

Significance
Uncoupling agents might be expected to be generally cytotoxic, but many US Food and Drug Administration (FDA)-approved drugs do have activity as uncouplers, in addition to targeting enzymes. There is therefore interest in the discovery of antibiotics that have such multitarget activity. Here, we show that some FDA-approved drugs, such as clofazimine, clomiphene, and bedaquiline, with antiinfective activity act as uncouplers. Using molecular dynamics-based in silico screening, we also discovered that the brain cancer drug lead vacquinol is an uncoupler that inhibits an enzyme involved in the formation of tuberculosis (TB) virulence factors, in addition to killing TB bacteria. Our results indicate strong drug–membrane interactions, and that screening for combined enzyme inhibition plus uncoupler activity will lead to new antibiotic leads.
Keywords: molecular dynamics simulations, clofazimine, bedaquiline, clomiphene, vacquinol
Abstract
There is a growing need for new antibiotics. Compounds that target the proton motive force (PMF), uncouplers, represent one possible class of compounds that might be developed because they are already used to treat parasitic infections, and there is interest in their use for the treatment of other diseases, such as diabetes. Here, we tested a series of compounds, most with known antiinfective activity, for uncoupler activity. Many cationic amphiphiles tested positive, and some targeted isoprenoid biosynthesis or affected lipid bilayer structure. As an example, we found that clomiphene, a recently discovered undecaprenyl diphosphate synthase inhibitor active against Staphylococcus aureus, is an uncoupler. Using in silico screening, we then found that the anti-glioblastoma multiforme drug lead vacquinol is an inhibitor of Mycobacterium tuberculosis tuberculosinyl adenosine synthase, as well as being an uncoupler. Because vacquinol is also an inhibitor of M. tuberculosis cell growth, we used similarity searches based on the vacquinol structure, finding analogs with potent (∼0.5–2 μg/mL) activity against M. tuberculosis and S. aureus. Our results give a logical explanation of the observation that most new tuberculosis drug leads discovered by phenotypic screens and genome sequencing are highly lipophilic (logP ∼5.7) bases with membrane targets because such species are expected to partition into hydrophobic membranes, inhibiting membrane proteins, in addition to collapsing the PMF. This multiple targeting is expected to be of importance in overcoming the development of drug resistance because targeting membrane physical properties is expected to be less susceptible to the development of resistance.
There is a need for new antibiotics, due to the increase in drug resistance (1, 2). For example, some studies report that by 2050, absent major improvements in drug discovery and use, more individuals will die from drug-resistant bacterial infections than from cancer, resulting in a cumulative effect on global gross domestic product of as much as 100 trillion dollars (3, 4). To discover new drugs, new targets, leads, concepts, and implementations are needed (5, 6).
Currently, one major cause of death from bacterial infections is tuberculosis (TB) (7), where very highly drug-resistant strains have been found (8). Therapy is lengthy, even with drug-sensitive strains, and requires combination therapies with four drugs. Two recent TB drugs/drug leads (9–11) are TMC207 [bedaquiline (1); Sirturo] and SQ109 (2) (Fig. 1). Bedaquiline (1) targets the Mycobacterium tuberculosis ATP synthase (9) whereas SQ109 (2) has been proposed to target MmpL3 (mycobacterial membrane protein large 3), a trehalose monomycolate transporter essential for cell wall biosynthesis (12). SQ109 (2) is a lipophilic base containing an adamantyl “headgroup” connected via an ethylene diamine “linker” to a geranyl (C10) “side chain,” and in recent work (13), we synthesized a series of 11 analogs of SQ109 (2) finding that the ethanolamine (3) was more potent than was SQ109 (2) against M. tuberculosis H37Rv [0.063 vs. 0.25 μg/mL minimal inhibitory concentration (MIC)], and that at least one protonatable nitrogen in the linker was essential for activity. The latter observation suggested to us that SQ109 (2) and ethanolamine (3) might have activity as uncouplers, collapsing the proton motive force (PMF; ∆P) used to drive ATP synthesis, because we had observed similar uncoupling effects for lipophilic bases, US Food and Drug Administration (FDA)-approved drugs, in trypanosomatid parasites (14, 15). The PMF is given by Mitchell (16, 17): ∆P = ∆ψ − Z∆pH, where ∆ψ is the electrical or membrane potential component of ∆P, ∆pH is the transmembrane pH gradient, and Z is 2.303RT/F where R is the gas constant, T is temperature (in kelvins), and F is the Faraday constant.
Fig. 1.

Structures of inhibitors/uncouplers and other compounds of interest. Common or previously used names are indicated.
We found with SQ109 and its analogs that the most potent M. tuberculosis cell growth inhibitors investigated did indeed collapse pH gradients and ∆ψ, as also observed with the lipophilic bases amiodarone (4) (14) and dronedarone (5) (15), antiarrhythmia drugs, in trypanosomatid parasites (18), and SQ109 (2) also acts as an uncoupler in Trypanosoma cruzi (19). Amiodarone (4) and dronedarone (5) had little uncoupling activity against host cells. In related work, Li et al. (20) found that other TB drug leads, BM212 (6), THPP-2 (7), Ro 48-8071 (8), the urea AU1235 (9), and the indolecarboxamide 2418 (10), most of which had been proposed to target MmpL3, likewise had activity as uncouplers, collapsing pH gradients, and in some cases were active against the nonreplicative bacteria found under hypoxic conditions. Several of these compounds also have enzyme targets. For example, SQ109 (2), ethanolamine (3), and Ro 48-8071 (8) have been found (13, 20) to inhibit enzymes involved in menaquinone biosynthesis, particularly the prenyl transferase 1,4-dihydroxy-2-naphthoate octaprenyltransferase (MenA) and human oxidosqualene cyclase (OSC) (21), and bedaquiline (1) is a potent ATP synthase inhibitor, indicating the possibility of multitarget activity for such compounds. These results are of interest because they show that several recently discovered M. tuberculosis drug leads can act as uncouplers in addition to targeting one or more enzymes that are essential for bacterial cell growth, with membrane targeting being of particular interest because it might be expected to be less susceptible to the development of resistance than is purely enzyme targeting, and SQ109 (2) does indeed have a low frequency of resistance in M. tuberculosis (∼2.55 × 10−11) (22). Targeting membrane lipids is also a reason for the low frequency of resistance found with, for example, amphotericin [which binds to ergosterol in fungi and protozoa (23)], as well as the recently discovered teixobactin, which binds to lipid II/III (24).
In other work by Goldman (25), it has been pointed out that most of the new TB drug leads that have been discovered by phenotypic screens and genome sequencing are highly lipophilic (logP ∼ 5.7) bases with membrane targets, which suggested to us the possibility that these drug leads might function by targeting the PMF, as well as membrane proteins. Although targeting the PMF might be expected to be purely mitotoxic, Stock et al. (26) have shown that compounds with logP > 6 have generally low mitotoxicity, which is due, they proposed, to low membrane permeability attributable to accumulation in lipophilic membranes.
