

Abstract
Yeast Mec1, the ortholog of human ATR, is the apical protein kinase that initiates the cell cycle checkpoint in response to DNA damage and replication stress. The basal activity of Mec1 kinase is activated by cell cycle phase-specific activators. Three distinct activators stimulate Mec1 kinase using an intrinsically disordered domain of the protein. These are the Ddc1 subunit of the 9-1-1 checkpoint clamp (ortholog of human and Schizosaccharomyces pombe Rad9), the replication initiator Dpb11 (ortholog of human TopBP1 and S. pombe Cut5), and the multifunctional nuclease/helicase Dna2. Here, we use small peptides to determine the requirements for Mec1 activation. For Ddc1, we identify two essential aromatic amino acids in a hydrophobic environment that when fused together are proficient activators. Using this increased insight, we have been able to identify homologous motifs in S. pombe Rad9 that can activate Mec1. Furthermore, we show that a 9-amino acid Dna2-based peptide is sufficient for Mec1 activation. Studies with mutant activators suggest that binding of an activator to Mec1 is a two-step process, the first step involving the obligatory binding of essential aromatic amino acids to Mec1, followed by an enhancement in binding energy through interactions with neighboring sequences.
Keywords: checkpoint control, DNA damage response, enzyme kinetics, peptides, serine/threonine protein kinase
Introduction
Our genome is constantly under assault from both intrinsic insults, such as oxidative damage or replication errors, and extrinsic factors, such as radiation or genotoxic agents. To preserve the integrity of our genetic material, eukaryotic organisms have evolved checkpoint mechanisms that slow down cell cycle progression to provide sufficient time for DNA repair or completion of DNA replication (1). In Saccharomyces cerevisiae, the DNA checkpoint relies on two major checkpoint kinases of the phosphatidyl-inositol 3-kinase-related (PIKK)3 kinase family, Tel1 (human ATM), and Mec1 (human ATR). Tel1 is primarily activated in response to DNA double-strand breaks (2, 3), whereas Mec1 is activated in response to Replication Protein A-coated single-stranded DNA (RPA-ssDNA) (4), which is a common intermediate in many DNA repair pathways as well as a signal of replication stress. The division between the checkpoint roles of Tel1 and Mec1 is not that clear-cut, however, and partial redundancy exists between these two checkpoint kinases (3).
Mec1/ATR forms an obligate heterodimer with the Ddc2 regulatory subunit (human ATRIP) (5, 6) and localizes to sites of DNA damage through the interaction of Ddc2 with RPA-coated ssDNA intermediates that occur during DNA repair processes (7). However, recruitment of Mec1-Ddc2 to RPA-coated ssDNA is not sufficient to trigger the checkpoint response, as this requires a stimulatory signal from an activator protein. The activator is recruited to the site of DNA damage independently of Mec1-Ddc2 and upon interaction with the kinase complex triggers its activation (8, 9). Active Mec1-Ddc2 complex phosphorylates numerous downstream substrates, including the effector kinase Rad53 that propagates the checkpoint signal.
In vertebrate cells, Mec1/ATR is activated by the essential replication initiation factor TopBP1 (10, 11). Similarly, Dpb11, the yeast ortholog of TopBP1, activates Mec1 through a mechanism that is dependent on two aromatic residues in its structurally disordered C-terminal tail (Fig. 1A) (12–14). Dpb11/TopBP1 is recruited to the site of DNA damage by interaction with the Ddc1/Rad9 subunit of the 9-1-1 checkpoint clamp that is loaded at 5′ ssDNA-dsDNA junctions (15–17). However, in S. cerevisiae, Ddc1 has a dual role in Mec1 activation, as it is also able to directly activate Mec1 (6, 18). A corresponding activity has not been demonstrated in Schizosaccharomyces pombe nor in higher eukaryotes, where Cut5/TopBP1 remains the only activator of Rad3/ATR identified to date (10, 15). Studies in a mouse knock-in model with an activation-defective TopBP1 mutant support the conclusion that TopBP1 is the critically important ATR activator for embryonic development and for cell proliferation (19).
FIGURE 1.

Three activators of S. cerevisiae Mec1. A, schematic presentation of the domain structure of Ddc1, Dpb11, and Dna2 of S. cerevisiae. The aromatic residues required for Mec1 activation are indicated. Structural disorder was calculated using IUPRED (40). Regions with disorder parameter >0.5 are considered to be intrinsically disordered. B, kinase assay comparing the ability of 20 nm full-length Dpb11, 9-1-1, or Dna2 to activate Mec1. Mec1 stimulation over basal activity is indicated as fold-activation for either GST-Rad53-kd or Rpa1; see “Experimental Procedures” for details. C, WEB logos derived from sequence comparisons as described in “Experimental Procedures.” The range of distances between conserved motifs in different organisms is shown. Numbering below motifs is for the S. cerevisiae proteins with the underlined numbers indicating the critical aromatics. D, structure of the Rad9 subunit in human 9-1-1 from (27), with comparable human and S. cerevisiae residues indicated. The start of the unstructured C-terminal tail is also indicated.
The lagging strand maturation factor Dna2 constitutes a third, S-phase specific, activator of Mec1 in S. cerevisiae (20). Neither its nuclease nor helicase activities are required for its checkpoint function, which is mediated by its unstructured N-terminal tail (Fig. 1A). In budding yeast, these three activators show partial redundancy in a manner that is dependent on cell cycle phase. Specifically, activation through the Ddc1 subunit of the 9-1-1 clamp mediates full Mec1 kinase activation in the G1 phase, whereas both Dpb11 and Ddc1 contribute to checkpoint activation in G2 (14, 18, 21). In contrast, Dna2, Dpb11 and Ddc1 all contribute to Mec1 activation in S-phase, and do so with partial redundancy, and the Mec1-dependent replication checkpoint response can only be abolished when the checkpoint functions of all three activators are eliminated by mutations (20).
Past work from our laboratory has allowed the identification of some common features of the Mec1 activators in S. cerevisiae. Mec1 stimulation by each of the three activators is dependent on two aromatic residues, the mutation of which abolishes Mec1 activation both in vitro and in vivo (14, 18, 20). Significantly, the aromatic residues in the activators tend to reside in an unstructured region of the protein, and, while the actual sequences are not well conserved, the disorder is (Fig. 1A). Furthermore, the spacing between the critical aromatic residues can also vary substantially. For the three S. cerevisiae activators, the aromatic residues are separated by as much as 192 amino acids (aa) in Ddc1, but only by 35 aa and 1 aa in Dpb11 and Dna2, respectively. Furthermore, this variability in inter-motif distance extends to the activators in other eukaryotic organisms (Fig. 1C). Supporting the model that the distance between the motifs is flexible is the observation that the two activation motifs of Ddc1 can be fused together in a single peptide that exhibits robust Mec1 stimulatory activity (18).
