

Abstract
Bacterial viruses of the P22-like family encode a specialized tail needle essential for genome stabilization after DNA packaging and implicated in Gram-negative cell envelope penetration. The atomic structure of P22 tail needle (gp26) crystallized at acidic pH reveals a slender fiber containing an N-terminal “trimer of hairpins” tip. Although the length and composition of tail needles vary significantly in Podoviridae, unexpectedly, the amino acid sequence of the N-terminal tip is exceptionally conserved in more than 200 genomes of P22-like phages and prophages. In this paper, we used x-ray crystallography and EM to investigate the neutral pH structure of three tail needles from bacteriophage P22, HK620, and Sf6. In all cases, we found that the N-terminal tip is poorly structured, in stark contrast to the compact trimer of hairpins seen in gp26 crystallized at acidic pH. Hydrogen-deuterium exchange mass spectrometry, limited proteolysis, circular dichroism spectroscopy, and gel filtration chromatography revealed that the N-terminal tip is highly dynamic in solution and unlikely to adopt a stable trimeric conformation at physiological pH. This is supported by the cryo-EM reconstruction of P22 mature virion tail, where the density of gp26 N-terminal tip is incompatible with a trimer of hairpins. We propose the tail needle N-terminal tip exists in two conformations: a pre-ejection extended conformation, which seals the portal vertex after genome packaging, and a postejection trimer of hairpins, which forms upon its release from the virion. The conformational plasticity of the tail needle N-terminal tip is built in the amino acid sequence, explaining its extraordinary conservation in nature.
Keywords: bacteriophage, hydrogen-deuterium exchange, infection, protein folding, x-ray crystallography, DNA packaging, tail needle, trimer of hairpins, viral genome ejection
Introduction
Podoviridae forms a family of bacterial viruses (bacteriophages) characterized by short and noncontractile tails (1). The tail complex is a sophisticated molecular machine, which is attached to a unique vertex of the icosahedral capsid and provides an entry through which the viral genome is packaged during replication and is ejected into the host during infection (2). In the prototypical Salmonella enterica phage P22 (3), the tail machine consists of a ∼2.8-MDa multisubunit complex (4, 5) that replaces a single penton of the icosahedral capsid (6–8). P22 binds to the Salmonella surface via tailspike proteins (gp9) that provide adsorption specificity by binding to the Salmonella O-antigen surface polysaccharide and by cleaving it (9). In addition to the tailspike, P22 virions contain a tail needle that is located at the distal tip of the tail axis (5–7), projecting ∼140 Å outwards from the virion. In P22, the tail needle protein is encoded by gene 26 (10, 11) and at acid pH forms a 240-Å-long trimeric coiled-coil fiber containing three domains: an N-terminal tip (NTT),3 a central α-helical coiled coil core, and a C-terminal tip (CTT) that folds as an inverted coiled-coil (12–14). Orthologues of gene 26 have been identified bioinformatically in hundreds of phage genomes and prophages (15). Significant sequence variability exists in the length and composition of the tail needle helical core and CTT, whereas the NTT is extraordinarily conserved throughout evolution. Based upon the structural and biochemical analysis of tail needles from four different P22-like phages, we divided the tail needle superfamily in two subgroups that differ primarily in the structure of their CTT (16). In P22-like bacteriophages, the C terminus folds into a helical trimer whose main chain is inverted with respect to the helical core, as seen in the previously published crystal structure of P22 gp26 (12, 13). In contrast, the Sf6-like tail needle C terminus folds into a globular knob-like structure (16, 17) that shares structural resemblance to the fiber knob of bacteriophage PRD1 (18) and adenovirus (19). Regardless of the C-terminal architecture, all tail needles analyzed experimentally display remarkable structural stability and unfold in solution with an apparent denaturing midpoint guanidine concentration (Cm) between 6.4 and 7.4 m (16, 20).
The tail needle serves several critical functions in the life cycle of bacteriophage P22. First, following genome packaging by the large and small terminase subunits (21–23), the tail needle is one of the three virus-encoded tail factors (gp4, gp10, and gp26) that bind to the portal vertex, stabilizing packaged DNA inside the virion (24). The N-terminal tip of gp26 plugs the portal vertex channel (25), and loss of gp26 caused by mutation results in viral particles that incorporate DNA efficiently but fail to retain it inside the capsid (“DNA leakage phenotype”) (24). Assembly studies using purified P22 tail components revealed that the N-terminal 60 amino acids of gp26 are essential for binding to hexameric gp10 (20, 26), which attaches to the dodecameric portal protein via 12 copies of the tail protein gp4 (27). In addition, the tail needle CTT is responsible for contacting the host outer membrane by a mechanism that remains poorly understood, and during the adsorption/DNA delivery process gp26 is released into the host, possibly followed by the three ejection proteins, gp7, gp16 and gp20 (9, 28), that are ejected before the P22 genome (29).
In vitro, aggregates of purified Salmonella LPS are sufficient to trigger slow ejection of P22 DNA, possibly by releasing gp26 (30). Although the kinetics of DNA release in vitro is much slower than the physiological rate of phage genome ejection, the signal for tail needle release and genome injection is likely transmitted from the tailspike to the tail needle through a series of conformational changes within the tail hub. Swapping the gp26 C-terminal tip with the corresponding domain of the tail needle knob (17) of bacteriophage Sf6 provided compelling evidence that this moiety does not confer host specificity between Escherichia coli, Shigella flexneri, and Salmonella enterica, at least under laboratory conditions (31). However, entirely replacing the P22 C-terminal tip spanning residues 141–233 with the short (∼27 residues) foldon domain of bacteriophage T4 fibritin (32) reduces infectivity and slows potassium release from the host during infection. Together, these findings support the idea that the tail needle tip plays a direct role in DNA delivery, possibly by controlling the kinetics of DNA release (31).
In this paper, we studied the structure and conformational dynamics of tail needle NTT in P22 and its close relatives HK620 and Sf6. Our data suggest that the NTT folds into a trimer of hairpins only after ejection into the host.
Experimental Procedures
Biochemical Techniques
Cloning, expression, and purification of the full-length (FL) tail needles from bacteriophages P22 (11–13), HK620 (16), and Sf6 (16) were previously described. Gp26–140 (spanning residues 1–140) was generated by introducing a stop codon at position 141, using plasmid pET-21b-gp26 as template. Gp26–70 (residues 1–70) and gp26–99 (residues 1–99) were generated by introducing a stop codon at positions 71 and 100, respectively, using plasmid pMAL-gp26 as template (33). Gp26–140 was expressed with and N-terminal His tag and purified as described for FL-gp26 (33). Gp26–70 and gp26–99 were purified fused to an N-terminal maltose-binding protein (MBP) as previously described (33). Limited proteolysis of purified, untagged HK620 tail needle was carried out as described (34). Analytical gel filtration chromatography was carried out on a Superpose 12 column equilibrated in buffer A (20 mm Tris, pH 8, 150 mm NaCl, 5 mm β-mercaptoethanol, 0.1 mm PMSF) as described (35).
