

Abstract
γ-Secretase is a multisubunit membrane protein complex containing presenilin (PS1) as a catalytic subunit. Familial Alzheimer disease (FAD) mutations within PS1 were analyzed in yeast cells artificially expressing membrane-bound substrate, amyloid precursor protein, or Notch fused to Gal4 transcriptional activator. The FAD mutations, L166P and G384A (Leu-166 to Pro and Gly-384 to Ala substitution, respectively), were loss-of-function in yeast. We identified five amino acid substitutions that suppress the FAD mutations. The cleavage of amyloid precursor protein or Notch was recovered by the secondary mutations. We also found that secondary mutations alone activated the γ-secretase activity. FAD mutants with suppressor mutations, L432M or S438P within TMD9 together with a missense mutation in the second or sixth loops, regained γ-secretase activity when introduced into presenilin null mouse fibroblasts. Notably, the cells with suppressor mutants produced a decreased amount of Aβ42, which is responsible for Alzheimer disease. These results indicate that the yeast system is useful to screen for mutations and chemicals that modulate γ-secretase activity.
Keywords: Alzheimer disease, amyloid-β (Aβ), γ-secretase, intramembrane proteolysis, Saccharomyces cerevisiae, yeast
Introduction
The γ-secretase is closely associated with familial Alzheimer disease (FAD)2 (1). Presenilin is the catalytic subunit of γ-secretase, which cleaves the intramembrane domain of amyloid precursor protein (APP), thus generating amyloid β (Aβ) peptides. More than 150 missense mutations within the two isoforms (PS1 or PS2) of presenilin are identified for the disease. These mutations decrease the generation of Aβ but increase the ratio between Aβ42 and Aβ40 (2–4), which contain 42 and 40 amino acid residues, respectively. The increased ratio leads to formation of senile plaques in the brains of patients and is responsible for the disease (1). γ-Secretase modulators also alter the ratios among Aβ species and are classified as Aβ42-lowering or -raising compounds (5). The former could represent promising therapeutic agents for the treatment of Alzheimer disease.
γ-Secretase, a member of the intramembrane-cleaving protease family, is composed of four membrane proteins as follows: presenilin (PS1 or PS2), nicastrin (NCT), Aph-1 (anterior pharynx-1), and Pen2 (6, 7). Crystal structures of the protease family members, including rhomboid, site-2 protease, and signal peptide peptidases PSH (8–10), have a membrane-embedded chamber structure, which harbors catalytic residues accessible for water molecules. Although the crystal structure of γ-secretase is currently unavailable, similar chamber-like structures have been observed by electron microscopy (11, 12).
Structural analysis, including cysteine-scanning assays, have suggested that PS1 contains nine transmembrane domains (TMD1–9) (13–15), and a hydrophilic “catalytic pore” is formed by TMD1, -6, -7, and -9 (16–20). The catalytic residues Asp-257 and Asp-385, within TMD6 and -7, respectively, are located in the chamber-like structure (16, 17). The catalytic pore resides in the convex side of a horseshoe-like transmembrane arrangement of γ-secretase, identified by cryo-electron microscopy (21).
The interactions between PS1 and other subunits have also been evaluated. Aph-1, Pen2, and NCT, which contain seven, three, and one transmembrane domains, respectively, interact with PS1. Pen2 associates directly with the WNF motif within the TMD4 of PS1 (22, 23). The NCT·Aph-1 subcomplex interacts with the carboxyl-terminal region of PS1, including TMD8 and TMD9 (24–26). Prior to entry into the catalytic site, the substrate interacts with the extracellular domain of NCT (27), as well as the recognition site of PS1, which is formed by TMD1 and the carboxyl terminus, including TMD9 (28, 29).
It is difficult to study the catalytic mechanism or interaction between subunits in γ-secretase, which is a transmembrane protease with multiple subunits. However, the assay system could be constructed in the yeast cell, which does not have functional γ-secretase homologue or APP. Through a yeast transcriptional activator Gal4 system using an artificial substrate containing APP segments, Edbauer et al. (7) demonstrated that the four subunits of γ-secretase are essential for its protease activity. We also established a system to evaluate the specificity of γ-secretase and to screen mutations (30–32). We have isolated constitutively active PS1 mutants that do not require NCT for proteolysis (30). We also observed a difference between PS1 and PS2 activities (32), and we detected the release of different Aβ species (Aβ40, Aβ42, and Aβ43) after cleavage in yeast microsomes (31). Thus, yeast cells are a useful model system to analyze γ-secretase.
In this study, the properties of PS1 FAD mutations were analyzed using the yeast system. When FAD mutations, L166P or G384A (Leu-166 to Pro or Gly-384 to Ala, respectively), were introduced into PS1, yeast cells could not grow by lack of APP cleavage. We identified amino acid substitutions in PS1 that suppress the original mutations. Critical secondary mutations were in a catalytic pore as follows: K380E, S384P, or L432M. Using PS1/PS2 double knock-out mouse embryonic fibroblasts (33), we could show that the secondary mutations increased the level of Aβ and the intracellular domain but decreased Aβ42. Furthermore, we studied individual secondary mutations, and we discuss their modulatory effects.
Experimental Procedures
γ-Secretase Reconstitution in Yeast
To reconstitute γ-secretasein yeast, human PS1, NCT, FLAG-Pen2, Aph-1aL-HA, APP fragment (C55), Notch-1 fragment (NotchTM), and Gal4 were cloned into respective vectors, as described previously (30). Briefly, PS1 and NCT were cloned into the KpnI and XbaI sites of pBEVY-T (34). FLAG-Pen2 and Aph1aL-HA were cloned into the KpnI and XbaI sites of pBEVY-L (34). APPC55-Gal4p and NotchTM-Gal4p were fused to the SUC2 signal peptide sequence and cloned into the BamHI and EcoRI sites of p426ADH (35). APPC55 and NotchTM represent amino acids 672–726 of human APP770 and 1703–1754 of mouse Notch-1, respectively. These recombinant plasmids were transformed into Saccharomyces cerevisiae strain PJ-69–4A (MATa trp1-901 leu2-3, 112 ura3-52 his3-200 gal4Δ gal80Δ LYS2::GAL1-HIS3 GAL2-ADE2 met2::GAL7-lacZ) (36). Reporter gene expression was monitored to assess substrate cleavage. The expression of HIS3 (His) and ADE2 (Ade) was estimated by colony growth on minimal SD agar medium, lacking Leu, Trp, His, Ura, and SD-LWHUAde (30). β-Galactosidase was assayed using o-nitrophenyl β-d-galactopyranoside, as described previously (30). Exponentially growing cells (1 × 107 cells) were lysed using glass beads in 30 μl of lysis buffer (20 mm Tris-HCl, pH 8.0, 10 mm MgCl2, 50 mm KCl, 1 mm EDTA, 5% glycerol, and 1 mm dithiothreitol (DTT)), including a protease inhibitor mixture (30). Cell lysates were centrifuged for 10 min at 15,000 × g, and β-galactosidase activities and protein concentrations (Bradford protein assay, Bio-Rad) were estimated in the resulting supernatants.