Currently, it remains unknown which structural and sequence features define a Mec1 activator and, secondly, how the binding of an activator causes stimulation of Mec1 protein kinase activity. In this study, we sought to better understand the common characteristics and requirements of a Mec1 activator. Through mutation analysis of small peptides that can function as activators, we identified critical residues within the Ddc1 activation motif necessary for Mec1 stimulation. Based on the insights derived from our Ddc1 peptide studies, we designed a peptide from S. pombe Rad9 sequences, which can activate S. cerevisiae Mec1, suggesting a conservation of both motifs and mechanism in both yeasts. We then focused our efforts on the Dna2 activator and showed that its Mec1 stimulatory potential depends solely on a small nine amino acid sequence. We also showed that binding of an activator to Mec1 is a two-step sequential process, the first step involving the obligatory binding of the aromatic amino acids to Mec1, followed by an enhancement in binding energy through interactions with neighboring sequences.
Experimental Procedures
Bioinformatic Analysis
Using PSI-BLAST, the Ddc1/Rad9 genes were identified in the fungi and metazoa kingdoms. The sequences were aligned using MSAProb from the MPI toolkit, reduced to maximal 43 most divergent sequences, and then re-aligned using MSAProb (22). WEB-logos were then generated for the consensus motifs (23). The same analysis was carried out for Dpb11/TopBP1. For Dna2, an N-terminal structural domain containing the activation motif could only be identified in the ascomycetes phylum of fungi. The other fungal phyla and metazoans largely lack an unstructured domain. Therefore, the analysis is solely based on 30 most divergent ascomycetes species.
Proteins and Peptides
Mec1-Ddc2, Rad53-kd, Rad24-RFC, RPA, Dpb11, 9-1-1, Ddc1, and Dna2 or Dna2(1–499) were expressed and purified as previously described (6, 13, 18, 20, 24). Mutant derivatives of Dna2(1–499) were expressed and purified similarly. Peptides were synthesized by GenScript (GenScript USA Inc.) or by the peptide synthesis facility at the School of Medicine, Washington University in St. Louis. They were dissolved in sterile water or 25–50% DMSO as 10 mm stock solutions, neutralized to pH 7.5–8, if necessary, and stored at −80 °C. Serial dilutions were made in the same solvent. Peptides that required extreme conditions for solubility, e.g. acid or base, were not used in this study.
Kinase Assays
Standard kinase assays with peptides were performed in a 10 μl volume of 25 mm Hepes-KOH pH 7.5, 8 mm magnesium acetate, 20 μg/ml BSA, 1 mm DTT, 50 mm NaCl (final concentration, including contributions made by protein storage buffers), 100 μm ATP, 0.5 μCi [γ-32P]ATP with 125 nm Rad53-kd as substrate, and peptides at the indicated concentrations. Reactions without activator contained water or up to 2.5% DMSO. Concentrations of DMSO up to 5% did not affect the kinase assay. Assays were started by addition of 5–10 nm Mec1-Ddc, incubated at 30 °C for 10–15 min and stopped by addition of 5 μl of 5×SDS-PAGE loading dye. After boiling, samples were separated on 8% SDS-PAGE gels, dried, and exposed to phosphor screens (GE Healthcare). For each peptide, a titration was performed, generally up to 50 μm peptide, and in each series of peptides, the efficiency of a given peptide as an activator was evaluated with respect to the parent peptide, either Ddc1-1 or Dna2-1. The efficiency of the peptides was evaluated using standard Michaelis-Menten kinetics: V = V0 + Vmax × X/(Km + X) in which V is the extent of Rad53-kd phosphorylation, V0 is phosphorylation without activator, and x is the peptide concentration. Examples of these fittings are given in Figs. 2A and 3A. To evaluate the relative efficiency of the peptides, the kinetics were fitted with the Vmax fixed at the value obtained with either the Ddc1-1 or Dna2-1 peptide as appropriate, and the resulting Vmax/Km values, relative to that of Ddc1-1 or Dna2-1 set to 100%, plotted in Figs. 2C and 3D. The assays in Fig. 1B also contained 2.5 nm deca-primed single-stranded SKII plasmid, 150 nm RPA, and 30 nm Rad24-RFC, and 100 mm NaCl.
FIGURE 2.

Activation of Mec1 by Ddc1- and Rad9-derived peptides. A, titration of full-length Ddc1 (at 50 mm NaCl) or the Ddc1-1 peptide (sequence given in B) at varying NaCl concentrations in the standard Mec1 assay with Rad53-kd as substrate (see “Experimental Procedures”). Data are modeled to standard Michaelis-Menten kinetics, and the Km values are shown. B, activity of wild-type Ddc1-1 peptide and the indicated variants. The Ddc1-1 peptide sequence is shown, with amino acid positions corresponding to the full-length protein. The two essential tryptophans are in bold. C, various residues or groups of residues in the Ddc1-1 peptide were deleted or mutated to alanine, as indicated in the graph, and their ability to stimulate Mec1 was quantified relative to the wild-type peptide. Peptide concentrations ranged from 5–50 μm. Data represent the average of at least three independent experiments for peptides that showed activity, and of two experiments for peptides that were inactive. Error bars are standard error of the mean. Pro-356 and Pro-359 were simultaneously mutated to alanines. * indicates that the ILMQMQ and RAD deletions were made in a peptide that also had ND346,347 deleted. D, S. pombe Rad9-derived wild-type and variant peptides and a human Rad9-derived peptide were tested in the same assay.
FIGURE 3.

A nine amino acid Dna2 peptide is sufficient for activation of Mec1. A, titration of the Dna2(1–499) domain or the Dna2-1 peptide (sequence given in B) in the standard Mec1 assay with Rad53-kd as substrate (see “Experimental Proceduress”). B, effect on peptide activity of substituting the Trp-128 residue with other aromatics. C, reduction of the size of the peptide results in gradual loss in affinity. Peptide size numbering is shown in B, and Km values determined. D, residues in the Dna2-1 peptide were mutated to alanine, as indicated in the graph, and their ability to stimulate Mec1 was quantified relative to the wild-type peptide. Peptide concentrations ranged from 1.5–50 μm. Data represent the average of at least three independent experiments for peptides that showed activity, and of two experiments for peptides that were inactive. Error bars are standard error of the mean. Pro-122 and Pro-132 were simultaneously mutated to alanines.