Perhaps the most well-known uncoupler is 2,4-dinitrophenol (DNP; 11). DNP functions as a protonophore, a proton-translocating molecule, and analogs such as niclosamide (12) and nitazoxanide (13) [active form, tizoxanide (14)] are used clinically: niclosamide (12) to treat tapeworm infections (27) and nitazoxanide (13) to treat infections due to Giardia lamblia (28) and Cryptosporidium parvum. Nitazoxanide (13) has also been in clinical trials for the treatment of Helicobacter pylori and Clostridium difficile infections. Interestingly, SQ109 (2) has similar activity against both organisms (29), and with H. pylori, SQ109 (2) once again has a very low (≈10−12) frequency of resistance (29). In addition, nitazoxanide (13) has been found to kill both replicating and nonreplicating M. tuberculosis (30–33), and Nathan and coworkers (30, 31) were unable to develop resistant colonies using up to 1012 cfu, proposing a dual “PMF + unknown target” mechanism of action. Niclosamide (12) has been proposed as a lead for the treatment of type II diabetes (34), and it is also an inhibitor of breast cancer stem-like cells (35) and an inhibitor of Pseudomonas aeruginosa quorum sensing (36). There has also been very recent interest in the development of DNP analogs such as DNP methyl ether (37), for treating diabetes, and of controlled-release DNP formulations (38) as mild hepatic mitochondrial uncouplers for treating hypertriglyceridemia, insulin resistance, hepatic steatosis, and diabetes. Niclosamide (12) and tizoxanide (14) are both FDA-approved, and closantel (15) is an anthelmintic uncoupler in veterinary use, and all could provide leads for new and improved inhibitors that target other pathogens. There has also been considerable renewed interest (39) in the use of pyrazinoic acid (16), which functions, at least in part, as a protonophore uncoupler, for treating TB (39, 40), stimulating our interest in discovering new TB drug leads with uncoupler activity.
In this work, we carried out three main types of investigation. First, we investigated the uncoupling effects of 21 compounds (primarily known drugs or drug leads) on uncoupling (∆pH/∆ψ collapse) in bacterial inverted membrane vesicles (IMVs) and in porcine mitochondria. Second, we investigated drug–membrane interactions using differential scanning calorimetry (DSC) and electron paramagnetic resonance (EPR) spectroscopy. Third, we used molecular dynamics (MD) structure-based in silico screening and structure-similarity searches to find prenyl synthase inhibitors with uncoupler activity, leading finally to a consideration of the future prospects for discovering new “enzyme + uncoupler” antiinfective drug leads.
Results and Discussion
Targeting the PMF.
We first investigated two TB drugs that seemed likely to act, at least in part, as protonophore uncouplers: clofazimine (17) (Fig. 1) and TMC207 (1), which have similar logP and pKa values to each other as well as to amiodarone (4, Table 1), a known uncoupler we worked on previously. Clofazimine (17) was originally developed as a TB drug (41) but later was used extensively (42) in treating leprosy, caused by another Mycobacterium, Mycobacterium leprae. There have been several mechanisms of action demonstrated or proposed for clofazimine (17), including a redox cycling reaction involving the generation of reactive oxygen species (43), and clofazimine is currently of interest for use in combination therapies with benzothiazinones (44). We used the sealed, inside-out IMV assay used previously to investigate SQ109 (2) (13) with 9-amino-6-chloro-2-methoxyacridine (ACMA) as a pH-sensitive fluorescence probe of the pH gradient, ΔpH (computed as illustrated in Fig. S1). Using ATP hydrolysis through the ATP synthase, protons are driven inside the membrane vesicles, protonated ACMA accumulates, and its fluorescence is self-quenched. The same effect is seen with addition of succinate/O2, where, again, H+ is pumped into the vesicles.
Table 1.
Uncoupling effects, computed molecular properties, and M. tuberculosis cell growth inhibition for compounds investigated
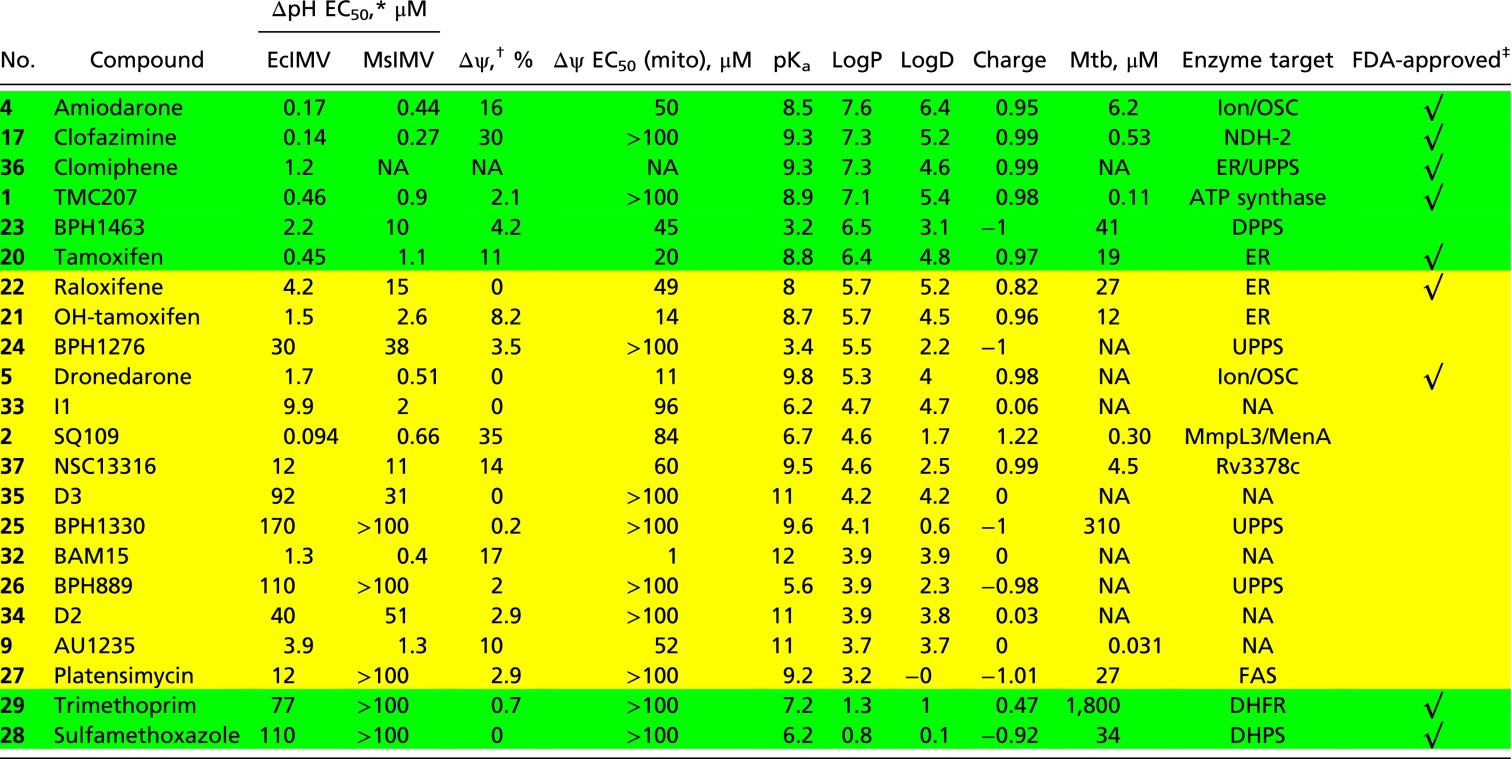 |
Compounds with a green background have logP > 6 or logP < 3; compounds with a yellow background have logP between 3 and 6. BAM15, N5,N6-bis(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine; DHFR, dihydrofolate reductase; DHPS, dihydropteroate synthetase; ER, estrogen receptors; FAS, fatty acid synthase; ion, ion channel; Mtb, M. tuberculosis; NA, not applicable (no results reported); NDH-2, NADH/quinone oxidoreductase II; OSC, oxidosqualene cyclase.