Random Mutagenesis by PCR
Random mutations were introduced into PS1 FAD mutants, L166P and G384A, by polymerase chain reaction (PCR), as described previously (30). Briefly, the 50-μl reaction contained 50 ng of template DNA, 0.2 mm dGTP, 1 mm dATP/dTTP/dCTP, 3 mm MgCl2, 0.5 mm MnCl2, 1.25 units of rTaq in 1× rTaq buffer, and 400 nm primers. Primer sequences are as follows: PS1S, 5′-TTCAAGCTATACCAAGCATACAATCAACTCCCCGGGTACCAAAAATGACAGAGTTACCTGCACCGTTG-3′, and PS1AS,5′-GATCCGCTTATTTAGAAGTGTCGAATTCGACCTCGGTACCATGCTAGATATAAAATTGATGGAATGC-3′. PCRs were performed under the following conditions: 94 °C for 5 min; 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 120 s (30 cycles); and 72 °C for 10 min. The error ratio of these conditions was ∼2.6 mutations per PS1 gene. Mutagenized PS1 cDNA fragments (4 μg) were then cotransformed with 4 μg of the KpnI fragment from NCTpBEVY-T into PJ69-4A. PS1 primers containing 40-bp regions from pBEVY-T (34) enable homologous recombination between two fragments (37). Approximately 106 transformants were screened on the selection medium SD-LWHUAde (Table 1). Plasmid DNAs were isolated from yeast colonies, and mutations were identified by DNA sequencing. Site-directed mutations were introduced using a QuikChange mutagenesis kit (Stratagene).
TABLE 1.
Growth of mutants that suppress FAD mutations of PS1 (L166P and G384A)
| Mutants | Growtha | No. of clones isolated |
|---|---|---|
| L166P, PCR mutation | ||
| Revertants | ||
| L166 (true revertant) | +++ | 104 |
| L166T | +++ | 34 |
| L166S | ++ | 60 |
| L166H | ++ | 2 |
| Second mutation | ||
| K380E/L232H/C419G | +++ | 1 |
| K380E/V272I | ++ | 1 |
| Directed mutation | ||
| L232H/C419G | − | |
| K380E/L232H | ++ | |
| K380E/C419G | ++ | |
| K380E | + | |
| C419G | − | |
| L232H | − | |
| V272I | − | |
| G384A, PCR mutation | ||
| Revertants | ||
| G384 (true revertant) | +++ | 2 |
| Second mutation | ||
| S438P | + | 12 |
| S438P/I287L | ++ | 1 |
| L432M/C158Y | ++ | 1 |
| Directed mutation | ||
| I287L | − | |
| L432M | + | |
| C158Y | − | |
a The growth of cells with PS1 mutations (L166P and G384A) were analyzed on SD-LWHUAde medium after 3 days at 30 °C. All cell contains NCT, Aph-1aL, Pen2, and APPC55-Gal4p. +++ represent wild-type growth, formed >1 mm colonies and − represents no growth. ++ or + represents partial growth, formed small colonies (>0.5 or <0.5 mm, respectively). 9.5 × 105 or 1.1 × 106 cells were screened for L166P or G384A suppressors, respectively.
Protein Expression in Mouse Embryonic Fibroblasts
For expression in mouse cells, wild-type or mutant PS1 cDNAs were inserted into the EcoRI site of pMXs-puro (38). A highly efficient retroviral infection was performed as reported previously (23, 38). Briefly, retroviral plasmids were transfected into the packaging cell line PLAT-E using FuGENE 6 (Roche Applied Science). After 48 h, conditioned media were filtered through a 0.45-μm pore and used as viral stocks. For infection, cells were cultured with the viral stock containing 5 μg/ml Polybrene. After 6 h, the virus stock was replaced with new medium lacking virus and analyzed further.
To measure the amount of secreted Aβ, recombinant retroviruses encoding each mutant PS1 were transiently infected into mouse fibroblasts from PS1/PS2 double knock-out (DKO) mice stably expressing APP-NL (KM670/671NL Swedish mutation) (23). After incubation for 24 h, conditioned media were collected and subjected to two-site ELISAs (i.e. BNT77/BA27 and BNT77/BC05 for Aβ40 and Aβ42, respectively) (39) or to immunoblot analysis using the urea/SDS-PAGE system (40–43). Cells were also recovered and subjected to immunoblotting. Media with increasing concentrations (50–150 μm) of sulindac sulfide (Sigma) were used to analyze the Aβ42-lowering effect of NSAIDs.
To measure the amount of the intracellular domains of APP and Notch, retrovirus with mutant PS1 were infected into 1210/DKO-expressing γ-substrates and cognate luciferase reporters (44). For the generation of 1210/DKO cells, retroviral expression vectors containing SPC99gvp-6myc (modified SPA4CT (45) fused with Gal4/VP16 and a hexa-Myc epitope tag) in pMXs-puro, NΔE-6myc (truncated mouse Notch (46) fused with a hexa-Myc epitope tag) in pLPCX (Clontech), UAS-firefly luciferase in pMXs-EGFPII, and TP1-Renilla luciferase in pMXs-II were constructed. pMXs-EGFPII is a derivative-encoding enhanced GFP, rather than the puromycin resistance gene in pMXs-puroII (47). pMXs-II was generated by excising the puromycin resistance gene in pMXs-puroII. 1210/DKO cells express truncated APP (SPC99gvp-6myc) and Notch (NΔE-6myc), which correspond to different luciferase reporter systems (i.e. Gal4/VP16-fused APP intracellular domain-driven UAS-firefly luciferase and Notch intracellular domain-driven TP1-Renilla luciferase, respectively). Following infection, luciferases were assayed as described previously (44).
Antibodies
Anti-G1L3, anti-G1Nr3, and PNT3 antibodies against the human PS1 loop, human PS1 amino terminus, and human Pen-2 amino terminus, respectively, were used as described previously (41, 48). The anti-PS1NT antibody was provided kindly by Dr. G. Thinakaran (49). Anti-Nct (N1660) and anti-α-tubulin (DM1A) were purchased from Sigma. Anti-APP carboxyl terminus (APP(c)) and anti-Aβ 1-x (82E1) were purchased from IBL.
Results
Secondary Mutations Suppressing PS1 FAD Mutations
Plasmids encoding γ-secretase and APP-based recombinant substrates (APPC55-Gal4p) were introduced into the yeast strain PJ69-4A, which possesses the HIS3, ADE2, and lacZ genes under Gal4p control. After cleavage of the substrate by γ-secretase, Gal4p is released from the APP transmembrane domain (APPC55) to activate the transcription of HIS3/ADE2 and lacZ. Thus, the protease activity could be monitored by cell growth and β-galactosidase activity (30, 32). When FAD mutants (L166P or G384A) of PS1 were introduced into cells with APPC55-Gal4p, severe loss-of-function was observed, indicated by the absence of growth in the media lacking adenine and histidine (Fig. 1, A and B, FAD mutation shown in red). To understand why the specificity of γ-secretase changed, secondary mutations that could suppress FAD mutations were isolated. After PCR-based mutagenesis of PS1 harboring FAD mutations, true revertants and secondary mutants were isolated by screening 1 × 106 cells (Table 1). Two L166P suppressors contained the K380E/L232H/C419G or K380E/V272I replacements (Table 1 and Fig. 1A). L166P in combination with the secondary mutations K380E/L232H and K380E/C419G were also able to grow but that with L232H/C419G (a combination without K380E) could not (Fig. 1A). When each replacement (K380E, C419G, L232H, or V272I) was introduced into L166P, no positive growth was observed, except that L166P/K380E formed small colonies (Fig. 1A). These results indicate the importance of the K380E replacement for suppression.
FIGURE 1.