Crystallographic Methods
Purified, untagged tail needles were concentrated to 7.5 mg/ml and crystallized using the hanging drop vapor diffusion technique. P22 gp26 crystals were obtained in the presence of 40% PEG 1000, 0.1 m MOPS, pH 7.0 (crystal form I, pH 7.0) and 40% PEG 4000, 0.1 m CAPS, pH 10.0 (crystal form II, pH 10.0). P22 Gp26–140 crystals were obtained in the presence of 20% PEG 8000, 0.05 m potassium phosphate monobasic, pH 3.9. HK620 tail needle was crystallized using 40% PEG 4000, 0.1 m TAPS, pH 9.0 (Crystal form I-pH 9.0 and Crystal Form II-pH 9.0) and 30% PEG 1000, 0.1 m Tris, pH 7.5 (crystal form III, pH 7.5). Crystal form III crystals were derivatized with xenon gas prior to data collection, as described (13). All crystallization drops were equilibrated at 20 °C, and crystals appeared within several weeks. 27.5% ethylene glycol was added as cryoprotectant before flash-freezing in a nitrogen stream at −170 °C. X-ray data were collected from frozen P22 and HK620 tail needle crystals at National Synchrotron Light Source Beamlines X6A and X29 and CHESS A1, F1, on ADSC Quantum q270, q315, and q4 CCD detectors. All data were indexed, integrated, and scaled using HKL-2000 (36). P22 crystals grown at pH 7 and 10 are hexagonal with two monomers in the asymmetric unit (AU), each generating a homotrimeric fiber by applying 3-fold crystallographic symmetry. Gp26–140 crystals are primitive monoclinic and contain two trimeric needles in the AU. Three crystal forms were identified for HK620 tail needle: crystal forms I and II in P1 space group with two tail needles per AU and crystal form III that contains only two protomers in the AU (like P22 gp26 crystal form I and II). With the exception of HK620 crystal form I and II, all crystals analyzed in this study suffered from twinning (see Table 1) with twinning fractions close to 0.5, as determined by PHENIX (37). The structure of P22 and HK620 tail needles were determined by molecular replacement in PHASER (38) using P22 gp26 residues 141–233 as search model (PDB code 2POH). Initial solution were refined by rigid body refinement, simulated annealing, and isotropic B-factor refinement with TLS domains using PHENIX (39). For twinned structures, refinement was carried out using a twin target function and a twin law, as reported in Table 1. The program Coot (40) was used for manual rebuilding. After modeling the solvent and bound ions, final models of P22 gp26 and HK620 tail needles were refined to an Rfree between 18 and 25% (see Table 1). All models have outstanding stereochemistry, with r.m.s.d. for bond and angles in line with their resolution and >90% of residues in the most favored regions of the Ramachandran plot and no outliers in disallowed regions (see Table 1).
TABLE 1.
Crystallographic data collection and refinement statistics
In parentheses are the highest resolution shell statistics.
|
P22 tail needle |
HK620 tail needle |
|||||
|---|---|---|---|---|---|---|
| Crystal form (I) pH 7.0 | Crystal form (II) pH 10.0 | Gp26–140 pH 3.9 | Crystal form (I) pH 9.0 | Crystal form (II) pH 9.0 | Crystal form (III) pH 7.5 | |
| Data collection | ||||||
| Space group | P3 | P3 | P21 | P1 | P1 | P3 |
| Cell dimensions | ||||||
| a (Å) | 43.8 | 42.3 | 58.9 | 53.0 | 57.0 | 44.0 |
| b (Å) | 43.8 | 42.3 | 94.1 | 73.3 | 73.8 | 44.0 |
| c (Å) | 272.3 | 271.2 | 97.6 | 77.4 | 77.6 | 271.0 |
| α (°) | 90.0 | 90.0 | 90.0 | 96.6 | 83.2 | 90.0 |
| β (°) | 90.0 | 90.0 | 90.01 | 89.8 | 66.4 | 90.0 |
| γ (°) | 120.0 | 120.0 | 90.0 | 111.2 | 70.6 | 120.0 |
| Resolution (Å) | 15–2.10 (2.18–2.10) | 15–2.50 (2.59–2.50) | 15–2.75 (2.85–2.75) | 15–1.95 (2.02–1.95) | 15–2.50 (2.59–2.50) | 15–2.20 (2.28–2.20) |
| Rsyma | 8.4 (20.6) | 9.9 (24.2) | 11.0 (48.7) | 13.6 (41.0) | 11.4 (40.0) | 11.7 (31.8) |
| I/σI | 16.3 (4.6) | 13.7 (4.4) | 20.4 (4.3) | 14.5 (2.5) | 9.0 (2.3) | 23.5 (7.9) |
| Reflections (unique/total) | 32,511/113,472 | 19,166/310,479 | 27,270/229,028 | 72,716/131,894 | 37,023/71,365 | 26,538/110,693 |
| Completeness (%) | 95.2 (70.4) | 96.9 (84.6) | 98.1 (97.9) | 93.1 (73.1) | 98.1 (95.0) | 90.5 (58.5) |
| Redundancy | 3.5 (1.9) | 3.1 (2.2) | 2.9 (2.7) | 1.8 (1.4) | 1.9 (1.8) | 4.2 (1.9) |
| Wilson B-factor (Å2) | 15.5 | 12.2 | 37.2 | 19.8 | 20.3 | 12.7 |
| Refinement | ||||||
| Resolution (Å) | 30–2.1 | 30–2.5 | 15.0–2.75 | 15.0–1.95 | 15.0–2.50 | 30–2.20 |
| No. reflections | 32,511 | 19,111 | 27,356 | 68,211 | 67,488 | 26,470 |
| Twin law | -h, -k, l | -h, -k, l | h, -k, -l | - | - | -h, -k, l |
| Rwork/Rfreeb | 15.3 /18.9 | 18.4 /22.5 | 21.5 /25.2 | 20.1 /22.3 | 20.9 /25.9 | 22.6 /25.1 |
| Protomers in AU | 2 | 2 | 6 | 6 | 6 | 2 |
| No. atoms (protein/ionc/water) | 2,684/4/764 | 2,684/4/466 | 6,318/3/495 | 7,920/4/926 | 8,178/4/347 | 2,805/8/300 |
| B-factors (protein/ionc/water) | 20.6/22.5/30.7 | 18.5/16.1/20.2 | 44.6/45.1/40.7 | 31.8/58.2/41.4 | 37.2/40.1/32.8 | 21.6/50.0/21.0 |
| r.m.s.d. | ||||||
| Bond lengths (Å) | 0.002 | 0.003 | 0.004 | 0.004 | 0.004 | 0.006 |
| Bond angles (°) | 0.58 | 0.66 | 0.93 | 0.88 | 0.78 | 0.90 |
| PDB code | 4ZKP | 4ZKU | 4ZXQ | 5BU5 | 5BVZ | 5BU8 |
a Rsym = Σi,h|I(i,h) − <I(h)>|/Σi,h|I(i,h)|, where I(i,h) and <I(h)> are the ith and mean measurement of intensity of reflection h.
b The Rfree value was calculated in thin resolution shells using ∼5% of reflections up to 2,000 reflections.
c It includes Cl−, Ca2+, and xenon atoms.