Yeast Strains and Genetic Analysis
The strains used are PY263 (MATa leu2 trp1 ura3–52 his3-Δ200 dna2Δ::HIS3 with pRS316-DNA2 (ARSH4 CEN6 URA3 DNA2)); PY270 (MATa can1 leu2 trp1 ura3–52 his3-Δ200 dna2Δ::HIS3 ddc1Δ::KanMx4 tel1Δ::NATMx4 with pRS316-DNA2 (ARSH4 CEN6 URA3 DNA2)); PY284 (MATa can1 leu2 trp1 ura3–52 his3-Δ200 dna2Δ::HIS3 ddc1Δ::KanMx4 with pRS316-DNA2 (ARSH4 CEN6 URA3 DNA2)). They were transformed with plasmid pBL584 (ARSH4 CEN6 TRP1 DNA2) or the dna2-WY128,130AA or dna2-PP122,132AA mutant plasmids, or pRS314 empty vector. Transformants were grown on SC-Trp medium, and 10-fold serial dilutions plated on SC-Trp medium containing 5-fluoroorotic acid (5-FOA) to allow growth of strains that had lost the pRS316-DNA2 plasmid. For Western analysis, the PY270 series cells were synchronized in G1 phase by treating exponentially growing cells with 5 μg/ml alpha factor, which was replenished again after 90 min for a total of 180 min. Washed cells were resuspended in medium with Pronase (0.1 mg/ml) and released into S phase 90 min with or without 200 mm hydroxyurea. Protein extracts were prepared by trichloroacetic acid precipitation. These extracts were probed with anti-Rad53 antibodies (SC-6749 from Santa Cruz Biotechnology). The blots were quantified using ImageJ software, and overlapping peaks were deconvoluted using the IGOR-Pro software package. In our analysis, the result obtained with wild-type yeast and no damage was set to 0% Rad53 phosphorylation.
Results
The S. cerevisiae PIKK kinase Mec1 can be stimulated by three different activators: the Ddc1 subunit of the 9-1-1 (Ddc1-Rad17-Mec3) checkpoint clamp, the replication initiator protein Dpb11, and the multifunctional nuclease/helicase Dna2 (6, 12, 13, 20, 25). We analyzed the effects of each of the three activators on Mec1 activity in a defined in vitro kinase assay with purified recombinant proteins (Fig. 1B). To ensure that all observed phosphorylation events were Mec1-dependent, a kinase-dead variant (K227A) of Rad53, the physiological target of Mec1, was used as phosphorylation substrate. Optimal activation of Mec1 by Ddc1 requires that the 9-1-1 clamp be loaded onto DNA by its loader (6). Therefore, the assay mixture for each activator also contained RPA-coated multiple-primed single-stranded DNA (ssDNA) and the Rad24-RFC clamp loader. All three activators could stimulate Mec1 kinase activity to a similar extent (Fig. 1B). The activators differed only slightly in promoting the phosphorylation of Rad53-kd versus RPA. Our peptide studies below will focus on Ddc1 and Dna2. As yet, we have not been able to generate small Dpb11-derived peptides that show robust stimulation of Mec1 kinase activity.
Identification of Critical Residues for Mec1 Stimulation in the Activation Motif of Ddc1
Previous studies have identified Trp-352 and Trp-544 as the two aromatic residues of Ddc1 that are required for Mec1 activation in vivo and in vitro (18). We carried out a phylogenetic analysis in the fungi and animalia kingdoms of the two sequence motifs surrounding the critical Trp-352 and Trp-544 in Ddc1 (Fig. 1C). This analysis showed strong conservation of the first motif in all organisms. This sequence conservation was not surprising because this motif resides in a β-strand-loop-β-strand structure of the conserved PCNA-like domain of Ddc1/Rad9 (Fig. 1C). However, the most highly conserved residue in this motif (hF240 = scF353) is buried inside the structure of hRad9, whereas the more variable residue (hH239 = scW352) is solvent exposed and therefore has an opportunity to directly interact with Mec1 (26–28). High sequence conservation continued 10–15 aa downstream of Trp-352, but while mutations in that region affected the structural integrity of Ddc1 and the 9-1-1 complex and, therefore its checkpoint activity in vivo, those mutations did not affect the in vitro Mec1 activation potential by the isolated subunit, or domains thereof (18). The second motif in Ddc1/Rad9 is conserved in fungi and metazoa, but the distance between the two motifs is highly variable, being 66 aa in S. pombe and 192 aa in S. cerevisiae. The alignment for the second motif is primarily based on the presence of an aromatic amino acid plus a long aliphatic amino acid in its −2 position, and confirmation with regard to the correct alignment has to depend on independent experimental verification (see below).
Previously, we showed that Ddc1(340–562), an N- and C-terminally truncated derivative of the 612 aa Ddc1 protein, has full activity, but that further N-terminal truncation to Ddc1(348–562) significantly reduced activity (18). In that study we also reported on the design of a 30-mer peptide consisting of a fusion of the two aromatic residue-containing motifs (346–365 + 537–546; sequence in Fig. 2B). The Ddc1–1 peptide triggered robust kinase activity by Mec1, suggesting that the intervening 171 aa appear to be largely dispensable. However, these intervening regions may provide auxiliary binding energy, because the apparent affinity of the peptide for Mec1 is about 100–1000-fold lower than that of full-length Ddc1 (Fig. 2A). In addition, the maximal extent of activation is also somewhat decreased. An increase in the NaCl concentration in the assay significantly reduced activation by the peptide: the peptide Km value increased from 1.2 μm at 50 mm NaCl to 8.2 μm at 110 mm NaCl (Fig. 2A). Therefore, to be able to detect lower activities of variant peptides, our peptide studies were carried out at 50 mm NaCl.
We previously determined that each of the two tryptophans in the Ddc1-1 peptide is essential for activity; substituting either Trp-352 or Trp-544 for Ala resulted in a complete loss of activity (18). However, changing the Trp-352 residue to Tyr, as occurs in S. pombe Rad9, or to His, as occurs in human Rad9, retained considerable activity (Fig. 2B). To identify additional features required for stimulation of Mec1, we deleted various residues or groups of residues within the Ddc1 peptide or replaced them with alanines, and analyzed their effect on Mec1 activity in the kinase assay (Fig. 2C). Deletion of the first two aa (ΔN346,D347) did not affect activation, and neither did deletion of the terminal six aa (Δ360-ILMQMQ-365) of motif 1. Since this latter sequence, which constitutes a β-strand in the PCNA-like fold, is highly conserved in Ddc1/Rad9, we conclude it is only so for structural reasons. A change of both prolines (P356A, P359A) that are in the loop of the β-strand-loop-β-strand secondary structure, had a minor effect on activity. Similarly, deletion of 537-RAD-539 in motif 2 showed only a minor decrease in activity.
