMA-induced endothelial inflammation via suppression of VCAM-1 and mitochondrial ROS production in endothelial cells.
Keywords: endothelial cells, PKC, ROS, TSPO, VCAM-1
INTRODUCTION
Endothelial activation is a pro-inflammatory state of the endothelial cells lining the lumen of blood vessels and is characterized by the elevated expression of cell-surface adhesion molecules such as vascular cell adhesion molecule-1 (VCAM-1) (Liao, 2013). This activation is a crucial event in the pathological process of vascular inflammation, the primary cause of cardiovascular diseases such as atherosclerosis (Ross, 1995). Various risk factors associated with vascular inflammation induce mitochondrial dysfunction via the generation of mitochondrial reactive oxygen species (ROS). The resulting dysfunctional mitochondria contribute to the pathogenesis of endothelial dysfunction, leading to the development of vascular diseases (Madamanchi and Runge, 2007). Due to the sensitivity of endothelial mitochondria to oxidative stress, they play a crucial role in mediating cellular responses (Davidson and Duchen, 2007; Kluge et al., 2013). Thus, endothelial mitochondria are critical targets for the regulation of endothelial activation, early state of vascular inflammation.
In the mitochondria, the 18-kDa translocator protein (TSPO), also known as the peripheral benzodiazepine receptor (PBR), is localized to the outer membrane (Anholt et al., 1986) and is part of the mitochondrial permeability transition pore (MPTP). It associates with various other proteins at the MPTP, including the voltage-dependent ion channel-1 (VDAC-1) and the adenine nucleotide transporter (McEnery et al., 1992). TSPO participates in several regulatory functions of the mitochondria, including modulation of apoptosis (Bono et al., 1999; Levin et al., 2005; Veenman et al., 2004), mitochondrial membrane potential (Galiegue et al., 2003; Leducq et al., 2003), and the mitochondrial respiratory chain (Zisterer et al., 1992).
TSPO expression levels have been correlated with certain inflammatory diseases. For instance, TSPO expression was found to be elevated in the arterial plaques of patients with atherosclerosis (Fujimura et al., 2008) and inflammation (Hardwick et al., 2005), suggesting that a high expression of TSPO is strongly correlated with modulating inflammatory processes. Although the evidence indicates a potential role of TSPO in the regulation of inflammatory processes, the mechanisms involved have yet to be elucidated. In the present study, we investigated the functional role of TSPO on PMA-induced endothelial activation in cultured endothelial cells.
MATERIALS AND METHODS
Reagents
Phorbol 12-myristate 13-acetate (PMA), cyclosporin A and methyl-triphenylphosphonium (TPP) were purchased from Sigma-Aldrich (USA). Triphenylphosphonium chloride (2-(2,2,6,6-tetramethylpiperidin-1-oxyl-4-ylamino)-2-oxoethyl) (Mito-TEMPO), a mitochondrion-specific antioxidant, was purchased from Enzo Life Sciences (USA). Midazolam was purchased from Bukwang Company (Korea). Specific antibodies against TSPO, β-actin, and FLAG (Sigma-Aldrich); VCAM-1, ICAM-1 (Santa Cruz Biotechnology, USA); and manganese superoxide dismutase (MnSOD, Enzo Life Sciences) were used in this study.
Adenoviral TSPO vector construction
Adenoviruses encoding the full-length human TSPO (AdTSPO) were generated by homologous recombination, as described previously (Joo et al., 2012). Human embryonic kidney 293A cells were cultured until an 80% cytopathic effect was observed, at which point they were harvested for the adenoviral stock through recombination with prepared adenoviruses per the manufacturer’s instructions (Life Technologies, USA). The adenovirus was propagated in 293A cells and subsequently purified using the CsCl2 gradient technique, as described previously (Jeon et al., 2004). To overexpress TSPO in endothelial cells, the cells were infected with a multiplicity of infection (MOI; particle forming units per cell) of 100 for 24 h. Adβgal was used as an adenoviral control.
Cell culture and transfection
Human umbilical vein endothelial cells (HUVECs) were purchased from Clonetics (Cambrex Bio Science, USA), and were cultured and maintained in endothelial growth medium. Cells were used between passages 3 and 6. Small interfering RNA (siRNA) oligonucleotides against human TSPO and a control with scrambled siRNA sequences were purchased from Bioneer Co. (Korea); the siRNA targeting human TSPO had the following gene-specific sense sequence: 5′-CGU AUG GCG GGA CAA CCA U-3′. HUVEC cells were transfected with either 20 nM TSPO-specific or scrambled siRNA. The siRNAs and Lipofectamine® 2000 (Invitrogen, USA) were diluted separately in OptiMEM medium (Life Technologies) and incubated for 5 min at room temperature. Next, siRNA/Lipofectamine® mixtures were incubated together for 20 min before use in the transfection of the HUVEC cells. Cells were incubated at 37°C in a 5% CO2 incubator for 48 h for gene knockdown.
Western blot analysis
To determine protein expression levels, 2 × 105 cells were harvested in 100 μl lysis buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, phosphatase-inhibitor cocktail, and protease-inhibitor cocktail)(Park et al., 2013). Cell homogenates (30 μg) were separated by 12% SDS-PAGE and electrotransferred onto polyvinylidene fluoride (PVDF) membranes. After blocking with 5% skim milk for 1 h at room temperature, blots were incubated overnight at 4°C with specific primary antibodies (1:1000 anti-VCAM-1, 1:1000 anti-ICAM-1, 1:5000 anti-FLAG, 1:1000 anti-MnSOD, 1:2000 anti-TSPO, and 1:5000 anti-β-actin), which were detected using horseradish peroxidase (HRP)-conjugated secondary antibodies. Blots were developed for visualization using an enhanced chemiluminescence western blotting substrate kit (Pierce, USA).