Structure Analysis, Fitting into EM Density and Illustrations
Interface surface area and solvation free energy (ΔiG) calculations were performed using the PISA server (41). All figures in the paper were prepared using the program PyMOL (42). Atomic models of P22 gp9 (PDB codes 3TH0 and 1LKT), gp1 (PDB codes 1VT0 and 3LJ4), gp4 (PDB codes 1VT0 and 3LJ4), and gp26 (PDB code 2POH and tail needles solved in this paper) were placed inside the 9.4 Å EM density (EMDB-5051) using MOLREP (43), and their atomic positions were refined using phenix.real_space_refine (44). A structural model of Sf6 tail needle was generated using I-TASSER (45) by providing the crystal structure of Sf6 knob (17) and the helical core of P22 gp26 as templates to guide I-TASSER modeling. Sf6 tail needle was manually fit into the asymmetric cryo-EM reconstructions of Sf6 virion (accession code EMDB-5730) (46).
Negative Stain Transmission Electron Microscopy
For negative stain electron microscopy, untagged Sf6 tail needles were applied to glow discharged carbon-coated copper grids at a concentration of ∼1 μg/ml and stained with 1% uranyl formate. Images were collected on an FEI Tecnai F20 transmission electron microscope operated at 200 kV and recorded on a charge-coupled device (CCD; Gatan Ultrascan 4000). Micrographs were collected at an electron optical magnification of 70,373× and ∼0.5–1 μm underfocus. The pixel size on the specimen scale was 1.99 Å. Individual particles were manually selected using the EMAN program boxer (47). Images were CTF corrected and subjected to reference-free image alignment and classification using EMAN2 software (47). To determine tail length distribution of individual Sf6 tail needle particles, reference images were first computationally assembled from a 45-Å sphere designed to represent the CTT connected to a rod of varying lengths to symbolize the tail. The final lengths of 16 computationally derived models ranged between 85 and 215 Å. Projections were computed for each of the references, and the data image projections were aligned to each reference, and the cross-correlation coefficient was calculated using SPIDER software (48). Each projection was binned into the tail length class that represented the highest cross-correlation coefficient for each image.
Hydrogen-Deuterium Exchange and Data Processing
Untagged gp26 was diluted 10-fold into a buffer (30 mm Tris, pH 8, 100 mm NaCl) prepared with D2O. At the indicated time points, the exchange reaction was quenched by the addition 150 mm phosphate buffer (pH 2.4). Samples were flash frozen in liquid nitrogen and stored at −80 °C until analysis. For analysis, the quenched reactions were rapidly thawed and injected into a liquid chromatography system held at 4 °C consisting of sequential immobilized Aspergillus saitoi protease type XIII and pepsin columns, a C4 peptide trap, and a Phenomenex Aeris 3.6 100-mm C18 analytical column. Peptides resulting from digestion were trapped and desalted on the C4 trap, captured by the C18 column, and eluted with an increasing gradient of AcN with 0.1% formic acid. Mass spectra were recorded on a Waters Quadrupole Time-of-Flight Premier Mass Spectrometer. Peptide identification was performed through repeated lock mass corrected data-dependent MS/MS runs analyzed using Protein Lynx global server (Micromass). Hydrogen-deuterium exchange data were analyzed, and heat maps were generated using HDExaminer (Sierra Analytics). Back exchange as estimated by averaged exchange end points was ∼20%.
Circular Dichroism
CD scans and thermal melting curves were acquired on a Jasco J-810 spectropolarimeter equipped with a Peltier temperature control system using a 0.1-cm quartz cuvette (Starna Cells, Inc.). Assays were carried out using purified, untagged gp26–99 dissolved at 6 μm final concentration in pH 5.5 MES buffer (50 mm MES, 150 mm NaCl) or pH 8.0 sodium phosphate buffer (50 mm NaH2PO4/Na2HPO4, 150 mm NaCl). CD scans were measured between 196 and 260 nm at 5 °C. Thermal denaturation assays were carried out by increasing the temperature from 5 to 60 °C and monitoring the resultant changes in ellipticity at 222 nm. All wavelength scans and thermal melting curves were baseline-subtracted using buffer scans. The apparent midpoint of the unfolding transition temperature (appTm value) was determined by fitting the experimental thermal profile to standard two-state unfolding equations using Origin 6.1.
Results
The N-terminal Tip of P22 Tail Needle Is Unstructured at Neutral and Alkaline pH
The crystal structure of full-length P22 gp26 (FL-gp26) was previously determined from crystals grown at pH 4.6 (12, 13, 33). In this structure, the NTT adopts a compact conformation, whereby residues 1–26, containing a 3/10 helix between amino acids 4–7, fold backwards onto the helical core to form a trimer of hairpins (Fig. 1A). This topology was also observed in a new crystal structure of gp26 helical core lacking the CTT (gp26–140), which we crystallized at pH 3.9 and refined to an Rwork/free of 21.5/25.2, at 2.75 Å resolution (Fig. 1B and Table 1). Gp26–140 and FL-gp26 are essentially identical between residues 1 and 140 (r.m.s.d ∼ 1.19 Å). Both of the FL-gp26 and gp26–140 constructs crystallized at acidic pH have an N-terminal polyhistidine tag that is partially visible in the electron density and that likely stabilizes crystal contacts by allowing the protonated imidazole side chains to bind surface-exposed residues in the gp26 helical core.
FIGURE 1.

The tail needle NTT is not visible in crystal structures obtained at neutral and alkaline pH. A–D, ribbon diagrams of P22 gp26 crystallized at pH 4.6 (A) (12, 13), pH 3.9 (only residues 1–140) (B), pH 7.0 (C), and pH 10.0 (D). E–G, structure of phage HK620 tail needle determined in two crystal forms at pH 9.0 (E and F) and at pH 7.5 bound to xenon gas (G). All tail needles are colored by B-factor with dark blue and red representing the lowest and highest refined B-factor, respectively. Calcium, chloride, and xenon atoms buried at the trimeric interface are shown as magenta, orange, and black spheres, respectively. res., residue.
To visualize the gp26 structure at physiological pH, we purified an untagged version of the tail needle and screened it for crystallization. We obtained two new crystal forms, one at pH 7 (crystal form I) and one at pH 10 (crystal form II), each of which diffracted x-rays to a resolution comparable with gp26 crystals grown at acidic pH (12, 13) (Table 1). Unable to solve these structures using a trimer or a protomer of FL-gp26 as search model, we obtained an unambiguous molecular replacement solution using a fragment of gp26 protomer encompassing the CTT (residues 141–233). Crystal form I was refined to an Rwork/free of 15.3/18.9% at 2.1 Å, and crystal form II was refined to an Rwork/free of 18.4/22.5% at 2.5 Å resolution (Table 1). Both gp26 structures revealed strong and continuous electron density for residues 55–233 but unexpectedly had no discernible density for the NTT (residues 1–54) (Fig. 1, C and D). The two new crystal forms are essentially identical (r.m.s.d. ∼0.29 Å) and differ minimally from FL-gp26 crystallized at pH 4.6 (r.m.s.d ∼1.59 and 1.60 Å, respectively), with the exception of the invisible NTT. In both refined models (Table 1), the B-factor increased significantly toward the fiber N termini, suggesting high flexibility above residue 60. The lack of electron density for the NTT suggested that this domain was either cleaved during crystallogenesis or averaged out in the final density because of structural differences among protomers. Unlike crystals grown at acidic pH that contain one or two trimers in the AU (12, 13, 33), both new crystal forms are hexagonal and contain two protomers in the AU (Table 1) that form homotrimeric fibers by applying 3-fold crystallographic symmetry. SDS-PAGE analysis of dissolved crystals ruled out proteolysis (data not shown), lending support to the notion that, under nonacidic conditions, NTT is flexible and does not conform to 3-fold crystallographic symmetry. Thus, crystallographic analysis of P22 gp26 crystallized at neutral and alkaline pH provides clues to an unexpected conformational dynamics in the genome-sealing NTT.
