

Abstract
The maintenance of genomic stability relies on the concerted action of DNA repair and DNA damage signaling pathways. The PIAS (protein inhibitor of activated STAT) family of SUMO (small ubiquitin-like modifier) ligases has been implicated in DNA repair, but whether it plays a role in DNA damage signaling is still unclear. Here, we show that the PIAS3 SUMO ligase is important for activation of the ATR (ataxia telangiectasia and Rad3 related)-regulated DNA damage signaling pathway. PIAS3 is the only member of the PIAS family that is indispensable for ATR activation. In response to different types of DNA damage and replication stress, PIAS3 plays multiple roles in ATR activation. In cells treated with camptothecin (CPT), PIAS3 contributes to formation of DNA double-stranded breaks. In UV (ultraviolet light)- or HU (hydroxyurea)-treated cells, PIAS3 is required for efficient ATR autophosphorylation, one of the earliest events during ATR activation. Although PIAS3 is dispensable for ATRIP (ATR-interacting protein) SUMOylation and the ATR-ATRIP interaction, it is required for maintaining the basal kinase activity of ATR prior to DNA damage. In the absence of PIAS3, ATR fails to display normal kinase activity after DNA damage, which accompanies with reduced phosphorylation of ATR substrates. Together, these results suggest that PIAS3 primes ATR for checkpoint activation by sustaining its basal kinase activity, revealing a new function of the PIAS family in DNA damage signaling.
Keywords: checkpoint control, DNA damage response, DNA repair, DNA replication, small ubiquitin-like modifier (SUMO)
Introduction
Multiple types of post-translational modifications (PTMs),2 including protein SUMOylation, are important for regulation of the DNA damage response (DDR) from yeast to human (1, 2). Several SUMO E3 ligases, particularly the members of the PIAS family, are shown to modify DDR proteins. In the budding yeast, the PIAS family member Siz1 SUMOylates PCNA during S phase, promoting the recruitment of Srs2 to replication forks to suppress unwanted recombination (3–6). Siz2, the second PIAS ligase in yeast, SUMOylates a large number of DNA replication and repair proteins in response to DNA damage, which may enhance the efficiency of DNA repair by stabilizing multiple protein interactions in repair complexes (7, 8). A recent study showed that Siz2 is recruited to sites of DNA damage through its interaction with RPA, a sensor of single-stranded DNA (ssDNA), explaining the regulation of protein SUMOylation by DNA damage (9). Together these findings in yeast clearly show that the PIAS family SUMO ligases are critical regulators of the DDR.
In human cells, four members of the PIAS family SUMO ligases have been identified: PIAS1, PIAS2 (PIASxα/β), PIAS3, and PIAS4 (PIASy) (10). Two of these SUMO ligases, PIAS1 and PIAS4, are recruited to sites of DNA damage (11). Both PIAS1 and PIAS4 contribute to the efficient recruitment of 53BP1, BRCA1, and RNF168 (11). A number of DDR proteins, such as 53BP1, BRCA1, MDC1, HERC2, RNF168, FNACI, and FANCD2, are SUMOylated by PIAS1 and/or PIAS4. The SUMOylation of these DDR proteins regulate their functions in several different ways. For example, BRCA1 SUMOylation increases its ubiquitin ligase activity (12). The SUMOylation of HERC2 enhances its interaction with RNF8 and stabilizes the RNF8-UBC13 complex (13). The SUMOylation of MDC1, FANCI, and FANCD2 triggers RNF4-mediated ubiquitylation of these proteins and their removal from DNA damage-induced foci (14–17), ensuring that these proteins accumulate at sites of DNA damage at appropriate levels and times. In addition to PIAS1 and PIAS4, PIAS3 has also been implicated in the DDR. In etoposide treated cells, PIAS3 binds and SUMOylates DBC1, which sequesters SIRT1 from p53 and promotes p53 activation (18). Furthermore, overexpression of PIAS3 was shown to enhance both homologous recombination (HR) and distal non-homologous end joining (NHEJ) (19), but the mechanisms underlying these observations remain unclear.
In addition to its role in DNA repair, protein SUMOylation is also important for DNA damage signaling. We recently showed that depletion of UBC9, the sole SUMO E2 in human cells, compromised the activation of the ATR-Chk1 DNA damage-signaling pathway (20). Furthermore, we showed that ATRIP, the regulatory partner of ATR, is SUMOylated at two specific lysine residues, which are important for ATRIP to interact with multiple proteins in the ATR-Chk1 pathway efficiently (20). Although our previous work has linked protein SUMOylation to DNA damage signaling, which SUMO ligases are involved in this process and how they function are yet to be determined.
Here, we investigated the role of PIAS family SUMO ligases in DNA damage signaling. We found that PIAS3, but not PIAS1 or PIAS4, is important for activation of the ATR checkpoint pathway. In the context of CPT-induced ATR activation, PIAS3 contributes to formation of double-stranded breaks (DSBs). In response to UV or HU, PIAS3 is required for the early step of ATR activation marked by ATR autophosphorylation. Importantly, we found that loss of PIAS3 reduces the basal kinase activity of ATR prior to DNA damage, suggesting that PIAS3 primes ATR for the DDR. These results establish a new role for the PIAS family in DNA damage signaling, and reveal priming as a new mode of action for SUMO in the DDR.
Experimental Procedures
Cell Culture and Small Interfering RNAs (siRNAs)
HeLa, U2OS, and HEK293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), l-glutamine, penicillin, and streptomycin. Plasmids expressing Flag-ATR or Flag-ATRΔC (21) were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. Silencer-select siRNAs targeting human PIAS family proteins were transfected using Lipofectamine RNAiMAX (Invitrogen). The sequences of the siRNAs are: PIAS1 (5′-GGAUCAUUCUAGAGCUUUA-3′), PIAS2 (5′-CUUGAAUAUUACAUCUUUA-3′), PIAS3 (5′-GGUCGAAGUUAUUGACUUG-3′), PIAS3-2 (5′-GGUGCAGCUAAGGUUCUGU-3′), PIAS3-3 (5′-GCCUAGGACCUAUGUUGUA-3′), PIAS4 (5′-GGAGUAAGAGUGGACUGAA-3′), and control siRNA (5′-GGGUAUCGACGAUUACAAA-3′).