Next, we mutated several conserved residues in the 30-mer peptide (Fig. 2C). In general, mutation of amino acids close to the critical tryptophans showed stronger defects. In particular, mutation of long-chain aliphatic and aromatic amino acids (NI348,349AA, L351A, F353A, V542A) resulted in strong activation defects, whereas mutation of hydrophilic/small residues (S350A, C353A, T543A, G545A) showed only minor defects. Only one variant (K546A) showed an increase in Mec1 activation activity. Consistent with these results, an 18-mer minimal peptide (348NISLWFCHPGD358–540TTVTWGK546) still showed 20% activity (data not shown). From these experimental results and the bioinformatics, we deduce a consensus motif of φxφαα-x63–190-φxα, in which φ is long-chain hydrophobic and α is aromatic. Interestingly, most Ddc1/Rad9 sequences (36 out of 43) have a Gly either directly before or after the aromatic residue in motif 2, although in our study, changing this Gly to Ala, reduced the activity only by 50%.
A Peptide Derived from S. pombe Rad9 Activates S. cerevisiae Mec1
Our Ddc1 peptide analysis has suggested specific motifs that are required for activation of Mec1. A comparison between S. cerevisiae Ddc1 and S. pombe Rad9 indicates that spRad9 may fulfill these requirements in the two motifs that are anchored by Tyr-271 and Trp-348. The Tyr-271 residue is predicted to reside at the same position in the 9-1-1 structure as human His-239 and scTrp-352, whereas Trp-348 is in the unstructured C-terminal tail. We fused the two S. pombe motifs into a 27-mer peptide, and found that this peptide showed robust stimulation of S. cerevisiae Mec1 (Fig. 2D). In addition, mutation of either Tyr-271 or Trp-348 to alanine abolished stimulatory activity. These data parallel those for S. cerevisiae Ddc1 (Fig. 2C). From these observations we can draw the conclusion that critical interaction motifs are conserved between S. cerevisiae Mec1/Ddc2 and S. pombe Rad3/Rad26. Secondly, our data suggest that in S. pombe, the 9-1-1 clamp has the potential to activate Rad3 kinase independent of the activation motifs on Cut5 (the ortholog of Dpb11/TopBP1). We attempted to extend this heterologous activation system with an analogous human Rad9 peptide, but observed no significant activity. Therefore, we can draw no conclusion with regard to the activation of human ATR/ATRIP by 9-1-1, but others have proposed that the sole function of human 9-1-1 is to recruit TopBP1 (15).
A Nine Amino Acid Peptide of Dna2 Is Sufficient for Mec1 Activation
The Mec1-stimulatory activity of S. cerevisiae Dna2 resides in its intrinsically disordered N-terminal tail (Fig. 1A), and the activity is independent of its nuclease and helicase activities. In fact, the isolated domain, Dna2(1–499) showed the same apparent affinity and activation kinetics as the 1522 aa full-length Dna2 (20). In contrast with the Ddc1/Rad9 and Dbp11/Cut5/TopBP1 activators, a potential conserved motif for Dna2 was only identified in the ascomycetes phylum of fungi, a large phylum that contains both budding and fission yeast; it was not found in other fungi nor in metazoa. Consistent with this finding, human Dna2 does not contain a long disordered N-terminal tail (29). A comprehensive mutational analysis of the aromatic residues in Dna2(1–499) showed that only the mutation of Trp-128 and of Tyr-130 compromised the stimulatory activity of Dna2 (20). This was a surprising deviation from the knowledge we had gained from our studies with Ddc1 and Dpb11, because the activation motifs in those two activators are bipartite and the two motifs are separated by a highly variable distance, but at least by 30 amino acids (Fig. 1C). Indeed, the 30-mer Dna2–1 peptide that surrounds these two close aromatics showed robust stimulatory activity (20), and its maximum stimulatory activity approached that of the entire 499 aa long intrinsically disordered domain (Fig. 3A). However, as before with the Ddc1–1 peptide, the apparent affinity of Dna2–1 for Mec1 is 100–1000-fold lower than that of Dna2(1–499). We first determined whether the critical Trp-128 residue in the peptide could be replaced by other aromatic residues with retention of activity. Substitution of Trp by Tyr resulted in almost complete retention of activity, however, substitution with Phe substantially compromised activity and substitution with His completely inactivated the peptide (Fig. 3B). Interestingly, S. pombe is the only one out of the thirty species in the Dna2 alignment that has a Phe rather than Trp at this central position.
We next took two approaches to defining a minimal consensus sequence. First, we truncated the peptide sequence to identify a minimal sequence with stimulatory activity. Progressive truncation of the peptide resulted in a gradual decrease in its affinity for Mec1 (Fig. 3C). However, the smallest peptide we tested still showed significant activity. This is a 9-mer, containing the central aromatics and adjacent hydrophobic aliphatic amino acids, but, surprisingly, lacking the highly conserved proline residues (Fig. 1C). We then mutated central residues within the 30-mer Dna2-1 peptide to identify critical determinants for activity (Fig. 3D). To a large extent, mutations of residues close to the critical Trp mirrored the results we found with the Ddc1 peptide: adjacent long aliphatic amino acids were crucial for activity, while charged residues were less so. This leads to a consensus of φφαxα that is much simpler than the bipartite Ddc1 consensus sequence (φxφαα-x63–190-φxα).
Considering that the two prolines on either side of the central Trp-128 residue are highly conserved in the ascomycetes phylum (Fig. 1C), we were surprised that neither the single P → A mutants (not shown), nor the double mutant showed major defects (Fig. 3D). We entertained two possibilities. First, we considered that the two prolines serve to prevent folding of the central Trp-containing sequence, however, that they have no function in our small peptides that are already unfolded. Therefore, we made the double PP-AA mutation in the entire (1–499) domain, but the mutant domain showed the same activity as wild-type (Fig. 4A). In addition, the far UV CD spectra of both Dna2(1–499) proteins showed the characteristics of that of disordered proteins with very minor α-helical content and no discernible β-strand content, as predicted by structural disorder algorithms (Figs. 1A and 4B). The α-helical content of the Dna2(1–499)PP-AA mutant was only slightly increased compared with wild-type.
FIGURE 4.