Crystal Structure of the Phage HK620 Tail Needle, a Close Relative of P22 gp26
To expand on the analysis of P22-like gp26 structures, we also crystallized the tail needle from the E. coli phage HK620, which shares 60% sequence identity with the P22 gp26. When considering the NTT alone, the sequence identity exceeds 85%, indicating that this domain is almost invariant among P22-like bacteriophages (49). We obtained structures from three crystal forms of the untagged HK620 tail needle. Each structure was solved by molecular replacement using the P22 gp26-CTT protomer as a search model, followed by refinement to Rwork/free values of 20.1/22.3%, 20.9/25.9%, and 22.6/25.1%, respectively (Table 1). The first crystal form was grown at pH 9.0 in space group P1 and contains two tail needles in the AU with no density to accommodate NTT residues 1–59 (Fig. 1E). The second crystal form, obtained from the same crystallization drop as crystal form I (pH 9.0) but with a slightly different triclinic unit cell, contains two tail needles and has no electron density for residues 1–53 (Fig. 1F). Finally, a third crystal form was grown at pH 7.5 and belongs to space group P3, with unit cell parameters similar to P22 crystal forms I and II (Table 1). This crystal was also subjected to derivatization with xenon gas for 2 h at 450 p.s.i., as described for P22 gp26 (13). The electron density of this crystal form had strong and clear features for residues 57–233 and only sparse density for N-terminal residues 1–57 (visible as elongated “tubes” after blurring the B-factor) that were modeled as an extended stretch of polyalanine (Fig. 1G). In addition, three well occupied xenon atoms were identified at discrete sites of the trimeric interface.
Both triclinic crystal forms had an entire trimer in the AU but invisible NTTs, ruling out the possibility that this domain is crystallographically invisible because it fails to obey 3-fold symmetry. Incubation of the purified HK620 tail needle with traces of chymotrypsin readily cleaved the NTT at amino acid 35, as determined by N-terminal sequencing (Fig. 2A). Thus, by analogy to P22 gp26, the NTT of phage HK620 tail needle is also highly flexible at neutral pH and is susceptible to proteolytic degradation in solution at neutral pH.
FIGURE 2.

Molecular determinants for tail needle trimerization. A, limited proteolysis analysis of purified HK620 tail needle in the presence of chymotrypsin at 20 °C. Aliquots of digestion mixture were collected after 10, 20, 30, 45, 60, 120, and 180 min; the reaction was blocked with 1 mm PMSF and resolved by 12.5% SDS-PAGE. Room temperature and 95 °C indicate FL tail needle incubated for 5 min at room temperature and 95 °C prior loading on to gel. B, stick representation of the tail needle helical core (residues 1–140 for P22 gp26 and residues 54–140 for HK620) with hydrophobic side chains at the trimerization interface shown as yellow spheres. Gp26 NTT is overlaid to a light gray box. A calcium ion (magenta) and a chloride ion (orange) are buried at the interior of P22 and HK620 tail needles. C, left panel, gel filtration analysis of purified MBP-gp26–70 (molecular mass, ∼53 kDa) and MBP-gp26–99 (molecular mass, ∼56 kDa). The Superose 12 gel filtration column was calibrated using molecular mass markers, whose elution volumes and relative molecular masses are indicated in the right panel. MBP-gp26–70 eluted after 69.6 ml, consistent with a species of a ∼85 kDa, reflecting an elongated monomer. In contrast, MBP-gp26–99 eluted after 58.4 ml, consistent with a 269-kDa species corresponding to a trimer. RT, room temperature.
The NTT Lacks Determinants for Trimerization
To rationalize the NTT structural dynamics (Fig. 1, A and B), we analyzed the trimerization interface of P22 and HK620 tail needles. The fibers from both species form SDS-resistant trimers (Fig. 2A). However, in folding studies, the HK620 tail needle is more stable than P22 gp26, unfolding with apparent Cm and Tm values of ∼7.4 m and 88–90 °C, respectively, versus lower Cm ∼6.4 m and Tm = 85 °C values for gp26 (16, 20). Tail needle stability is promoted by a spine of hydrophobic residues (Fig. 2B) that are organized in 11 contiguous heptads, spanning most of the interior α-helical core that forms between residues 70 and 140. Compared with the P22 needle, the HK620 tail needle appears to have a preference for bulkier hydrophobic residues (Leu/Phe over Val) that fill the trimeric interface even more densely than in gp26. This difference reduces the number of internal cavities of HK620 tail needle compared with that of P22, which possibly accounts for the higher stability measured in vitro. This is supported by the fact that only three xenon atoms are trapped inside the HK620 tail needle, as compared with seven in P22 needle under comparable derivatization conditions (13). Interestingly, both fibers have only one trimerization heptad in the NTT that resides between residues 42–48, with four Ala (at positions 37, 44, 46, 49) and three Gly (at 39, 43, 47) surrounding the heptad and a tyrosine cluster at the far N-terminal tip (Fig. 2B). Hydrophobic contacts mediated by these two motifs are unlikely to provide sufficient energy to completely stabilize the NTT trimeric interface. In addition, both tail needles have a buried calcium ion and chloride ion at the trimeric interface. In P22, these ions make polar contacts with the side chains of residues Asn63/Gln66 and Asn94, whereas in HK620 tail needle, the chloride ion coordinates with Asn80 (Fig. 2B).
Consistent with the paucity of contacts in the NTT, the trimeric helical portion of P22 gp26 NTT (residues 30–60) has a calculated free energy of assembly dissociation (ΔGdiss) of only ∼6.4 kcal/mol. This value, which corresponds to the free energy difference between dissociated and associated states (41), is very low compared with an equivalent stretch of 30 residues in the gp26 helical core (e.g. residues 100–130), which has a ΔGdiss >15 kcal/mol, suggesting a marginal ability to trimerize. To validate this finding experimentally, we generated two fusion proteins containing the MBP fused to gp26 residues 1–70 (gp26–70) and 1–99 (gp26–99) and asked whether these constructs are able to trimerize in solution. As expected from our above results, MBP-gp26–70 remained monomeric in solution at all concentrations tested (Fig. 2C), confirming that the NTT alone is not sufficient to promote stable trimerization. In contrast, the longer fragment MBP-gp26–99 that contains the NTT, along with four additional trimerization heptads (Fig. 2B), readily formed trimers in solution (Fig. 2C). Thus, the tail needle NTT is most likely not oligomeric on its own but can be forced to trimerize if fused to additional C-terminal heptads.