Antibodies
The antibodies used in this work include: Chk1 pSer317, PIAS1, PIAS3, PIAS4, and γH2AX antibodies from Cell Signaling; ATR, ATM, RPA70, RPA32, TopBP1, MRE11, RPA32 pSer33, and RPA32 pSer4/8 antibodies from Bethyl; Chk1 antibody from Santa Cruz; SUMO-2/3 and Rad9 antibodies from Abcam; RPA32 antibody from Thermo; GAPDH antibody from Millipore; HA antibodies from Convance and Santa Cruz; and Flag antibody from Sigma. ATR pT1989 antibody from GeneTex; ATRIP antibody was previously described (22).
Immunoprecipitation
Immunoprecipitation of endogenous ATRIP and analysis of ATRIP SUMOylation were previously described (20). For immunoprecipitation of endogenous ATR, cell extracts were prepared in the NETN buffer (20 mm Tris-HCl pH 8.0, 0.5 mm EDTA, 150 mm NaCl, 0.5% Igepal CA-630, and 10% glycerol) supplemented with protease inhibitor mixture. Endogenous ATR was immunoprecipitated with ATR polyclonal antibodies followed by incubation with Dynabeads Protein G (Novex).
Cell Viability Assay
U2OS cells transfected with control and PIAS3 siRNA for 48 h were seeded in 96-well plates (1,600 cells/well). On the next day, cells were treated with vehicle, HU, PARP inhibitor, or ATR inhibitor, and the treatments were continued for 4 days. Cell survival was analyzed using CellTiter-Glo (Promega) according to the manufacturer's protocol. Cell survival rates were normalized to vehicle-treated samples and presented as mean ± S.D. (n = 3).
Immunofluorescence and Laser Micro-irradiation
HeLa and U2OS cells transfected with control or PIAS3 siRNA were seeded on coverslips in 6-well plates. To detect RPA, γH2AX, and ATRIP at DNA damage sites, cells were treated with CPT or irradiate with UV through 5-μm filter (Millipore). Subsequently cells were pre-extracted with 0.5% Triton X-100 in PBS buffer for 3 min on ice, fixed with 3% paraformaldehyde/2% sucrose for 15 min, and then extracted again with 0.5% Triton X-100 in PBS buffer for 3 min on ice. Cells were incubated with primary antibodies to RPA, γH2AX, ATRIP diluted in 1× PBS containing 3% BSA/0.05% Tween 20 for >2 h at room temperature. After 3 washes with 1× PBS containing 0.05% Tween 20, cells were incubated with Cy3-conjugated anti-rabbit antibody and Alexa Fluor 488-conjugated anti-mouse antibody at room temperature for 1 h. Cells were then washed three times with 1× PBS containing 0.05% Tween 20, and DNA was stained by DAPI (4′,6-diamidino-2-phenylindole).
To test if PIAS3 localizes to laser-induced DNA damage stripes, U2OS cells were micro-irradiated with UV laser as previously described (20). The pre-extraction step of immunostaining was skipped to avoid potential loss of PIAS3 from DNA damage stripes.
In Vitro Kinase Assay
HEK293T cells were treated with control, PIAS3, or PIAS1 siRNA for 24 h followed by transfection of Flag-ATR plasmids. Two days after plasmid transfection, Flag-ATR and Flag-ATRΔC were immunoprecipitated and tested with in vitro kinase assay as previously described (21).
Analysis of the UV-induced Replication Inhibition
HeLa and U2OS cells transfected with control or PIAS3 siRNA were either irradiated with UV (10 J/m2) or left untreated. At 1 h after UV or mock treatment, cells were labeled with 10 μm EdU (5-ethynyl-2′-deoxyuridine) for 30 min, trypsinized, washed with 1× PBS, and fixed in 75% ethanol at −20 °C. EdU-labeled cells were processed using a Click-iT EdU Alexa Fluor 647 Flow Cytometry Assay kit according to the manufacturer's instructions (Invitrogen). Data acquisition was performed on a FACS LSRII apparatus and analyzed with Kaluza software (Beckman Coulter).
RT-Quantitative PCR (RT-qPCR) of PIAS2 mRNA
Total RNA of HeLa cells transfected with control siRNA or siRNA targeting each of the PIAS family member was isolated using PureLink RNA mini kit (Invitrogen). cDNA was synthesized using the (dT)16 primer and TaqMan Reverse Transcriptase Reagents (Life Technologies). Two primer pairs that specifically amplify PIAS2 (#1 forward primer 5′-GCTATTTCCTTTGCCTGGCTAT-3′; #1 reverse primer 5′-TTCTTCCCAATTTCTGATGCC-3′; #2 forward primer 5′-CCAAGTTCAGTTGAGACTTTGC-3′; #2 reverse primer 5′-GTGGTGCATAGCCAGGCAA-3′) were used in qPCR, which was performed using PowerUp SYBR Green Master Mix (Applied Biosystems) according to the manufacturer's protocol. Reactions were analyzed by LightCycler 480 (Roche) and the relative PIAS2 mRNA levels were normalized to GAPDH (forward primer 5′-CGGATTTGGTCGTATTGGGC-3′ and reverse primer 5′-TGGAAGATGGTGATGGGATTTC-3′).
Results
PIAS3 Is the Only PIAS SUMO Ligase Indispensable for ATR Activation
While members of the PIAS family of SUMO ligases have been implicated in the DDR, whether and how they contribute to DNA damage signaling is still unclear. To address this question, we used siRNAs to knock down all 4 members of the PIAS family in HeLa cells and analyzed the effects on the CPT-induced, ATR-mediated Chk1 phosphorylation at Ser317. The knockdown of PIAS1, PIAS3, and PIAS4 was confirmed by Western blot (Fig. 1A). Because of the lack of PIAS2-specific antibodies, we confirmed the knockdown of PIAS2 using RT-qPCR (Fig. 1B). Knockdown of PIAS1 and PIAS4, which are involved in DNA repair, did not affect Chk1 phosphorylation significantly (Fig. 1A). Depletion of PIAS2 did not affect Chk1 phosphorylation either. In contrast, the CPT-induced Chk1 phosphorylation was clearly reduced in PIAS3 knockdown cells (Fig. 1A). The reduction of Chk1 phosphorylation by PIAS3 knockdown was not attributed to alterations of the cell cycle (Fig. 1C). Similar to the CPT-induced Chk1 phosphorylation, the UV and HU-induced Chk1 phosphorylation was also reduced by PIAS3 knockdown (Fig. 1D, also see 2C). The effects of PIAS3 knockdown on Chk1 activation were seen not only in HeLa cells, but also in U2OS cells (Fig. 2A). These results suggest that PIAS3 is the only PIAS family member that plays a non-redundant role in ATR activation, although they do not exclude the possibility that other PIAS ligases function redundantly in this pathway.