∆pH EC50 is calculated as shown in Fig. S1 using different compound concentrations.
∆ψ collapse of EcIMV measured at 1 μM compound.
Drugs that are currently approved for use in humans by the United States Food and Drug Administration.
Fig. S1.

Calculation of percentage of ∆pH recovery. Percentage of ∆pH recovery is defined as I1/I0 at a given compound concentration, where I0 is the fluorescence intensity decrease on IMV energization and I1 the increase after compound addition. The ∆pH recovery percentages from different compound concentrations were used to obtain EC50 values using a standard rectangular hyperbolic dose–response function.
Addition of clofazimine (17) caused rapid increases in ACMA fluorescence in both succinate-oxidation and ATP-powered assays, as shown in Fig. 2 A and B. These results are very similar to the results we reported previously for SQ109 (2) (13), as well as to the results we found for TMC207 (1) in the same assays (Fig. 2 C and D). TMC207 (1) is thought to target the ATP synthase in M. tuberculosis, but in recent work, it has also been proposed to act as an uncoupler, targeting again the ATP synthase (45). However, there is expected to be a significant protonophore contribution to its activity because clofazimine (17) (not thought to target the ATP synthase) and TMC207 (1) have almost identical logP, pKa, logD, and computed charge values (at pH 7.4) (Table 1), even though the chemical structures are completely different. For clofazimine (17), the values are logP = 7.3, pKa = 9.29, logD = 5.23, and charge = 0.99; for TMC207 (1), the values are logP = 7.13, pKa = 8.91, logD = 5.42, and charge = 0.98 (Table 1). It thus seems likely that clofazimine (17), as well as TMC207 (1), can act, at least in part, in a similar manner to the potent anionic protonophores, such as carbonyl cyanide m-chlorophenyl hydrazone (CCCP) (20), which has an EC50 of ∼400 nM in this assay, about the same as clofazimine (17). We found similar results with both Escherichia coli IMVs (EcIMVs) and Mycobacterium smegmatis IMVs (MsIMVs) (Table 1), so it is unlikely that there are specific drug–membrane interactions with the mycobacterial inner membrane lipids. What is also of interest with clofazimine (17) is that it is an example of a cationic protonophore (46) with extensive charge delocalization, as found in the 4-piperidinopyridine uncoupler AU1421 (18) (47) and the 2-(6-ethylamino-3-ethylimino-2,7-dimethyl-3H-xanthen-9-yl)-benzoates (19) (46). This extensive charge delocalization is likely to contribute to membrane solubility, as will a large surface area, leading to a low charge density.
Fig. 2.

Effects of drugs/drug leads on the PMF. ACMA assays with EcIMV: clofazimine (17), succinate/O2 substrate (A); clofazimine (17)/ATP-powered PMF (B); TMC207 (1)/succinate (C); and TMC207 (1)/ATP (D). Methods used are the same as in the study by Li et al. (13). EC50 results for all compounds investigated are given in Table 1. Au, arbitrary units. (E) Heat map of correlation coefficients between uncoupling activities [∆pH in EcIMV and MsIMV, ∆ψ in EcIMV, ∆ψ in mitochondria (mito)], molecular properties (logD, pKa, charge), and cell growth inhibition [M. tuberculosis (Mtb)]. (F) Heat map of uncoupling activities (∆pH in EcIMV and MsIMV, ∆ψ in EcIMV), molecular properties (logD, charge), and cell growth inhibition (Mtb) for Mtb inhibitors shown in Table 1. High activities refer to small ΔpH/Mtb EC50 values, and high Δψ collapse/logD/charge. Molecular property calculations were carried out using Marvin ChemAxon (https://www.chemaxon.com/marvin/sketch/index.php) and Chemicalize (www.chemicalize.org/). All structures are shown in Fig. 1. AMIO, amiodarone; CFZ, clofazimine; PLAT, platensimycin; RAL, raloxifene; SMX, sulfamethoxazole; TAM, tamoxifen; TMP, trimethoprim.
We next investigated several anticancer and antiarrhythmia drugs, some of which have activity against M. tuberculosis: tamoxifen (20), 4-hydroxytamoxifen (21), raloxifene (22), amiodarone (4), and dronedarone (5), with all but 4-hydroxytamoxifen (21) being FDA-approved drugs for use in humans. Four of these compounds have moderate activity against M. tuberculosis: amiodarone (4), 4 μg/mL (25); tamoxifen (20), 7.1 μg/mL (48); 4-hydroxytamoxifen (21), 4.6 μg/mL (48); and raloxifene (22), 13 μg/mL (48). As can be seen in Table 1, tamoxifen (20) is the most potent uncoupler, followed by 4-hydroxytamoxifen (21) and then raloxifene (22), in both the succinate/O2 and ATP-driven assays. All five compounds are also very lipophilic (logP ∼ 5.7–7.6, logD ∼ 4.5–6.4; Table 1) weak bases (pKa ∼ 8–8.8; Table 1) with computed charges (pH 7.4) in the range of 0.82–0.97, with raloxifene (22) being the least hydrophobic and having the smallest positive charge (+0.82; Table 1). There are, therefore, several FDA-approved drugs active against M. tuberculosis that act as cationic protonophore uncouplers and collapse Δψ. It is also possible, however, that there could be many anionic protonophore uncouplers, as well as neutral species (e.g., AU1235), so we next investigated several such compounds in the IMV assays.
In recent work, we (49) and others (50, 51) identified several anionic, bacterial cell growth inhibitors that, in addition to inhibiting bacteria-specific enzyme targets, might have activity as uncouplers, as expected for lipophilic, weak acid, classic uncouplers like CCCP and DNP. We first investigated seven compounds with known antibacterial activity and a diverse range of proposed protein targets. In all cases, we anticipated a negative net charge (at pH 7.4). The compounds were the benzoic acid BPH1463 (23) (Fig. 1), which inhibits undecaprenyl diphosphate synthase (UPPS) (52); the benzoic acid BPH1276 (24) developed by Pharmacia, which has been proposed to target transcription/translation (50) but also inhibits UPPS (49); the diketoacid BPH1330 (25) that inhibits S. aureus UPPS, S. aureus dehydrosqualene synthase (CrtM) (52), and S. aureus cell growth, as well as inducing formation of neutrophil extracellular traps (52); the dihydropyridin-2-one-3-carboxamide BPH889 (26) developed by Novartis, which inhibits Streptococcus pneumoniae UPPS, S. pneumoniae, and S. aureus cell growth (51); platensimycin (27); and sulfamethoxazole (28). The phenol platensimycin (27) is an antiinfective that inhibits fatty acid biosynthesis (53), as well as having antidiabetic activity (54). There is also renewed interest in sulfamethoxazole (28)/trimethoprim (29) combined activity against M. tuberculosis (55), and sulfamethoxazole has a sulfonamide that can act as a weak acid. However, of these compounds, only the benzoate (23) had significant activity as a protonophore (Table 1), due perhaps to relatively unfavorable interactions of most species with anionic membrane lipids.