Growth and β-galactosidase activity of FAD mutants with secondary suppressor mutations. Growth of FAD mutants with secondary mutations (A and B). Four γ-secretase subunits were reconstituted in yeast with APPC55-Gal4p; Gal4p cleaved from APPC55-Gal4p activates HIS3 and ADE2 genes, which leads to growth on selection medium lacking His and adenine. Growth of cells expressing wild-type (WT) PS1, FAD mutants (L166P or G384A), and secondary suppressors was examined after incubation at 30 °C for 3 days in selection media (SD-LWHUAde). Three independent clones were evaluated for each strain. Growth of the mutants in medium containing His and adenine (SD-LWU) was essentially the same as that of WT (data not shown). FAD and secondary mutations are indicated in red and black, respectively. β-Galactosidase activity of the FAD mutants and their suppressors (C). β-Galactosidase activity was assayed for each cell expressing either WT, FAD mutants, or FAD plus secondary suppressor mutations. One unit of β-galactosidase activity corresponds to 1 nmol of o-nitrophenyl β-galactoside hydrolyzed per min and is expressed as units/mg lysate protein. Activity was normalized to that of the control cells without nicastrin (35 units/mg protein). Representative results from three independent assays are shown with standard deviations. Statistical analysis was performed by one-way analysis of variance followed by Dunnett's multiple comparison test. Asterisks indicate p < 0.01 (**) compared with the FAD mutations (L166P or G384A).
β-Galactosidase activity semi-quantitatively determined the cleavage of APPC55-Gal4p by the mutants. L166P/K380E/L232H/C419G mutant possessed the highest activity, which was equivalent to 40% that of wild type (Fig. 1C). L166P/K380E/L232H, L166P/K380E/C419G, and L166P/K380E/V272I showed low but significant β-galactosidase activity (9, 8.5 and 4% of wild type, respectively). However, L166P/L232H/C419G (combination of secondary mutations from the original suppressor but without K380E) showed diminished activity (0.8% of wild type).
Three G384A suppressors contained the secondary mutations, S438P, S438P/I287L, and L432M/C158Y restored the growth of G384A (Table 1 and Fig. 1B). Effect of S438P/I287L was stronger than S438P alone, whereas I287L showed no effect on the cell growth (Fig. 1B). Similarly, L432M partially restored cell growth but C158Y had no effect (Fig. 1B). These results indicate that two replacements were necessary for suppression, and S438P and L432M are essential.
β-Galactosidase activities of the G384A suppressors correlated with the growth. G384A/S438P/I287L exhibited the highest activity (8% that of wild type, Fig. 1C), followed by G383A/S438P, G384A/L432M/C156Y, and G384A/L432M, which exhibited 5, 2.5, and 1.5% wild-type activity, respectively (Fig. 1C).
These results indicate that multiple secondary mutations are required for suppression of FAD mutants. In contrast to L166P located within TMD3, the suppressor mutations L232H, V272I, K380E, and C419G map to TMD5, TMD7, TMD8, and to the sixth loop, respectively (Fig. 2A). In contrast, G384A is located within TMD7, and the suppressor mutations map to TMD9 (L432M and S438P) and to the second (C158Y) and sixth loops (I287L, Fig. 2B).
FIGURE 2.
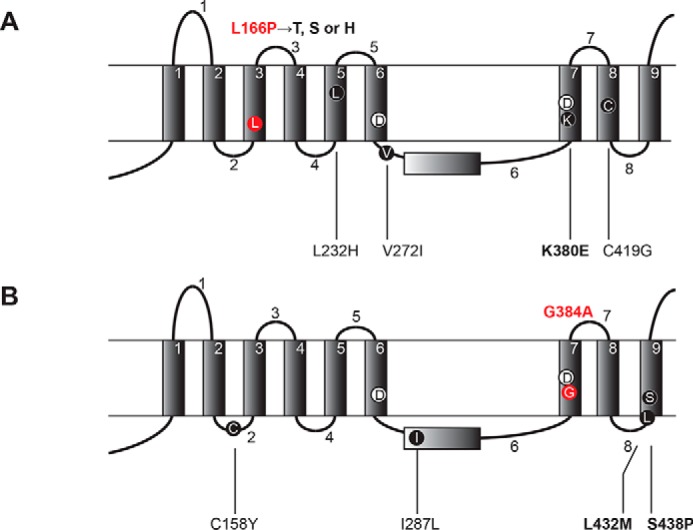
FAD mutations and secondary suppressor mutations map to the transmembrane and loop domains of PS1. FAD mutation (L166P or G384A) and secondary suppressors (A and B) are shown. PS1 models with the nine transmembrane domains (TMD) 1–9 and the eight loop domains 1–8 (13–15) are shown. The suppressor mutations for L166P (red) include the primary mutation K380E together with three variable mutations (L232H, V272I and C419G) (A). The suppressor mutations for G384A (red) include the primary mutation L432M or S438P with secondary mutation C158Y or I287L, respectively (B). The critical secondary mutations are indicated in boldface and the FAD mutations are in red. Catalytic Asp residues in TMD6 and TMD7 (Asp-257 and Asp-385) are also indicated.
Suppressor Mutations Restored Notch Cleavage
To further understand the suppression of PS1 FAD mutations, we tested the cleavage of NotchTM-Gal4p (transmembrane domain of Notch-1 fused to Gal4p) in yeast. The γ-secretase containing the FAD mutation was able to release Notch fragments from NotchTM-Gal4p at reduced efficiency; L166P and G384A mutants exhibited 25 and 50% of the β-galactosidase activity as that of the wild type, respectively (Fig. 3A). When the suppressor mutants were examined, the activities induced by L166P/K380E/L232H/C419G, G384A/L432M/C158Y and G384A/S438P/I287L become similar to that by the wild type, indicating that the Notch-cleaving activity of FAD mutants was recovered to wild-type levels by the second mutations. Immunoblotting confirmed that the expression of mutant PS1s was at a similar level to that of wild type (Fig. 3B).
FIGURE 3.

β-Galactosidase activity of FAD suppressors or PS1 with the secondary mutations alone. β-Galactosidase activity of wild-type (WT), FAD mutations (L166P, G384A), their suppressor mutants (A), or the secondary mutants (C and D). APPC55-Gal4p or NotchTM-Gal4p was used for each assay and indicated as APP (C) or Notch (A and D), respectively. One unit of β-galactosidase activity corresponds to 1 nmol of o-nitrophenyl β-galactoside hydrolyzed per min and is expressed as units/mg lysate protein. Activity was normalized to that of the control cells without nicastrin (35 units/mg protein for APPC55-Gal4p and 95 units/mg protein for NotchTM-Gal4p). Asterisks (**) indicate p < 0.01. Expression of WT and mutant PS1 in yeast (B). Cell lysates from WT or mutant PS1s were analyzed by immunoblotting using an antibody against the carboxyl terminus of PS1. Positions of PS1 and its amino terminus (PS1CTF) are indicated. An asterisk in the immunoblot indicates nonspecific band.
Second Mutations Activate γ-Secretase Activities
Next, we tested the cleavage of APP and Notch by PS1 carrying second mutations alone. K380E/L232H/C419G and S438P/I287L showed higher β-galactosidase activities for APP and Notch cleavage (Fig. 3, C and D); APP cleavage by the two mutants was 2.2- and 1.9-fold higher than wild type, respectively (Fig. 3C). In contrast, L432M/C158Y mutant showed wild-type activity. Essentially the same results were obtained for Notch cleavage (Fig. 3D). These results suggest that K380E/L232H/C419G and S438P/I287L enhance protease activity for APP and Notch.
Secondary Mutations Suppressed L166P and G384A in Mouse Fibroblasts
We further analyzed the PS1 FAD mutation in embryonic fibroblasts (DKO cells) from PS1/PS2 double knock-out mice (Fig. 4). Wild-type and mutant PS1 cDNAs were cloned into a transfer vector to generate recombinant retroviruses, which were then infected into DKO cells.