Detection of mitochondrial ROS
Mitochondrial superoxide production was assessed using MitoSOX™ Red (Molecular Probes, USA), a mitochondrion-specific, hydroethidine-derivative fluorescent dye that undergoes oxidation to form the DNA-binding, red fluorophore ethidium bromide. After 6 h of exposure to 250 nM PMA, cells were incubated with 5 μM MitoSOX™ Red for 10 min at 37°C in 5% CO2 incubator. Fluorescence was measured at 590 nm using a plate reader (Thermo Fisher Scientific, USA). Fluorescence images were obtained by fluorescence microscopy (Carl Zeiss Inc., USA). Cells were stained with Mitosox Red (red fluorescence) for mitochondrial ROS detection and 4′,6-diamidino-2-phenylindole (DAPI, blue fluorescence) for nucleus staining. The data are presented as fold-changes of the mean intensity of MitoSOX™ fluorescence relative to that of the un-treated controls.
Statistical analysis
The statistical significance of differences in the measured variables for control and PMA-treated groups was determined using one-way ANOVA followed by a Tukey’s post-hoc test, and p < 0.05 was considered statistically significant. Values are expressed as means ± SEM.
RESULTS
PMA increased endogenous TSPO expression
To examine if endogenous TSPO expression was affected by exposure of PMA, we investigated the changes in TSPO expression in HUVECs exposed to PMA at various dose for 18 h. As shown in Fig. 1A, the exposure of PMA increased TSPO expression in a dose-dependent manner. For quantitative analysis of TSPO expression, the data was plotted as folds increase over basal value (Fig. 1B). We also investigated the time-dependent change of TSPO expression in the response to PMA in HUVECs. As shown in Fig. 1C, TSPO expression was significantly up-regulated in 6 h in the response to 250 nM of PMA. Quantitative analysis was plotted as folds increase over basal value (Fig. 1D). This data suggested that TSPO expression levels were correlated with PMA-induced endothelial activation.
Fig. 1.
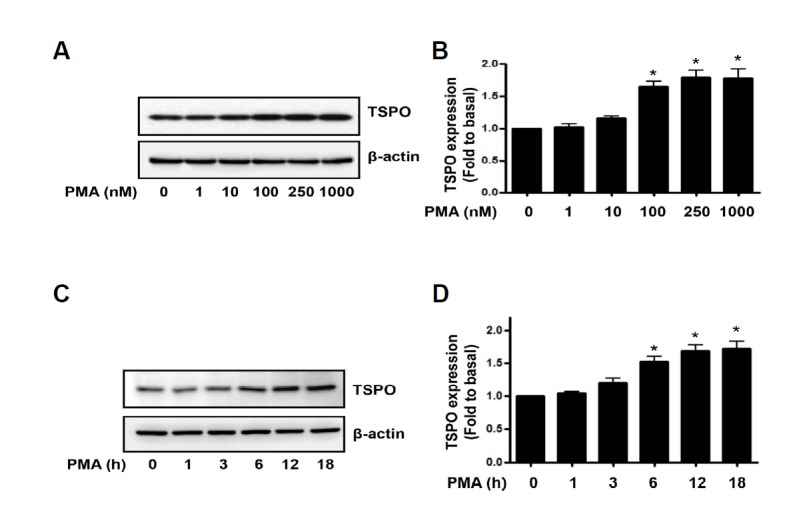
PMA increased endogenous TSPO expression in endothelial cells. (A) HUVECs were treated with 1–1000 nM PMA for 18 h, followed by Western blot analysis of the cell lysates. The β-actin was used as a loading control. (C) HUVECs were exposed for 1–18 h at 250 nM PMA, followed by Western blot analysis of the cell lysates. The β-actin was used as a loading control. (B, D) Densitometric analysis of Western blots. Data are expressed as fold values of basal TSPO expression levels. Each bar represents mean ± SEM (n = 3). *p < 0.05 vs. baseline value.
Overexpression of TSPO inhibited PMA-induced VCAM-1 and ICAM-1 expression
To investigate whether TSPO regulates PMA-induced endothelial activation, we examined the effects of TSPO overexpression on PMA-induced adhesion molecule expression. As shown by Western blot analysis with anti-VCAM-1 and ICAM-1 in Fig. 2A, VCAM-1 and ICAM-1 were minimally detected in the un-stimulated condition, whereas treatment with PMA resulted in a marked increase in VCAM-1 and ICAM-1 expression. Adenoviral FLAG-tagged TSPO gene transfer in an MOI range of 10–100 successfully induced TSPO expression, as assessed by Western blot analysis with the anti-TSPO antibody. Adenoviral TSPO overexpression decreased PMA-induced VCAM-1 and ICAM-1 expression with MOI dependency, compared with the Adβgal-infected cells. For quantitative analysis of VCAM-1 and ICAM-1 expression, the data was plotted as a percentage of that in PMA alone (Figs. 2B and 2C). These results suggest that TSPO overexpression prevented PMA-induced endothelial activation.
Fig. 2.