In addition to cationic and anionic uncouplers, there are several known neutral uncouplers. In early work, it was found that in the industrial preparation of the herbicide Diuron [3-(3,4-dichlorophenyl)-1,1-dimethylurea], there was an impurity that had potent activity as an uncoupler: N,N′-bis(3,4-dichlorophenyl)urea (30) (56). In later work (57), it was found that N,N′-bis(para-trifluoromethylphenyl)urea (31) was an even more potent uncoupler (in plant mitochondria and thylakoid membranes), and this compound displayed no titration behavior between pH 2 and pH 9 (57), as expected for a urea. More recently, the analog 1-(2-adamantyl)-3-(2,3,4-trifluorophenyl)urea [AU1235 (9)] has been found to kill replicating but not nonreplicating M. tuberculosis (58) and to collapse the pH gradient in M. smegmatis (20). The detailed molecular mechanism of action of the neutral uncouplers is not known but has been proposed to be related to either conformational changes in membrane proteins or changes in phospholipid bilayer organization (57). We find that AU1235 (9) collapses the pH gradient in the EcIMV assay, consistent with the NMR observations (20). In addition, we find that N5,N6-bis(2-fluorophenyl)-[1,2,5]oxadiazolo[3,4-b]pyrazine-5,6-diamine (BAM15; 32), a mitochondrial uncoupler that does not depolarize the plasma membrane (59), also acts as a (neutral) uncoupler in the EcIMV assay. Clearly, further work is needed to clarify the molecular mechanism of action of the neutral uncouplers, and we briefly investigate below the possible effects of AU1235 on membrane bilayer structure. Finally, we investigated several other compounds that have been reported to act as uncouplers in S. aureus (60): pyrazinocarbazole (33), D2 (34), and D3 (35). Only the pyrazinocarbazole (33) showed uncoupler activity and although it has previously been reported (60) to collapse Δψ selectively in S. aureus, it appears to act here as a protonophore, consistent with its computed pKa (6.2) and logP (4.7) values.
Mechanisms of Action of Uncoupling.
We next tested all compounds for their effects on the membrane potential, Δψ, in EcIMVs using oxonol VI as the fluorescence probe and on Δψ in porcine mitochondria using 3,3-dipropylthiadicarbocyanine iodide [DiSC3(5)] fluorescence as the probe (61) (Table 1). Most compounds tested were active in Δψ collapse in EcIMVs, but very few were active in porcine mitochondria (Table 1).
When examining the results on ∆pH/∆ψ collapse in IMVs, it can be seen from the heat map shown in Fig. 2E that ∆pH collapse (∆pH EcIMV and ∆pH MsIMV) and EcIMV ∆ψ collapse (∆ψ) are highly correlated (red, Pearson R values are shown on the heat map), which suggests that these compounds mainly affect the proton gradient across the membrane and are not highly bacteria-specific. The R value for EcIMV ∆pH and ∆ψ collapse is ∼0.8, comparable to the ∼0.89 reported previously (13) for a highly homologous series of compounds, all SQ109 analogs. However, the ∆pH/∆ψ collapse in bacterial IMVs (∆pH EcIMV, ∆pH MsIMV, and ∆ψ) is not highly correlated with Δψ collapse in mitochondria (mito). Reasons are unknown but could be due, in part, to differences in lipid composition, lipid–protein interactions, and the possible roles of transporters. The ∆pH/∆ψ collapse in the bacterial IMVs has a linear correlation with logD and charge, but not with pKa [because a parabolic pKa dependence is expected (62)]. These results indicate that logD (computed from logP, pKa, and pH) and charge are both important descriptors for uncoupling activity. More importantly, the ∆pH/∆ψ collapse found with bacterial IMVs is correlated with the IC50 for M. tuberculosis cell growth inhibition (Mtb), as would be expected if uncoupling activity contributes to the inhibitory potency of many of these compounds. The R values are not high (R = 0.6–0.7) because, of course, in many if not most instances, enzyme targeting will dominate. Nevertheless, as shown in Fig. 2F, the compounds with good M. tuberculosis activity are the ones with the most potent activities in ∆pH/∆ψ collapse of bacterial IMVs, high logD, and positive charge (as indicated by the black square in Fig. 2F): clofazimine, TMC207, and SQ109. So, in general, the lipophilic, cationic species are the best uncouplers, as well as the best M. tuberculosis cell growth inhibitors. The mycobacterial cell wall is, of course, an exceptionally strong barrier for drug penetration (63), and, in addition, mycobacteria produce many active efflux pumps. However, although a highly hydrophobic cell wall is a strong barrier for most drugs, for highly hydrophobic species [e.g., SQ109, clofazimine, TMC207] targeting inner membrane proteins, it could actually act as a drug “reservoir,” enabling effective enzyme targeting. In addition, efflux pumps that are powered by the PMF will be inhibited by such species.
The results discussed above suggested that it might be of interest to investigate drug–membrane interactions with neutral (zwitterionic) and anionic model lipid bilayer membranes. We show in Fig. 3 A–E the DSC results for drugs binding to the saturated, zwitterionic lipid, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine [DPPC, having a gel-to-liquid crystal phase transition temperature (Tm) ∼ 42 °C], and in Fig. S2 the results for the anionic lipid, 1,2-dimyristoyl-sn-glycero-3-phosphatidic acid (DMPA; Tm ∼ 46 °C at pH 7). We chose to investigate three types of inhibitors: the cationic species SQ109 (2), amiodarone (4), and TMC207 (1), all of which are expected to have relatively localized charges; the cationic species clofazimine (17), where the charge is expected to be delocalized over the π-system; and the neutral species, AU1235 (9).
Fig. 3.

DSC thermograms and EPR spectra for five compounds binding to DPPC or DMPA. DSC thermograms for five compounds binding to DPPC: SQ109 (A), amiodarone (B), TMC207 (C), clofazimine (D), and AU1235 (E). Cp, the heat capacity at constant pressure. (F) pH dependence of DSC thermograms of DPPC-SQ109 (2) (20 mol%) at pH 5, pH 7, and pH 9. (G) Comparison of pH dependence of logD, ∆Tm, IC50s of ΔpH, and M. smegmatis cell growth inhibition of SQ109. Drug concentration is 20 mol% total lipids, excess water. (H) Variable temperature (VT) X-band (9.14 GHz) continuous wave (CW)-EPR spectra for DPPC lipid with 0.1% 5-DOXYL stearate methyl ester (5-DSME) spin label, with or without 20 mol% SQ109 (2), amiodarone (4), clofazimine (17), or AU1235 (9). (I) VT X-band (9.14 GHz) CW-EPR spectra for DMPA lipid with 0.1% 5-DOXYL stearic acid (5-DSA) spin label, with or without compound SQ109 (2), amiodarone (4), clofazimine (17), or AU1235 (9). The red boxes in H and I indicate phase transitions of lipids. The red arrows in H indicate evidence for more than a single spectral component.
Fig. S2.

DSC thermograms for five compounds binding to DMPA: SQ109 (A), amiodarone (B), TMC207 (C), clofazimine (D), and AU1235 (E). Cp, the heat capacity at constant pressure. (F) pH dependence of DSC thermograms of DMPA-SQ109 (2) (20 mol%) at pH 5, pH 7, and pH 9.