Visualization of the Sf6 Tail Needle by EM Reveals an Unstructured NTT
We next sought to visualize the conformation of the tail needle of bacteriophage Sf6, which is only ∼19% identical to P22 gp26 but contains ∼85% identical residues in the NTT domain (16). Because we were unable to obtain diffracting crystals of the whole Sf6 tail needle, we used negative stain transmission EM to determine whether its NTT is structured at physiological pH. Unlike P22 and HK620 needles that appear as featureless sticks by negative stain transmission EM with N- and C-terminal tips that are indistinguishable from each other (16), the Sf6 tail needle presents a large knob at its CTT, a structure that has independently been solved by x-ray crystallography (17). The knob provides a useful C-terminal marker that is ∼40 Å in diameter and is clearly distinguishable in negative stain electron micrographs, providing an exceptional reference for measuring fiber lengths. We incubated purified Sf6 tail needles in a physiological buffer and, after staining in 1% uranyl acetate, imaged the tail needle by transmission EM. Altogether 1,394 individual particles were selected and grouped into class averages. Although the various class averages differed in tail length, all revealed a blurring at the end of the needle that is opposite the CTT (Fig. 3A). To determine the distribution of individual Sf6 tail needle lengths, computational models were generated to represent different tail lengths that ranged between 85 and 215 Å in 10-Å increments (distances include the CTT knob). The 1,394 projections were compared with projections of each of these models and distributed into the class with the highest cross-correlation coefficient. This analysis revealed that the majority of Sf6 tail needles has a helical core of ∼140–160 Å in length, with a comparable number of significantly shorter and longer needles (Fig. 3B). The full-length Sf6 tail needle bearing a trimer of hairpins NTT should measure ∼215 Å in length, or ∼50 Å longer than the average length of ∼165 Å measured for fibers by EM (Fig. 3C). The EM analysis of fiber lengths combined with the visualization of blurry NTT ends in all class averages further supports the notion that the majority of Sf6 fibers have unstructured NTTs. Thus, P22, HK620, and Sf6 tail needles all share an unstructured conformation of the NTT at physiological pH.
FIGURE 3.

Negative stain electron microscopy visualization of Sf6 tail needle. A, six representative projection class averages of Sf6 tail needle stained with 1% uranyl formate and obtained at pH 8.0. The class averages differ by tail needle length. B, histogram of average fiber length sorted in increments of 10 Å between 85 and 215 Å. C, a ribbon model of the Sf6 tail needle assuming a trimer of hairpins NTT (∼215 Å) or an unstructured NTT (∼165 Å).
Hydrogen-Deuterium Exchange MS and CD Spectroscopy
We next employed hydrogen-deuterium exchange MS to characterize the dynamics of P22 gp26 in solution at room temperature and pH 8.0. Purified P22 gp26 was incubated with a deuterated solution to allow free exchange of hydrogen with deuterium. The exchange reaction was quenched at different times (0.5, 1, 3, 10, 30, 90, and 300 s), followed by digestion with two acidic proteases, namely the A. saitoi protease type XIII and pepsin. Digested peptides were purified by reverse phase HPLC chromatography, and mass spectra were recorded on a Waters Quadrupole Time-of-Flight Premier Mass Spectrometer. The heat map in Fig. 4A provides a visual representation of hydrogen-deuterium exchange as a function of time using color gradients. Two regions of the protein display particularly noteworthy and diametrically opposed exchange kinetics. The region from residues 1 to 40 displays very rapid exchange that is essentially complete at the earliest time point of 0.5 s as expected for a completely unprotected peptide in solution. In contrast, the region spanning residues 45–75 is highly protected because it undergoes very limited exchange over the time course of the experiment, which is indicative of a well ordered and stable region.
FIGURE 4.

Hydrogen-deuterium exchange analysis of the tail needle structural flexibility. A, the kinetics of hydrogen-deuterium exchange of gp26 at pH 8.0 and 20 °C were monitored by mass spectrometry-based analysis of proteolytic fragments obtained following quenching at time points ranging from 0.5 to 300 s. The shaded bars above the sequence represent the boundaries of the peptides identified and their average degree of deuteration over the time course of the experiment. The shaded bars below the sequence represent a heat map of exchange, which depicts the extent of exchange at the indicated time point. HDExaminer (Sierra Analytics) performs a least squares-based analysis of the deuteration level of overlapping peptides to estimate local exchange, thereby increasing the resolution of the map. Hence, color boundaries in the heat map do not necessarily correspond directly to the peptides identified. The color key is common to both representations. B, the far UV-CD spectra of untagged gp26–99 dissolved at 6 μm final concentration in solutions buffered at pH 5.5 (red curve) and pH 8.0 (green curve). C, thermal unfolding curves of untagged gp26–99 dissolved at 6 μm final concentration in solutions buffered at pH 5.5 (red curve) and pH 8.0 (green curve). Melting curves were recorded by measuring changes in the ellipticity at 222 nm. The arrows indicate the appTm.
We also used CD spectroscopy to investigate structural rearrangements in gp26 NTT. For this analysis, we employed the shorter construct gp26–99 that forms stable trimers in solution (Fig. 2C) but has reduced helical content as compared with FL-gp26 (∼70 versus 145 residues in α-helical conformation at acidic pH; Fig. 1, A and B). Reducing the α-helical “background” allowed to accurately measure differences in gp26 ellipticity caused by variations in pH. Wavelength scans using 6 μm untagged gp26–99 revealed a ∼20% gain in α-helical signal at pH 5.5 versus pH 8.0 (Fig. 4B), supporting the idea of a more structured NTT at acidic pH. Assuming that gp26–99 helicity measured at pH 5.5 corresponds to the continuous α-helix spanning residues 29–99 (∼70 amino acids) seen in gp26 crystallized at acidic pH (Fig. 1, A and B), the reduced ellipticity at pH 8.0 is consistent with loss of helical folding for ∼15 residues. This positions the beginning of gp26 α-helix at neutral pH roughly at residue 44 (i.e. residues 29 + 15 ∼44), in good agreement with the folded boundary probed by hydrogen-deuterium exchange (Fig. 4A). Likewise, the thermal stability of gp26–99 was slightly increased at pH 5.5 as compared with pH 8.0 (appTm = ∼42° versus 45 °C) (Fig. 4C), also supporting the idea of a more folded conformation of the NTT at acidic pH.
Fitting the Tail Needle into the Cryo-EM Map of P22 Tail Apparatus: a Reassessment
A 9.4 Å cryo-EM reconstruction of P22 tail apparatus (5) and a 7.8 Å asymmetric reconstruction of the mature virion (8) have been determined. Together, these reconstructions provide a subnanometer description of the gp26 structure visualized in the physiological context of the mature virion. Previous attempts to fit the structure of gp26, solved under acidic pH conditions, into the tail cryo-EM map modeled the NTT trimer of hairpins inside the gp10 channel (5, 8, 25) with the CTT positioned in a region lacking experimental density (Fig. 5, A and B). We reanalyzed this fitting in light of the neutral pH structure of gp26 presented in this paper, using the 9.4 Å cryo-EM reconstruction of P22 tail that has better resolved density for gp26. To avoid bias introduced by manual docking, we placed all atomic models of P22 tail components solved crystallographically (e.g. tailspike, portal protein, gp4, and FL-gp26) inside the EM density using molecular replacement in a phased map, followed by real space refinement. This reanalysis suggests that the previous fitting (5, 6, 25) of gp26 NTT inside gp10 channel is likely incorrect for three reasons. First, P22 gp26 NTT trimer of hairpins is too wide (∼35 Å van der Waals diameter) to fit into the gp10 channel of the cryo-EM map (shown by an asterisk in Fig. 5B). Although an atomic model for gp10 is not available, the exact diameter of the gp10 channel can be estimated from the cryo-EM map by adjusting the map contour at a sigma level that covers homogeneously the atomic models of tailspike and portal protein, which are exceptionally well resolved. This exercise suggests that the gp10 channel has a maximum diameter of ∼25 Å, comparable with the van der Waals diameter of the gp26 helical core and significantly smaller than the P22 gp26 NTT trimer of hairpins. Accordingly, placing the NTT in the first portion of the gp26 cryo-EM density results in a poor fitting, with the majority of gp26 atoms being unaccounted for in the EM-density (Fig. 5C). Second, no density is visible for gp26 residues 1–26 that fold back onto the helical core and, if present, should enlarge the cryo-EM density for the gp26 NTT (indicated by arrows in Fig. 5B). Third, placing the gp26 trimer of hairpins inside the first portion of the cryo-EM density map places the CTT in a region of no density (Fig. 5B). This is unlikely, because the CTT is well structured in all available crystal structures of P22 gp26 (Fig. 1, C and D, and PDB entries 2POH and 3C9I) and should also be visible in a subnanometer cryo-EM reconstruction. Instead, a more convincing fitting of gp26 inside the tail of the cryo-EM map was obtained by docking the crystal structure of gp26 solved at neutral pH that lacks NTT residues 1–55 (Fig. 5D). In this case, the narrow trimeric coiled-coil portion of gp26 helical core starting around residue 55 fits perfectly inside the first part of the cryo-EM density (Fig. 5F), placing most of gp26 CTT in the observed cryo-EM density (Fig. 5E). This fitting has no clashing of NTT residues inside the gp10 channel because the gp26 van der Waals diameter at residues 55 is close to the estimated channel diameter (Fig. 5E). In agreement with this docking model, a boundary can be observed around residue 140 (see arrow in Fig. 5E) between the smooth and continuous cryo-EM density for gp26 α-helical coiled coil and the CTT, visible at higher contour as “disks” of density, as expected for a β-stranded domain at this resolution.
