

Abstract
Pro-inflammatory cytokines secreted by adipose tissue macrophages (ATMs) contribute to chronic low-grade inflammation and obesity-induced insulin resistance. Recent studies have shown that adipose tissue hypoxia promotes an inflammatory phenotype in ATMs. However, our understanding of how hypoxia modulates the response of ATMs to free fatty acids within obese adipose tissue is limited. We examined the effects of hypoxia (1% O2) on the pro-inflammatory responses of human monocyte-derived macrophages to the saturated fatty acid palmitate. Compared with normoxia, hypoxia significantly increased palmitate-induced mRNA expression and protein secretion of IL-6 and IL-1β. Although palmitate-induced endoplasmic reticulum stress and nuclear factor κB pathway activation were not enhanced by hypoxia, hypoxia increased the activation of JNK and p38 mitogen-activated protein kinase signaling in palmitate-treated cells. Inhibition of JNK blocked the hypoxic induction of pro-inflammatory cytokine expression, whereas knockdown of hypoxia-induced transcription factors HIF-1α and HIF-2α alone or in combination failed to reduce IL-6 and only modestly reduced IL-1β gene expression in palmitate-treated hypoxic macrophages. Enhanced pro-inflammatory cytokine production and JNK activity under hypoxia were prevented by inhibiting reactive oxygen species generation. In addition, silencing of dual-specificity phosphatase 16 increased normoxic levels of IL-6 and IL-1β and reduced the hypoxic potentiation in palmitate-treated macrophages. The secretome of hypoxic palmitate-treated macrophages promoted IL-6 and macrophage chemoattractant protein 1 expression in primary human adipocytes, which was sensitive to macrophage JNK inhibition. Our results reveal that the coexistence of hypoxia along with free fatty acids exacerbates macrophage-mediated inflammation.
Keywords: cytokine, fatty acid, hypoxia, inflammation, macrophage
Introduction
Adipose tissue inflammation is a major driver of obesity-induced insulin resistance and associated metabolic diseases (1, 2). Its development is attributed to the accumulation of macrophages and other immune cell types and the overproduction of pro-inflammatory cytokines (3, 4). Macrophage accumulation is mediated by recruitment of blood monocytes as well as by in situ proliferation of resident macrophages (5, 6). Additionally, the phenotype of ATM3 subpopulations shifts from a non-inflammatory phenotype toward a pro-inflammatory phenotype characterized by the production of pro-inflammatory cytokines, which impair adipocyte function and promote insulin resistance (7, 8).
Of the factors promoting adipose tissue inflammation, elevated FFAs are considered to be of most relevance. Adipose tissue FFA levels rise within 3 days of consuming a high-fat diet (9). Particularly important in the context of inflammation are saturated fatty acids, which induce the secretion of pro-inflammatory cytokines in macrophages (10, 11). Saturated fatty acids engage inflammatory signaling pathways through various mechanisms, including direct activation of Toll-like receptor 2 and Toll-like receptor 4 (11, 12); disruption of endoplasmic reticulum (ER) homeostasis, causing the unfolded protein response (13); and nucleotide-binding oligomerization domain-like receptor family, pyrin domain-containing 3 (NLRP3) inflammasome activation, causing maturation and release of IL-1β (14).
Adipose tissue is poorly oxygenated in the obese state (4, 15, 16). Development of adipose tissue hypoxia is multifactorial and is thought to be driven by adipocyte hypertrophy, compromised vascularization (4, 17), and increased adipocyte oxygen consumption through FFA-induced uncoupling (9). Recent studies have suggested hypoxia as a potential cause of the inflammatory changes occurring in obese adipose tissue (9, 18, 19). Hypoxia has been shown to increase inflammatory cytokine secretion from macrophages in stromal vascular fractions (19). Furthermore, macrophages derived from hypoxic obese adipose tissue expressed higher levels of the hypoxia-related markers Hif-1α and Glut1, the inflammatory marker genes Cd11c and Nos2, and the cytokines Il-6 and Il-1β compared with non-hypoxic macrophages (18). These results indicate that adipose tissue hypoxia promotes a pro-inflammatory ATM phenotype. However, the mechanisms by which mediators within the obese adipose tissue initiate this phenotype are not completely understood.
Although saturated FFA-induced pro-inflammatory signaling pathways in macrophages are well documented (11), it is unclear how hypoxia modulates this response. To address this issue, we treated primary human macrophages with palmitate under both normoxia and hypoxia and assessed their pro-inflammatory response. Our results showed that decreased oxygen tension potentiated palmitate-induced pro-inflammatory cytokine production through enhanced JNK activity. Furthermore, the secretome of hypoxic palmitate-treated macrophages promoted inflammatory shifts in adipokine expression in primary human adipocytes.
Experimental Procedures
Human Macrophage and Adipocyte Differentiation
Human monocytes were isolated from commercially obtained buffy coats from anonymous donors (Deutsches Rotes Kreuz Blutspendedienst Baden-Württemberg-Hessen, Institut für Transfusionsmedizin und Immunhämatologie, Frankfurt, Germany) using Ficoll density centrifugation followed by magnetic separation with positive selection (CD14 MicroBeads, Miltenyi Biotec, Bergisch Gladbach, Germany). Monocytes were differentiated into macrophages by culture in macrophage serum-free medium (Life Technologies) containing 50 ng/ml recombinant human macrophage colony-stimulating factor (Immunotools, Friesoythe, Germany) for 7 days. Following differentiation, cells were cultured in RPMI 1640 medium supplemented with 10% FCS (PAA Laboratories, Cölbe, Germany), 100 units/ml penicillin, and 100 mg/ml streptomycin and maintained at 37 °C in a 5% CO2/air environment. Hypoxic incubations were performed using a hypoxic work station with 1% O2, 94% N2, and 5% CO2 at 37 °C (Invivo2 400, Ruskinn Technology, Leeds, UK).
Human primary preadipocytes were obtained from subcutaneous fat from anonymous donors undergoing elective cosmetic surgery (Rosenparkklinik, Darmstadt, Germany) and prepared according to an established protocol (20). In brief, adipose tissue was digested by collagenase solution (Collagenase NB 4G proved grade, Serva Electrophoresis, Heidelberg, Germany) and then centrifuged to remove mature adipocytes. The stromal vascular fraction (SVF) was incubated with erythrocyte lysing buffer and then filtered through 100-, 70-, and 40-μm cell strainers. The resulting preadipocytes were seeded in DMEM/Ham's F12 (1:1) medium (Life Technologies) containing 10% FCS (PAA Laboratories), 33 μm biotin, and 17 μm panthotenate. To induce adipocyte differentiation, the cells were cultured in DMEM/Ham's F12 (1:1) supplemented with 33 μm biotin, 17 μm panthotenate, 0.01 mg/ml transferrin, 20 nm insulin, 100 nm cortisol, 200 pm T3, 25 nm dexamethasone, 250 μm isobutylmethylxanthine and 2 μm rosiglitazone for 4 days, followed by incubation in differentiation medium without dexamethasone, isobutylmethylxanthine, and rosiglitazone for an additional 10 days. Following differentiation, adipocytes were cultured in DMEM/Ham's F12 (1:1) containing biotin and panthotenate. This investigation conformed to the ethical principles outlined in the Declaration of Helsinki and was approved by the ethics committee of Goethe University.