With the cationic species [SQ109 (2), amiodarone (4), and TMC207 (1)], there are 2.4–9.4 °C decreases in Tm [the maximum in the heat capacity at constant pressure (Cp)-vs.-T thermogram] with DPPC, with amiodarone (4) also broadening the thermal transition, which means that with amiodarone, there are both fluid and ordered lipid domains present in the range of the broad transition (Fig. 3 A–C). However, there is essentially no change in Tm (∆Tm ∼ 0.8 °C) with clofazimine (17), only a slight broadening of the transition (Fig. 3D). With the neutral uncoupler, AU1235 (9), there is likewise only a small change in the transition, with ∆Tm = −1.2 °C (Fig. 3E). Thus, the largest effects on the gel-to-liquid crystal phase transition with DPPC are found with the two relatively elongated molecules, SQ109 (2) and amiodarone (4), in which the cationic charge center is located at one end of the molecule. TMC207 (1) is more spherical or globular, and this shape is likely to affect its interaction with the lipid hydrocarbon chains. Clofazimine (17) is essentially planar, and the positive charge is delocalized over N3 and N5, and it has essentially no effect on lipid chain packing, the exception being that the pretransition (associated with ripple phase formation) is no longer present. With the uncharged AU1235 (9), the negative ∆Tm is certainly a surprising result and suggests slightly stronger chain packing, due perhaps to urea–polar headgroup interactions. With the anionic lipid DMPA, which might be expected to undergo strong electrostatic interactions with the protonated drug molecules, the effects on the phase transition (Fig. S2 A–E) are, in most cases, quite similar to the effects seen with DPPC (which is zwitterionic), suggesting that the effects that are seen with both lipids are due, primarily, to a direct perturbation of chain packing. The exception is found with SQ109 (2), which causes a more extensive broadening and shifting of the phase transition with DMPA (Fig. S2) than is found when it interacts with DPPC.
The effects of protonophore uncouplers on lipid (bilayer) membranes are expected to be pH-dependent not only because, for the most part, uncouplers have protonatable groups but also because in many cases the lipids themselves contain titratable headgroups. We thus conducted a DSC investigation of two model systems: DPPC-SQ109 (2) and DMPA-SQ109 (2), at three pH values: pH 5, pH 7, and pH 9. As can be seen in Fig. 3F, the main gel-to-liquid crystal phase transition in pure DPPC is independent of pH, but in the presence of SQ109 (2), the transition temperature decreases as the pH increases. The pH effects on ∆Tm are positively correlated with logD and negatively correlated with the EC50 values for ∆pH collapse and M. smegmatis cell growth inhibition by SQ109, as shown in the radar plot in Fig. 3G. Lipophilicity and the pKa of the uncoupler thus both have a significant effect on the interaction with membrane lipids, and hence uncoupling activity and cell growth inhibition, consistent with the observation that the ∆pH/∆ψ collapse of bacterial IMVs correlates with logD of the uncoupler (Fig. 2E). Moreover, at pH 5, the ethylenediamine SQ109 is predicted to be diprotonated, and it has the smallest effect on Tm, whereas the singly protonated form (at pH 9) has the largest effect, due, perhaps, to enhanced chain packing with the singly protonated form, although more work is needed to pin-down the details of such drug–membrane interactions. With pure DMPA, the main transition is pH-dependent, and at pH 7 and pH 9, where the DMPA is deprotonated but SQ109 (2) remains (positively) charged, there are large shifts in Tm (Fig. S2F). In both the DPPC and DMPA systems, however, there are decreases in the thermal transition temperatures in the presence of SQ109 (2), which may be expected to increase membrane lipid disorder/fluidity and, arguably, uncoupler activity.
We also investigated the effects of four drug molecules, SQ109 (2), amiodarone (4), clofazimine (17), and AU1235 (9), on lipid order/dynamics, using the EPR spin label approach we used previously to investigate the effects of cholesterol on lipid membranes (64). Fig. 3 H and I shows variable temperature results for DPPC and DMPA. With DPPC at 25 °C, all spectra correspond to immobilized spin labels, characteristic of a gel-like phase (64). As temperatures increase, DPPC + SQ109 has an EPR-detected phase transition between 30 °C and 35 °C, whereas pure DPPC and DPPC + clofazimine/AU1235 show phase transitions between 35 °C and 40 °C, as indicated by the red boxes in Fig. 3H. With DPPC + amiodarone (4), there is evidence for more than a single spectral component, consistent with the considerable broadening of the transition seen in DSC. Similar results are seen with DMPA with the four compounds, as shown in Fig. 3I. The DSC and EPR spin label results therefore both indicate that the largest effects on membrane structure are seen with the localized charge uncouplers, whereas the delocalized charge and neutral species have much smaller effects on bilayer structure.
Overall then, the results described here and in the previous sections indicate that many TB drugs and drug leads are quite potent uncouplers. In some cases, as discussed in the next section, these compounds also act as enzyme inhibitors, providing the possibility of multisite targeting. This possibility is, of course, of importance because most antibiotics that have been relatively resistant to the development of drug resistance over time have more than one target. We next consider one class of enzymes that might be particularly good targets for inhibitors that are also uncouplers: the isoprenoid or prenyl biosynthesis enzymes.
Targeting Prenyl Synthases.
As a class, prenyl synthases are important drug targets. In addition, several protonophore uncouplers are known to act as prenyl synthase inhibitors. A plausible reason for this uncoupler/inhibitor relation is that most prenyl synthases use either cationic (transition state/reactive intermediate) or anionic (substrate/product) headgroups and they have substrates/products with large hydrophobic side chains. Therefore, cationic or anionic uncouplers with charged-hydrophobic structural characteristics can be well accommodated by the polar-nonpolar active site pockets of prenyl synthases, and may have high activity as competitive inhibitors. By way of some examples, we show in Fig. 4 the structures of six prenyl synthase drug targets, together with known enzyme inhibitors that are also uncouplers [several of which are FDA-approved drugs (4, 36) or are in clinical trials (2)]. All six proteins have polar-nonpolar active-site pockets that correlate with the involvement of charged-hydrophobic substrates/products or transition state/reactive intermediates, as shown in Fig. 4.
Fig. 4.

Structures of prenyl synthases that are targets for dual PMF/enzyme inhibitors and some inhibitors. (A) Phyre2 model (65) prediction of MenA. (B) CrtM (PDB ID code 4EA1). (C) OSC (PDB ID code 1W6J). (D) UPPS (PDB ID code 4H2J). (E) DPPS (PDB ID code 2VG3). (F) Rv3378c (PDB ID code 3WQM). The polarity surfaces for the enzyme pockets are also shown, with orange indicating a polar surface and white indicating a nonpolar surface.