FIGURE 4.

Production of Aβ and the intracellular domains of APP and Notch by PS1 with FAD mutations and secondary suppressors in mouse embryonic fibroblasts. Using the retroviral infection system, PS1/PS2 DKO cells, stably expressing APP-NL, were transfected with WT or mutant PS1, L166P, G384A, L166P/K380E/L232H/C419G, G384A/S438P/I287L, or G384A/L432M/C158Y (A–D). Cells were harvested and analyzed by immunoblotting using antibodies specific to NCT (N1660), PS1 (anti-PS1NT for NTF and G1L3 for CTF), Pen2 (PNT3), or tubulin (DM1A) (A). Secretion of Aβ peptides including Aβ40 and Aβ42 (B and C). After transfection, media were recovered, and the secreted Aβ peptides were quantified by ELISA using antibodies against Aβ40 (gray) or Aβ42 (black) (B). Values for 10 μm DAPT (+), N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester, are shown as the controls (inset) (B). Media were also analyzed using urea/SDS-PAGE followed by immunoblotting with an antibody specific to Aβ (82E1). Synthetic Aβ37, Aβ38, Aβ39, Aβ40, and Aβ42 were loaded as indicated (C). APP and APP CTF in cell lysates were analyzed by immunoblotting (D). Effects of FAD and suppressor mutations on ϵ-cleavage of APP and Notch in a cell-based γ-secretase assay are shown (E). 1210/DKO cells expressing APP and Notch luciferase reporters were infected transiently with recombinant retrovirus encoding PS1 mutants. Relative levels of intracellular domains of APP (black) and Notch (gray) are indicated (n = 3; means ± S.E. of the mean). Values for 10 μm DAPT (+) are shown as a control (inset) (E). Data represent the mean ± S.D.; n = 3. Statistical analyses were performed by one-way analysis of variance followed by Dunnett's multiple comparison test. Asterisks indicate p < 0.01 (**) compared with PS1 L166P or G384A (B and E). FAD and secondary mutations are indicated in red and black, respectively. Schematic model of processive intramembrane cleavage by γ-secretase is shown (F). γ-Secretase cleaves APP in a manner similar to that of endopeptidase (ϵ-cleavage), followed by a mechanism similar to that of carboxypeptidase (γ-cleavage). Decreased total Aβ/APP intracellular domain (AICD) and increased longer Aβ peptides by PS1 FAD mutations can be explained by partial loss of both ϵ- and γ-cleavage activities.
In DKO cells with wild-type or mutant PS1, we detected similar amounts of mature NCT and Pen-2 (Fig. 4A, lanes 2–7). The amounts of PS1, full-length and amino- and carboxyl-terminal fragments (PS1NTF and PS1CTF, respectively), were also similar, except L166P/K380E/L232H/C419G mutant (Fig. 4A, lanes 2–7). NTF bands of L166P/K380E/L232H/C419G were more intense, and PS1 full-length band of L166P/K380E/L232H/C419G mutant migrated faster than the wild type (Fig. 4A, lane 5). These results suggest that this mutant has different post-translational modification and stability/turnover.
Cells harboring L166P and G384A FAD mutants secreted a significantly increased amount of Aβ42 compared with the wild type (2.3- and 3.8-fold higher, respectively) but a decreased amount of total Aβ (Fig. 4B, lanes 3 and 4). γ-Secretase activity was confirmed by the addition of N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester (DAPT), a specific γ-secretase inhibitor (Fig. 4B, inset). As shown previously, γ-secretase cleaves APP by two different proteolytic activities (Fig. 4F) (5, 50). The initial cleavage is similar to that of endopeptidase (ϵ-cleavage), leading to a separation of the intracellular domains and long Aβ peptides. Next, long Aβ peptides (Aβ48 or Aβ49) are processed in a similar manner to that by carboxypeptidase-like γ-cleavage, producing different Aβ species (Aβ37 to Aβ46). Thus, decreased total Aβ and increased longer Aβ42 species among FAD mutants suggest that ϵ-cleavage and trimming activities are damaged (Fig. 4F).
In contrast to the FAD mutants, the decreased level of Aβ42 was detected with suppressor mutants (Fig. 4B). G384A/L432M/C158Y resulted in a decreased level of Aβ42 and an increased level of Aβ40 (44 and 350% of G384A levels, respectively; Fig. 4B, lane 6). L166P/K380E/L232H/C419G and G384A/S438P/I287L were associated with a decrease of both Aβ40 and Aβ42 (Fig. 4B, lanes 5 and 7). Because these suppressor mutant cells may secrete shorter Aβ peptides, we analyzed the levels of Aβ using immunoblotting (Fig. 4C). Compared with G384A, G384A/L432M/C158Y caused secretion of increased levels of shorter Aβ37, Aβ38, and Aβ40 peptides (Fig. 4C, lanes 4 and 6), suggesting higher ϵ-cleavage and trimming activity induced by the suppressor FAD mutant. However, G384A/S438P/I287L was associated with increased levels of Aβ37 and Aβ38 but decreased levels of Aβ40 and Aβ42 (Fig. 4C, lanes 4 and 7), suggesting that the suppressor mutation G384A/S438P/I287L leads to increased trimming activity of γ-secretase. Essentially no Aβ peptides were detectable in the media from cells expressing the L166P/K380E/L232H/C419G (Fig. 4C, lane 5). Next, we detected APP carboxyl-terminal fragments in DKO cells (Fig. 4D). In contrast to the FAD mutants, G384A/L432M/C158Y and G384A/S438P/I287L showed significant reduction of APP CTFs (Fig. 4D, lanes 6 and 7), confirming the increased cleavage. However, L166P/K380E/L232H/C419G results in accumulation of APPCTF (Fig. 4D, lanes 5). Considering the somewhat different metabolism and/or the post-translational modification of PS1 (Fig. 4A), we did not pursue the function of L166P/K380E/L232H/C419G further, and instead we focused on the G384A suppressors in this study.
To further confirm the γ-secretase activity of the mutants, we evaluated the release of intracellular APP and Notch domains fused to luciferase reporters (44). In this assay, luciferase activity is correlated to that of ϵ-cleavage of APP and Notch. G384A mutants showed decreased levels of the APP intracellular domain (70% that of wild type), but G384A/L432M/C158Y mutants showed increased APP intracellular domain levels (230% that of G384A; Fig. 4E, lanes 2–4), suggesting that the ϵ-cleaving activity is highly enhanced in the suppressor mutant cells. In contrast, G384A/S438P/I287L mutant cells produced a similar amount of the APP intracellular domain as did G384A cells (Fig. 4E, lanes 3 and 5), indicating that ϵ-cleaving activity was not affected. This result supports the notion that G384A/S438P/I287L specifically affects the trimming activity of Aβ in DKO cells. Notch intracellular domain produced by G384A and the suppressed cells were similar, albeit slightly decreased by G384A/S438P/I287L cells (Fig. 4E), suggesting that the effects of FAD and secondary mutations have little effect on Notch cleavage in DKO cells.
Second Mutations Modulate γ-Secretase Activities
Next, we analyzed PS1 carrying second mutations alone in PS1/PS2 DKO cells. Cells harboring L432M/C158Y and S438P/I287L mutants secreted a significantly low amount of Aβ42 (22 and 13% wild type, respectively) (Fig. 5A). S438P/I287L was also associated with decreased Aβ40 levels. Notably, immunoblot analysis revealed that the ratio of shorter Aβ species (i.e. Aβ37, Aβ38, and Aβ39) to the Aβ40 mutant was increased in DKO cells with L432M/C158Y or S438P/I287L (Fig. 5B, lanes 3 and 4). Thus, L432M/C158Y and S438P/I287L altered trimming activity of γ-secretase activity to reduce Aβ42. These results suggest that γ-secretase is affected by the second mutations alone. Aβ42/Aβ40 ratios of wild type, L432M/C158Y, and S438P/I287L were 0.070, 0.019, 0.065, respectively. Robust reduction of the ratio was observed with the L432M/C158Y mutant.