Maintenance of Mice
C57BL/6J mice were purchased from Charles River Laboratories (Sulzfeld, Germany) and bred in the animal housing facility at Frankfurt University. From the age of 8 weeks, animals were fed a high-fat diet consisting of 60% of calories from fat (catalog no. D12492, ssniff Spezialdiäten GmbH, Soest, Germany) for 20 weeks. Mice were housed under conditions that conform to the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (publication no. 85-23). All experiments were approved by government authorities (Regierungspräsidium Darmstadt: FU1012).
Isolation and Culture of Stromal Vascular Fractions
Epididymal fat pads from male C57BL/6J mice fed a high-fat diet were excised and minced in PBS with 0.5% BSA. Tissue suspensions were centrifuged to remove erythrocytes and free leukocytes. Adipose tissue was digested with collagenase (Serva Electrophoresis) for 30 min at 37 °C with constant shaking. The cell suspension was filtered through a 100-μm filter and then centrifuged to separate floating adipocytes from the SVF pellet.
For cell culture, the SVF pellet was resuspended at 1 million SVF cells/ml of RPMI 1640 medium supplemented with 10% FCS and cultured under standard culture conditions at 37 °C in a 5% CO2/air environment. After 1 h of incubation, medium containing non-adherent cells was aspirated and replaced with fresh complete medium. For flow cytometry analysis of macrophage content in SVF, cultured SVF cells were transferred to FACS tubes, and nonspecific antibody binding to FC-γ receptors was blocked using mouse BD Fc block (BD Biosciences). Next, cells were incubated with antibodies for CD45, F4/80, and CD11b on ice for 30 min. Samples were acquired using an LSRII/Fortessa flow cytometer (BD Biosciences) and analyzed using FlowJo software Vx.0.7 (TreeStar, Ashland, OR). Dead cells were excluded with 7-amino actinomycin D. Macrophages were identified as CD45-positive/F4/80-positive/CD11b-positive.
Fatty Acid Preparations
Palmitate (catalog no. P5585, Sigma-Aldrich, Steinheim, Germany), stearate (catalog no. S4751, Sigma-Aldrich), and myristate (catalog no. M3128, Sigma-Aldrich) were prepared by diluting 100 mm stock solution in 70% ethanol/0.1 M NaOH into 10% fatty acid-free, low-endotoxin BSA solution (catalog no. A-8806, Sigma-Aldrich, adjusted to pH 7.4) to obtain a fatty acid:BSA molar ratio of 6:1. BSA was used in control incubations.
Macrophage-conditioned Medium
Macrophages were treatedwith palmitate in hypoxia for 24 h in serum-free medium. Two hours after treatment, the palmitate-containing medium was replaced with fresh serum-free medium without palmitate and incubated for an additional 22 h under hypoxia. Supernatants were collected, clarified by centrifugation, and used to stimulate adipocytes in a 1:4 dilution with culture medium for 24 h.
HIF-1α, HIF-2α, DUSP16, and DUSP1 Knockdown in Primary Human Macrophages
Knockdown of HIF-1α, HIF-2α, DUSP16, and DUSP1 was achieved using siRNA (siGENOME human SMARTpool, Thermo Scientific, Karlsruhe, Germany) at 50 nm and Hyperfect transfection reagent (Qiagen, Hilden, Germany) according to the recommendations of the manufacturer. Cells were treated 72 h post-transfection.
RNA Extraction and Quantitative Real-time PCR
Total RNA from human macrophages and adipocytes was isolated using PeqGold RNA Pure reagent (PeqLab Biotechnology, Erlangen, Germany), quantified using a NanoDrop spectrophotometer (NanoDrop), and transcribed using the Maxima first-strand cDNA synthesis kit (Fermentas, St. Leon-Rot, Germany). Total RNA from SVFs was isolated using PeqGold RNA Pure reagent and the RNeasy MinElute cleanup kit (Qiagen). Quantitative real-time PCR was performed using iQ SYBR Green Supermix (Bio-Rad) and the Bio-Rad CFX96 system. Primer sequences for quantitative PCR are available upon request. β2 microglobulin was used as an endogenous control for human macrophages, Actin as an endogenous control for human adipocytes, and 18S as an endogenous control for murine SVF cells.
Immunoblots
Macrophage lysates were resolved on 10% polyacrylamide gels followed by transfer onto nitrocellulose membranes. Membranes were incubated with antibodies against phospho-IRE1 (Ser(P)724, Abcam, Cambridge, UK); phospho-c-Jun (Ser-73, catalog no. 3270), total c-Jun (catalog no. 2315), phospho-p38 (Thr-180/Tyr-182, catalog no. 9216), total p38 (catalog no. 8690), phospho-ERK 1/2 (Thr-202/Tyr-204, catalog no. 9101), phospho-IκBα (Ser-32/Ser-36, catalog no. 8219), and IκBα (catalog no. 4814) (Cell Signaling Technology); and Actin (catalog no. A-2066, Sigma-Aldrich) followed by IRDye 680 and IRDye 800-coupled secondary antibodies (LI-COR Biosciences, Bad Homburg, Germany). Blots were visualized and quantified using the Odyssey imaging system (LI-COR Biosciences).
Cytokine Detection in Supernatants
Macrophage supernatants were analyzed for IL-6, IL-1β, IL-8, IL-10, and TNF-α using a cytometric bead array (BD Biosciences) according to the recommendations of the manufacturer. Samples were measured using a LSRII/Fortessa flow cytometer (BD Biosciences) and analyzed by FCAP array software v1.0 (Soft Flow Inc.).
ROS Measurements
Mitochondrial ROS generation in macrophages was assessed by MitoSOX (Life Technologies), a red mitochondrial superoxide indicator. When in mitochondria, MitoSOX is oxidized by superoxide and exhibits red fluorescence. Macrophages were incubated with 5 μm MitoSOX for 30 min at 37 °C. Cells were then placed under hypoxia or left under normoxia for 30 min before treatment with or without palmitate for 1 h. Fluorescence was analyzed using a LSRII/Fortessa flow cytometer (BD Biosciences).
Statistical Analysis
Graphical data are presented as mean ± S.E. of at least three independent experiments. Gene expression and cytokine concentrations were analyzed by one-way analysis of variance with Bonferroni post hoc means comparisons with the significance level set at 0.05. Student's t test was used to determine the significance of Western blot densitometry. p < 0.05 was considered statistically significant (GraphPad, La Jolla, CA).
Results
Hypoxia Enhances Palmitate-induced Pro-inflammatory Cytokine Production in Primary Human Macrophages
Recently, adipose tissue hypoxia has been reported in obese subjects and obese animal models (9, 17). Because macrophages are a primary source of inflammatory cytokine production in the expanding adipose tissue, we questioned how hypoxia modulates saturated fatty acid-induced, pro-inflammatory signaling in macrophages (8). Under normoxic conditions (20% O2), palmitate increased the mRNA expression and protein secretion of TNF-α, IL-6, and IL-8 compared with BSA alone. Under hypoxic conditions (1% O2), palmitate enhanced the expression of IL-6, IL-8, and IL-1β as well as the secretion of IL-6 and IL-1β compared with normoxic palmitate treatments. In contrast, the expression and secretion of the anti-inflammatory cytokine IL-10 were reduced by palmitate treatment under both normoxia and hypoxia (Fig. 1, A–J).