Fig. 4A is a Phyre2 (65) model prediction for MenA [based on UbiA (66); Protein Data Bank (PDB) ID code 4OD5]. SQ109 analog 3 inhibits M. smegmatis MenA with an IC50 of 4 μM and E. coli MenA with an IC50 of 400 nM (13). Fig. 4B shows the structure of S. aureus dehydrosqualene synthase, CrtM (PDB ID code 4EA1) and its inhibitor SQ109 (67), and Fig. 4C shows the structure of human OSC [a model for the trypanosomatid OSC drug targets; PDB ID code 1W6J (21)]. MenA and CrtM both have typical all α-helical structures found in trans-prenyl synthases, whereas OSC has the two-domain structure found in the class II terpene cyclases. The proteins shown in Fig. 4 D–F all contain the cis-isoprenoid biosynthesis enzyme fold: UPPS [PDB ID code 2E98 (68)] in Fig. 4D; decaprenyl diphosphate synthase [DPPS; PDB ID code 2VG3 (69)] in Fig. 4E; and Rv3378c, tuberculosinyl adenosine (TbAd) synthase [PDB ID code 3WQM (70)], in Fig. 4F. Rv3378c is of interest because it is a target for antivirulence-based therapeutics for TB. As one example of a UPPS uncoupler-inhibitor, we reported in recent work that the fertility drug clomiphene also had activity against S. aureus and that a major target was UPPS (71). What is interesting about the clomiphene (36) structure is that it is remarkably similar to the structure of tamoxifen (20), which itself has antiinfective activity. We find an EC50 of 1.2 μM for clomiphene (in the EcIMV assay; Table 1) in comparison to 0.39 μM for the potent uncoupler CCCP and 0.45 μM for tamoxifen. Clomiphene also has an 8 μg/mL MIC against Enterococcus faecium (72) and a 0.22 μM IC50 against liver stage malaria parasites (73), suggesting that there may be dual target activity in several pathogens. So, uncoupler-inhibitors are known for MenA, CrtM, OSC, DPPS, and UPPS, whereas uncoupler-inhibitors for Rv3378c have yet to be discovered.
Rv3378c catalyzes the formation of TbAd (74) (Fig. 5A) and related compounds (75). Transposon mutants of Rv3378c show inhibited phagosomal acidification, and Rv3378c is necessary for production of TbAd and related metabolites. Therefore, it is likely that one of the TbAd compounds controls intravacuolar pH (76). Here, we initially sought to discover inhibitors of Rv3378c that were also uncouplers, reasoning that the combination of direct uncoupling (as with SQ109) and antivirulence activity would be a good approach to killing intracellular M. tuberculosis. We thus carried out an in silico screen of a library of 1,013 compounds [from National Cancer Institute (NCI) diversity set III] using the MD-based structures reported previously (77). The X-ray structure of Rv3378c with a bound inhibitor is shown in Fig. 5B, together with one snapshot from an MD trajectory.
Fig. 5.

Vacquinol and its analogs as inhibitors for Rv3378c activity and bacterial cell growth. (A) Reaction catalyzed by Rv3378c. (B) MD simulation/in silico screening approach used to identify Rv3378c inhibitors, showing the X-ray structure (Left), MD snapshot (Center), and in silico hit (Right). (C) Heat map of correlation coefficients between uncoupling activity (ΔpH EcIMV) and antibacterial (Mtb, MtbE, Ms, Sa)/antifungal (Sc)/anticancer (GBM) activities of the vacquinol series. Structures are shown in Fig. S5. Ms, M. smegmatis; Sa, S. aureus; Sc, S. cerevisiae.
We tested 39 compounds (Fig. S3) from the in silico screen for Rv3378c inhibition activity using tuberculosinyl diphosphate and 3H-adenosine as substrates. The structures of the compounds tested are shown, together with their IC50 values against Rv3378c, in Fig. S3. Compounds that failed the pan-assay interference compounds (PAINS) (78) test are shown in red in Fig. S3. The most active compound was the ethanolamine NSC13316 (37), which had an IC50 of 2.7 μM in Rv3378c inhibition (Fig. S4). Surprisingly, NSC13316 (37) has already been reported (79) to potently inhibit the growth of M. tuberculosis, with an MIC of 1.6 μg/mL (4.5 μM). This effect is not expected for a virulence-targeting drug lead. Moreover, NSC13316 (37), now known as vacquinol-1, has activity against glioblastoma multiforme (GBM) cancer cells (80), both in vitro and in vivo, and is thought to kill these tumor cells by an unusual mechanism involving a decrease in ATP levels and extensive vacuolization (80). NSC13316 (37) is, therefore, a potentially interesting new antiinfective multitarget drug lead.
Fig. S3.

Thirty-nine compounds from the in silico screen tested against Rv3378c. IC50 values obtained are shown. Compounds in red failed the pan-assay interference compounds (PAINS) test (78). The compounds were obtained from the NIH, and their National Service Center code numbers are shown.
Fig. S4.

Rv3378c inhibition and uncoupling activity of vacquinol (EcIMV assay).
Vacquinol Analogs as Antiinfective Drug Leads.
The observation that vacquinol is active against M. tuberculosis growth, as well as the antivirulence target Rv3378c, was of interest, so we next carried out a structure similarity search based on NSC13316 (37) and obtained 13 analogs from the NCI/Developmental Therapeutics Program Open Chemical Repository (dtp.cancer.gov/). We then tested NSC13316 (37) and these analogs (Fig. S5) for uncoupling activity in the EcIMV and MsIMV assays, finding that vacquinol (37) had an ∼12–13 μM IC50 (Fig. S4). Results for the analogs are summarized in Table S1.
Fig. S5.

Thirteen analogs of NSC13316 (37) found by using a similarity search.
Table S1.
Uncoupling and antibacterial activities for vacquinol and its analogs
| ∆pH, μM | Cell growth IC50, μg/mL | |||||||
| NSC no. | EcIMV | MsIMV | Mtb | MtbE | Ms | Sa | Sc | GBM* |
| 2450 | 3.0 | 3.5 | 1.5 | 3.1 | 1.7 | 2.8 | 3.4 | N.T. |
| 4377 | 4.1 | 3.1 | 1.2 | 2.3 | 1.1 | 1.4 | 3.4 | 0.40 |
| 13097 | 7.3 | 6.8 | 1.1 | 2.1 | 2.7 | 26 | 35 | N.T. |
| 13316 | 12 | 13 | 2.1 | 8.5 | 4.6 | 41 | 35 | 1.1 |
| 13466 | 42 | 21 | 3.2 | 7.8 | 10.6 | 65 | 75 | 4.1 |
| 13480 | 11 | N.T. | 0.55 | 1.5 | 0.67 | 1.7 | 6.6 | 0.14 |
| 14224 | 6.8 | 12 | 0.70 | 2.8 | 1.4 | 28 | 5.6 | 0.75 |
| 16001 | 6.2 | 6.9 | 0.53 | 2.8 | 1.4 | 9.0 | 13 | N.T. |
| 23924 | 14 | 5.8 | 2.3 | 6.2 | 4.1 | 27 | 38 | N.T. |
| 23925 | 32 | N.T. | N.T. | N.T. | 7.8 | 60 | 350 | 2.6 |
| 146028 | 300 | N.T. | N.T. | N.T. | 120 | 84 | 84 | 25 |
| 157571 | 6.6 | N.T. | N.T. | N.T. | 2.4 | 3.6 | 15 | 0.27 |
| 305758 | 4.6 | 7.7 | 1.2 | 1.5 | 1.1 | 4.0 | 3.4 | 0.42 |
| 305821 | 31 | 100 | N.E. | N.E. | 310 | 430 | 430 | N.T. |
Ms, M. smegmatis (100 μM); Mtb, M. tuberculosis (50 μM); N.E., no effect at highest concentration tested; NSC, National Service Center; N.T., not tested; Sa, S. aureus (100 μM); Sc, S. cerevisiae (150 μM).
From Kitambi et al. (80).
The vacquinol class of compounds, 2-piperidinyl-4-quinolinemethanols, were originally developed as antimalarials (81, 82), and in addition to their activity against M. tuberculosis and brain cancer cells, they can inhibit efflux pumps and are of interest in combating multidrug resistance in cancer cells (79, 83, 84), suggesting the possibility that their uncoupling effects could contribute to a diverse range of activities. The fact that vacquinol also has promising in vivo activity against GBM, a good in vivo pharmacokinetic profile, oral bioavailability, and favorable overall preclinical characteristics (80), in addition to killing M. tuberculosis, makes it an interesting lead for antiinfective development.