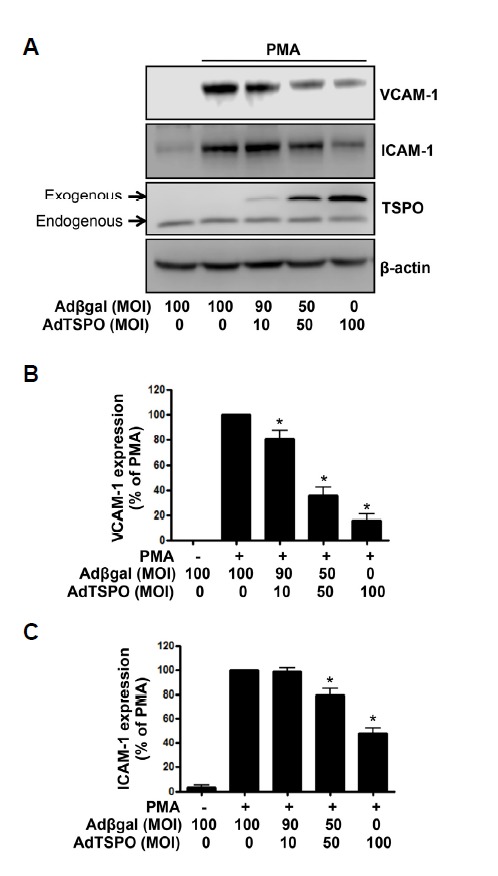
TSPO overexpression inhibited PMA-induced VCAM-1 and ICAM-1 expression in endothelial cells. (A) HUVECs transfected with AdTSPO were treated with 250 nM PMA for 6 h, followed by Western blot analysis of the cell lysates. The total adenoviral MOI of 100 was balanced in the Adβgal control. FLAG-tagged TSPO over-expression was confirmed by anti-TSPO antibody detection. The β–actin was used as a loading control. (B) Densitometric analysis of Western blots. Data are expressed as percent densitometric values of VCAM-1 expression induced by PMA. Each bar represents mean ± SEM (n = 4). *p < 0.05 vs. PMA alone. (C) Densitometric analysis of Western blots. Data are expressed as percent densitometric values of ICAM-1 expression induced by PMA. Each bar represents mean ± SEM (n = 3). *p < 0.05 vs. PMA alone.
Mitochondrial ROS inhibitors reduced PMA-induced VCAM-1 expression
To examine the involvement of mitochondrial ROS in endothelial cell activation, we assessed the effect of Mito-TEMPO, a specific mitochondrial antioxidant (Dikalova et al., 2010), in PMA-induced VCAM-1 expression. As shown in Fig. 3A, pre-treatment of HUVECs with 0.1–0.5 μM Mito-TEMPO significantly inhibited PMA-induced VCAM-1 expression in a dose-dependent manner. For quantitative analysis of VCAM-1 expression, the data was plotted as a percentage of that in PMA alone (Fig. 3B). Whereas methyltriphenylphosphonium (TPP), used as control, did not affected PMA-induced VCAM-1 expression (Data not shown). Cyclosporin A, a known cyclophilin-D inhibitor, has been shown to attenuate MPTP opening (Basso et al., 2005), caused by mitochondrial ROS. We investigated whether a MPTP inhibitor affects PMA-induced VCAM-1 expression. As shown in Fig. 3C, the increase in the expression of VCAM-1 in response to PMA was effectively suppressed by pretreatment with cyclosporin A (1–5 μM), suggesting that the closing of MPTP inhibited PMA-induced VCAM-1 expression in endothelial cells. For quantitative analysis of VCAM-1 expression, the data was plotted as a percentage of that in PMA alone (Fig. 3D). Together, these results suggest that PMA-induced VCAM-1 expression is regulated by mitochondrial ROS, leading to the opening of the MPTP.
Fig. 3.
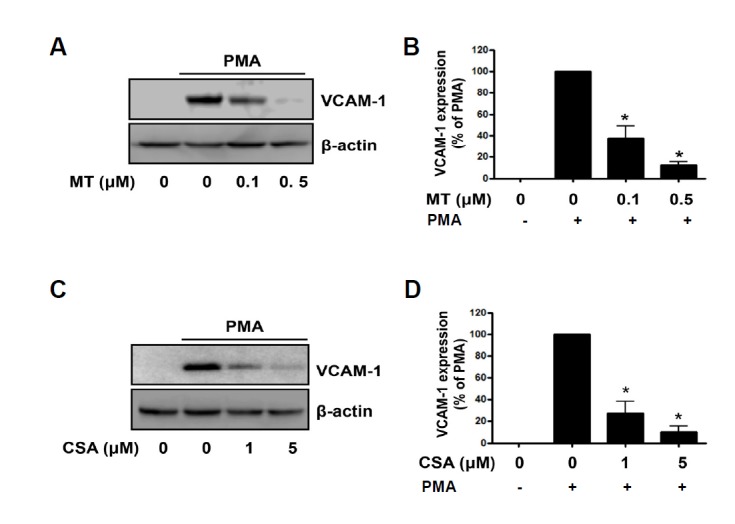
Mitochondrial ROS inhibitors reduced PMA-induced VCAM-1 expression in endothelial cells. The effects of (A) Mito-TEMPO (MT) and (C) cyclosporin A (CSA) on VCAM-1 expression by PMA are shown via Western blot. After pretreatment of HUVECs with Mito-TEMPO or cyclosporin A, cells were exposed to 250 nM PMA for 6 h. Following PMA treatment, cell lysates were analyzed by Western blots for VCAM-1 expression with the anti-VCAM-1 antibody. The β-actin was used as a loading control. (B, D) Densitometric analysis for Figs. 3A and 3C. Data are expressed as percent densitometric values of VCAM-1 expression induced by PMA. Each bar represents mean ± SEM (n = 4). *p < 0.05 vs. PMA alone.