FIGURE 1.

Hypoxia potentiates palmitate-induced, pro-inflammatory cytokine expression and secretion in primary human macrophages. A–E, mRNA expression of TNF-α (A), IL-6 (B), IL-1β (C), IL-8 (D), and IL-10 (E) in primary human macrophages treated with 500 μm palmitate (C16:0) or BSA alone for 9 h under normoxia (20% O2) or hypoxia (1% O2). F–J, protein concentrations of TNF-α (F), IL-6 (G), IL-1β (H), IL-8 (I), and IL-10 (J) in cell culture supernatants after 24-h treatment with C16:0 or BSA alone under normoxia or hypoxia. K–O, mRNA expression of Tnfa (K), Il6 (L), Il1b (M), Nos2 (N), and Il10 (O) in murine SVFs treated with 500 μm C16:0 or BSA alone for 9 h under normoxia (20% O2) or hypoxia (1% O2). Results are presented as mean ± S.E. of at least four independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant.
To determine whether hypoxia also potentiates palmitate-induced pro-inflammatory activation in adipose tissue, we performed in vitro experiments using cultures of SVFs isolated from murine epididymal adipose tissue. Our flow cytometry analysis showed that 34.57% ± 3.15% of viable cultured SVF cells were CD45+/F4/80+/CD11b+ (data not shown). Under normoxic conditions (20% O2), palmitate increased the expression of Tnfa, Il6, and Il1b in SVF cells compared with BSA alone. Under hypoxic conditions, palmitate significantly enhanced the expression of Il1b and Nos2 compared with normoxic palmitate treatments. Il10 was also reduced by palmitate treatment in both normoxic and hypoxic SVF cells (Fig. 1, K–O).
Reduced Oxygen Levels Dose-dependently Potentiate Palmitate-induced, Pro-inflammatory Cytokine Expression
To determine whether moderate levels of hypoxia also enhance palmitate-induced, pro-inflammatory cytokine expression, we treated human macrophages with palmitate under 2.5% and 5.0% O2. At 2.5% O2, IL-6, IL-8, and IL-1β expression levels were enhanced by palmitate compared with normoxic palmitate treatments. At 5.0% O2, both IL-8 and IL-1β expression levels were enhanced by palmitate compared with normoxic palmitate treatments (Fig. 2, A–D), suggesting that moderate hypoxia (>1% O2) also potentiates palmitate-induced, pro-inflammatory cytokine expression. Furthermore, to determine whether the pro-inflammatory potentiation of palmitate under hypoxia is unique to palmitate, we treated macrophages with the longer-chain saturated fatty acid stearate (18-carbon chain) or the shorter-chain saturated fatty acid myristate (14-carbon chain). Under normoxic conditions, stearate increased the mRNA expression of TNF-α, IL-6, and IL-8 compared with BSA alone, whereas, under hypoxic conditions (1% O2), stearate further enhanced the expression of IL-6, IL-8, and IL-1β compared with normoxic stearate treatments. In contrast, myristate failed to induce cytokine expression under either normoxic or hypoxic conditions (Fig. 2, E–H). Collectively, these results imply that palmitate- and stearate-induced pro-inflammatory activation of macrophages are potentiated by hypoxia.
FIGURE 2.

Reduced oxygen levels dose-dependently potentiate palmitate-induced, pro-inflammatory cytokine expression. A–D, mRNA expression of TNF-α (A), IL-6 (B), IL-1β (C), and IL-8 (D) in primary human macrophages treated with 500 μm palmitate (C16:0) or BSA alone for 9 h under normoxia (20% O2) or moderate hypoxia (5% and 2.5% O2). E–H, mRNA expression of TNF-α (E), IL-6 (F), IL-1β (G), and IL-8 (H) in primary human macrophages treated with 500 μm myristate (C14:0), 500 μm stearate (C18:0), or BSA alone for 9 h under normoxia (20% O2) or hypoxia (1% O2). Results are presented as mean ± S.E. of at least four independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Hypoxia Does Not Enhance Palmitate-induced ER Stress
To elucidate how hypoxia augments palmitate-induced, pro-inflammatory activation of macrophages, we first focused on mechanisms whereby lipids and hypoxia are known to induce inflammatory signaling. Both saturated fatty acids and hypoxia have been shown to induce the unfolded protein response, which can activate the MAPK and NF-κB signaling pathways (21, 22). We questioned whether hypoxia heightens palmitate-triggered ER stress. For this, we treated macrophages with palmitate under normoxia and hypoxia and analyzed the mRNA expression of the ER stress markers GRP78 and CHOP as well as phosphorylation of the ER stress sensor inositol-requiring protein 1 (IRE1). Treatment with palmitate under normoxia increased expression of GRP78 and CHOP as well as the phosphorylation levels of IRE1 at 9 h (Fig. 3, A–C) and 24 h (data not shown). Interestingly, under hypoxic conditions, palmitate did not further increase the ER stress markers. Furthermore, there were no differences in GRP78 or CHOP transcript levels or IRE1 phosphorylation between the normoxic and hypoxic control-treated (BSA alone) macrophages, suggesting that 1% O2 does not induce the unfolded protein response in primary human macrophages. Collectively, these results suggest that augmented palmitate-induced inflammation in hypoxia is probably not mediated by increased levels of ER stress. Because ER stress and inflammatory signals may cause macrophage apoptosis, we tested the effects of palmitate and hypoxia treatments on macrophage viability using Annexin V/propidium iodide staining. Our results show that, under normoxia (20% O2), 24-h palmitate treatments did not induce apoptosis in human macrophages. Although hypoxia (1% O2) alone did not enhance apoptosis, the combination of palmitate and hypoxia did increase the levels of apoptosis in human macrophages (data not shown).
FIGURE 3.