FIGURE 5.

Aβ production by PS1 with secondary mutations alone. Using the retroviral infection system, PS1/PS2 DKO cells, stably expressing APP-NL, were transfected with WT or the second mutants (L432M/C158Y or S438P/I287L) (A and B). After transfection, the secreted Aβ peptides were quantified by ELISA using antibodies against Aβ40 (gray) or Aβ42 (black) (A) and analyzed by immunoblotting (B). Asterisks (* or **) indicate p < 0.05 or p < 0.01, respectively (A). Asterisks in the immunoblot indicate nonspecific bands (B).
Effect of Second Mutations on PS1 Mutants and NSAIDs
We tested the effects of G384A suppressors on different mutations at Gly-384 (i.e. G384P or G384K) (51). Consistent with previous results, G384P and G384K mutations failed to restore the β-galactosidase activities in yeast (Fig. 6, A and B) and Aβ production from DKO cells (Fig. 6C) (51). Among the suppressor mutants, G384P/L432M/C158Y and G384P/S438P/I287L had increased activity (13 and 36% wild type, respectively) with NotchTM-Gal4p (Fig. 6B), indicating partial activation of Notch cleavage by the suppressors, although APP cleavage in yeast and Aβ production in DKO cells were not restored (Fig. 6, A–C).
FIGURE 6.

Suppression of G384P, G384K, or L166P mutants by the secondary mutations. Wild-type (WT) PS1, G384P, G384K, L166P or the suppressor mutants were examined for β-galactosidase activity in yeast (A and B) or Aβ production in PS1/PS2 DKO cells stably expressing APP-NL (C). β-Galactosidase activity corresponds to APPC55-Gal4p (A) or NotchTM-Gal4p cleavage (B). Secreted Aβ peptides were quantified by ELISA using antibodies against Aβ40 (gray) or Aβ42 (black) (C). Asterisks (**) indicate p < 0.01.
Next, we tested the effect of G384A suppressor mutation on L166P mutation. L166P/L432M/C158Y and L166P/S438P/I287L showed higher β-galactosidase activities than L166P mutant (Fig. 6, A and B), suggesting that FAD mutations were suppressed. In DKO cells, L166P/L432M/C158Y produced increased levels of Aβ40 and decreased levels of Aβ42 compared with L166P mutants (Fig. 6C). The level of Aβ40 was close to wild type, suggesting that the suppressor mutations are more effective on L166P. In contrast, L166P/S438P/I287L produced decreased levels of both Aβ40 and Aβ42 (Fig. 6C). These results suggest that the secondary mutations, L432M/C158Y and S438P/I287L, could restore the γ-secretase activities of different FAD mutants effectively.
Finally, we tested the effect of the second mutation on γ-secretase modulator. Consistent with the previous results (52), NSAID sulindac sulfide showed Aβ42-lowering effect on cells with wild type PS1, but the effect was diminished with the G384A mutant (Table 2). When the L432M/C158Y mutation was combined with G384A, cells displayed a significant Aβ42-lowering effect (Table 2), indicating that the second mutations restored the NSAID insensitivity of the G384A mutant.
TABLE 2.
The diminished Aβ42 response of PS1-G384A to sulindac sulfide was restored by the second mutation L432M/C158Y
| PS1 |
Aβ42 (% of Aβ40 + Aβ42) |
||
|---|---|---|---|
| 0 μm | 50 μm | 150 μm | |
| WT | 10.15 ± 1.92 | 9.91 ± 3.20 | 3.37 ± 0.55a |
| G384A | 35.5 ± 7.99 | 33.25 ± 6.41 | 47.95 ± 8.70 |
| G384A/L432M/C158Y | 29.05 ± 4.89 | 23.59 ± 6.15 | 18.42 ± 3.95a |
a PS1/PS2 DKO cells with PS1-WT or mutant PS1 were treated with increasing concentrations of Aβ42-lowering NSAID sulindac sulfide and Aβ40 and Aβ42 levels in conditioned media were quantified as Fig. 4B (n = 4; p < 0.05 compared with 0 μm control).
Discussion
FAD mutations affecting Aβ production have been analyzed extensively (1). However, the role of FAD mutations on the function and structure of PS1 is poorly understood. To address this problem, we examined γ-secretase with PS1 FAD mutation in yeast (30–32). Utilizing the advantages of yeast for genetic screening, we identified suppressor mutations for G384A and L166P FAD mutants. The protease activity of γ-secretase consists of endopeptidase-like cleavage (ϵ-cleavage) and carboxypeptidase-like trimming (γ-cleavage) (Fig. 4F) (5, 50). The FAD mutations severely damaged ϵ-cleavage in yeast, and the suppressor mutations activated this cleavage of FAD mutants or wild-type PS1. In mouse fibroblasts (PS1/PS2 DKO cell), FAD mutation mildly damaged ϵ-cleavage of APP, and the main outcome is reduced trimming (γ-cleavage) and thus increased pathogenic Aβ species, Aβ42. The suppressor mutations activated the trimming and reduce the Aβ42 production in fibroblasts. These finding led us to study the effect of each mutation on γ-secretase as discussed below.
With respect to the G384A suppressors, two second mutations, L432M/C158Y and S438P/I287L, showed Aβ42-lowering modulatory effects on FAD mutants (L166P and G384A). Unfortunately, however, the L166P suppressor, L166P/K380E/L232H/C419G, suffered post-translational modification and reduction of total Aβ production in fibroblasts. Further biochemical and cell biological studies are needed to study the structure-function relation of L166P and the secondary mutation. L432M/C158Y and S438P/I287L were found to change the γ-secretase of wild-type PS1 in DKO cells, reducing the amount of Aβ42. It is intriguing that the second mutations alone activated γ-secretase and lowered the amount of Aβ42. These two mutations are the “Aβ42-lowering modulatory mutation” of PS1, which caused the conformational changes of γ-secretase. Such mutations may be included in the rare protective variant of PS1 against Alzheimer disease, similar to the APP A673T mutation in the elderly Icelandic population (53).
We mapped the locations of mutations on the PS1 structure predicted from the crystal structure of signal peptide peptidase PSH (Fig. 7) (10). L432M/C158Y and S438P/I287L possess critical secondary mutations in TMD9 (L432M and S438P, respectively), which are more important than those in the second (C158Y) and sixth loops (I287L) (Fig. 2B). Leu-432 and Ser-438 residues are conserved throughout PS1/PS2 in all species and are located close to the following sequence: 432LPALPIS438. This sequence includes the PAL motif (Pro-Ala-Leu, underlined), which is completely conserved among PS1, PS2, and signal peptide peptidase. A mutation within PAL motif abolishes γ-secretase activity (26). Leu-432 and Ser-438 are also located close to the catalytic Asp-257 residue in TMD6 (15). Because S438P is an active-state mutant, active in the absence of nicastrin (30), these mutations may affect the structure of the PAL motif and the catalytic pore. Thus, mutations near the PAL motif may increase the accessibility of the substrate to the catalytic pore.
FIGURE 7.