Overexpression of TSPO inhibited the PMA-induced mitochondrial ROS generation
PMA induces mitochondrial ROS production, leading to mitochondrial dysfunction in endothelial cells (Joo et al., 2014). Next, we investigated the effects of TSPO on PMA-induced mitochondrial ROS generation. After exposing to PMA for 6 h, the fluorescent signals of MitoSOX™ Red were analyzed. Interestingly, adenoviral TSPO overexpression significantly inhibited PMA-induced mitochondrial ROS generation (Fig. 4A). In TSPO-overexpressed cells, the PMA-induced mitochondrial ROS production was reduced by 57.4%, compared with that of Adβgal-infected cells. The representative fluorescence images were shown in Fig. 4B.
Fig. 4.
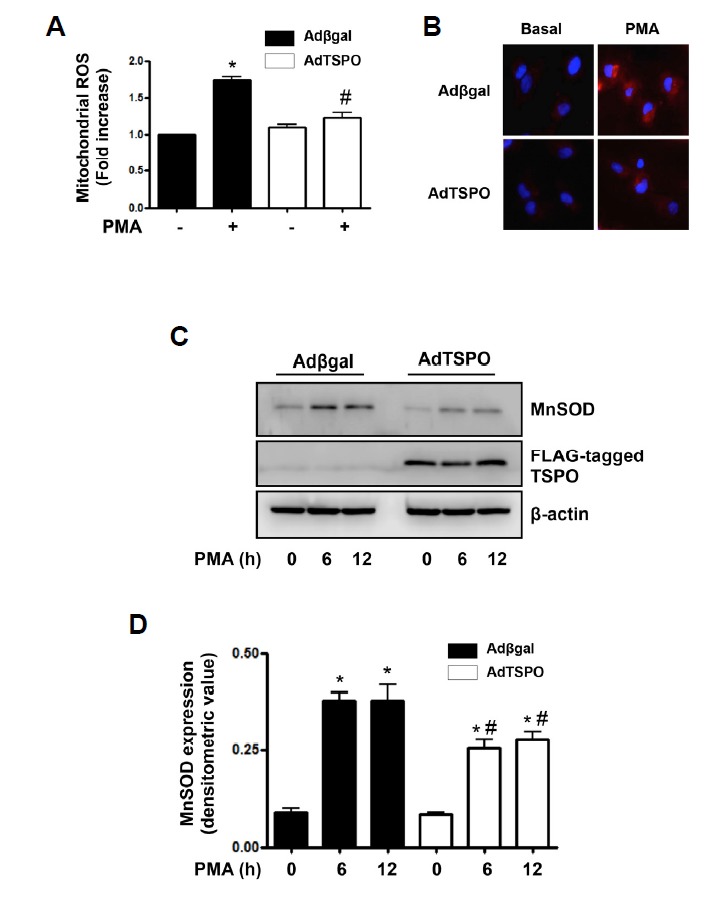
TSPO overexpression inhibited PMA-induced mitochondrial ROS generation and MnSOD expression in endothelial cells. (A) Overexpression of TSPO inhibited PMA-induced mitochondrial ROS generation. AdTSPO-infected cells were exposed to 250 nM PMA for 6 h. Relative mitochondrial ROS generation was determined from MitoSOX™ fluorescence assays. Fluorescence emission was measured at 590 nm after excitation at 530 nm. Values are expressed as mean fold-change in arbitrary fluorescence values over baseline values. Each bars show mean ± S.E.M., n = 5. *p < 0.05 vs. baseline value, #p < 0.05 vs. PMA alone. (B) Representative fluorescent images show PMA-induced mitochondrial ROS level in AdTSPO-infected cells. Cells were stained with Mitosox Red (red fluorescence) for mitochondrial ROS detection and 4′,6-diamidino-2-phenylindole (DAPI, blue fluorescence) for nucleus staining. (C) Overexpression of TSPO inhibited PMA-induced MnSOD expression. AdTSPO-infected cells were exposed to 250 nM PMA for 6 and 12 h. MnSOD expression was detected with the anti-MnSOD antibody in Western blots. TSPO-overexpression was confirmed by anti-FLAG antibody. The β-actin was used as a loading control. The displayed results are representative of three independent experiments. (D) Densitometric analysis for Fig. 4C. Each bar represents mean ± SEM (n = 3). *p < 0.05 vs. baseline value, #p < 0.05 vs. Adβgal.
MnSOD is up-regulated by mitochondrial oxidative stress (Rogers et al., 2001). As established TSPO reduced mitochondrial ROS production, we investigated that TSPO overexpression can affect PMA-induced MnSOD expression in cultured endothelial cells. As shown in Fig. 4C, the exposure of PMA increased MnSOD expression in Adβgal-infected cells, however, PMA-induced MnSOD expression was decreased in TSPO-overexpressed cells. For quantitative analysis of MnSOD expression, the data was plotted as a densitometric value (Fig. 4D). These results suggested that the reduced mitochondrial ROS generation by TSPO lead to decreased MnSOD expression.