FIGURE 1.

PIAS3 is the only PIAS ligase indispensable for ATR activation. A, HeLa cells were treated with control siRNA or siRNAs targeting each of the PIAS family member for 3 days. Levels of the indicated proteins were analyzed by Western blot 1 h after CPT (1 μm) treatment. B, HeLa cells were treated with siRNAs and CPT as in A. Relative PIAS2 mRNA levels were determined by RT-qPCR. PIAS2#1 and #2, two primer pairs used in RT-qPCR. C, HeLa cells were transfected with control siRNA or siRNAs targeting the PIAS family members for 3 days. DNA synthesis and DNA contents were analyzed by EdU incorporation and PI (propidium iodide) staining, respectively. D, HeLa cells transfected with control siRNA or siRNAs targeting the PIAS family members. Levels of the indicated proteins were analyzed by Western blot 30 min after UV (50 J/m2) treatment.
FIGURE 2.

Knockdown of PIAS3 by multiple independent siRNAs leads to reduced ATR activation. A, U2OS cells were transfected with control siRNA or three independent siRNAs targeting PIAS3 for 3 days. Levels of the indicated proteins were analyzed by Western blot. B, U2OS cells were transfected with control or three independent siRNAs targeting PIAS3. DNA synthesis and DNA content were analyzed by EdU incorporation and PI staining, respectively. C, HeLa cells were treated with control or PIAS3–3 siRNA, and subsequently transfected with HA-PIAS3-expressing plasmids as indicated. HU-induced Chk1 phosphorylation was analyzed.
To ascertain that the reduction of Chk1 phosphorylation by PIAS3 knockdown was not due to off target effects of the siRNA, we used 3 independent siRNAs to knock down PIAS3 in U2OS cells (Fig. 2A). All 3 siRNAs efficiently depleted PIAS3 and reduced Chk1 phosphorylation. None of the PIAS3 siRNAs significantly altered the cell-cycle distribution of cells (Fig. 2B). The levels of a number of DDR proteins involved in ATR activation, including ATR, ATRIP, RPA70, RPA32, Rad9, TopBP1, MRE11, and BRCA1, were not affected by PIAS3 knockdown (Fig. 3, A–C). Importantly, in cells treated with a PIAS3 siRNA targeting the 3′-UTR (siPIAS3-3), expression of HA-tagged wild-type PIAS3 suppressed the defect in Chk1 activation (Fig. 2C). Together, these results suggest that PIAS3 has a specific and possibly direct role in ATR activation.
FIGURE 3.

PIAS3 knockdown does not affect the levels of known ATR regulators. A–C, HeLa or U2OS cells were transfected with PIAS3 and other siRNAs as indicated. Levels of a number of proteins involved in ATR activation, including ATR, ATRIP, RAP70, RPA32, TopBP1, RAD9, MRE11, and BRCA1, were analyzed by Western blot.
PIAS3 Is Required for DNA Damage-induced Replication Inhibition
To investigate if PIAS3 is functionally required for the ATR-mediated DNA damage response, we tested its involvement in the inhibition of DNA synthesis after DNA damage. HeLa and U2OS cells were treated with control or two independent PIAS3 siRNAs (Fig. 4, A–C). Newly synthesized DNA was labeled with EdU after UV irradiation or mock treatment. In control cells, DNA synthesis was significantly reduced by UV treatment. The UV-induced reduction in DNA synthesis was less pronounced in PIAS3 knockdown cells, suggesting that this checkpoint-mediated response is compromised by the loss of PIAS3. These results are consistent with the role of PIAS3 as a regulator of the ATR checkpoint pathway (23).
FIGURE 4.

PIAS3 is required for UV-induced replication inhibition. A and B, HeLa cells were transfected with control or 2 independent PIAS3 siRNAs. Levels of the indicated proteins were analyzed by Western blot (A). Relative levels of DNA synthesis (EdU Incorporation) were quantified (B). C, U2OS cells were transfected with control or 2 independent PIAS3 siRNAs. Relative levels of DNA synthesis (EdU Incorporation) were quantified. Similar results were obtained at 1 h (this panel) or 1.5 h (not shown) after UV treatment.
PIAS3 Knockdown Is Synthetic Lethal with PARP and ATR Inhibition
ATR plays critical roles in the replication stress response and HR. PIAS3 knockdown cells were more sensitive to HU and Olaparib, a PARP inhibitor, than control cells (Fig. 5, A and B), suggesting that the contribution of PIAS3 to ATR activation is indispensable for normal replication stress response and HR. Furthermore, PIAS3 knockdown cells were more sensitive to VE-821 and AZ20, two ATR inhibitors, than control cells (Fig. 5, C and D). A previous study suggested that cells with partial defects in the ATR pathway are particularly sensitive to ATR inhibition (24). Our results are consistent with this finding, showing that the defect of ATR activation in PIAS3 knockdown cells is functionally significant.
FIGURE 5.

PIAS3 knockdown cells are sensitive to HU, PARP inhibitor, and ATR inhibitor. A, U2OS cells were transfected with control or 2 independent PIAS3 siRNAs, and then treated with HU for 4 days. *, p < 0.05. B, U2OS cells were transfected with control, PIAS3, or BRCA2 siRNA, and then treated with Olaparib for 4 days. *, p < 0.05. C and D, U2OS cells were transfected with control or PIAS3 siRNA, and then treated with VE-21 or AZ20 for 4 days. The cell survival at 1 μm AZ20 and 5 μm VE-821 is shown in D. *, p < 0.05.