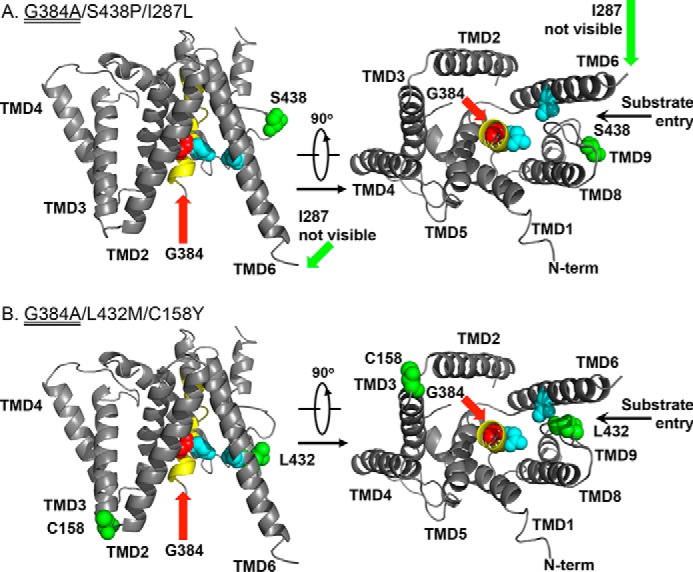
Secondary mutations suppressing the FAD mutants in the structure-based PS1 model. A structure-based model of the transmembrane core of human PS1 (10) (Protein Data Bank code 4HYG) is shown with a sphere containing the following residues: two catalytic Asp (blue), the site of FAD mutant (G384A) (red), and suppressor mutations (green). TMD7 containing the FAD mutation is shown in yellow.
Comparison of the two substrates (APP or Notch) in yeast showed that the effects of mutations are substrate-specific. APP cleavage was completely lost with L166P or G384A, and APP cleavage by the suppressors was up to 40% of wild type. However, Notch cleavage was weakly damaged (25 or 50% wild type) in FAD mutants and similar to the wild type in suppressors. As shown in Fig. 3, NotchTM-Gal4p cleavage consistently produced 5-fold more β-galactosidase than did APPC55-Gal4p, suggesting that Notch was cleaved at a higher efficiency than that of APP in yeast. It has been shown that γ-secretase easily captures a short extracellular domain of Notch but has decreased interaction with the long extracellular domain of APPC55 (54). Therefore, a substrate-specific effect of these mutations may be due to substrate preference.
Substitutions of G384, G384P, and G384K significantly reduced Aβ and Notch intracellular domain generation in a similar manner to that by D385A mutant (51). Consistent with this, neither G384P nor G384K recovered the γ-secretase activity in yeast and DKO cells. The G384A suppressors, L432M/C158Y and S438P/I287L, only restored the Notch cleavage in the G384P mutation but had no effect on APP cleavage nor Aβ production and had no effect on G384K. Theses results support the notion that the G384K mutation caused a loss-of-function effect (51), although G384P severely affected the γ-secretase activity that can be slightly restored by suppressor mutation. The G384A suppressors restored decreased L166P activity. These results indicate that secondary mutations generally activate γ-secretase and that the specific effect to each FAD mutation is small.
γ-Secretase is a prominent drug target for Alzheimer disease. However, a growing list of substrates (55, 56), including developmentally indispensable Notch, has rendered inhibitor design quite challenging. γ-Secretase modulators have been found to reduce Aβ42 production without affecting Notch processing (5). The target of GSM-1 was determined to interact with the luminal side of TMD1 of PS1 and modulate γ-secretase activity (40). Similar to these modulators, secondary mutations also modulate the protease function and reduce Aβ42 without affecting Notch cleavage in DKO cells. Considering the development of new modulators, TMD9 (Leu-432 and Ser-438) may serve as potential target regions. TMD9 (PAL motif) was previously shown to interact with γ-secretase inhibitors.
In this study, we tested the effect of NSAIDs (sulindac sulfides), which modulate the cleavage reaction lowering Aβ42 (Table 2) (57). It has been known that PS1 FAD mutations, including G384A, become insensitive to NSAIDs (52). However, we showed that L432M/C158Y restored the sensitivity to sulindac sulfide. Our results suggest that the second mutations activate PS1 by recovering the conformational flexibility of PS1 that was locked by G384A mutation and act synergistically with NSAIDs to reduce Aβ42. Structural analysis of the PS1 protein with the second mutations will provide a novel mechanistic insight of Aβ42 generation and trimming activity of γ-secretase. Taken together, our results indicate that the yeast system can be used to screen for mutations and chemicals that modulate γ-secretase activity and block Aβ42 production specifically.
Author Contributions
E. F. and T. T. conceived and coordinated the study and wrote the paper. E. F. designed, performed, and analyzed the experiments shown in Figs. 1–3 and 6 and Table 1. T. T., S. O., and T. C. designed, performed, and analyzed the experiments shown in Figs. 4–6 and Table 2. T. F. performed the experiments in Fig. 3. S. I. provided technical assistance and contributed to the preparation of the paper. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We are grateful to Dr. Raphael Kopan (University of Cincinnati) for providing the mNotch1 clone; Dr. Bart De Strooper (Katholieke Universiteit Leuven) for the DKO cells; Dr. Toshio Kitamura (The University of Tokyo) for the retroviral infection system; Dr. Gopal Thinakaran (University of Chicago) for the PS1-NT antibody; Drs. Tohru Fukuyama and Satoshi Yokoshima (Nagoya University) for DAPT; Dr. Yigong Shi (Tsinghua University) for molecular data of PS1 model; Takeda Pharmaceutical Co. (Osaka, Japan) for the Aβ ELISA; and Dr. Philip James (University of Wisconsin) for the PJ-69-4A yeast strain. We also thank Dr. Noboru Sasagawa (Tokai University) for discussions and technical suggestions. Special thanks to the members of our laboratory at Tohoku University and the University of Tokyo.
This work was supported by research grants from the Ministry of Education, Culture, Sports, Science, and Technology (to E. F., T. T., and S. I.), a research grant from Japanese Foundation for Applied Enzymology (to E. F.), the Tokyo Biochemical Research Foundation (to T. T.), Nagase Science and Technology Foundation (to T. T.), and Daiichi Sankyo Foundation of Life Science (to T. T.). The authors declare that they have no conflicts of interest with the contents of this article.
- FAD
- familial Alzheimer disease
- Aβ
- amyloid β peptide
- APP
- amyloid precursor protein
- CTF
- carboxyl-terminal fragment
- NCT
- nicastrin
- NTF
- amino-terminal fragment
- PS1
- presenilin 1
- PS2
- presenilin 2
- Pen2
- presenilin enhancer 2
- NSAID
- nonsteroidal anti-inflammatory drug
- TMD
- transmembrane domain
- DKO
- double knock-out
- DAPT
- N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester.