Gene silencing of TSPO increased PMA-induced VCAM-1 expression and mitochondrial ROS generation
Having determined that TSPO overexpression inhibited PMA-induced VCAM-1 expression, we investigated whether down-regulation of TSPO affected PMA-induced VCAM-1 expression. As shown in Fig. 5A, PMA-induced VCAM-1 expression was compared in cells transfected with a TSPO-specific siRNA or a control siRNA. Incubation with TSPO-targeting siRNA (20 nM) for over 48 h completely suppressed endogenous TSPO expression in endothelial cells. Moreover, VCAM-1 induction in response to PMA was increased in TSPO-targeting siRNA-transfected cells when compared with control siRNA. For quantitative analysis of VCAM-1 expression, the data was plotted as a densitometric value (Fig. 5B). Furthermore, we investigated whether the increased VCAM-1 expression by the gene silencing of TSPO would be mediated by mitochondrial ROS. As results, Mito-TEMPO and cyclosporin A, mitochondrial ROS inhibitors, inhibited the increased PMA-induced VCAM-1 expression to control levels in TSPO-targeting siRNA- transfected cells (Supplementary Fig. 1).
Fig. 5.
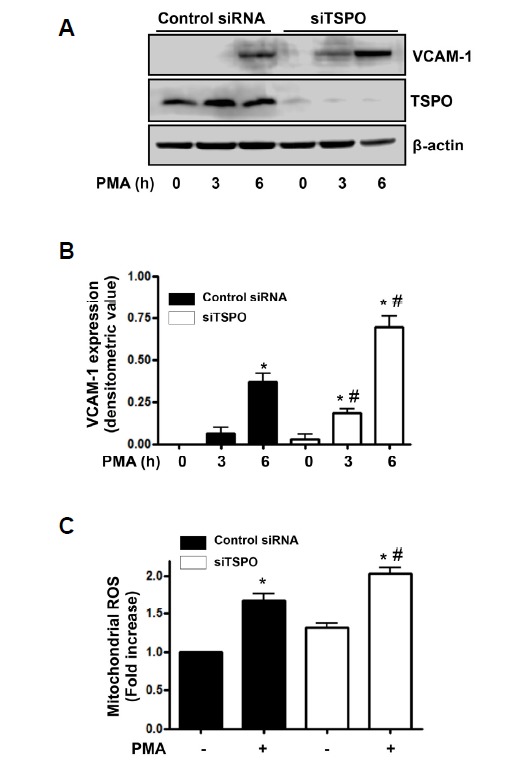
Gene silencing of TSPO increased PMA-induced VCAM-1 expression and mitochondrial ROS generation in endothelial cells. (A) After transfection (48 h) with scrambled siRNA or TSPO-targeting siRNA (20 nM), cells were exposed to 250 nM PMA for 3 and 6 h. Down-regulation of TSPO using TSPO-specific siRNA was confirmed by anti-TSPO antibodies in Western blots. The β-actin was used as a loading control. The displayed results are representative of three independent experiments. (B) Densitometric analysis for Fig. 5A. Each bar represents mean ± SEM (n = 3). *p < 0.05 vs. baseline value, #p < 0.05 vs. control siRNA. (C) Cells transfected with scrambled or TSPO-specific siRNA were exposed to 250 nM PMA for 6 h, and the relative mitochondrial ROS levels were determined via MitoSOX™ fluorescence. Values are expressed as mean fold-change in arbitrary fluorescence values over baseline values. Each error bar shows mean ± SEM, (n = 4). *p < 0.05 vs. baseline value, #p < 0.05 vs. control siRNA.
We also measured the mitochondrial ROS generation in response to PMA in endothelial cells transfected with the TSPO-targeting or control siRNA. As shown in Fig. 5C, the gene silencing of TSPO significantly increased PMA-induced mitochondrial ROS generation. Fluorescence intensity, indicative of the mitochondrial ROS levels, was elevated 1.68-fold in control siRNA-transfected cells exposed to PMA, relative to that in un-stimulated cells. In comparison, in TSPO-targeting siRNA-transfected cells, PMA-induced mitochondrial ROS increased 2.02-fold relative to that in un-stimulated cells.
Midazolam, TSPO ligand, inhibited PMA-induced VCAM-1 expression and mitochondrial ROS generation
To explore whether TSPO ligand can regulate VCAM-1 expression, we examined the effect of midazolam on the VCAM-1 expression in PMA-stimulated HUVECs. After pretreatment with midazolam at 1–50 μM, the cells were treated with 250 nM PMA for 6 h. As shown in Fig. 6A, the pretreatment with midazolam suppressed PMA-induced VCAM-1 expression in a dose-dependent manner. For quantitative analysis of VCAM-1 expression, the data was plotted as a percentage of that in PMA alone (Fig. 6B). Also pretreatment of midazolam (50 μM) significantly inhibited PMA-induced mitochondrial ROS generation (Fig. 6C), suggesting of anti-inflammatory role of midazolam in endothelial cells.
Fig. 6.