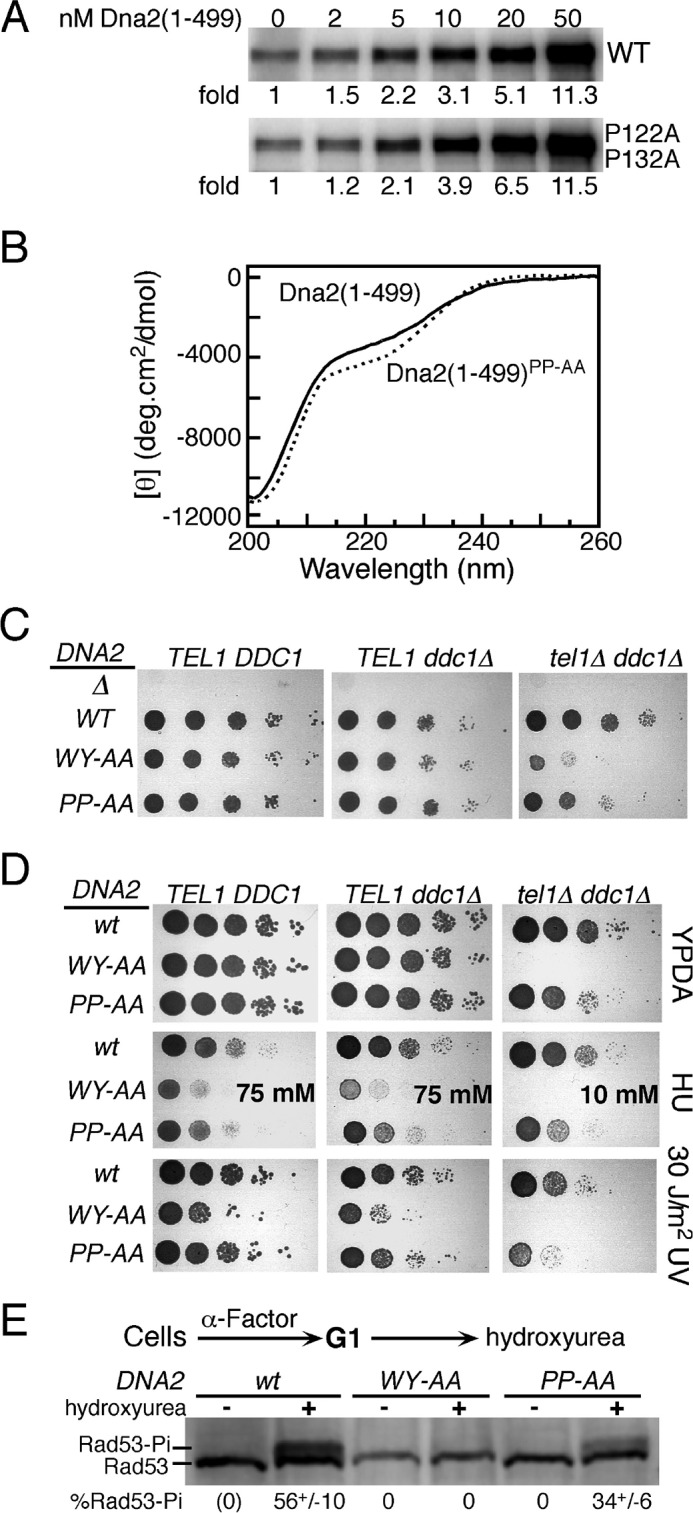
A role for Dna2 prolines 122 and 132 in checkpoint function. A, titration of the Dna2(1–499) domain or the PP122,132AA mutant domain in the standard Mec1 assay with Rad53-kd as substrate, but with 100 mm NaCl (see “Experimental Procedures”). B, far UV CD spectra of the two domains at 2 μm, in 50 mm Hepes 7.5, 150 mm NaCl buffer at 20 °C. C, 10-fold serial dilutions of strains containing both plasmid pRS316-DNA2 (URA3 DNA2) and the indicated DNA2 mutant plasmid or empty vector (Δ) were plated on 5-FOA medium to evict the complementing URA3 plasmid, and growth scored after 2 days at 30 °C. The relevant genotypes are shown. See “Experimental Procedures” for details. D, 10-fold serial dilutions of strains containing either wild-type DNA2 or the indicated mutants were plated on YPD medium and irradiated with 30 J/m2 of UV254 where indicated, or plated on YPD containing either 75 mm or 10 mm hydroxyurea (HU). Growth was scored after 2 days at 30 °C. E, strains PY270 (tel1Δ ddc1Δ) containing the indicated DNA2 mutant were synchronized in G1 and released into S phase with or without hydroxyurea. Phosphorylation of Rad53 was measured by Western analysis. See “Experimental Procedures” for details.
The second possibility we considered is that the prolines do serve a function in checkpoint activation, however, this function is not recapitulated in our simple in vitro assay. We have previously shown that the Mec1 stimulation-defective dna2-WY128,130AA mutant shows minimal checkpoint defects in an otherwise wild-type yeast due to partial redundancy for activators, but the mutant allele shows very strong growth defects when the checkpoint is more severely compromised in a ddc1Δ tel1Δ deletion strain (20). The ddc1Δ mutant simultaneously incapacitates the functions of both Ddc1 and Dpb11, since Dpb11's function depends on an intact 9-1-1 (18), and tel1Δ is a deletion of the ATM-like kinase Tel1, which in yeast can partially substitute for Mec1 in the DNA replication checkpoint (3, 30). The poor viability of tel1Δ ddc1Δ dna2-WY128,130AA has been attributed to a vast increase in gross chromosomal rearrangements, which are suppressed either by the presence of Tel1 or by either Mec1 activator Ddc1 or Dna2 (31).
We repeated the analysis for both the dna2-WY128,130AA and the double proline mutant dna2-PP122,132AA (henceforth referred to as dna2-WY-AA and dna2-PP-AA, respectively) alleles in three strains, an otherwise wild-type strain, a ddc1Δ mutant, and a ddc1Δ tel1Δ double mutant. The wild-type DNA2 gene, present on a URA3 centromere plasmid, was evicted from these strains that were heterozygous for DNA2/dna2 by growth on 5-fluoroorotic acid (5-FOA). The analysis in Fig. 4C shows that DNA2 is an essential gene in all strain backgrounds since the empty vector (Δ) did not complement. However, while growth defects were not apparent in a wild-type or minimal in a ddc1Δ strain, severe defects were observed in the ddc1Δ tel1Δ double mutant, with dna2-WY-AA showing a more severe phenotype than dna2-PP-AA (Fig. 4C). We also tested the sensitivity of these strains to the replication inhibitor hydroxyurea, and to UV irradiation (Fig. 4D). Again, both dna2-WY-AA and dna2-PP-AA showed sensitivity to hydroxyurea in all strains with dna2-WY-AA displaying a more severe phenotype. However, sensitivity of the double proline dna2-PP-AA mutant to UV irradiation was only displayed in the most severely checkpoint-defective ddc1Δ tel1Δ double mutant. To determine whether the increased sensitivity of the dna2-PP-AA mutant to hydroxyurea is actually the result of checkpoint defects, we released cells that were arrested at G1/S with α-Factor mating pheromone into S phase in the presence of 200 mm hydroxyurea. Cells were harvested after 90 min, and the degree of activation of the replication checkpoint was assessed by determining the phosphorylation status of the effector kinase Rad53. A 1.5–2-fold reduction in Rad53 phosphorylation was observed (Fig. 4E). In comparison, Rad53 phosphorylation after hydroxyurea treatment in the dna2-WY-AA mutant was completely abrogated, as determined previously (20). These data indicate that the two prolines surrounding the central motif in Dna2 serve a checkpoint function that, while not as critical as the aromatic residues, is important for proper genome maintenance.