PIAS3 Is Dispensable for ATRIP SUMOylation
Our previous studies showed that ATRIP is increasingly SUMOylated after UV treatment, and that ATRIP SUMOylation is required for efficient activation of the ATR pathway (20). To test if PIAS3 or another PIAS ligase is responsible for ATRIP SUMOylation, we knocked down all 4 PIAS ligases individually and immunoprecipitated endogenous ATRIP under a denaturing condition (Fig. 6). The UV-induced increase of SUMOylated ATRIP was readily detected by SUMO-2/3 antibodies in cells treated with control siRNA. Surprisingly, none of the siRNAs targeting the PIAS family SUMO ligases, including PIAS3, reduced the UV-induced ATRIP SUMOylation. Thus, although PIAS3 is a regulator of the ATR pathway, it is not the key SUMO ligase for ATRIP. ATRIP may be SUMOylated by multiple PIAS ligases redundantly, or by one or more SUMO ligases outside of the PIAS family.
FIGURE 6.
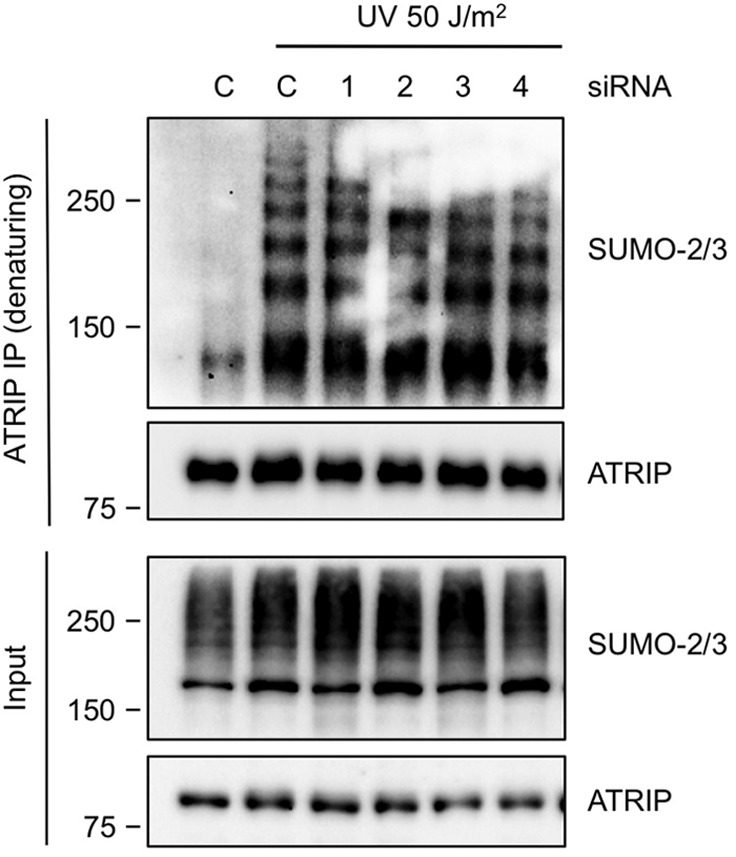
PIAS3 is dispensable for ATRIP SUMOylation. HeLa cells were transfected with control siRNA or siRNAs targeting the PIAS family ligases. Endogenous ATRIP was immunoprecipitated under a denaturing condition. The unmodified and SUMOylated forms of ATRIP were detected with ATRIP and SUMO-2/3 antibodies, respectively.
PIAS3 Differentially Regulates the Activation of ATR and ATM by CPT
To understand how PIAS3 regulates ATR activation, we next sought to identify the PIAS3-dependent event(s) in the ATR pathway. The topoisomerase I inhibitor CPT is known to induce replication-associated DNA DSBs, which activate both ATM and ATR (25–27). Notably, ATM and ATR are activated by CPT in a sequential manner: ATM is activated within a few minutes, whereas ATR is gradually activated through a resection-dependent process over a period of 30–60 min (26). To test if PIAS3 regulates both ATM and ATR during the CPT response, we analyzed the effects of PIAS3 knockdown on the ATR-regulated phosphorylation of Chk1 and RPA32, as well as the ATM-mediated phosphorylation of Chk2 and ATM itself (Fig. 7A). At 1 h after CPT treatment, the phosphorylation of Chk1 and RPA32 was clearly reduced by PIAS3 knockdown, whereas the phosphorylation of ATM and Chk2 was not. These results suggest that PIAS3 is important for ATR activation but not ATM activation. However, a more detailed analysis of the kinetics of Chk2 phosphorylation revealed that this ATM-mediated event was slightly delayed in PIAS3 knockdown cells (see Fig. 7C). Thus, PIAS3 appears to play a major role in ATR activation and a minor role in ATM activation during the CPT response, raising the possibility that PIAS3 may have more than one function in this process.
FIGURE 7.

PIAS3 promotes formation of CPT-induced DSBs. A, HeLa and U2OS cells were transfected with control or PIAS3 siRNA, and treated with 1 μm CPT for 1 h or left untreated. Levels of the indicated proteins were analyzed by Western blot. B and C, HeLa cells were transfected with control or PIAS3 siRNA, and treated with 1 μm CPT for the indicated lengths of time. Levels of the indicated proteins were analyzed by Western blot.
PIAS3 Regulates CPT-induced DSB Formation but Not Resection
The effects of PIAS3 knockdown on the activation of ATR and ATM during the CPT response prompted us to test if it participates in the formation and/or resection of DSBs in this context. Consistent with a defect in DSB formation or resection, the CPT-induced phosphorylation of Chk1 and RPA32 was significantly reduced during the 60 min after CPT treatment (Fig. 7B). Interestingly, the phosphorylation of H2AX, which is triggered by DSB formation directly, was also diminished (Fig. 7C). Immunofluorescence analysis of γH2AX confirmed that this phosphorylation event was compromised in PIAS3 knockdown cells (Fig. 8A). The reduction of CPT-induced γH2AX by PIAS3 is reminiscent to the observations in cells depleted of MUS81 or SLX4, which are implicated in the cleavage of CPT-induced aberrant replication structures (28, 29). Interestingly, we recently found that the function of SLX4 in processing CPT-induced DNA structures is regulated by its binding to SUMO (29). Together, these findings suggest that PIAS3 contributes to the generation of DSBs in CPT-treated cells, possibly working in concert with the SLX4-MUS81 endonuclease complex.