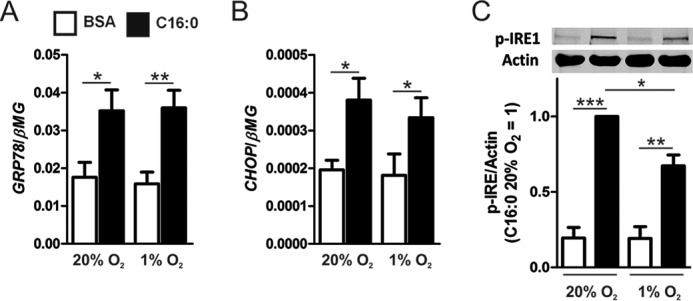
Hypoxia does not induce or potentiate palmitate-induced markers of ER stress. A and B, mRNA expression of GRP78 (A) and CHOP (B) at 9 h in macrophages treated with 500 μm palmitate (C16:0) under normoxia (20% O2) or hypoxia (1% O2). C, phosphorylation of IRE1 9 h after treatment with C16:0 or BSA alone under normoxia or hypoxia. Results are presented as mean ± S.E. of at least four independent experiments. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Palmitate-induced Cytokine Production under Hypoxia Is HIF-1α- and HIF-2α-independent
HIFs are transcription factors that regulate cellular adaptation to reduced oxygen tension (23). In addition to regulating metabolism, HIFs have been shown to increase pro-inflammatory gene expression in macrophages (24). Our previous studies have shown that HIF protein levels increase in macrophages cultured in 1% O2 (25). To determine the role of HIFs in palmitate-treated macrophages, we used siRNA to silence both HIF-1α and HIF-2α individually or in combination (Fig. 4, A and B) and assessed changes in mRNA expression of a known HIF target gene, GLUT1, as well as pro-inflammatory cytokines. As expected, GLUT1 mRNA expression was reduced significantly in palmitate- or BSA-treated hypoxic macrophages upon HIF knockdown (KD) (Fig. 4C). Interestingly, neither HIF-1α- nor HIF-2α-targeting siRNA or a combination of HIF-1α and HIF-2α siRNA reduced mRNA expression or protein levels of IL-6 (Fig. 4, D and G) or IL-8 (Fig. 4, F and I) compared with hypoxic palmitate-treated control siRNA-transfected macrophages. Only a combination of HIF-1α- and HIF-2α-targeting siRNAs modestly reduced the mRNA expression levels of IL-1β, which translated to a slight but non-significant reduction of IL-1β protein compared with (Fig. 4, E and H) hypoxic palmitate-treated control siRNA-transfected macrophages. These results suggest that palmitate-induced production of IL-6 and IL-1β under hypoxia is predominately independent of both HIF isoforms.
FIGURE 4.

Enhanced palmitate-induced, pro-inflammatory cytokine production under hypoxia is HIF-1α- and HIF-2α-independent. A–F, mRNA expression levels of HIF-1α (A), HIF-2α (B), the HIF target gene GLUT1 (C), IL-6 (D), IL-1β (E), and IL-8 (F) in macrophages transfected with control, HIF-1α, HIF-2α, or HIF-1α/HIF-2α siRNA and treated with 500 μm palmitate (C16:0) for 9 h under normoxia (20% O2) or hypoxia (1% O2). G–I, protein concentrations of IL-6 (G), IL-1β (H), and IL-8 (I) in macrophages transfected with control, HIF-1α, HIF-2α, or HIF-1α/HIF-2α siRNA and treated with 500 μm C16:0 for 24 h under normoxia (20% O2) or hypoxia (1% O2). Results are presented as mean ± S.E. of at least three independent experiments. C–F, the expression in hypoxic C16:0-treated control siRNA macrophages was set to 1. *, p < 0.05.
Hypoxia Enhances Palmitate-induced Cytokine Production through MAPK but Not NF-κB Signaling
The MAPK and NF-κB signaling cascades are major inflammatory pathways activated by palmitate (11). We determined whether either of these pathways contributed to enhanced palmitate-induced cytokine production in hypoxia. Evaluation of the phosphorylation levels of the JNK substrates c-Jun or p38 MAPK revealed time-dependent increases of phospho-p38 and phospho-c-Jun upon palmitate treatment (Fig. 5A). Palmitate-induced phosphorylation of p38 and c-Jun was increased under hypoxia compared with normoxia (Fig. 5, B and C). Stearate-induced phosphorylation of p38 and c-Jun was also increased under hypoxia compared with normoxia, whereas myristate failed to induce a robust MAPK response under both normoxia and hypoxia (Fig. 5D). BSA alone, under both normoxia and hypoxia, did not induce phosphorylation of p38 and c-Jun. Unlike p38 and c-Jun, ERK 1/2 phosphorylation levels were not enhanced by palmitate treatment under hypoxia. Severe hypoxia, less than 1% O2, has been shown to activate the NF-κB signaling pathway in macrophages (26, 27). However, in our experiments, exposure to 1% O2 did not enhance the levels of palmitate-induced p-IκBα compared with normoxia (data not shown). To further assess the contribution of p38 and JNK to IL-6 and IL-1β expression, we pretreated cells with inhibitors of p38 (SB203580) and JNK (SP600125). Under normoxia, neither inhibitor reduced the palmitate-induced expression levels of IL-6, IL-1β, or IL-8 (Fig. 5, E–G). However under hypoxia, both inhibitors suppressed the expression of palmitate-induced IL-6 and IL-8 (Fig. 5, E and G). SP600125 also suppressed IL-1β expression under hypoxia, whereas SB203580 enhanced its expression (Fig. 5F). These results suggest that palmitate-enhanced cytokine induction under hypoxia is not mediated by enhanced NF-κB signaling but is mediated, at least in part, through enhanced JNK activity. p38 MAPK activity may contribute to enhanced expression of some, but not all, cytokines under hypoxia.
FIGURE 5.

Hypoxia enhances palmitate-induced cytokine production through MAPK. A–C, phosphorylation of p38, c-Jun, and ERK 1/2 1, 3, and 9 h after treatment with 500 μm palmitate (C16:0) in normoxic (20% O2) and hypoxic (1% O2) macrophages. D, phosphorylation of p38 and c-Jun following treatment with 500 μm myristate (C14:0), 500 μm stearate (C18:0), or BSA alone for 3 h under normoxia or hypoxia. E–G, mRNA expression of IL-6 (E), IL-1β (F), and IL-8 (G) in macrophages pretreated with JNK inhibitor (SP600125, 10 μm) or p38 inhibitor (SB203580, 10 μm) and then co-incubated for 9 h with 500 μm C16:0. Results are presented as mean ± S.E. of at least three independent experiments. *, p < 0.05.
NAC and MitoTEMPO Abolish Palmitate-induced c-Jun and p38 Phosphorylation and Cytokine Expression in Hypoxic Macrophages
Hypoxia has been shown to induce cellular and mitochondrial ROS production through mechanisms not entirely understood (28–32). To determine whether incubation under hypoxic conditions (1% O2) induces ROS production in macrophages, we measured mitochondrially derived ROS using MitoSOX. Our data show that ROS was increased after 1-h incubation under hypoxia but not altered by palmitate treatment (Fig. 6A). Next, because tissue hypoxia has been shown to activate JNK in a ROS-dependent manner, which can be attenuated by the antioxidant N-acetylcysteine (NAC) (33, 34), we determined whether NAC and the mitochondria-targeted antioxidant MitoTEMPO could inhibit palmitate-induced, pro-inflammatory cytokine expression under hypoxia. Although NAC failed to reduce palmitate-induced expression of IL-6, IL-1β, and IL-8 under normoxia, both NAC and MitoTEMPO significantly reduced expression levels of these cytokines under hypoxia (Fig. 6, B–D). Furthermore, both NAC and MitoTEMPO pretreatment reduced palmitate-induced phosphorylation of c-Jun and p38 in hypoxic macrophages compared with palmitate alone (Fig. 6, E–H). These results suggest that hypoxia-induced intracellular ROS contribute to enhanced c-Jun and p38 phosphorylation, augmenting cytokine production in palmitate-treated macrophages.
FIGURE 6.