A Two-step Mechanism for Activator Binding to Mec1
The aromatic residues that are critical for Mec1 activation by the three different activators have been identified, and mutation of both residues together leads to complete loss of stimulation of Mec1 kinase in vitro and in vivo. However, how these essential amino acids and the hundreds of amino acid long neighboring domains contribute to Mec1 binding has not been determined. While the short peptides derived from Ddc1 and Dna2 activate Mec1 nearly as efficiently as the full-length proteins, their apparent affinity for Mec1 is 100–1000-fold lower.
We considered two distinct models for binding of the structurally disordered domains to Mec1 (Fig. 5A). In the first model, both the aromatics and neighboring domains bind independently to Mec1, each contributing binding energy that, when combined results in low nm affinity. In the second model, the aromatics bind first in the activation site of Mec1 with low affinity, followed by an overall increase in complex stability through binding of the neighboring sequences. Here we provide data that support the latter, sequential binding model.
FIGURE 5.
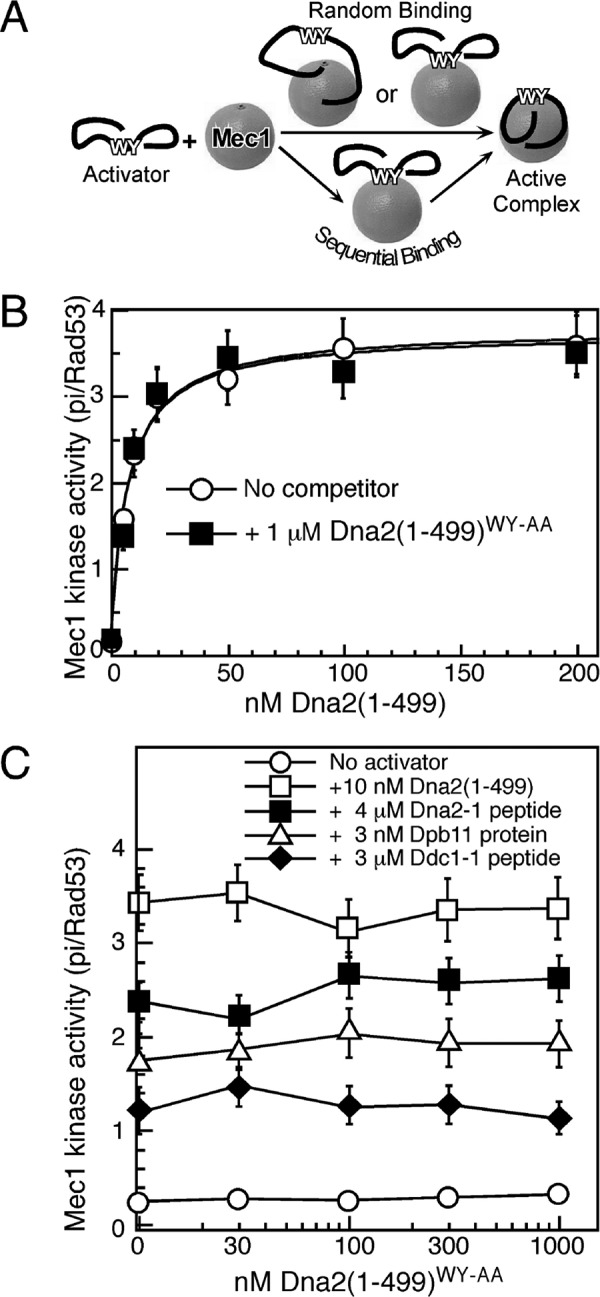
A model for sequential binding of Dna2 to Mec1. A, scheme depicting random or sequential binding of Dna2 activator to Mec1. The random pathway is inconsistent with the data below. B, titration of Dna2(1–499) in an assay with or without Dna2(1–499)WY-AA. C, titration of Dna2(1–499)WY-AA in assays with subsaturating concentrations of the indicated proteins, or the Ddc1-1 or Dna2-1 peptide.
If tight binding consisted of contributions by multiple independent modules, then one would expect that the inactive Dna2(1–499)WY-AA mutant protein would interfere with binding of the wild-type protein to Mec1. However, this is not observed. The apparent affinity of Dna2(1–499) for Mec1 remains unchanged when 1 μm Dna2(1–499)WY-AA was added to the assay (Fig. 5B). Conversely, increasing concentrations of Dna2(1–499)WY-AA failed to inhibit activation by either the Dna2 or Dpb11 activators, or by the Dna2–1 or Ddc1–1 peptides (Fig. 5C). In this assay, the activators were present at subsaturating levels.
We therefore conclude that Dna2(1–499)WY-AA cannot compete with the activator proteins and peptides for Mec1 binding, and therefore Trp-128 and Tyr-130 are essential determinants for initiating activator binding to Mec1.
Discussion
The importance of a functional checkpoint pathway is underscored by its role in human disease. Mutations in ATM, the human homolog of Tel1, lead to the neurodegenerative and cancer predisposition disorder ataxia telangiextasia (32). ATR, on the other hand, is essential for replicating cells (33–35). Mutations in ATR can lead to the rare Seckel syndrome that is characterized by growth retardation and microcephaly as well as genome instability (36). In this study, we have focused on Mec1, the yeast homolog of ATR, and sought to better understand the initial steps in its activation. In mammals, the only ATR activator described to date is the replication protein TopBP1 (19, 37, 38), whereas three different activators contribute to Mec1 activation in S. cerevisiae, both in vitro and in vivo (6, 12, 13, 20). These three activators, Ddc1 of the 9-1-1 checkpoint clamp, the Dpb11 replication initiation protein, and Dna2 nuclease-helicase, have very different functions in DNA metabolism. However, they all contain an unstructured region with key aromatic residues that are absolutely required for Mec1 activation.