FIGURE 5.
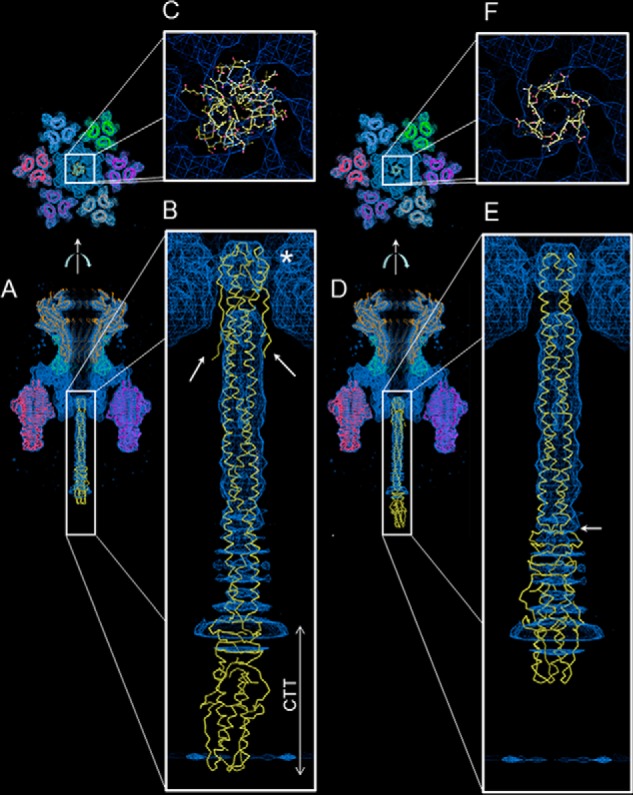
Fitting gp26 structure inside P22 tail apparatus. A–C, fitting the acidic pH structure of P22 gp26 (residues 1–233) inside the cryo-EM reconstruction of P22 tail in side (A and B) and top (C) view. D–F, fitting the neutral pH structure of P22 gp26 (residues 55–233) inside the cryo-EM reconstruction of P22 tail in side (D and E) and top (F) view.
We also fit a modeled structure of the Sf6 tailed needle into the asymmetric cryo-EM reconstruction of the phage Sf6 mature virion (46). Although determined at lower resolution (∼16 Å), the presence of a well defined C-terminal knob allowed unambiguous docking of the CTT into the cryo-EM density, providing a register to place the tail needle inside gp10 channel. Notably, this fitting positions residue 55 at the outside entrance of the gp10 channel and the trimer of hairpins inside the virion, in a region lacking EM density (Fig. 6A). In contrast, a modeled structure of the Sf6 tail needle lacking residues 1–55 and similar to gp26 conformation observed crystallographically at neutral pH (Fig. 1, C and D) gave the best fit for both the C-terminal knob and the N-terminal helical core (Fig. 6B). Thus, the structure adopted by P22 and Sf6 tail needles in the context of the mature virions is very consistent with an unstructured NTT.
FIGURE 6.
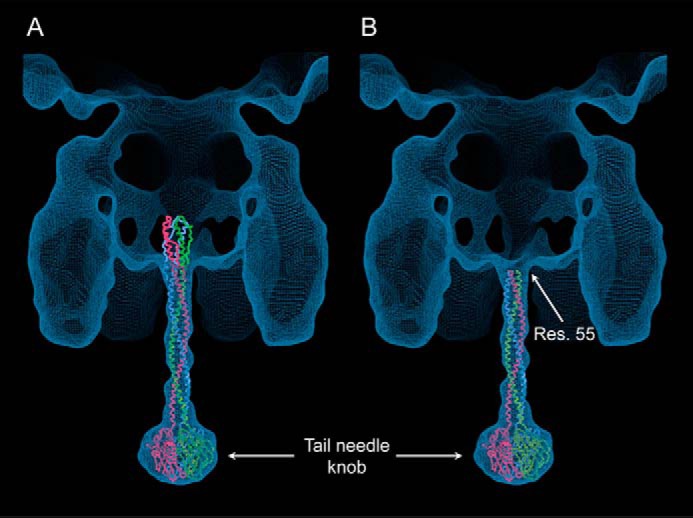
Fitting Sf6 tail needle structure inside the asymmetric reconstruction of Sf6 virion. FL (A) and truncated (residues 55–265) (B) Sf6 tail needle were docked inside the EM density (46) using as a reference point the structure of the C-terminal knob.
Discussion
We used hybrid structural methods to explore the structure and conformational dynamics of the genome-sealing NTT of P22-like tail needles, which is extraordinarily conserved in nature (16). Comparing tail needles from P22, HK620, and Sf6, we determined that the trimer of hairpins observed in gp26 crystallized at acidic pH is not likely to exist at neutral pH. In the absence of other tail components, we found the tail needle NTT adopts a poorly structured, possibly extended conformation (Figs. 1 and 3) that is susceptible to proteolysis (Fig. 2A) and exchanges hydrogens rapidly in solution (Fig. 4A). We will refer to this conformation of the tail needle NTT as “pre-ejection.” Cryo-EM modeling (Fig. 5 and 6) supports the notion the pre-ejection conformation also exists in mature virions, when the tail needle is assembled in the context of the tail apparatus. The paucity of contacts among gp26 protomers between residues 30 and 60, which contain only one trimerization heptad (Fig. 2B) and are insufficient to promote stable trimerization (Fig. 2C), provides a structural explanation to rationalize the loss of a trimer of hairpins fold at physiological pH. Given the remarkable conservation of the NTT in over 200 genomes of P22-like phages and prophages (16), we reasoned that such a flexible structure must confer an evolutionary advantage to the virus of paramount importance. We propose the NTT structural plasticity aids the tail needle in its function as molecular plug. This idea is supported by several lines of evidence. First, an NTT folded as trimer of hairpins is too wide to fit inside gp10 (Fig. 5C). If stably folded as a trimer of hairpins, like a static “cork,” the NTT could pose topological problems during assembly, when a tip larger than the portal vertex channel may result in poor assembly and thus inefficient stabilization of packaged DNA (24). Second, an extended conformation of the NTT could extend inside the gp10 channel and make contact with tailspike and/or gp4 (Fig. 6B). This would make the NTT a prime candidate to sense the signal for ejection initiated by tailspike and/or needle CTT interaction with LPS that is propagated to the tail needle NTT by an as yet unknown mechanism (30). Finally, an extended NTT may provide a surface of interaction with DNA and/or the ejection protein (gp20, gp16, and gp7), whose position inside the virion remains unknown but that could be poised for ejection inside the portal protein channel (28, 50) prior to genome ejection (29). All these factors would justify NTT remarkable conservation throughout evolution, which goes hand in hand with the conservation of gp10 (51), gp26's main binding partner in the tail apparatus.