Mitochondrial ROS in palmitate-induced inflammation under hypoxia. A, mitochondrial ROS measured by MitoSOX fluorescence in primary human macrophages treated with 500 μm palmitate (C16:0) or BSA alone for 1 h under normoxia (20% O2) or hypoxia (1% O2). B–D, mRNA expression of IL-6 (B) IL-1β (C), and IL-8 (D) in normoxic and hypoxic macrophages pretreated with 5 mm NAC or 1 μm MitoTEMPO (MT) and then co-incubated for 9 h with 500 μm C16:0. E–H, phosphorylation of c-Jun (E and G) and p38 (F and H) in hypoxic macrophages pretreated with NAC or MitoTEMPO and then co-incubated with 500 μm C16:0 for 3 h. Results are presented as mean ± S.E. of at least three independent experiments. MitoSOX fluorescence in normoxic C16:0-treated macrophages was set to 1. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Dual-specificity Phosphatase 16 (DUSP16) Negatively Regulates c-Jun Phosphorylation and Pro-inflammatory Cytokine Production
MAPK inactivation occurs primarily through dephosphorylation of the threonine and tyrosine residues of the shared TXY motif by DUSPs, which dephosphorylate their substrates using a catalytic cysteine located in the conserved enzyme active center (35). The catalytic cysteine is highly sensitive to oxidation and inactivation under conditions of elevated ROS production, such as hypoxia (36, 37). Therefore, we questioned whether enhanced palmitate-induced JNK activity and cytokine production under hypoxia were mediated through reduced JNK-specific MAPK phosphatase activity. For this reason, we focused on DUSP16, because of its reported specificity for JNK in macrophages (37, 38), and on DUSP1, a well characterized component of inflammatory signaling in immune cells (39, 40). To examine their roles, we transfected macrophages with DUSP16- or DUSP1-specific siRNA. Our data show that DUSP1 was up-regulated markedly after 9-h treatment with palmitate under both normoxia (7.7-fold) and hypoxia (13.3-fold) compared with DUSP16, which was up-regulated only modestly after treatment under hypoxia (1.8-fold). Both DUSP1 and DUSP16 remained low in DUSP siRNA-targeted cells (Fig. 7, A and F). KD of DUSP16 or DUSP1 enhanced JNK activity, as assessed by c-Jun phosphorylation (Fig. 7, B and H), but only KD of DUSP1 enhanced phosphorylation of p38 (Fig. 7, C and I). Furthermore, both DUSP16- and DUSP1-silenced cells showed significantly increased mRNA expression of IL-6 (DUSP16, 3.5-fold versus 1.7-fold in control cells; DUSP1, 3.4-fold versus 1.9-fold in control cells) and IL-1β (DUSP16, 3.7-fold versus 2.2-fold in control cells; DUSP1, 5.3-fold versus 2.6-fold in control cells) after palmitate treatment under normoxia. However, under hypoxia, only DUSP1-silenced cells showed significantly increased mRNA expression of IL-6 (7.4-fold versus 3.3-fold in control cells) and IL-1β (3.8-fold versus 2.3-fold in control cells) after palmitate treatment because hypoxia failed to further increase palmitate-mediated expression levels of IL-6 (3.6-fold versus 4.6-fold in control cells) or IL-1β (2.9-fold versus 3.4-fold in control cells) in DUSP16 KD macrophages (Fig. 7, D, E, J, and K). The potentiation of palmitate in DUSP1-silenced but not DUSP16-silenced hypoxic macrophages suggests that DUSP16 may be inactivated under hypoxia.
FIGURE 7.

Attenuating DUSP expression increases c-Jun phosphorylation and potentiates palmitate-induced, pro-inflammatory cytokine expression. A, mRNA expression of DUSP16 in macrophages transfected with DUSP16 siRNA and then treated with 500 μm palmitate (C16:0) for 9 h under normoxia and hypoxia. B and C, phosphorylation of c-Jun (B) and p38 (C) following transfection with DUSP16 siRNA. D and E, mRNA expression of IL-6 (D) and IL-1β (E) in macrophages transfected with DUSP16 siRNA and then treated with 500 μm C16:0 for 9 h under normoxia and hypoxia. F and G, mRNA expression (F) and protein (G) of DUSP1 in macrophages transfected with DUSP1 siRNA and then treated with 500 μm C16:0 for 9 h under normoxia and hypoxia. H and I, phosphorylation of c-Jun (H) and p38 (I) following transfection with DUSP1 siRNA. J and K, mRNA expression of IL-6 (J) and IL-1β (K) in macrophages transfected with DUSP1 siRNA and then treated with 500 μm C16:0 for 9 h under normoxia and hypoxia. Results are presented as mean ± S.E. of at least three independent experiments. D, E, J, and K, the expression in normoxic C16:0-treated control siRNA macrophages was set to 1. *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant.
Macrophage-conditioned Medium Induces an Inflammatory Response in Primary Human Adipocytes
Dysregulated adipocytokine production is associated with the pathophysiology of obesity-related metabolic diseases (2). Therefore, we questioned how the palmitate-induced, pro-inflammatory response in hypoxic macrophages affects adipocytokine production in primary human adipocytes. For this, we treated human primary adipocytes with conditioned medium from palmitate- and hypoxia-exposed macrophages. The direct effects of palmitate on adipocyte gene expression were minimized by replacing palmitate-containing medium with fresh serum-free medium without palmitate after 2 h and incubating macrophages for an additional 22 h under hypoxia. Our data show that normoxic adipocyte cultures supplemented with macrophage-conditioned medium from hypoxic, palmitate-treated macrophages increased the mRNA expression levels of IL-6 and macrophage chemoattractant protein 1 (MCP-1) (Fig. 8, A and B). Adiponectin transcript levels were reduced in adipocytes cultured with macrophage-conditioned medium from palmitate-treated macrophages under hypoxia (Fig. 8C). Furthermore, supplementation of macrophage-conditioned medium from hypoxic, palmitate-treated macrophages pre-exposed to the JNK inhibitor SP600125, but not the p38 inhibitor SB203580, reduced mRNA expression levels of IL-6 and MCP-1 in normoxic human adipocytes (Fig. 8D), further supporting the significance of JNK in augmenting the palmitate-induced, pro-inflammatory response in hypoxic primary human macrophages. Macrophage-conditioned medium did not alter insulin signaling in adipocytes, as assessed by Akt or AS160 phosphorylation and insulin-stimulated glucose uptake (data not shown). Collectively, these results indicate that hypoxia contributes to a pro-inflammatory milieu of adipose tissue by potentiating inflammatory macrophage polarization.
FIGURE 8.

Conditioned media from hypoxic, palmitate-treated human macrophages induce an inflammatory response in primary human adipocytes. A–C, mRNA expression levels of IL-6 (A), MCP-1 (B), and Adiponectin (C) in normoxic adipocytes following treatment with macrophage-conditioned media. D, mRNA expression levels of IL-6 and MCP-1 in normoxic adipocytes following treatment with conditioned media prepared from hypoxic, palmitate-treated macrophages preincubated with 10 μm SP600125 or 10 μm SB203580. Expression in adipocytes incubated with conditioned media from C16:0-, vehicle-treated hypoxic macrophages was set to 1. E, proposed model by which decreased oxygen tension potentiates palmitate-induced, pro-inflammatory cytokine production in macrophages through enhanced JNK activity. Results are presented as mean ± S.E. of at least five independent experiments. *, p < 0.05.