FIGURE 8.

PIAS3 is not critical for resection of DSBs. A and B, HeLa cells were transfected with control or PIAS3 siRNA, and treated with 1 μm CPT for 1 h or left untreated. The fractions of cells with γH2AX foci were quantified in A, and the fractions of γH2AX foci-positive cells with RPA foci were quantified in B. C, intensity of RPA32 immunofluorescence was quantified in control and PIAS3 knockdown cells. Cells with background RPA32 staining were colored green. Cells with CPT-induced RPA32 staining were colored black. Mean RPA32 intensities of the latter cell populations were determined. Error bars: interquartile range (I.Q.R).
While the CPT-induced H2AX phosphorylation was significantly reduced by PIAS3 knockdown, the phosphorylation of Chk2 was only slightly delayed (Fig. 7C). This observation suggests that the activation of ATM is only modestly affected by PIAS3 loss, raising the possibility that the efficient phosphorylation of Chk2 by ATM requires fewer DSBs than the extensive phosphorylation of H2AX by ATM and DNA-PK. Alternatively, PIAS3 may be involved in the spreading of ATM on the chromatin flanking DSBs, which may be more important for H2AX phosphorylation than Chk2 phosphorylation. To assess if PIAS3 is important for the resection of DNA ends following DSB generation, we analyzed the formation of RPA foci in γH2AX-positive cells 1 h after CPT treatment (Fig. 8, B and C). RPA foci formed similarly in the presence or absence of PIAS3, showing that PIAS3 is not critical for resection once DSBs are formed. Hence, although DSB formation is reduced in PIAS3 knockdown cells, ATM activation is only modestly affected and resection still occurs efficiently.
PIAS3 Regulates ATR Autophosphorylation
Although the role of PIAS3 in the formation of CPT-induced DSBs contributes to ATR activation in this specific context, it is unlikely the only function of PIAS3 in ATR regulation. The UV and HU-induced phosphorylation of Chk1 and RPA32 was also reduced in PIAS3 knockdown cells (Figs. 1C and 9A), suggesting that PIAS3 promotes ATR activation even in the absence of DSBs. Our previous studies suggested that ATR autophosphorylation at Thr-1989 is one of the earliest events during ATR activation (30). The autophosphorylation of ATR is dependent on the accumulation of ATR-ATRIP on RPA-coated ssDNA at sites of DNA damage and ATR basal kinase activity. To investigate if PIAS3 is involved in the initial phase of ATR, we tested the effects of PIAS3 knockdown on ATR autophosphorylation. Both HU and UV-induced ATR autophosphorylation was reduced by PIAS3 knockdown (Fig. 9, B and C), suggesting that PIAS3 is required for this early event.
FIGURE 9.

PIAS3 promotes ATR autophosphorylation. A and B, HeLa cells were transfected with control or PIAS3 siRNA, and treated with 1 mm HU for the indicated lengths of time. Levels of the indicated proteins were analyzed by Western blot. C, HeLa cells were transfected with control siRNA or two independent PIAS3 siRNAs. Cells were irradiated with UV or mock treated. Levels of the indicated proteins were analyzed by Western blot.
To test if PIAS3 regulates the localization of ATR-ATRIP to sites of DNA damage, we stained endogenous ATRIP with specific antibodies in cells irradiated with UV through filters (Fig. 10A). ATRIP colocalized with γH2AX in both control cells and cells treated with PIAS3 siRNA, but the efficiency of ATRIP localization to DNA damage sites was slightly reduced by PIAS3 knockdown (Fig. 10, A and B). Although PIAS3 may have a minor role in ATRIP localization, this role seems unlikely to be solely responsible for the pronounced effects of PIAS3 on ATR auto-phosphorylation. Furthermore, PIAS3 did not localize to sites of DNA damage in cells irradiated with UV through filters and cells microirradiated with UV laser (Fig. 10, C and D). Together, these findings hinted that PIAS3 might prime ATR for activation even before DNA damage.
FIGURE 10.

The localization of PIAS3 and its effects on ATRIP recruitment. A and B, HeLa cells transfected with control or PIAS3 siRNA were irradiated with UV (50 J/m2) through filters. The nuclear areas exposed to UV were visualized by γH2AX immunofluorescence. The localization of endogenous ATRIP was analyzed by ATRIP immunofluorescence. Representative images of cells are shown in A. The fractions of irradiated cells (γH2AX-positive cells) with ATRIP spots were quantified (B). *, p < 0.05. C and D, HeLa cells were transfected with control or PIAS3 siRNA. Cells were either irradiated with UV through filters (C) or with UV laser (D). Sites of DNA damage were visualized by γH2AX immunofluorescence. The localization of endogenous PIAS3 was analyzed by PIAS3 immunofluorescence. The staining specificity of the PIAS3 antibody was confirmed by PIAS3 knockdown in C.
PIAS3 Is Required for the Basal Kinase Activity of ATR
PIAS3 may prime ATR for activation through several possible mechanisms. For example, PIAS3 may be required for the proper interaction between ATR and ATRIP, which may be necessary for the trans interaction among ATR-ATRIP complexes on RPA-ssDNA. Alternatively, PIAS3 may regulate the basal kinase activity of ATR, which is needed for ATR autophosphorylation. To test if PIAS3 regulates the interaction between ATR and ATRIP, we immunoprecipitated endogenous ATR from cells treated with control or PIAS3 siRNA before and after UV damage (Fig. 11A). As expected, the interaction between ATR and ATRIP was not affected by DNA damage. Moreover, the ATR-ATRIP interaction was not affected by PIAS3 knockdown, suggesting that PIAS3 does not regulate ATR by stabilizing the ATR-ATRIP complex. Interestingly, low levels of PIAS3 were coprecipitated by ATR before and after UV treatment in control cells but not PIAS3 knockdown cells (Fig. 11A), suggesting that PIAS3 interacts with ATR independently of DNA damage. Consistent with this result, endogenous ATR was coprecipitated by HA-PIAS3 in undamaged cells (Fig. 11B).
FIGURE 11.