To study the effect of the residues surrounding these essential aromatics, we made use of 30-mer peptides that exhibit robust Mec1 activation (18, 20). The Ddc1-1 peptide is a fusion of the two activation motifs of Ddc1. Several hydrophobic residues within the Ddc1 activation motifs are required for efficient Mec1 activation in vitro (Fig. 2C). Based on the crystal structure of the human 9-1-1 checkpoint clamp, the N-terminal activation motif of Ddc1 that is centered around the essential tryptophan residue (Trp-352) forms a conserved β-strand-loop-β-strand motif (26–28). Also the C-terminal motif of the Ddc1 peptide may fold into a short β-strand (39). Earlier work from our lab has shown that the first β-strand in motif 1 is both required for Mec1 stimulation and for correct assembly of the 9-1-1 complex while the second β-strand has only a structural function (18). According to the GOR V protein secondary structure prediction algorithm (39), point mutations that abolish Mec1 stimulatory activity are predicted to attenuate the propensity to form the first β-strand (NI348–349, L351, F353, Fig. 2C), whereas the PP356/359AA, S350A, and C354A mutations, which retain activity, do not affect the potential for β-strand formation. Similarly, the V542A mutation in motif 2 affects both the activity of the peptide and its potential for β-strand formation, while T543A and G545A did neither. Therefore, we suggest that folding into a β-strand onto the stimulatory site of Mec1 may be an important feature of the activation process.
Because the structures of Dna2 and Dpb11 have not been solved, we cannot reliably know whether all of our Mec1 activators contain β-strands in their activation domains, and interestingly, the central motif of the Dna2-1 peptide is predicted to have a higher propensity to form an α-helix than a β-strand (39). However, the CD spectrum of Dna2(1–499) shows that the protein is largely unfolded, and so is the Dna2(1–499)PP-AA mutant in which the helix-destabilizing prolines have been mutated (Fig. 4B). And while we detected no significant defect of the mutant domain to activate Mec1 kinase (Fig. 4A), the mutant showed substantial checkpoint defects in yeast (Fig. 4D). We speculate that the Dna2 NTD folds more readily in the crowded nucleoplasm of the cell, and that the helix-destabilizing function of the two proline residues do serve a important destabilizing function in vivo to present the essential aromatics to the activation site of Mec1. However, that the prolines are part of a binding motif to other protein(s) that function in the checkpoint pathway cannot be excluded.
We have used the novel insights of Mec1 activators to probe the mechanism of Mec1 activation. The presence of a large excess of the Dna2(1–499)WY-AA mutant that is deficient for Mec1 stimulation did not affect activation of Mec1 by wild-type Dna2(1–499) (Fig. 5, B and C). This is consistent with a model where the two aromatics within the activation motif are the primary determinant of activator binding to Mec1. However, they do not appear to be the sole determinant, because the apparent affinity of Ddc1- and Dna2-derived short activator peptides to Mec1 is two-three orders of magnitude lower than that of the respective full-length activator. Our studies therefore support a model where the key aromatic residues within the activation motif of the stimulating protein first bind the activation site of Mec1. Sequences surrounding the aromatics can then bind Mec1, increasing the overall binding affinity and the stability of the activator-Mec1 complex. This would explain why the activation motif in the context of the full-length protein is more potent in activating Mec1.
Our in-depth analysis of the Mec1 activation motifs in Ddc1 and Dna2 has provided us with detailed insight into the requirements for Mec1 stimulation. Future studies will reveal whether the activation motifs of all three Mec1 activators really do form structural domains that show similarities in conformational display to Mec1, and whether this feature is conserved in other eukaryotic species.
Author Contributions
P. H. W. and P. M. B. designed the study, performed the experiments in Figs. 1–3 and 5, analyzed the data and wrote the paper. E. T. designed, performed, and analyzed the experiments shown in Fig. 4. V. N. P. and S. K. initially conceived the study and carried out the experiments in Figs. 2 and 3.
Acknowledgments
We thank Carrie Stith for expert technical support, Bonnie Yoder for strain construction, James Havranek for the use of the CD spectrophotometer and advise in the interpretation, Robert Obermann for peptide synthesis, and John Majors for fruitful discussions during the course of work.
This work was supported in part by National Institutes of Health Grant GM083970 (to P. M. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- PIKK
- phosphatidyl-inositol 3-kinase-related
- aa
- amino acid
- ssDNA
- single-stranded DNA
- ATR
- ataxia telangiectasia and Rad3-related
- RPA
- replication protein A
- 9-1-1
- Ddc1-Rad17-Mec3 checkpoint clamp.
References
- 1.Hartwell L. H., and Weinert T. A. (1989) Checkpoints: controls that ensure the order of cell cycle events. Science. 246, 629–634 [DOI] [PubMed] [Google Scholar]
- 2.Bakkenist C. J., and Kastan M. B. (2003) DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421, 499–506 [DOI] [PubMed] [Google Scholar]
- 3.Craven R. J., Greenwell P. W., Dominska M., and Petes T. D. (2002) Regulation of genome stability by TEL1 and MEC1, yeast homologs of the mammalian ATM and ATR genes. Genetics 161, 493–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zou L., and Elledge S. J. (2003) Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes.[see comment]. Science 300, 1542–1548 [DOI] [PubMed] [Google Scholar]
- 5.Cortez D., Glick G., and Elledge S. J. (2004) Minichromosome maintenance proteins are direct targets of the ATM and ATR checkpoint kinases. Proc. Natl. Acad. Sci. U.S.A. 101, 10078–10083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Majka J., Niedziela-Majka A., and Burgers P. M. (2006) The checkpoint clamp activates Mec1 kinase during initiation of the DNA damage checkpoint. Mol. Cell 24, 891–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zou L., Liu D., and Elledge S. J. (2003) Replication protein A-mediated recruitment and activation of Rad17 complexes. Proc. Natl. Acad. Sci. U.S.A. 100, 13827–13832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kondo T., Wakayama T., Naiki T., Matsumoto K., and Sugimoto K. (2001) Recruitment of Mec1 and Ddc1 checkpoint proteins to double-strand breaks through distinct mechanisms. Science 294, 867–870 [DOI] [PubMed] [Google Scholar]