What happens to gp26 after its release from the virion? We propose that the tail needle NTT undergoes a conformational change possibly in response to the lower pH in the periplasm to adopt a postejection conformation, with the NTT folded as trimer of hairpins. In Gram-negative bacteria, a proton motive force is maintained across the cytoplasmic membrane, and in a typical E. coli cell, for instance, the periplasmic pH is reported to be about 1.7 pH units lower than the cytoplasmic compartment (52, 53). Similarly, lower pH values may exist in the local proximity of proton pumps (e.g. ATPase) possibly stabilizing the trimer of hairpins conformation as seen in all crystal structured determined at acidic pH (Fig. 1, A and B) (12, 13). Spectroscopic studies using CD revealed that mild acidification to pH 5.5 increases gp26 α-helical content and enhances thermal stability (Fig. 4, B and C), pointing to a pH-dependent stabilization of the NTT. It is obviously difficult to reconcile this in vitro observation with the in vivo setting of a phage infection, which is infinitely more complex. However, in light of the putative role of gp26 as a cell envelope-penetrating needle (12), it can be speculated that a more compact and folded NTT aids the tail needle to pass through the bacterial cell envelope during genome ejection. The mechanisms of this complex reaction remain, however, unknown. In conclusion, the current study expands our understanding of phage tail needle biology and presents a new framework to investigate the cascade of events accompanying packaging and ejection of viral genomes though bacterial membranes.
Author Contributions
G. C. conceived and coordinated the study and wrote the paper. A. B., R. S. S., and A. S. O. purified and crystallized tail needle proteins, determined x-ray structures in Fig. 1 with the help of GC, and carried out biochemical experiments in Fig. 2. D. J. T. performed and analyzed the electron microscopy experiments shown in Fig. 3. D. B. and P. E. P. designed, performed, and analyzed the deuterium-exchange MS experiments shown in Fig. 4A. A. B. carried out all CD studies in Fig. 4 (B and C). G. C. did the cryo-EM modeling in Figs. 5 and 6. S. R. C. provided clones for tail needles, provided technical assistance with the project, and contributed to the writing of the paper. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We are grateful to National Synchrotron Light Source staff at Beamlines X26A and X29 and to the macCHESS staff for beam time and assistance in data collection.
This work was supported by National Institutes of Health Grants GM100888 (to G. C.) and GM114817 (to S. R. C.). Research in this publication includes work carried out at the Sidney Kimmel Cancer Center X-ray Crystallography and Molecular Interaction Facility, which is supported in part by NCI Cancer Center Support Grant P30 CA56036. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- NTT
- N-terminal tip
- CTT
- C-terminal tip
- AU
- asymmetric unit
- MBP
- maltose-binding protein
- r.m.s.d.
- root mean square deviation
- FL
- full-length
- CAPS
- 3-(cyclohexylamino)propanesulfonic acid
- TAPS
- 3-{[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]amino}-1-propanesulfonicacid
- PDB
- Protein Data Bank.
References
- 1.Ackermann H. W. (2003) Bacteriophage observations and evolution. Res. Microbiol. 154, 245–251 [DOI] [PubMed] [Google Scholar]
- 2.Bhardwaj A., Olia A. S., and Cingolani G. (2014) Architecture of viral genome-delivery molecular machines. Curr. Opin. Struct. Biol. 25, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Teschke C. M., and Parent K. N. (2010) “Let the phage do the work”: using the phage P22 coat protein structures as a framework to understand its folding and assembly mutants. Virology 401, 119–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang L., Marion W. R., Cingolani G., Prevelige P. E., and Johnson J. E. (2005) Three-dimensional structure of the bacteriophage P22 tail machine. EMBO J. 24, 2087–2095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lander G. C., Khayat R., Li R., Prevelige P. E., Potter C. S., Carragher B., and Johnson J. E. (2009) The P22 tail machine at subnanometer resolution reveals the architecture of an infection conduit. Structure 17, 789–799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lander G. C., Tang L., Casjens S. R., Gilcrease E. B., Prevelige P., Poliakov A., Potter C. S., Carragher B., and Johnson J. E. (2006) The structure of an infectious p22 virion shows the signal for headful DNA packaging. Science 312, 1791–1795 [DOI] [PubMed] [Google Scholar]
- 7.Chang J., Weigele P., King J., Chiu W., and Jiang W. (2006) Cryo-EM asymmetric reconstruction of bacteriophage P22 reveals organization of its DNA packaging and infecting machinery. Structure 14, 1073–1082 [DOI] [PubMed] [Google Scholar]
- 8.Tang J., Lander G. C., Olia A. S., Li R., Casjens S., Prevelige P. Jr., Cingolani G., Baker T. S., and Johnson J. E. (2011) Peering down the barrel of a bacteriophage portal: the genome packaging and release valve in p22. Structure 19, 496–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Israel V. (1978) A model for the adsorption of phage P22 to Salmonella typhimurium. J. Gen. Virol. 40, 669–673 [DOI] [PubMed] [Google Scholar]
- 10.Berget P. B., and Poteete A. R. (1980) Structure and functions of the bacteriophage P22 tail protein. J. Virol. 34, 234–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Andrews D., Butler J. S., Al-Bassam J., Joss L., Winn-Stapley D. A., Casjens S., and Cingolani G. (2005) Bacteriophage P22 tail accessory factor GP26 is a long triple-stranded coiled-coil. J. Biol. Chem. 280, 5929–5933 [DOI] [PubMed] [Google Scholar]
- 12.Olia A. S., Casjens S., and Cingolani G. (2007) Structure of phage P22 cell envelope-penetrating needle. Nat. Struct. Mol. Biol. 14, 1221–1226 [DOI] [PubMed] [Google Scholar]
- 13.Olia A. S., Casjens S., and Cingolani G. (2009) Structural plasticity of the phage P22 tail needle gp26 probed with xenon gas. Protein Sci. 18, 537–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhardwaj A., Casjens S. R., and Cingolani G. (2014) Exploring the atomic structure and conformational flexibility of a 320 A long engineered viral fiber using x-ray crystallography. Acta Crystallogr. D Biol. Crystallogr. 70, 342–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leavitt J. C., Gilcrease E. B., Wilson K., and Casjens S. R. (2013) Function and horizontal transfer of the small terminase subunit of the tailed bacteriophage Sf6 DNA packaging nanomotor. Virology 440, 117–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhardwaj A., Walker-Kopp N., Casjens S. R., and Cingolani G. (2009) An evolutionarily conserved family of virion tail needles related to bacteriophage P22 gp26: correlation between structural stability and length of the alpha-helical trimeric coiled coil. J. Mol. Biol. 391, 227–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhardwaj A., Molineux I. J., Casjens S. R., and Cingolani G. (2011) Atomic structure of bacteriophage Sf6 tail needle knob. J. Biol. Chem. 286, 30867–30877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Merckel M. C., Huiskonen J. T., Bamford D. H., Goldman A., and Tuma R. (2005) The structure of the bacteriophage PRD1 spike sheds light on the evolution of viral capsid architecture. Mol. Cell 18, 161–170 [DOI] [PubMed] [Google Scholar]