PIAS3 is not required for the ATR-ATRIP interaction. A, HEK293T cells were transfected with control or PIAS3 siRNA. Endogenous ATR was immunoprecipitated. Levels of the indicated proteins in input extracts and ATR immunoprecipitates were analyzed by Western blot. Arrow: the PIAS3 coprecipitated by ATR. B, HEK293T cells were transfected with a HA-PIAS3-expressing plasmid or a control plasmid. Levels of the indicated proteins in input extracts and HA immunoprecipitates were analyzed by Western blot.
To address if the kinase activity of ATR is regulated by PIAS3, we immunoprecipitated Flag-ATR from control and PIAS3 knockdown cells and tested it using in vitro kinase assay (Fig. 12, A and B). The Flag-ATR isolated from control cells underwent autophosphorylation in vitro in the presence of 32P-labeled ATP. In contrast, an ATR mutant lacking part of the C-terminal kinase domain (Flag-ATRΔC) failed to autophosphorylate. The Flag-ATR isolated from PIAS3 knockdown cells autophosphorylated less efficiently than the Flag-ATR from control cells both before and after UV damage, suggesting that the basal kinase activity of ATR was compromised by PIAS3 loss (Fig. 12A). In vitro, a GST fusion protein containing an ATR substrate site derived from Rad17 (GST-Rad17 SQ) was phosphorylated by ATR efficiently (Fig. 12B). Consistent with the in vitro autophosphorylation of ATR, the Flag-ATR from PIAS3 knockdown cells phosphorylated GST-Rad17 SQ less efficiently than the Flag-ATR from control cells (Fig. 12B). In contrast, the Flag-ATR from PIAS1 knockdown cells phosphorylated GST-Rad17 SQ as efficiently as the Flag-ATR from control cells. A weak kinase activity toward GST-Rad17 SQ was detected in the immunoprecipiate of Flag-ATRΔC, possibly because of the low level of endogenous ATR coprecipitated with the ATR mutant. Together, these results demonstrate that PIAS3, but not PIAS1, is important for the basal kinase activity of ATR prior to DNA damage.
FIGURE 12.

PIAS3 is required for sustaining the kinase activity of ATR before and after DNA damage. A, Flag-ATR or Flag-ATRΔC was transiently expressed in HEK293T cells transfected with control or PIAS3 siRNA. Cells were treated with UV or left untreated. Flag-ATR and Flag-ATRΔC were immunoprecipitated and analyzed for autophosphorylation in vitro. Levels of Flag-ATR and Flag-ATRΔC used in the kinase assay were analyzed by Western blot. The ratios of phosphorylated ATR and ATR bands were quantified and shown in the lower half. B, Flag-ATR or Flag-ATRΔC was transiently expressed in HEK293T cells transfected with control, PIAS1, or PIAS3 siRNA. Flag-ATR and Flag-ATRΔC were immunoprecipitated and analyzed for kinase activity using GST-Rad17 SQ as a substrate. Levels of the indicated proteins in the input extracts and immunoprecipitates were analyzed by Western blot.
Discussion
Members of the PIAS family of SUMO ligases have been implicated in DNA repair (1, 2). In this study, we have extended the roles of PIAS family to DNA damage signaling. Surprisingly, the PIAS ligase important for ATR activation is not PIAS1 or PIAS4, whose functions in DNA repair have been extensively studied. Instead, PIAS3, which is not well characterized in the context of DDR, plays an important role in ATR activation. PIAS3 is required for efficient Chk1 phosphorylation in multiple cancer cell lines in response to different types of DNA damage and replication stress, suggesting that it is a general regulator of the ATR pathway. Our results also reveal that PIAS3 has both context-specific and general roles in ATR regulation. CPT induces reversal of replication forks even at low concentrations (31). During the CPT response, PIAS3 promotes formation of DSBs, suggesting that one or more of its substrates may be involved in generating or processing the aberrant replication structures induced by CPT. Top1 is SUMOylated in human cells in a CPT stimulated manner. Two SUMO ligases, TOPORS and PIAS1, have been implicated in Top1 SUMOylation (32, 33). Furthermore, the efficient formation of CPT-induced DSBs requires the SLX4-MUS81 endonuclease complex, which recognizes SUMO through SLX4 (28, 29). It is tempting to speculate that PIAS3 contributes to the SUMOylation of Top1 and/or other replication or repair factors that recruit SLX4, promoting the generation or cleavage of reversed forks.
Apart from its role in generating CPT-induced DSBs, PIAS3 is required for efficient Chk1 phosphorylation after UV or HU treatment, suggesting that it has a general role in ATR activation even in the absence of DSBs. Importantly, PIAS3 is required for ATR autophosphorylation, pinpointing its role to the initial event of ATR activation. Our results reveal that PIAS3 interacts with ATR and elevates its basal kinase activity even before DNA damage, showing that PIAS3 primes ATR for efficient checkpoint activation. In the budding yeast, Siz1 and Siz2, two members of the PIAS family, function in different ways to protect genomic stability. Whereas Siz2 is recruited to sites of DNA damage and SUMOylates DNA repair proteins in a DNA damage-induced manner (9), Siz1 SUMOylates PCNA during normal S phase and prevents unwanted recombination at replication forks (4, 6). Our findings suggest that the PIAS ligases in human cells may also function in different ways in the DDR. Whereas PIAS1 and PIAS4 are recruited to sites of DNA damage to promote SUMOylation of repair proteins (11), PIAS3 may SUMOylate specific proteins in the absence of DNA damage, priming them for the DDR. Multiple priming events of the ATR-ATRIP kinase complex are known to promote checkpoint activation. For example, the Tel2 co-chaperone complex is important for the proper interaction between ATR and ATRIP (34). Nek1, a kinase that interacts with ATR and ATRIP, is also required for the proper interaction between ATR and ATRIP and the basal kinase activity of the ATR-ATRIP complex (21). Furthermore, loss of ATRIP SUMOylation sites reduced the interaction between ATR and ATRIP (20). The results of this study suggest that PIAS3 is not critical for the ATR-ATRIP interaction, but is required for the basal kinase activity of ATR, suggesting a new priming mechanism of ATR. Although PIAS3 is dispensable for ATRIP SUMOylation, it may SUMOylate ATR or its associated proteins, thereby enhancing the kinase activity of ATR. Consistent with this possibility, both ATR and its yeast homologue Mec1 may be SUMOylated in the absence of DNA damage (7, 35). Whether ATR is SUMOylated by PIAS3 and whether the E3 ligase activity of PIAS3 is required for ATR activation remains to be tested.