We therefore next tested the 13 vacquinol analogs for growth inhibition activity against M. tuberculosis H37Rv and M. tuberculosis Erdman, as well as against another bacterium, S. aureus, and against the yeast Saccharomyces cerevisiae, basically to see if there were general growth inhibitory effects against bacteria and a fungus. We found that vacquinol and its analogs were quite potent in cell growth inhibition against each of these microorganisms, with the best IC50s for M. tuberculosis H37Rv, M. tuberculosis Erdman, S. aureus, and S. cerevisiae being 0.53, 1.5, 1.4, and 3.4 μg/mL, respectively (Table S1). Moreover, we found that the uncoupling activity of these compounds (EcIMV assay) correlated well with their bacterial and yeast cell growth inhibition potency, as well as with the growth inhibition of GBM cancer cells (Fig. 5C), with coefficients of cross-correlation larger than 0.7, suggesting the possibility that uncoupling is a contributor to the antibacterial/antifungal/anticancer activity of the vacquinol series.
Future Prospects for Enzyme/Uncoupler Drug Leads.
The results we have presented above show that numerous FDA-approved drugs and other drug leads have activity as uncouplers. Pure uncouplers (without enzyme targets) are generally not expected to be good drug leads, although, as noted in the Introduction, some antiparasitics function in this way, plus there is considerable interest in the development of DNP prodrugs/formulations for treating diabetes, insulin resistance, and hepatic steatosis. Because we find that TB drugs like clofazimine, bedaquiline, and SQ109 all have protonophore uncoupler activity, it seems likely that one route to finding new leads will be to investigate analogs of the types of compounds we have discussed here, for antiinfective, enzyme inhibition, and uncoupler activity.
It is also possible that prodrug uncouplers can be produced in some cases in much the same way that DNP-methyl ether is being developed to treat diabetes, or the fact that nitazoxanide (13) is a prodrug for tizoxanide (14). As an example, with M. tuberculosis, it has recently been shown that the heartburn/proton pump inhibitor drug lansoprazole is metabolized to lansoprazole sulfide (structures in Fig. 6), which has potent activity against M. tuberculosis inside macrophages (85), and similar antibacterial effects are seen with omeprazole (which is reduced to the active sulfide) in Helicobacter pylori (86), as well as with rabeprazole (87, 88). Lansoprazole, omeprazole, and rabeprazole are all proton pump inhibitors that contain benzimidazole groups with sulfoxide substituents. They can be reduced to sulfides, and this reduction correlates with a predicted large increase in benzimidazole pKa values (from ∼1 for the highly electron-withdrawing sulfoxide to ∼4.2 for the electron-donating sulfides), and this increased basicity would increase uncoupling activity (as would an increase in logP of ∼1 unit). We tested lansoprazole, lansoprazole sulfide, and rabeprazole sulfide for uncoupling activity in the EcIMV assay. Results are shown in Fig. S6. Clearly, lansoprazole itself has essentially no (∼1 mM) activity as an uncoupler, but the two sulfides have measurable activity, ∼70 μM for lansoprazole sulfide. The large changes in pKa predicted for benzimidazoles with sulfide/sulfoxide substituents, as well as the observation of uncoupling activity for the sulfides, are consistent with earlier work on sulfoxide/sulfide proton pump inhibitors (89), where uncoupling was observed with the sulfides, and suggests possible routes to new (pro)drug leads with multitarget (enzyme and uncoupler) activity. This prodrug activity involving the host is also a contributor to the activity of a standard TB drug, pyrazinamide, which is now thought to act, in large part, as pyrazinoic acid, a modest uncoupler that has multiple targets (39, 90). A pictorial summary of a selection of such enzyme/uncoupler multitarget inhibitors is shown in Fig. 6.
Fig. 6.

Schematic illustration of antiinfective drugs/drug leads that target both enzymes and the PMF. CFZ, clofazimine; CLO, clomiphene; LPZ, lansoprazole; LPZS, lansoprazole sulfide; PZA, pyrazinamide; PZOA, pyrazinoic acid; ROS, reactive oxygen species; VAC, vacquinol.
Fig. S6.

Uncoupler activity of CCCP, lansoprazole (Ipz), lansoprazole sulfide (Ipzs), and rabepazole sulfide (rabs) as a function of pH.
Conclusions
We tested a series of cationic, neutral, and anionic compounds for uncoupler activity in M. smegmatis and E. coli IMV assays and in porcine liver mitochondria. The most active compounds in the IMV assays were cationic amphipathic drugs. These drugs also inhibited M. tuberculosis and M. smegmatis cell growth. Clofazimine (17) and TMC207 (1) were particularly active uncouplers, comparable to CCCP. We investigated drug–membrane interactions using DSC and EPR, finding that lipophilic cations with localized charges had large effects on the lipid phase transition, whereas delocalized charge species [clofazimine (17) and the neutral uncoupler AU1235 (9)] had essentially no effects. Several uncouplers were inhibitors of isoprenoid biosynthesis enzymes, so we then used in silico screening to discover new inhibitors of TbAd synthase that were also uncouplers. We found that vacquinol (37) was one such compound, a result of interest because vacquinol is a new drug lead for treating GBM and also has direct killing activity against M. tuberculosis. These observations led to the discovery of more potent analogs with activity against M. tuberculosis and S. aureus that also likely function, at least in part, as uncouplers. We also discovered that the new S. aureus growth inhibitor clomiphene, known to target cell wall biosynthesis by inhibiting UPPS, was an uncoupler, expected to lead to resistance-resistance. Overall, the results are of broad general interest because we find that many lipophilic, cationic species have activity against bacteria and that they act, at least in part, as uncouplers. In addition, many of the vacquinol class of GBM cell growth inhibitors are also uncouplers, and some have promising activity against M. tuberculosis and S. aureus.
The fact that the new M. tuberculosis drugs/drug leads bedaquiline and SQ109, as well compounds such as clofazimine (where there is renewed interest in treating M. tuberculosis), are protonophore uncouplers that also have activity against (or are activated by) enzyme targets makes it likely that this multitargeting will contribute to overall activity and resistance-resistance, making the further development of such multitarget leads of interest.
Methods
M. smegmatis, S. cerevisiae, S. aureus, and M. tuberculosis growth inhibition assays; the porcine liver mitochondria Δψ assay; ΔpH and Δψ assays with IMV; MD simulation of Rv3378c; Rv3378c inhibition; DSC; and EPR were performed as described previously (13, 70, 77, 91, 92), with full details given in SI Methods.
SI Methods
General Methods.
All chemicals were reagent grade. 1H NMR and 13C NMR spectra were obtained on Varian Unity spectrometers at 400 and 500 MHz for 1H and at 100 and 125 MHz for 13C. Elemental analyses were carried out in the University of Illinois Microanalysis Laboratory. HPLC/MS analyses were performed by using an Agilent LC/MSD Trap XCT Plus system (Agilent Technologies) with an 1100 series HPLC system, including a degasser, an autosampler, a binary pump, and a multiple wavelength detector. All compounds were ≥95% pure as determined by elemental analysis or analytical HPLC/MS analysis and were also characterized by 1H nuclear magnetic resonance spectroscopy (NMR) and high resolution mass spectrometry (HRMS).