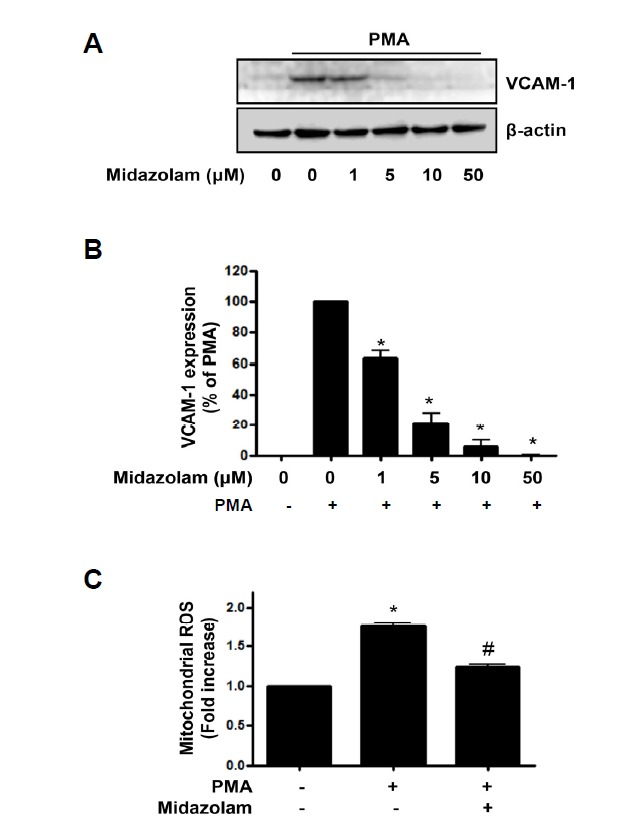
Midazolam inhibited PMA-induced VCAM-1 expression and mitochondrial ROS generation in endothelial cells. (A) After pre-treatment of midazolam (1–50 μM), cells were exposed to 250 nM PMA for 6 h, and the VCAM-1 expression levels were analyzed with western blots. β-actin was used as a loading control; results are representative of four independent experiments. (B) Densitometric analysis for Fig. 6A. Data are expressed as percent densitometric values of VCAM-1 expression induced by PMA. Each bar represents mean ± SEM (n = 4). *p < 0.05 vs. PMA alone. (C) Midazolam-pretreated cells were exposed to 250 nM PMA for 6 h, and the relative mitochondrial ROS levels were determined by MitoSOX™ fluorescence. Values are expressed as mean fold-change in arbitrary fluorescence values over baseline values. Each error bar shows mean ± SEM (n = 4). *p < 0.05 vs. baseline value, #p < 0.05 vs. PMA alone.
DISCUSSION
The present study demonstrates that TSPO has an anti-inflammatory role in endothelial activation through the regulation of mitochondrial function in endothelial cells. Our results showed that adenoviral overexpression of TSPO in endothelial cells significantly inhibited PMA-induced VCAM-1 expression, mitochondrial ROS production, and MnSOD expression. Conversely, gene silencing of TSPO resulted in the opposite effect.
TSPO regulates a wide range of biological functions in cardiovascular system such as atherosclerosis and is a promising therapeutic target and diagnostic tools for cardiovascular diseases (Qi et al., 2012). However, the role of TSPO in vascular inflammation still has unknown. Endothelial activation, an early state of vascular disease, can be defined by the expression of cell-surface adhesion molecules, such as VCAM-1 (Liao, 2013). PKC activation up-regulates the expression of adhesion molecules, suggesting the importance of PKC signaling in the endothelial activation (Ross, 1995). In the present study, adenoviral TSPO overexpression inhibited PKC-induced VCAM-1 expression that is used as a marker of endothelial cell activation. Therefore, increased TSPO expression during vascular inflammation might be an adaptive compensatory response to reduce vascular inflammation. Also anti-inflammatory activity of TSPO can be supported by pharmacological studies. Midazolam as TSPO ligands inhibited lipopolysaccharide-induced macrophage activation via the suppression of the pro-inflammatory response (Kim et al., 2006) and it also inhibited TNF-α-induced VCAM-1 expression in endothelial cells (Joo et al., 2009).
PKC activation induced the generation of mitochondrial ROS in endothelial cells (Joo et al., 2014). In the present study, TSPO overexpression inhibited the PKC activation-induced mitochondrial ROS generation. The overexpression of VDAC-1 triggers MPTP opening, in contrast, gene silencing VDAC-1 expression inhibits the opening of MPTP caused by oxidative stress (Tomasello et al., 2009). We have previously reported that VDAC-1 expression was suppressed by TSPO overexpression in endothelial cells. This result suggests that TSPO-mediated inhibition of mitochondrial ROS production might be related to its ability to inhibit VDAC-1 expression.
Mitochondria are considered the main source of ROS in the cell (Bae et al., 2011). Numerous previous reports showed that increased mitochondrial ROS production is associated with an increase in the expression of adhesion molecules an early state of vascular disease (Chen et al., 1999; Kim et al., 2007; Zinovkin et al., 2014). The potential implications of a TSPO-mediated suppression in endothelial activation might be related to a decrease in mitochondrial ROS production. Moreover, PMA-induced VCAM-1 expression was significantly inhibited by Mito-TEMPO, a mitochondrial ROS inhibitor, and cyclosporin A, an MPTP inhibitor. These data suggest that the regulation of mitochondrial activities, such as mitochondrial ROS generation and MPTP opening, can modulate inflammatory processes in response to an inflammatory stimulator, such as PMA, in endothelial cells.
Among the benzodiazepines, midazolam is the most widely used anxiolytic and sedative drugs for short procedures and in intensive care (Kanto, 1985). Midazolam as TSPO ligands inhibited lipopolysaccharide-induced macrophage activation via the suppression of the pro-inflammatory response (Kim et al., 2006) and it also inhibited TNF-α-induced VCAM-1 expression in endothelial cells (Joo et al., 2009). In the present study, the binding of midazolam to TSPO inhibited PMA-induced VCAM-1 expression and mitochondrial ROS generation in endothelial cells, suggesting that midazolam plays an anti-inflammatory action in PMA-induced vascular endothelial activation.