- 19.van Raaij M. J., Mitraki A., Lavigne G., and Cusack S. (1999) A triple beta-spiral in the adenovirus fibre shaft reveals a new structural motif for a fibrous protein. Nature 401, 935–938 [DOI] [PubMed] [Google Scholar]
- 20.Bhardwaj A., Olia A. S., Walker-Kopp N., and Cingolani G. (2007) Domain organization and polarity of tail needle GP26 in the portal vertex structure of bacteriophage P22. J. Mol. Biol. 371, 374–387 [DOI] [PubMed] [Google Scholar]
- 21.Roy A., and Cingolani G. (2012) Structure of p22 headful packaging nuclease. J. Biol. Chem. 287, 28196–28205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roy A., Bhardwaj A., Datta P., Lander G. C., and Cingolani G. (2012) Small terminase couples viral DNA binding to genome-packaging ATPase activity. Structure 20, 1403–1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McNulty R., Lokareddy R. K., Roy A., Yang Y., Lander G. C., Heck A. J., Johnson J. E., and Cingolani G. (2015) Architecture of the complex formed by large and small terminase subunits from bacteriophage P22. J. Mol. Biol. 427, 3285–3299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Strauss H., and King J. (1984) Steps in the stabilization of newly packaged DNA during phage P22 morphogenesis. J. Mol. Biol. 172, 523–543 [DOI] [PubMed] [Google Scholar]
- 25.Olia A. S., Prevelige P. E. Jr., Johnson J. E., and Cingolani G. (2011) Three-dimensional structure of a viral genome-delivery portal vertex. Nat. Struct. Mol. Biol. 18, 597–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olia A. S., Bhardwaj A., Joss L., Casjens S., and Cingolani G. (2007) Role of gene 10 protein in the hierarchical assembly of the bacteriophage P22 portal vertex structure. Biochemistry 46, 8776–8784 [DOI] [PubMed] [Google Scholar]
- 27.Olia A. S., Al-Bassam J., Winn-Stapley D. A., Joss L., Casjens S. R., and Cingolani G. (2006) Binding-induced stabilization and assembly of the phage P22 tail accessory factor gp4. J. Mol. Biol. 363, 558–576 [DOI] [PubMed] [Google Scholar]
- 28.Israel V. (1977) E proteins of bacteriophage P22: I. identification and ejection from wild-type and defective particles. J. Virol. 23, 91–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin Y., Sdao S. M., Dover J. A., Porcek N. B., Knobler C. M., Gelbart W. M., and Parent K. N. (2015) Bacteriophage P22 ejects all of its internal proteins before its genome. Virology 485, 128–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andres D., Hanke C., Baxa U., Seul A., Barbirz S., and Seckler R. (2010) Tailspike interactions with lipopolysaccharide effect DNA ejection from phage P22 particles in vitro. J. Biol. Chem. 285, 36768–36775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leavitt J. C., Gogokhia L., Gilcrease E. B., Bhardwaj A., Cingolani G., and Casjens S. R. (2013) The tip of the tail needle affects the rate of DNA delivery by bacteriophage p22. PLoS One 8, e70936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tao Y., Strelkov S. V., Mesyanzhinov V. V., and Rossmann M. G. (1997) Structure of bacteriophage T4 fibritin: a segmented coiled coil and the role of the C-terminal domain. Structure 5, 789–798 [DOI] [PubMed] [Google Scholar]
- 33.Cingolani G., Andrews D., and Casjens S. (2006) Crystallogenesis of bacteriophage P22 tail accessory factor gp26 at acidic and neutral pH. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 62, 477–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cingolani G., Lashuel H. A., Gerace L., and Müller C. W. (2000) Nuclear import factors importin alpha and importin beta undergo mutually induced conformational changes upon association. FEBS Lett. 484, 291–298 [DOI] [PubMed] [Google Scholar]
- 35.Lokareddy R. K., Bhardwaj A., and Cingolani G. (2013) Atomic structure of dual-specificity phosphatase 26, a novel p53 phosphatase. Biochemistry 52, 938–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Otwinowski Z., and Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 37.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., and Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCoy A. J. (2007) Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 63, 32–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., and Terwilliger T. C. (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 40.Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 41.Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 42.DeLano W. L. ( 2012) The PyMOL Molecular Graphics System, version 1.5.0.1, Schroedinger, LLC, New York [Google Scholar]
- 43.Vagin A., and Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 [DOI] [PubMed] [Google Scholar]
- 44.Afonine P. V., Headd J. J., Terwilliger T. C., and Adams P. D. (2013) New Tool: phenix.real_space_refine. Comput. Crystallogr. Newsletter 4, 43–44 [Google Scholar]
- 45.Roy A., Kucukural A., and Zhang Y. (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parent K. N., Gilcrease E. B., Casjens S. R., and Baker T. S. (2012) Structural evolution of the P22-like phages: comparison of Sf6 and P22 procapsid and virion architectures. Virology 427, 177–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tang G., Peng L., Baldwin P. R., Mann D. S., Jiang W., Rees I., and Ludtke S. J. (2007) EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 157, 38–46 [DOI] [PubMed] [Google Scholar]
- 48.Frank J., Radermacher M., Penczek P., Zhu J., Li Y., Ladjadj M., and Leith A. (1996) SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J. Struct. Biol. 116, 190–199 [DOI] [PubMed] [Google Scholar]
- 49.Clark A. J., Inwood W., Cloutier T., and Dhillon T. S. (2001) Nucleotide sequence of coliphage HK620 and the evolution of lambdoid phages. J. Mol. Biol. 311, 657–679 [DOI] [PubMed] [Google Scholar]
- 50.Poteete A. R., and King J. (1977) Functions of two new genes in Salmonella phage P22 assembly. Virology 76, 725–739 [DOI] [PubMed] [Google Scholar]
- 51.Casjens S. R., and Thuman-Commike P. A. (2011) Evolution of mosaically related tailed bacteriophage genomes seen through the lens of phage P22 virion assembly. Virology 411, 393–415 [DOI] [PubMed] [Google Scholar]
- 52.Kashket E. R. (1982) Stoichiometry of the H+-ATPase of growing and resting, aerobic Escherichia coli. Biochemistry 21, 5534–5538 [DOI] [PubMed] [Google Scholar]
- 53.Cingolani G., and Duncan T. M. (2011) Structure of the ATP synthase catalytic complex (F(1)) from Escherichia coli in an autoinhibited conformation. Nat. Struct. Mol. Biol. 18, 701–707 [DOI] [PMC free article] [PubMed] [Google Scholar]