References
- 1.Selkoe D. J. (2011) Alzheimer disease. Cold Spring Harbor Perspect. Biol. 3, a004457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shen J., and Kelleher R. J. 3rd (2007) The presenilin hypothesis of Alzheimer disease: evidence for a loss-of-function pathogenic mechanism. Proc. Natl. Acad. Sci. U.S.A. 104, 403–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De Strooper B. (2007) Loss-of-function presenilin mutations in Alzheimer disease. EMBO Rep. 8, 141–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wolfe M. S. (2007) When loss is gain: reduced presenilin proteolytic function leads to increased Aβ42/Aβ40. EMBO Rep. 8, 136–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tomita T. (2014) Molecular mechanism of intramembrane proteolysis by γ-secretase. J. Biochem. 156, 195–201 [DOI] [PubMed] [Google Scholar]
- 6.Takasugi N., Tomita T., Hayashi I., Tsuruoka M., Niimura M., Takahashi Y., Thinakaran G., and Iwatsubo T. (2003) The role of presenilin cofactors in the γ-secretase complex. Nature 422, 438–441 [DOI] [PubMed] [Google Scholar]
- 7.Edbauer D., Winkler E., Regula J. T., Pesold B., Steiner H., and Haass C. (2003) Reconstitution of γ-secretase activity. Nat. Cell Biol. 5, 486–488 [DOI] [PubMed] [Google Scholar]
- 8.Wu Z., Yan N., Feng L., Oberstein A., Yan H., Baker R. P., Gu L., Jeffrey P. D., Urban S., and Shi Y. (2006) Structural analysis of a rhomboid family intramembrane protease reveals a gating mechanism for substrate entry. Nat. Struct. Mol. Biol. 13, 1084–1091 [DOI] [PubMed] [Google Scholar]
- 9.Feng L., Yan H., Wu Z., Yan N., Wang Z., Jeffrey P. D., and Shi Y. (2007) Structure of a site-2 protease family intramembrane metalloprotease. Science 318, 1608–1612 [DOI] [PubMed] [Google Scholar]
- 10.Li X., Dang S., Yan C., Gong X., Wang J., and Shi Y. (2013) Structure of a presenilin family intramembrane aspartate protease. Nature 493, 56–61 [DOI] [PubMed] [Google Scholar]
- 11.Lazarov V. K., Fraering P. C., Ye W., Wolfe M. S., Selkoe D. J., and Li H. (2006) Electron microscopic structure of purified, active γ-secretase reveals an aqueous intramembrane chamber and two pores. Proc. Natl. Acad. Sci. U.S.A. 103, 6889–6894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Y., Lu S. H., Tsai C. J., Bohm C., Qamar S., Dodd R. B., Meadows W., Jeon A., McLeod A., Chen F., Arimon M., Berezovska O., Hyman B. T., Tomita T., Iwatsubo T., et al. (2014) Structural interactions between γ-secretase inhibitor and initial substrate docking sites give insight into mechanisms of human PS-1 complexes. Structure 22, 125–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laudon H., Hansson E. M., Melén K., Bergman A., Farmery M. R., Winblad B., Lendahl U., von Heijne G., and Näslund J. (2005) A nine-transmembrane domain topology for presenilin 1. J. Biol. Chem. 280, 35352–35360 [DOI] [PubMed] [Google Scholar]
- 14.Oh Y. S., and Turner R. J. (2005) Topology of the C-terminal fragment of human presenilin 1. Biochemistry 44, 11821–11828 [DOI] [PubMed] [Google Scholar]
- 15.Spasic D., Tolia A., Dillen K., Baert V., De Strooper B., Vrijens S., and Annaert W. (2006) Presenilin-1 maintains a nine-transmembrane topology throughout the secretory pathway. J. Biol. Chem. 281, 26569–26577 [DOI] [PubMed] [Google Scholar]
- 16.Tolia A., Chávez-Gutiérrez L., and De Strooper B. (2006) Contribution of presenilin transmembrane domain 6 and 7 to a water-containing cavity in the γ-secretase complex. J. Biol. Chem. 281, 27633–27642 [DOI] [PubMed] [Google Scholar]
- 17.Sato C., Morohashi Y., Tomita T., and Iwatsubo T. (2006) Structure of the catalytic pore of γ-secretase probed by the accessibility of substituted cysteines. J. Neurosci. 26, 12081–12088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sato C., Takagi S., Tomita T., and Iwatsubo T. (2008) The C-terminal PAL motif and transmembrane domain 9 of presenilin 1 are involved in the formation of the catalytic pore of the γ-secretase. J. Neurosci. 28, 6264–6271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tolia A., Horré K., and De Strooper B. (2008) Transmembrane domain 9 of presenilin determines the dynamic conformation of the catalytic site of γ-secretase. J. Biol. Chem. 283, 19793–19803 [DOI] [PubMed] [Google Scholar]
- 20.Takagi S., Tominaga A., Sato C., Tomita T., and Iwatsubo T. (2010) Participation of transmembrane domain 1 of presenilin 1 in the catalytic pore structure of the γ-secretase. J. Neurosci. 30, 15943–15950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu P., Bai X.-C., Ma D., Xie T., Yan C., Sun L., Yang G., Zhao Y., Zhou R., Scheres S. H., and Shi Y. (2014) Three-dimensional structure of human γ-secretase. Nature 512, 166–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim S. H., and Sisodia S. S. (2005) Evidence that the “NF” motif in transmembrane domain 4 of presenilin 1 is critical for binding with PEN-2. J. Biol. Chem. 280, 41953–41966 [DOI] [PubMed] [Google Scholar]
- 23.Watanabe N., Tomita T., Sato C., Kitamura T., Morohashi Y., and Iwatsubo T. (2005) Pen-2 is incorporated into the γ-secretase complex through binding to transmembrane domain 4 of presenilin 1. J. Biol. Chem. 280, 41967–41975 [DOI] [PubMed] [Google Scholar]
- 24.Kaether C., Capell A., Edbauer D., Winkler E., Novak B., Steiner H., and Haass C. (2004) The presenilin C-terminus is required for ER-retention, nicastrin-binding and γ-secretase activity. EMBO J. 23, 4738–4748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bergman A., Laudon H., Winblad B., Lundkvist J., and Näslund J. (2004) The extreme C terminus of presenilin 1 is essential for γ-secretase assembly and activity. J. Biol. Chem. 279, 45564–45572 [DOI] [PubMed] [Google Scholar]
- 26.Wang J., Beher D., Nyborg A. C., Shearman M. S., Golde T. E., and Goate A. (2006) C-terminal PAL motif of presenilin and presenilin homologues required for normal active site conformation. J. Neurochem. 96, 218–227 [DOI] [PubMed] [Google Scholar]
- 27.Shah S., Lee S. F., Tabuchi K., Hao Y. H., Yu C., LaPlant Q., Ball H., Dann C. E. 3rd, Südhof T., and Yu G. (2005) Nicastrin functions as a γ-secretase-substrate receptor. Cell 122, 435–447 [DOI] [PubMed] [Google Scholar]
- 28.Kornilova A. Y., Bihel F., Das C., and Wolfe M. S. (2005) The initial substrate-binding site of γ-secretase is located on presenilin near the active site. Proc. Natl. Acad. Sci. U.S.A. 102, 3230–3235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Annaert W. G., Esselens C., Baert V., Boeve C., Snellings G., Cupers P., Craessaerts K., and De Strooper B. (2001) Interaction with telencephalin and the amyloid precursor protein predicts a ring structure for presenilin. Neuron 32, 579–589 [DOI] [PubMed] [Google Scholar]
- 30.Futai E., Yagishita S., and Ishiura S. (2009) Nicastrin is dispensable for γ-secretase protease activity in the presence of specific mutations. J. Biol. Chem. 284, 13013–13022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yagishita S., Futai E., and Ishiura S. (2008) In vitro reconstitution of γ-secretase activity using yeast microsomes. Biochem. Biophys. Res. Commun. 377, 141–145 [DOI] [PubMed] [Google Scholar]