Discussion
Recent data suggest that hypoxia in expanding adipose tissue promotes a pro-inflammatory ATM phenotype (18). Although saturated FFA-induced pro-inflammatory signaling pathways in macrophages are well documented (11, 14), our understanding of how hypoxia modulates the response of ATMs to saturated FFAs is limited. To answer this question, we explored the role of hypoxia in palmitate-induced inflammation in primary human macrophages. Our data demonstrate that hypoxia potentiates the production of the palmitate-induced, pro-inflammatory cytokines IL-6 and IL-1β. Enhanced macrophage pro-inflammatory cytokine production causes increased inflammatory responses in adipocytes exposed to macrophage-conditioned medium, suggesting that hypoxia may potentiate a pro-inflammatory adipocyte-macrophage cross-talk.
Our major finding is that activation of JNK signaling is critical for the hypoxic enhancement of fatty acid-triggered inflammatory responses. We found that hypoxia enhanced palmitate-stimulated p38 phosphorylation and JNK activity but not IκBα phosphorylation. This is interesting considering that 1% O2 has been shown to modestly induce phosphorylation of IκBα in HeLa cells (41), whereas severe hypoxia (0.1% O2) stimulated the expression and phosphorylation of various proteins in the NF-κB signaling pathway in macrophages (26). Inhibiting p38 reduced IL-6 expression but enhanced IL-1β, whereas JNK inhibition reduced the expression of both IL-6 and IL-1β. These data imply that, unlike 0.1% O2, 1% O2 failed to activate NF-κB signaling in macrophages and that JNK activity is predominantly responsible for the hypoxic potentiation of palmitate-induced, pro-inflammatory activation of macrophages, whereasp38 plays a more complex role.
Considering the mechanism by which hypoxia enhances palmitate-induced JNK activity in macrophages, we initially addressed the role of ER stress. Both saturated fatty acids and hypoxia have been shown to induce the unfolded protein response (21, 22), which activates JNK in an IRE1-dependent manner (42, 43). Although palmitate increased phosphorylation of IRE1 and transcript levels of GRP78 and CHOP in macrophages, neither of these were enhanced further under hypoxia.
Given that both NAC and MitoTEMPO reduced palmitate-induced, pro-inflammatory cytokine expression and JNK activity under hypoxia (Fig. 6, B–D), we questioned how hypoxia-induced ROS enhanced JNK activity. Because the catalytic cysteine in DUSPs have an unusually low pKa, they are highly sensitive to oxidation and inactivation (37). Therefore, many stimuli that trigger ROS production in cells can transiently induce the oxidation and inactivation of DUSPs, leading to enhanced MAPK signaling. ROS-induced oxidation of DUSP1 and DUSP10 has been shown to enhance JNK activation in endothelial cells and mesangial cells, respectively (36, 44). These findings indicate that hypoxia-induced ROS may enhance JNK activity in macrophages through DUSP inactivation. In macrophages, DUSP16 has been identified, along with DUSP1 and DUSP2, as the most strongly induced MAPK phosphatase (35). However, of these, only DUSP16 exclusively dephosphorylates JNK (38). Our data show that KD of both DUSP1 and DUSP16 in normoxic macrophages enhanced palmitate-induced, pro-inflammatory activation of IL-6 and IL-1β, which approached or exceeded levels expressed in palmitate-treated hypoxic cells. In contrast, KD of DUSP1 but not DUSP16 enhanced the potentiation of palmitate in hypoxic macrophages, suggesting that DUSP16 is likely inactivated under hypoxia.
How DUSP16 activity is reduced under hypoxia remains a question for further studies. In addition to potential oxidation of the catalytic cysteine, various DUSPs use a non-catalytic regulatory cysteine to bind the catalytic cysteine, thereby reversibly inactivating the enzyme and protecting it from irreversible oxidation (37). Amino acid alignment of DUSP16 shows that it contains a putative redox-regulatory cysteine in proximity to the catalytic cysteine (37), suggesting that hypoxia may modulate DUSP16 activity through either direct oxidation of the catalytic cysteine or through a non-catalytic redox-regulatory mechanism. Although ROS-induced oxidation of DUSP1 has been reported in endothelial cells (36), our data showed a 13.3-fold induction of DUSP1 compared with only a 1.8-fold induction of DUSP16 following hypoxic palmitate treatment. This, together with our KD results, which showed enhanced potentiation of IL-6 and IL-1β by palmitate in DUSP1-silenced but not DUSP16-silenced hypoxic macrophages, suggests that the robust induction of DUSP1 could act to counteract ROS-mediated inactivation. In addition to ROS-mediated oxidation, DUSPs have also been shown to be regulated through non-redox mechanisms, including ubiquitination/proteasome pathway-mediated degradation (45) and altered subcellular distribution (46). Our attempts to question the nature of DUSP16 modification under hypoxia so far failed because of the unsuitability of commercially available antibodies for immunoprecipitation and Western blotting of DUSP16 in human macrophages (data not shown).
HIFs have been implicated in adipose tissue inflammation and insulin resistance (9, 47). For this reason, we speculated that the potentiation of palmitate-induced, pro-inflammatory cytokines in hypoxic macrophages may be mediated by HIF-1α and HIF-2α. Although siRNAs targeting both HIF isoforms reduced expression of the HIF target gene GLUT1, transcript and protein levels of IL-6 were not reduced, and IL-1β was only modestly reduced in double KD macrophages. Considering the variations in cell type, stimulus, and hypoxic treatment length, our results are mostly consistent with a recent report showing that hypoxia-enhanced LPS-induced IL-1β expression was reduced in Hif1α-deficient murine macrophages (24). Furthermore, that report showed that TNF-α was not affected, whereas IL-6 expression was inhibited by HIF-1α. A possible explanation for the discrepancy with our results regarding IL-1β levels is that we used siRNA to knock down HIF-1α rather than knocking it out, which did not completely repress the HIF-1α transcript. Furthermore, our results could reflect the differences between acute and chronic hypoxia. Examining the role of HIF in ATMs has shown that in vitro exposure to hypoxia increased the expression of IL-6 and IL-1β but not TNF-α, which strongly supports our findings (18). Additionally, Choe et al. (47) have shown that knocking down HIF-2α enhanced palmitate-induced IL-6 expression, which is also in agreement with our findings. Collectively, although HIFs modulate various pro-inflammatory pathways, including IL1β expression, our data indicate that, during acute exposure to 1% O2, HIFs are not primarily responsible for potentiating the production of palmitate-induced, pro-inflammatory cytokines in macrophages.
Our results show that palmitate-induced, pro-inflammatory cytokines from hypoxic macrophages altered the expression of MCP-1, IL-6, and Adiponectin in primary human adipocytes. In addition to storing lipids, adipocytes possess an exceptionally active secretory system releasing many endocrine factors to facilitate cross-talk with other metabolic organs. These secreted factors are central to the dynamic control of energy metabolism but also display a strong immune regulatory function. In addition to recruiting blood monocytes to adipose tissue, MCP-1 has been reported recently to enhance obesity-associated adipose tissue inflammation by stimulating in situ ATM proliferation (5). Although we did not detect changes in insulin signaling in adipocytes, changes in IL-6 and Adiponectin are significant considering that dysregulated secretion of these adipokines adversely effects insulin sensitivity in the liver and fatty acid oxidation in both the liver and skeletal muscle, which can result in dysfunctional metabolic homeostasis (48).