In conclusion, our data showed that TSPO can inhibit PMA-induced endothelial activation and that this effect was related to its ability to inhibit mitochondrial ROS generation in endothelial cells. Mitochondrial regulation by TSPO may be a therapeutic target for the treatment of vascular diseases.
Acknowledgments
This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2013R1A1A2060250 (HKJ), NRF-2014R1A6A1029617 (BHJ), 2007-0054932 (BHJ) and the research fund of Chungnam National University. English language usage has been checked by a professional editor who is a native English speaker.
Footnotes
Note: Supplementary information is available on the Molecules and Cells website (www.molcells.org).
REFERENCES
- Anholt R.R., Pedersen P.L., De Souza E.B., Snyder S.H. The peripheral-type benzodiazepine receptor. Localization to the mitochondrial outer membrane. J. Biol. Chem. 1986;261:576–583. [PubMed] [Google Scholar]
- Bae Y.S., Oh H., Rhee S.G., Yoo Y.D. Regulation of reactive oxygen species generation in cell signaling. Mol. Cells. 2011;32:491–509. doi: 10.1007/s10059-011-0276-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso E., Fante L., Fowlkes J., Petronilli V., Forte M.A., Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J. Biol. Chem. 2005;280:18558–18561. doi: 10.1074/jbc.C500089200. [DOI] [PubMed] [Google Scholar]
- Bono F., Lamarche I., Prabonnaud V., Le Fur G., Herbert J.M. Peripheral benzodiazepine receptor agonists exhibit potent antiapoptotic activities. Biochem. Biophys. Res. Commun. 1999;265:457–461. doi: 10.1006/bbrc.1999.1683. [DOI] [PubMed] [Google Scholar]
- Chen K.H., Reece L.M., Leary J.F. Mitochondrial glutathione modulates TNF-alpha-induced endothelial cell dysfunction. Free Radic. Biol. Med. 1999;27:100–109. doi: 10.1016/s0891-5849(99)00059-3. [DOI] [PubMed] [Google Scholar]
- Davidson S.M., Duchen M.R. Endothelial mitochondria: contributing to vascular function and disease. Circ. Res. 2007;100:1128–1141. doi: 10.1161/01.RES.0000261970.18328.1d. [DOI] [PubMed] [Google Scholar]
- Dikalova A.E., Bikineyeva A.T., Budzyn K., Nazarewicz R.R., McCann L., Lewis W., Harrison D.G., Dikalov S.I. Therapeutic targeting of mitochondrial superoxide in hypertension. Circ. Res. 2010;107:106–116. doi: 10.1161/CIRCRESAHA.109.214601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimura Y., Hwang P.M., Trout H., Iii, Kozloff L., Imaizumi M., Innis R.B., Fujita M. Increased peripheral benzodiazepine receptors in arterial plaque of patients with atherosclerosis: an autoradiographic study with [(3)H]PK 11195. Atherosclerosis. 2008;201:108–111. doi: 10.1016/j.atherosclerosis.2008.02.032. [DOI] [PubMed] [Google Scholar]
- Galiegue S., Tinel N., Casellas P. The peripheral benzodiazepine receptor: a promising therapeutic drug target. Curr. Med. Chem. 2003;10:1563–1572. doi: 10.2174/0929867033457223. [DOI] [PubMed] [Google Scholar]
- Hardwick M.J., Chen M.K., Baidoo K., Pomper M.G., Guilarte T.R. In vivo imaging of peripheral benzodiazepine receptors in mouse lungs: a biomarker of inflammation. Mol. Imaging. 2005;4:432–438. doi: 10.2310/7290.2005.05133. [DOI] [PubMed] [Google Scholar]
- Jeon B.H., Gupta G., Park Y.C., Qi B., Haile A., Khanday F.A., Liu Y.X., Kim J.M., Ozaki M., White A.R., et al. Apurinic/apyrimidinic endonuclease 1 regulates endothelial NO production and vascular tone. Circ. Res. 2004;95:902–910. doi: 10.1161/01.RES.0000146947.84294.4c. [DOI] [PubMed] [Google Scholar]
- Joo H.K., Oh S.C., Cho E.J., Park K.S., Lee J.Y., Lee E.J., Lee S.K., Kim H.S., Park J.B., Jeon B.H. Midazolam inhibits tumor necrosis factor-alpha-induced endothelial activation: involvement of the peripheral benzodiazepine receptor. Anesthesiology. 2009;110:106–112. doi: 10.1097/ALN.0b013e318190bc69. [DOI] [PubMed] [Google Scholar]
- Joo H.K., Lee Y.R., Lim S.Y., Lee E.J., Choi S., Cho E.J., Park M.S., Ryoo S., Park J.B., Jeon B.H. Peripheral benzodiazepine receptor regulates vascular endothelial activations via suppression of the voltage-dependent anion channel-1. FEBS Lett. 2012;586:1349–1355. doi: 10.1016/j.febslet.2012.03.049. [DOI] [PubMed] [Google Scholar]
- Joo H.K., Lee Y.R., Park M.S., Choi S., Park K., Lee S.K., Kim C.S., Park J.B., Jeon B.H. Mitochondrial APE1/Ref-1 suppressed protein kinase C-induced mitochondrial dysfunction in mouse endothelial cells. Mitochondrion. 2014;17:42–49. doi: 10.1016/j.mito.2014.05.006. [DOI] [PubMed] [Google Scholar]
- Kanto J.H. Midazolam: the first water-soluble benzodiazepine. Pharmacology, pharmacokinetics and efficacy in insomnia and anesthesia. Pharmacotherapy. 1985;5:138–155. doi: 10.1002/j.1875-9114.1985.tb03411.x. [DOI] [PubMed] [Google Scholar]