- 32.Yonemura Y., Futai E., Yagishita S., Suo S., Tomita T., Iwatsubo T., and Ishiura S. (2011) Comparison of presenilin 1 and presenilin 2 γ-secretase activities using a yeast reconstitution system. J. Biol. Chem. 286, 44569–44575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herreman A., Serneels L., Annaert W., Collen D., Schoonjans L., and De Strooper B. (2000) Total inactivation of γ-secretase activity in presenilin-deficient embryonic stem cells. Nat. Cell Biol. 2, 461–462 [DOI] [PubMed] [Google Scholar]
- 34.Miller C. A. 3rd, Martinat M. A., and Hyman L. E. (1998) Assessment of aryl hydrocarbon receptor complex interactions using pBEVY plasmids: expression vectors with bi-directional promoters for use in Saccharomyces cerevisiae. Nucleic Acids Res. 26, 3577–3583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mumberg D., Müller R., and Funk M. (1995) Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156, 119–122 [DOI] [PubMed] [Google Scholar]
- 36.James P., Halladay J., and Craig E. A. (1996) Genomic libraries and a host strain designed for high efficient two-hybrid selection in yeast. Genetics 144, 1425–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ono Y., Torii F., Ojima K., Doi N., Yoshioka K., Kawabata Y., Labeit D., Labeit S., Suzuki K., Abe K., Maeda T., and Sorimachi H. (2006) Suppressed disassembly of autolyzing p94/CAPN3 by N2A connectin/titin in a genetic reporter system. J. Biol. Chem. 281, 18519–18531 [DOI] [PubMed] [Google Scholar]
- 38.Kitamura T., Koshino Y., Shibata F., Oki T., Nakajima H., Nosaka T., and Kumagai H. (2003) Retrovirus-mediated gene transfer and expression cloning: powerful tools in functional genomics. Exp. Hematol. 31, 1007–1014 [PubMed] [Google Scholar]
- 39.Asami-Odaka A., Ishibashi Y., Kikuchi T., Kitada C., and Suzuki N. (1995) Long amyloid β-protein secreted from wild-type human neuroblastoma IMR-32 cells. Biochemistry 34, 10272–10278 [DOI] [PubMed] [Google Scholar]
- 40.Ohki Y., Higo T., Uemura K., Shimada N., Osawa S., Berezovska O., Yokoshima S., Fukuyama T., Tomita T., and Iwatsubo T. (2011) Phenylpiperidine-type γ-secretase modulators target the transmembrane domain 1 of presenilin 1. EMBO J. 30, 4815–4824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tomita T., Takikawa R., Koyama A., Morohashi Y., Takasugi N., Saido T. C., Maruyama K., and Iwatsubo T. (1999) C terminus presenilin is required for overproduction of amyloidogenic Aβ42 through stabilization and endoproteolysis of presenilin. J. Neurosci. 19, 10627–10634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tomita T., Maruyama K., Saido T. C., Kume H., Shinozaki K., Tokuhiro S., Capell A., Walter J., Grünberg J., Haass C., Iwatsubo T., and Obata K. (1997) The presenilin 2 mutation (N141I) linked to familial Alzheimer disease (Volga German families) increases the secretion of amyloid β protein at the 42nd (or 43rd) residue. Proc. Natl. Acad. Sci. U.S.A. 94, 2025–2030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qi-Takahara Y., Morishima-Kawashima M., Tanimura Y., Dolios G., Hirotani N., Horikoshi Y., Kametani F., Maeda M., Saido T. C., Wang R., and Ihara Y. (2005) Longer forms of amyloid β protein: implications for the mechanism of intramembrane cleavage by γ-secretase. J. Neurosci. 25, 436–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Imamura Y., Watanabe N., Umezawa N., Iwatsubo T., Kato N., Tomita T., and Higuchi T. (2009) Inhibition of γ-secretase activity by helical β-peptide foldamers. J. Am. Chem. Soc. 131, 7353–7359 [DOI] [PubMed] [Google Scholar]
- 45.Lichtenthaler S. F., Multhaup G., Masters C. L., and Beyreuther K. (1999) A novel substrate analyzing Alzheimer disease γ-secretase. FEBS Lett. 453, 288–292 [DOI] [PubMed] [Google Scholar]
- 46.Kopan R., Schroeter E. H., Weintraub H., and Nye J. S. (1996) Signal transduction by activated mNotch: Importance of proteolytic processing and its regulation by the extracellular domain. Proc. Natl. Acad. Sci. U.S.A. 93, 1683–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kamura T., Hara T., Matsumoto M., Ishida N., Okumura F., Hatakeyama S., Yoshida M., Nakayama K., and Nakayama K. I. (2004) Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27(Kip1) at G1 phase. Nat. Cell Biol. 6, 1229–1235 [DOI] [PubMed] [Google Scholar]
- 48.Isoo N., Sato C., Miyashita H., Shinohara M., Takasugi N., Morohashi Y., Tsuji S., Tomita T., and Iwatsubo T. (2007) Aβ42 overproduction associated with structural changes in the catalytic pore of γ-secretase. J. Biol. Chem. 282, 12388–12396 [DOI] [PubMed] [Google Scholar]
- 49.Leem J. Y., Vijayan S., Han P., Cai D., Machura M., Lopes K. O., Veselits M. L., Xu H., and Thinakaran G. (2002) Presenilin 1 is required for maturation and cell surface accumulation of nicastrin. J. Biol. Chem. 277, 19236–19240 [DOI] [PubMed] [Google Scholar]
- 50.Takami M., Nagashima Y., Sano Y., Ishihara S., Morishima-Kawashima M., Funamoto S., and Ihara Y. (2009) γ-Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of β-carboxyl terminal fragment. J. Neurosci. 29, 13042–13052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Steiner H., Kostka M., Romig H., Basset G., Pesold B., Hardy J., Capell A., Meyn L., Grim M. L., Baumeister R., Fechteler K., and Haass C. (2000) Glycine 384 is required for presenilin-1 function and is conserved in bacterial polytopic aspartyl proteases. Nat. Cell Biol. 2, 848–851 [DOI] [PubMed] [Google Scholar]
- 52.Page R. M., Baumann K., Tomioka M., Pérez-Revuelta B. I., Fukumori A., Jacobsen H., Flohr A., Luebbers T., Ozmen L., Steiner H., and Haass C. (2008) Attenuated Aβ42 responses to low potency γ-secretase modulators can be overcome for many pathogenic presenilin mutants by second-generation compounds. J. Biol. Chem. 283, 677–68317962197 [Google Scholar]
- 53.Jonsson T., Atwal J. K., Steinberg S., Snaedal J., Jonsson P. V., Bjornsson S., Stefansson H., Sulem P., Gudbjartsson D., Maloney J., Hoyte K., Gustafson A., Liu Y., Lu Y., Bhangale T., et al. (2012) A mutation in APP protects against Alzheimer disease and age-related cognitive decline. Nature 488, 96–99 [DOI] [PubMed] [Google Scholar]
- 54.Funamoto S., Sasaki T., Ishihara S., Nobuhara M., Nakano M., Watanabe-Takahashi M., Saito T., Kakuda N., Miyasaka T., Nishikawa K., Saido T. C., and Ihara Y. (2013) Substrate ectodomain is critical for substrate preference and inhibition of γ-secretase. Nat. Commun. 4, 2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kopan R., and Ilagan M. X. (2004) γ-Secretase: proteasome of the membrane. Nat. Rev. Mol. Cell Biol. 5, 499–504 [DOI] [PubMed] [Google Scholar]
- 56.Haapasalo A., and Kovacs D. M. (2011) The many substrate of presenilin/γ-secretase. J. Alzheimers Dis. 25, 3–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Weggen S., Eriksen J. L., Das P., Sagi S. A., Wang R., Pietrzik C. U., Findlay K. A., Smith T. E., Murphy M. P., Bulter T., Kang D. E., Marquez-Sterling N., Golde T. E., and Koo E. H. (2001) A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature 414, 212–216 [DOI] [PubMed] [Google Scholar]