ATR is a key regulator of HR (36). The role of PIAS3 as a modulator of ATR may explain its implication in HR (19). PIAS3 expression is low in significant fractions of squamous cell lung cancer (SCC), non-small cell lung cancer (NSCLC), gastric cancer (GC), and mesothelioma (37–40). The low expression of PIAS3 correlates with STAT3/5 activation and poor patient survival. Our results raise the possibility that low expression of PIAS3 in cancer cells may also compromise HR, rendering them genetically unstable and sensitive to PARP inhibitor. Furthermore, cancer cells that express PIAS3 poorly may be sensitive to ATR inhibitors. It would be interesting to test if inhibitors of PARP or ATR exert synergistic effects with STAT inhibitor in the treatment of low-PIAS3 tumors. Future studies will reveal more details of how PIAS3 promotes the basal activity of ATR, and determine whether low expression of PIAS3 in tumors offers a new opportunity for targeted therapy with PARP and ATR inhibitors.
Author Contributions
C. S. W. and L. Z. conceived the research plan. C. S. W. carried out all the experiments. C. S. W. and L. Z. prepared the manuscript.
Acknowledgments
We thank members of the Zou laboratory for helpful discussions.
This work was supported by NIH Grants GM076388 and CA197779 (to L. Z.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- PTM
- post-translational modification
- PIAS
- protein inhibitor of activated STAT
- SUMO
- small ubiquitin-like modifier
- CPT
- camptothecin
- HU
- hydroxyurea
- DDR
- DNA damage response
- ATM
- ataxia telangiectasia-mutated
- ATR
- ATM and Rad3 related
- ATRIP
- ATR-interacting protein
- DSB
- double-stranded break
- qPCR
- quantitative PCR.
References
- 1.Jackson S. P., and Durocher D. (2013) Regulation of DNA damage responses by ubiquitin and SUMO. Mol. Cell 49, 795–807 [DOI] [PubMed] [Google Scholar]
- 2.Sarangi P., and Zhao X. (2015) SUMO-mediated regulation of DNA damage repair and responses. Trends Biochem. Sci. 40, 233–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoege C., Pfander B., Moldovan G. L., Pyrowolakis G., and Jentsch S. (2002) RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419, 135–141 [DOI] [PubMed] [Google Scholar]
- 4.Pfander B., Moldovan G. L., Sacher M., Hoege C., and Jentsch S. (2005) SUMO-modified PCNA recruits Srs2 to prevent recombination during S phase. Nature 436, 428–433 [DOI] [PubMed] [Google Scholar]
- 5.Papouli E., Chen S., Davies A. A., Huttner D., Krejci L., Sung P., and Ulrich H. D. (2005) Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Mol. Cell 19, 123–133 [DOI] [PubMed] [Google Scholar]
- 6.Stelter P., and Ulrich H. D. (2003) Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature 425, 188–191 [DOI] [PubMed] [Google Scholar]
- 7.Psakhye I., and Jentsch S. (2012) Protein group modification and synergy in the SUMO pathway as exemplified in DNA repair. Cell 151, 807–820 [DOI] [PubMed] [Google Scholar]
- 8.Cremona C. A., Sarangi P., Yang Y., Hang L. E., Rahman S., and Zhao X. (2012) Extensive DNA damage-induced sumoylation contributes to replication and repair and acts in addition to the mec1 checkpoint. Mol. Cell 45, 422–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chung I., and Zhao X. (2015) DNA break-induced sumoylation is enabled by collaboration between a SUMO ligase and the ssDNA-binding complex RPA. Genes Dev. 29, 1593–1598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yunus A. A., and Lima C. D. (2009) Structure of the Siz/PIAS SUMO E3 ligase Siz1 and determinants required for SUMO modification of PCNA. Mol. Cell 35, 669–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galanty Y., Belotserkovskaya R., Coates J., Polo S., Miller K. M., and Jackson S. P. (2009) Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature 462, 935–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morris J. R., Boutell C., Keppler M., Densham R., Weekes D., Alamshah A., Butler L., Galanty Y., Pangon L., Kiuchi T., Ng T., and Solomon E. (2009) The SUMO modification pathway is involved in the BRCA1 response to genotoxic stress. Nature 462, 886–890 [DOI] [PubMed] [Google Scholar]
- 13.Danielsen J. R., Povlsen L. K., Villumsen B. H., Streicher W., Nilsson J., Wikström M., Bekker-Jensen S., and Mailand N. (2012) DNA damage-inducible SUMOylation of HERC2 promotes RNF8 binding via a novel SUMO-binding Zinc finger. J. Cell Biol. 197, 179–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gibbs-Seymour I., Oka Y., Rajendra E., Weinert B. T., Passmore L. A., Patel K. J., Olsen J. V., Choudhary C., Bekker-Jensen S., and Mailand N. (2015) Ubiquitin-SUMO circuitry controls activated fanconi anemia ID complex dosage in response to DNA damage. Mol. Cell 57, 150–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luo K., Zhang H., Wang L., Yuan J., and Lou Z. (2012) Sumoylation of MDC1 is important for proper DNA damage response. EMBO J. 31, 3008–3019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yin Y., Seifert A., Chua J. S., Maure J. F., Golebiowski F., and Hay R. T. (2012) SUMO-targeted ubiquitin E3 ligase RNF4 is required for the response of human cells to DNA damage. Genes Dev. 26, 1196–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galanty Y., Belotserkovskaya R., Coates J., and Jackson S. P. (2012) RNF4, a SUMO-targeted ubiquitin E3 ligase, promotes DNA double-strand break repair. Genes Dev. 26, 1179–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park J. H., Lee S. W., Yang S. W., Yoo H. M., Park J. M., Seong M. W., Ka S. H., Oh K. H., Jeon Y. J., and Chung C. H. (2014) Modification of DBC1 by SUMO2/3 is crucial for p53-mediated apoptosis in response to DNA damage. Nat. Commun. 5, 5483. [DOI] [PubMed] [Google Scholar]