- Kim S.N., Son S.C., Lee S.M., Kim C.S., Yoo D.G., Lee S.K., Hur G.M., Park J.B., Jeon B.H. Midazolam inhibits proinflammatory mediators in the lipopolysaccharide-activated macrophage. Anesthesiology. 2006;105:105–110. doi: 10.1097/00000542-200607000-00019. [DOI] [PubMed] [Google Scholar]
- Kim H.J., Park K.G., Yoo E.K., Kim Y.H., Kim Y.N., Kim H.S., Kim H.T., Park J.Y., Lee K.U., Jang W.G., et al. Effects of PGC-1alpha on TNF-alpha-induced MCP-1 and VCAM-1 expression and NF-kappaB activation in human aortic smooth muscle and endothelial cells. Antioxid Redox Signal. 2007;9:301–307. doi: 10.1089/ars.2006.1456. [DOI] [PubMed] [Google Scholar]
- Kluge M.A., Fetterman J.L., Vita J.A. Mitochondria and endothelial function. Circ. Res. 2013;112:1171–1188. doi: 10.1161/CIRCRESAHA.111.300233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leducq N., Bono F., Sulpice T., Vin V., Janiak P., Fur G.L., O'Connor S.E., Herbert J.M. Role of peripheral benzodiazepine receptors in mitochondrial, cellular, and cardiac damage induced by oxidative stress and ischemia-reperfusion. J. Pharmacol. Exp. Ther. 2003;306:828–837. doi: 10.1124/jpet.103.052068. [DOI] [PubMed] [Google Scholar]
- Levin E., Premkumar A., Veenman L., Kugler W., Leschiner S., Spanier I., Weisinger G., Lakomek M., Weizman A., Snyder S.H., et al. The peripheral-type benzodiazepine receptor and tumorigenicity: isoquinoline binding protein (IBP) antisense knockdown in the C6 glioma cell line. Biochemistry. 2005;44:9924–9935. doi: 10.1021/bi050150s. [DOI] [PubMed] [Google Scholar]
- Liao J.K. Linking endothelial dysfunction with endothelial cell activation. J. Clin. Invest. 2013;123:540–541. doi: 10.1172/JCI66843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madamanchi N.R., Runge M.S. Mitochondrial dysfunction in atherosclerosis. Circ. Res. 2007;100:460–473. doi: 10.1161/01.RES.0000258450.44413.96. [DOI] [PubMed] [Google Scholar]
- McEnery M.W., Snowman A.M., Trifiletti R.R., Snyder S.H. Isolation of the mitochondrial benzodiazepine receptor: association with the voltage-dependent anion channel and the adenine nucleotide carrier. Proc. Natl. Acad. Sci. USA. 1992;89:3170–3174. doi: 10.1073/pnas.89.8.3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park M.S., Kim C.S., Joo H.K., Lee Y.R., Kang G., Kim S.J., Choi S., Lee S.D., Park J.B., Jeon B.H. Cytoplasmic localization and redox cysteine residue of APE1/Ref-1 are associated with its anti-inflammatory activity in cultured endothelial cells. Mol. Cells. 2013;36:439–445. doi: 10.1007/s10059-013-0195-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi X., Xu J., Wang F., Xiao J. Translocator protein (18 kDa): a promising therapeutic target and diagnostic tool for cardiovascular diseases. Oxid. Med. Cell. Longev. 2012;2012:162934. doi: 10.1155/2012/162934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers R.J., Monnier J.M., Nick H.S. Tumor necrosis factor-alpha selectively induces MnSOD expression via mitochondria-to-nucleus signaling, whereas interleukin-1beta utilizes an alternative pathway. J. Biol. Chem. 2001;276:20419–20427. doi: 10.1074/jbc.M008915200. [DOI] [PubMed] [Google Scholar]
- Ross R. Cell biology of atherosclerosis. Annu. Rev. Physiol. 1995;57:791–804. doi: 10.1146/annurev.ph.57.030195.004043. [DOI] [PubMed] [Google Scholar]
- Tomasello F., Messina A., Lartigue L., Schembri L., Medina C., Reina S., Thoraval D., Crouzet M., Ichas F., De Pinto V., et al. Outer membrane VDAC1 controls permeability transition of the inner mitochondrial membrane in cellulo during stress-induced apoptosis. Cell Res. 2009;19:1363–1376. doi: 10.1038/cr.2009.98. [DOI] [PubMed] [Google Scholar]
- Veenman L., Levin E., Weisinger G., Leschiner S., Spanier I., Snyder S.H., Weizman A., Gavish M. Peripheral-type benzodiazepine receptor density and in vitro tumorigenicity of glioma cell lines. Biochem. Pharmacol. 2004;68:689–698. doi: 10.1016/j.bcp.2004.05.011. [DOI] [PubMed] [Google Scholar]
- Zinovkin R.A., Romaschenko V.P., Galkin II, Zakharova V.V., Pletjushkina O.Y., Chernyak B.V., Popova E.N. Role of mitochondrial reactive oxygen species in age-related inflammatory activation of endothelium. Aging. 2014;6:661–674. doi: 10.18632/aging.100685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zisterer D.M., Gorman A.M., Williams D.C., Murphy M.P. The effects of the peripheral-type benzodiazepine acceptor ligands, Ro 5-4864 and PK 11195, on mitochondrial respiration. Methods Find. Exp. Clin. Pharmacol. 1992;14:85–90. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.