It is well established that adipose tissue inflammation is a key component in the development of obesity-induced insulin resistance and associated metabolic diseases. Although the accumulation of macrophages in adipose tissue is the initial event in obesity-induced inflammation, the early events that initiate the inflammatory response are less well understood. Our results demonstrate that hypoxia enhances palmitate-induced, pro-inflammatory cytokine production in primary human macrophages through ROS-dependent modulation of JNK activity (Fig. 8E), which is capable of modulating adipocytokine production in primary human adipocytes. Therefore, the coexistence of low oxygen levels along with FFA in obese adipose tissue may promote an inflammatory shift of the ATM phenotype.
Author Contributions
R. G. S. designed, performed, and analyzed the experiments and wrote the paper. M. B., E. Z., and A. W. assisted with some of the experiments. N. D. and I. F. provided critical reagents. B. B. and D. N. participated in the study design, supervised the study, and wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
This study was supported by Deutsche Forschungsgemeinschaft Grants NA429/2-1, SFB1039 Teilprojekt, A05, B04, and Z01, and SFB815, Teilprojekt A08) and by the Else Kröner Fresenius Foundation (Translational Research Innovation—Pharma, TRIP). The authors declare that they have no conflicts of interest with the contents of this article.
- ATM
- adipose tissue macrophage
- FFA
- free fatty acid
- ER
- endoplasmic reticulum
- SVF
- stromal vascular fraction
- ROS
- reactive oxygen species
- HIF
- hypoxia-inducible factor
- KD
- knockdown
- NAC
- N-acetylcysteine
- DUSP
- dual-specificity phosphatase.
References
- 1.Chawla A., Nguyen K. D., and Goh Y. P. (2011) Macrophage-mediated inflammation in metabolic disease. Nat. Rev. Immunol. 11, 738–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McNelis J. C., and Olefsky J. M. (2014) Macrophages, immunity, and metabolic disease. Immunity 41, 36–48 [DOI] [PubMed] [Google Scholar]
- 3.Han J. M., and Levings M. K. (2013) Immune regulation in obesity-associated adipose inflammation. J. Immunol. 191, 527–532 [DOI] [PubMed] [Google Scholar]
- 4.Sun K., Kusminski C. M., and Scherer P. E. (2011) Adipose tissue remodeling and obesity. J. Clin. Invest. 121, 2094–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amano S. U., Cohen J. L., Vangala P., Tencerova M., Nicoloro S. M., Yawe J. C., Shen Y., Czech M. P., and Aouadi M. (2014) Local proliferation of macrophages contributes to obesity-associated adipose tissue inflammation. Cell Metab. 19, 162–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haase J., Weyer U., Immig K., Klöting N., Blüher M., Eilers J., Bechmann I., and Gericke M. (2014) Local proliferation of macrophages in adipose tissue during obesity-induced inflammation. Diabetologia 57, 562–571 [DOI] [PubMed] [Google Scholar]
- 7.Morris D. L., Singer K., and Lumeng C. N. (2011) Adipose tissue macrophages: phenotypic plasticity and diversity in lean and obese states. Curr. Opin. Clin. Nutr. Metab. Care 14, 341–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Osborn O., and Olefsky J. M. (2012) The cellular and signaling networks linking the immune system and metabolism in disease. Nat. Med. 18, 363–374 [DOI] [PubMed] [Google Scholar]
- 9.Lee Y. S., Kim J. W., Osborne O., Oh da Y., Sasik R., Schenk S., Chen A., Chung H., Murphy A., Watkins S. M., Quehenberger O., Johnson R. S., and Olefsky J. M. (2014) Increased adipocyte O2 consumption triggers HIF-1α, causing inflammation and insulin resistance in obesity. Cell 157, 1339–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Håversen L., Danielsson K. N., Fogelstrand L., and Wiklund O. (2009) Induction of proinflammatory cytokines by long-chain saturated fatty acids in human macrophages. Atherosclerosis 202, 382–393 [DOI] [PubMed] [Google Scholar]
- 11.Huang S., Rutkowsky J. M., Snodgrass R. G., Ono-Moore K. D., Schneider D. A., Newman J. W., Adams S. H., and Hwang D. H. (2012) Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J. Lipid Res. 53, 2002–2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Snodgrass R. G., Huang S., Choi I. W., Rutledge J. C., and Hwang D. H. (2013) Inflammasome-mediated secretion of IL-1β in human monocytes through TLR2 activation; modulation by dietary fatty acids. J. Immunol. 191, 4337–4347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hotamisligil G. S. (2005) Role of endoplasmic reticulum stress and c-Jun NH2-terminal kinase pathways in inflammation and origin of obesity and diabetes. Diabetes 54, S73–78 [DOI] [PubMed] [Google Scholar]
- 14.Wen H., Gris D., Lei Y., Jha S., Zhang L., Huang M. T., Brickey W. J., and Ting J. P. (2011) Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 12, 408–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trayhurn P. (2013) Hypoxia and adipose tissue function and dysfunction in obesity. Physiol. Rev. 93, 1–21 [DOI] [PubMed] [Google Scholar]
- 16.Ye J. (2011) Adipose tissue vascularization: its role in chronic inflammation. Curr. Diab. Rep. 11, 203–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pasarica M., Sereda O. R., Redman L. M., Albarado D. C., Hymel D. T., Roan L. E., Rood J. C., Burk D. H., and Smith S. R. (2009) Reduced adipose tissue oxygenation in human obesity: evidence for rarefaction, macrophage chemotaxis, and inflammation without an angiogenic response. Diabetes 58, 718–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fujisaka S., Usui I., Ikutani M., Aminuddin A., Takikawa A., Tsuneyama K., Mahmood A., Goda N., Nagai Y., Takatsu K., and Tobe K. (2013) Adipose tissue hypoxia induces inflammatory M1 polarity of macrophages in an HIF-1α-dependent and HIF-1alpha-independent manner in obese mice. Diabetologia 56, 1403–1412 [DOI] [PubMed] [Google Scholar]
- 19.O'Rourke R. W., White A. E., Metcalf M. D., Olivas A. S., Mitra P., Larison W. G., Cheang E. C., Varlamov O., Corless C. L., Roberts C. T. Jr., and Marks D. L. (2011) Hypoxia-induced inflammatory cytokine secretion in human adipose tissue stromovascular cells. Diabetologia 54, 1480–1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hauner H., Skurk T., and Wabitsch M. (2001) Cultures of human adipose precursor cells. Methods Mol. Biol. 155, 239–247 [DOI] [PubMed] [Google Scholar]