- 9.Melo J. A., Cohen J., and Toczyski D. P. (2001) Two checkpoint complexes are independently recruited to sites of DNA damage in vivo. Genes Dev. 15, 2809–2821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumagai A., Lee J., Yoo H. Y., and Dunphy W. G. (2006) TopBP1 activates the ATR-ATRIP complex. Cell 124, 943–955 [DOI] [PubMed] [Google Scholar]
- 11.Mordes D. A., Glick G. G., Zhao R., and Cortez D. (2008) TopBP1 activates ATR through ATRIP and a PIKK regulatory domain. Genes Dev. 22, 1478–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mordes D. A., Nam E. A., and Cortez D. (2008) Dpb11 activates the Mec1-Ddc2 complex. Proc. Natl. Acad. Sci. U.S.A. 105, 18730–18734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Navadgi-Patil V. M., and Burgers P. M. (2008) Yeast DNA replication protein Dpb11 activates the Mec1/ATR checkpoint kinase. J. Biol. Chem. 283, 35853–35859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Navadgi-Patil V. M., Kumar S., and Burgers P. M. (2011) The unstructured C-terminal tail of yeast Dpb11 (human TopBP1) protein is dispensable for DNA replication and the S phase checkpoint but required for the G2/M checkpoint. J. Biol. Chem. 286, 40999–41007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Delacroix S., Wagner J. M., Kobayashi M., Yamamoto K., and Karnitz L. M. (2007) The Rad9-Hus1-Rad1 (9–1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev. 21, 1472–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Furuya K., Poitelea M., Guo L., Caspari T., and Carr A. M. (2004) Chk1 activation requires Rad9 S/TQ-site phosphorylation to promote association with C-terminal BRCT domains of Rad4TOPBP1. Genes Dev. 18, 1154–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Majka J., Binz S. K., Wold M. S., and Burgers P. M. (2006) Replication protein A directs loading of the DNA damage checkpoint clamp to 5′-DNA junctions. J. Biol. Chem. 281, 27855–27861 [DOI] [PubMed] [Google Scholar]
- 18.Navadgi-Patil V. M., and Burgers P. M. (2009) The unstructured C-terminal tail of the 9–1-1 clamp subunit Ddc1 activates Mec1/ATR via two distinct mechanisms. Mol Cell 36, 743–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou Z. W., Liu C., Li T. L., Bruhn C., Krueger A., Min W., Wang Z. Q., and Carr A. M. (2013) An essential function for the ATR-activation-domain (AAD) of TopBP1 in mouse development and cellular senescence. PLoS Genet 9, e1003702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kumar S., and Burgers P. M. (2013) Lagging strand maturation factor Dna2 is a component of the replication checkpoint initiation machinery. Genes Dev. 27, 313–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pfander B., and Diffley J. F. (2011) Dpb11 coordinates Mec1 kinase activation with cell cycle-regulated Rad9 recruitment. EMBO J. 30, 4897–4907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biegert A., Mayer C., Remmert M., Söding J., and Lupas A. N. (2006) The MPI Bioinformatics Toolkit for protein sequence analysis. Nucleic Acids Res. 34, W335–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crooks G. E., Hon G., Chandonia J. M., and Brenner S. E. (2004) WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Majka J., and Burgers P. M. (2003) Yeast Rad17/Mec3/Ddc1: a sliding clamp for the DNA damage checkpoint. Proc. Natl. Acad. Sci. U.S.A. 100, 2249–2254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma J. L., Lee S. J., Duong J. K., and Stern D. F. (2006) Activation of the checkpoint kinase Rad53 by the phosphatidyl inositol kinase-like kinase Mec1. J. Biol. Chem. 281, 3954–3963 [DOI] [PubMed] [Google Scholar]
- 26.Doré A. S., Kilkenny M. L., Rzechorzek N. J., and Pearl L. H. (2009) Crystal Structure of the Rad9-Rad1-Hus1 DNA Damage Checkpoint Complex- Implications for Clamp Loading and Regulation. Mol. Cell 34, 735–745 [DOI] [PubMed] [Google Scholar]
- 27.Sohn S. Y., and Cho Y. (2009) Crystal Structure of the Human Rad9-Hus1-Rad1 Clamp. J. Mol. Biol. 390, 490–502 [DOI] [PubMed] [Google Scholar]
- 28.Xu M., Bai L., Gong Y., Xie W., Hang H., and Jiang T. (2009) Structure and functional implications of the human rad9-hus1-rad1 cell cycle checkpoint complex. J. Biol. Chem. 284, 20457–20461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wanrooij P. H., and Burgers P. M. (2015) Yet another job for Dna2: Checkpoint activation. DNA Repair 32, 17–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanchez Y., Desany B. A., Jones W. J., Liu Q., Wang B., and Elledge S. J. (1996) Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science 271, 357–360 [DOI] [PubMed] [Google Scholar]
- 31.Bastos de Oliveira F. M., Kim D., Cussiol J. R., Das J., Jeong M. C., Doerfler L., Schmidt K. H., Yu H., and Smolka M. B. (2015) Phosphoproteomics reveals distinct modes of Mec1/ATR signaling during DNA replication. Mol. Cell 57, 1124–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Savitsky K., Bar-Shira A., Gilad S., Rotman G., Ziv Y., Vanagaite L., Tagle D. A., Smith S., Uziel T., Sfez S., Ashkenazi M., Pecker I., Frydman M., Harnik R., Patanjali S. R., Simmons A., Clines G. A., Sartiel A., Gatti R. A., Chessa L., Sanal O., Lavin M. F., Jaspers N. G., Taylor A. M., Arlett C. F., Miki T., Weissman S. M., Lovett M., Collins F. S., and Shiloh Y. (1995) A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 268, 1749–1753 [DOI] [PubMed] [Google Scholar]
- 33.Brown E. J., and Baltimore D. (2000) ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 14, 397–402 [PMC free article] [PubMed] [Google Scholar]
- 34.de Klein A., Muijtjens M., van Os R., Verhoeven Y., Smit B., Carr A. M., Lehmann A. R., and Hoeijmakers J. H. (2000) Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr. Biol. 10, 479–482 [DOI] [PubMed] [Google Scholar]
- 35.Cortez D., Guntuku S., Qin J., and Elledge S. J. (2001) ATR and ATRIP: partners in checkpoint signaling. Science 294, 1713–1716 [DOI] [PubMed] [Google Scholar]
- 36.O'Driscoll M., Ruiz-Perez V. L., Woods C. G., Jeggo P. A., and Goodship J. A. (2003) A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat. Genet. 33, 497–501 [DOI] [PubMed] [Google Scholar]
- 37.Kumagai A., and Dunphy W. G. (2006) How cells activate ATR. Cell Cycle 5, 1265–1268 [DOI] [PubMed] [Google Scholar]
- 38.Choi J. H., Lindsey-Boltz L. A., and Sancar A. (2007) Reconstitution of a human ATR-mediated checkpoint response to damaged DNA. Proc. Natl. Acad. Sci. U.S.A. 104, 13301–13306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sen T. Z., Jernigan R. L., Garnier J., and Kloczkowski A. (2005) GOR V server for protein secondary structure prediction. Bioinformatics 21, 2787–2788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dosztányi Z., Csizmok V., Tompa P., and Simon I. (2005) IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 21, 3433–3434 [DOI] [PubMed] [Google Scholar]