- 19.Liu S., Fan Z., Geng Z., Zhang H., Ye Q., Jiao S., and Xu X. (2013) PIAS3 promotes homology-directed repair and distal non-homologous end joining. Oncol. Lett. 6, 1045–1048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu C. S., Ouyang J., Mori E., Nguyen H. D., Maréchal A., Hallet A., Chen D. J., and Zou L. (2014) SUMOylation of ATRIP potentiates DNA damage signaling by boosting multiple protein interactions in the ATR pathway. Genes Dev. 28, 1472–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu S., Ho C. K., Ouyang J., and Zou L. (2013) Nek1 kinase associates with ATR-ATRIP and primes ATR for efficient DNA damage signaling. Proc. Natl. Acad. Sci. U.S.A. 110, 2175–2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cortez D., Guntuku S., Qin J., and Elledge S. J. (2001) ATR and ATRIP: partners in checkpoint signaling. Science 294, 1713–1716 [DOI] [PubMed] [Google Scholar]
- 23.Heffernan T. P., Simpson D. A., Frank A. R., Heinloth A. N., Paules R. S., Cordeiro-Stone M., and Kaufmann W. K. (2002) An ATR- and Chk1-dependent S checkpoint inhibits replicon initiation following UVC-induced DNA damage. Mol. Cell. Biol. 22, 8552–8561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mohni K. N., Kavanaugh G. M., and Cortez D. (2014) ATR pathway inhibition is synthetically lethal in cancer cells with ERCC1 deficiency. Cancer Res. 74, 2835–2845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sartori A. A., Lukas C., Coates J., Mistrik M., Fu S., Bartek J., Baer R., Lukas J., and Jackson S. P. (2007) Human CtIP promotes DNA end resection. Nature 450, 509–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shiotani B., Nguyen H. D., Håkansson P., Maréchal A., Tse A., Tahara H., and Zou L. (2013) Two distinct modes of ATR activation orchestrated by Rad17 and Nbs1. Cell Rep. 3, 1651–1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pommier Y. (2004) Camptothecins and topoisomerase I: a foot in the door. Targeting the genome beyond topoisomerase I with camptothecins and novel anticancer drugs: importance of DNA replication, repair and cell cycle checkpoints. Curr. Med. Chem. Anticancer Agents 4, 429–434 [DOI] [PubMed] [Google Scholar]
- 28.Regairaz M., Zhang Y. W., Fu H., Agama K. K., Tata N., Agrawal S., Aladjem M. I., and Pommier Y. (2011) Mus81-mediated DNA cleavage resolves replication forks stalled by topoisomerase I-DNA complexes. J. Cell Biol. 195, 739–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ouyang J., Garner E., Hallet A., Nguyen H. D., Rickman K. A., Gill G., Smogorzewska A., and Zou L. (2015) Noncovalent interactions with SUMO and ubiquitin orchestrate distinct functions of the SLX4 complex in genome maintenance. Mol. Cell 57, 108–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu S., Shiotani B., Lahiri M., Maréchal A., Tse A., Leung C. C., Glover J. N., Yang X. H., and Zou L. (2011) ATR autophosphorylation as a molecular switch for checkpoint activation. Mol. Cell 43, 192–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ray Chaudhuri A., Hashimoto Y., Herrador R., Neelsen K. J., Fachinetti D., Bermejo R., Cocito A., Costanzo V., and Lopes M. (2012) Topoisomerase I poisoning results in PARP-mediated replication fork reversal. Nat. Struct. Mol. Biol. 19, 417–423 [DOI] [PubMed] [Google Scholar]
- 32.Hammer E., Heilbronn R., and Weger S. (2007) The E3 ligase Topors induces the accumulation of polysumoylated forms of DNA topoisomerase I in vitro and in vivo. FEBS Lett. 581, 5418–5424 [DOI] [PubMed] [Google Scholar]
- 33.Li M., Pokharel S., Wang J. T., Xu X., and Liu Y. (2015) RECQ5-dependent SUMOylation of DNA topoisomerase I prevents transcription-associated genome instability. Nat. Commun. 6, 6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takai H., Xie Y., de Lange T., and Pavletich N. P. (2010) Tel2 structure and function in the Hsp90-dependent maturation of mTOR and ATR complexes. Genes Dev. 24, 2019–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schimmel J., Eifler K., Sigurðsson J. O., Cuijpers S. A., Hendriks I. A., Verlaan-de Vries M., Kelstrup C. D., Francavilla C., Medema R. H., Olsen J. V., and Vertegaal A. C. (2014) Uncovering SUMOylation dynamics during cell-cycle progression reveals FoxM1 as a key mitotic SUMO target protein. Mol. Cell 53, 1053–1066 [DOI] [PubMed] [Google Scholar]
- 36.Wang H., Wang H., Powell S. N., Iliakis G., and Wang Y. (2004) ATR affecting cell radiosensitivity is dependent on homologous recombination repair but independent of nonhomologous end joining. Cancer Res. 64, 7139–7143 [DOI] [PubMed] [Google Scholar]
- 37.Li J., Cui J., Zhang J., Liu Y., Han L., Jia C., Deng J., and Liang H. (2015) PIAS3, an inhibitor of STAT3, has intensively negative association with the survival of gastric cancer. Int. J. Clin. Exp. Med. 8, 682–689 [PMC free article] [PubMed] [Google Scholar]
- 38.Abbas R., McColl K. S., Kresak A., Yang M., Chen Y., Fu P., Wildey G., and Dowlati A. (2015) PIAS3 expression in squamous cell lung cancer is low and predicts overall survival. Cancer Med. 4, 325–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dabir S., Kluge A., Kresak A., Yang M., Fu P., Groner B., Wildey G., and Dowlati A. (2014) Low PIAS3 expression in malignant mesothelioma is associated with increased STAT3 activation and poor patient survival. Clin. Cancer Res. 20, 5124–5132 [DOI] [PubMed] [Google Scholar]
- 40.Pastuszak-Lewandoska D., Domańska D., Czarnecka K. H., Kordiak J., Migdalska-Sęk M., Nawrot E., Kiszałkiewicz J., Antczak A., Górski P., and Brzeziańska E. (2014) Expression of STAT5, COX-2 and PIAS3 in correlation with NSCLC histhopathological features. PLoS ONE 9, e104265. [DOI] [PMC free article] [PubMed] [Google Scholar]