- 21.Ishiyama J., Taguchi R., Akasaka Y., Shibata S., Ito M., Nagasawa M., and Murakami K. (2011) Unsaturated FAs prevent palmitate-induced LOX-1 induction via inhibition of ER stress in macrophages. J. Lipid Res. 52, 299–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koumenis C., Naczki C., Koritzinsky M., Rastani S., Diehl A., Sonenberg N., Koromilas A., and Wouters B. G. (2002) Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2α. Mol. Cell. Biol. 22, 7405–7416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dehne N., and Brüne B. (2009) HIF-1 in the inflammatory microenvironment. Exp. Cell Res. 315, 1791–1797 [DOI] [PubMed] [Google Scholar]
- 24.Tannahill G. M., Curtis A. M., Adamik J., Palsson-McDermott E. M., McGettrick A. F., Goel G., Frezza C., Bernard N. J., Kelly B., Foley N. H., Zheng L., Gardet A., Tong Z., Jany S. S., Corr S. C., Haneklaus M., Caffrey B. E., Pierce K., Walmsley S., Beasley F. C., Cummins E., Nizet V., Whyte M., Taylor C. T., Lin H., Masters S. L., Gottlieb E., Kelly V. P., Clish C., Auron P. E., Xavier R. J., and O'Neill L. A. (2013) Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496, 238–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dehne N., Tausendschön M., Essler S., Geis T., Schmid T., and Brüne B. (2014) IL-4 reduces the proangiogenic capacity of macrophages by down-regulating HIF-1α translation. J. Leukocyte Biol. 95, 129–137 [DOI] [PubMed] [Google Scholar]
- 26.Fang H. Y., Hughes R., Murdoch C., Coffelt S. B., Biswas S. K., Harris A. L., Johnson R. S., Imityaz H. Z., Simon M. C., Fredlund E., Greten F. R., Rius J., and Lewis C. E. (2009) Hypoxia-inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood 114, 844–859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rius J., Guma M., Schachtrup C., Akassoglou K., Zinkernagel A. S., Nizet V., Johnson R. S., Haddad G. G., and Karin M. (2008) NF-κB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1α. Nature 453, 807–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brunelle J. K., Bell E. L., Quesada N. M., Vercauteren K., Tiranti V., Zeviani M., Scarpulla R. C., and Chandel N. S. (2005) Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab. 1, 409–414 [DOI] [PubMed] [Google Scholar]
- 29.Chandel N. S., McClintock D. S., Feliciano C. E., Wood T. M., Melendez J. A., Rodriguez A. M., and Schumacker P. T. (2000) Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1α during hypoxia: a mechanism of O2 sensing. J. Biol. Chem. 275, 25130–25138 [DOI] [PubMed] [Google Scholar]
- 30.Guzy R. D., Hoyos B., Robin E., Chen H., Liu L., Mansfield K. D., Simon M. C., Hammerling U., and Schumacker P. T. (2005) Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 1, 401–408 [DOI] [PubMed] [Google Scholar]
- 31.Mansfield K. D., Guzy R. D., Pan Y., Young R. M., Cash T. P., Schumacker P. T., and Simon M. C. (2005) Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-α activation. Cell Metab. 1, 393–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Srinivasan S., and Avadhani N. G. (2007) Hypoxia-mediated mitochondrial stress in RAW264.7 cells induces osteoclast-like TRAP-positive cells. Ann. N.Y. Acad. Sci. 1117, 51–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Portella A. O., Montero E. F., Poli de Figueiredo L. F., Bueno A. S., Thurow A. A., and Rodrigues F. G. (2004) Effects of N-acetylcysteine in hepatic ischemia-reperfusion injury during hemorrhagic shock. Transplant. Proc. 36, 846–848 [DOI] [PubMed] [Google Scholar]
- 34.Wang C., Chen K., Xia Y., Dai W., Wang F., Shen M., Cheng P., Wang J., Lu J., Zhang Y., Yang J., Zhu R., Zhang H., Li J., Zheng Y., Zhou Y., and Guo C. (2014) N-acetylcysteine attenuates ischemia-reperfusion-induced apoptosis and autophagy in mouse liver via regulation of the ROS/JNK/Bcl-2 pathway. PLoS ONE 9, e108855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lang R., Hammer M., and Mages J. (2006) DUSP meet immunology: dual specificity MAPK phosphatases in control of the inflammatory response. J. Immunol. 177, 7497–7504 [DOI] [PubMed] [Google Scholar]
- 36.Kamata H., Honda S., Maeda S., Chang L., Hirata H., and Karin M. (2005) Reactive oxygen species promote TNFα-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 120, 649–661 [DOI] [PubMed] [Google Scholar]
- 37.Ríos P., Nunes-Xavier C. E., Tabernero L., Köhn M., and Pulido R. (2014) Dual-specificity phosphatases as molecular targets for inhibition in human disease. Antioxid. Redox Signal. 20, 2251–2273 [DOI] [PubMed] [Google Scholar]
- 38.Arthur J. S., and Ley S. C. (2013) Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 13, 679–692 [DOI] [PubMed] [Google Scholar]
- 39.Chi H., Barry S. P., Roth R. J., Wu J. J., Jones E. A., Bennett A. M., and Flavell R. A. (2006) Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc. Natl. Acad. Sci. U.S.A. 103, 2274–2279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huang G., Wang Y., Shi L. Z., Kanneganti T. D., and Chi H. (2011) Signaling by the phosphatase MKP-1 in dendritic cells imprints distinct effector and regulatory T cell fates. Immunity 35, 45–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cummins E. P., Berra E., Comerford K. M., Ginouves A., Fitzgerald K. T., Seeballuck F., Godson C., Nielsen J. E., Moynagh P., Pouyssegur J., and Taylor C. T. (2006) Prolyl hydroxylase-1 negatively regulates IκB kinase-β, giving insight into hypoxia-induced NFκB activity. Proc. Natl. Acad. Sci. U.S.A. 103, 18154–18159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hetz C. (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 13, 89–102 [DOI] [PubMed] [Google Scholar]
- 43.Ozcan U., Cao Q., Yilmaz E., Lee A. H., Iwakoshi N. N., Ozdelen E., Tuncman G., Görgün C., Glimcher L. H., and Hotamisligil G. S. (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457–461 [DOI] [PubMed] [Google Scholar]
- 44.Wu J., Mei C., Vlassara H., Striker G. E., and Zheng F. (2009) Oxidative stress-induced JNK activation contributes to proinflammatory phenotype of aging diabetic mesangial cells. Am. J. Physiol. Renal Physiol. 297, F1622–1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chan D. W., Liu V. W., Tsao G. S., Yao K. M., Furukawa T., Chan K. K., and Ngan H. Y. (2008) Loss of MKP3 mediated by oxidative stress enhances tumorigenicity and chemoresistance of ovarian cancer cells. Carcinogenesis 29, 1742–1750 [DOI] [PubMed] [Google Scholar]
- 46.Berdichevsky A., Guarente L., and Bose A. (2010) Acute oxidative stress can reverse insulin resistance by inactivation of cytoplasmic JNK. J. Biol. Chem. 285, 21581–21589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Choe S. S., Shin K. C., Ka S., Lee Y. K., Chun J. S., and Kim J. B. (2014) Macrophage HIF-2α ameliorates adipose tissue inflammation and insulin resistance in obesity. Diabetes 63, 3359–3371 [DOI] [PubMed] [Google Scholar]
- 48.Harwood H. J., Jr. (2012) The adipocyte as an endocrine organ in the regulation of metabolic homeostasis. Neuropharmacology 63, 57–75 [DOI] [PubMed] [Google Scholar]