M. smegmatis, S. cerevisiae, S. aureus, and M. tuberculosis Growth Inhibition Assay.
IC50 values for M. smegmatis growth inhibition were determined by using a microbroth dilution method. A stationary starter culture of M. smegmatis was diluted to an OD600 of ∼0.001 with fresh 7H9 medium supplemented with 10% (vol/vol) albumin dextrose catalase (ADC) and 4% (vol/vol) glycerol to give a working solution; 200 μL of working solution was then transferred to each well of a 96-well, flat-bottom culture plate (Corning, Inc.). Inhibitors were then added at 0.2 mM and threefold serial-diluted to 30 nM. Plates were incubated for 36 h at 37 °C, and the A at 600 nm was determined. A nonlinear regression analysis was carried out using Origin 6.1 (OriginLab Corporation) to obtain the IC50 values. S. cerevisiae and S. aureus IC50 values were obtained similarly, except for different growth media and incubation times: yeast peptone dextrose (YPD) medium and 36 h of growth for S. cerevisiae and tryptic soy broth (TSB) medium and 16 h of growth for S. aureus. The M. tuberculosis growth inhibition assay was performed as described previously (91).
ΔpH and Δψ Assay with IMV.
The preparation of IMVs, assay for ATP- or succinate-driven proton translocation, and determination of Δψ collapse in IMVs were as reported previously (13).
Porcine Liver Mitochondria Δψ Assay.
Mitochondria were prepared from fresh porcine liver according to a reported protocol (92), with 225 mM mannitol, 75 mM sucrose, 10 mM KCl, 10 mM Tris⋅3-(N-morpholino)propanesulfonic acid (pH 7.5), 5 mM sodium phosphate (pH 7.5), 0.5 mM EDTA, 0.5 mM EGTA as washing buffer, washing buffer with 1 mg/mL BSA, and protease inhibitors as an isolation buffer. The effects of inhibitors on mitochondrial Δψ were determined by fluorescence quenching of the potential-sensitive probe 3,3′-dipropylthiadicarbocyanine iodide DiSC3(5). Mitochondria were mixed with washing buffer with 1 mg/mL BSA, 5 mM MgSO4 and 5 μM DiSC3(5), and the baseline was monitored for 5 min. The reaction was then initiated by adding 5 mM succinate. When the signal had stabilized, compounds were added and proton translocation was measured fluorometrically. Different concentrations of compounds were added to the suspension, and changes in fluorescence due to the disruption of Δψ were continuously monitored with a fluorescence spectrophotometer (FLUOstar Omega; BMG LABTECH) using an excitation wavelength of 643 nm and an emission wavelength of 666 nm.
MD Simulation of Rv3378c.
MD simulation of Rv3378c was performed as described previously (71). A total of 12 receptor structures from crystal structures and MD simulations were used in the in silico screening with a library of 1,013 compounds (from NCI diversity set III).
Rv3378c Inhibition.
Rv3378 was expressed and purified as described previously (70). The IC50 values for the Rv3378c inhibitors were determined by radiometric assay as follows. Thirteen microliters of 100 nM Rv3378c and inhibitors in the assay buffer (25 mM Tris⋅HCl, 1 mM MgCl2, 0.01% Triton X-100) was incubated for 10 min at 4 °C. Eighteen microliters of 20 μM tuberculosinyl diphosphate and 40 μM adenosine [0.18% (mole/mole) [2,8-3H] adenosine, 1 mCi/mL; Moravek Biochemicals, Inc.] were then added. The reaction was incubated at 37 °C for ∼16–20 h before quenching with 500 μL of saturated NaCl solution. The adenosine in the saline solution was extracted with 500 μL of butanol by vortexing, and 300 μL of the organic layer was transferred into a scintillation vial for counting. IC50 values were obtained from dose–response curves by using Origin 9.0 software.
DSC.
The DPPC and DMPA were obtained from Avanti Polar Lipids, and 20 mol% compounds were codissolved with DPPC (DMPA) in chloroform (chloroform/methanol/water). Solvent was removed under a stream of dry nitrogen, and remaining solvent traces were removed in vacuo overnight. Ten microliters of deionized water or sodium phosphate buffer (pH 5, pH 7, or pH 9) was added, and the mixtures were homogenized by hand. Weighed amounts of the mixtures were then sealed into solid sample inserts (stainless-steel tubes). DSC experiments were performed by using a Microcal VP-DSC instrument. Scans covered a 10–70 °C range at a scan rate of 60 °C per hour. The DSC thermograms were analyzed using Origin 7.1. Water vs. water scans were used for baseline correction.
EPR Sample Preparation.
Each EPR sample contained DPPC or DMPA lipid with or without a drug (4:1 lipid/drug molar ratio) for a total weight of 3.5 mg. The lipids and drugs were dissolved in either chloroform or 65:35:8 chloroform/methanol/water. A total of 0.05 mg of 5-DOXYL stearic acid or 5-DOXYL stearate methyl ester (both from Sigma-Aldrich) spin label was dissolved in methanol and added to each sample. The solutions were mixed, dried under a nitrogen gas stream until no liquid phase was visible, and then placed under vacuum for 3 h to remove residual solvent. To make a pH-controlled aqueous suspension sample, 500 μL of phosphate buffer (145 mM, pH 7.0) was added. The sample was heated to 65 °C in a water bath, vortexed to form a suspension, and then cooled on ice. Lipids were pelleted by centrifugation at 16,873 × g for 2 min and then cooled on ice again, and 450 μL of supernatant was removed, yielding a 50-μL sample with 7% (wt/vol) lipids and 0.1% spin label. The sample was again heated to 65 °C and mixed thoroughly to form a suspension. The final suspension was injected into a glass tube with an internal diameter of 1.1 mm and stored at −20 °C before spectroscopic experiments.
EPR Spectroscopy.
Continuous-wave EPR spectroscopy was performed at X-band (9.14 GHz) using a Varian E-Line Century Series spectrometer with a Varian E-102 microwave bridge. A stable flow (25 L⋅h−1) of nitrogen gas was passed through a cold trap and heated with a Varian 906790-07 variable temperature controller before passing through the EPR sample cavity. The temperature of the sample cavity was monitored with an Omega thermocouple immediately before and after data acquisition; temperature was maintained within ±0.3 °C of the desired value. First-derivative EPR data were obtained with the following parameters: magnetic field scan range = 3,160–3,360 G; field modulation = 2.5 G, modulation frequency = 100 kHz, time constant = 64 μs; total scan time = 6–15 min for each spectrum.
Acknowledgments
We thank Prof. Tsutomu Hoshino for providing tuberculosinyl diphosphate and Prof. David B. Moody for his helpful comments. This work was supported by the US Public Health Service (NIH Grant GM065307), by a Harriet A. Harlin Professorship (to E.O.), and by the University of Illinois Foundation/Oldfield Research Fund. Work at the University of California San Diego was supported, in part, by the NIH, National Science Foundation, Howard Hughes Medical Institute, the National Biomedical Computation Resource (NBCR), and the San Diego Supercomputer Center (SDSC).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1521988112/-/DCSupplemental.
References
- 1.Anonymous . Dictionary of Natural Products on DVD. Chapman & Hall/CRC; London: 2013. [Google Scholar]
- 2.Oldfield E, Lin FY. Terpene biosynthesis: Modularity rules. Angew Chem Int Ed Engl. 2012;51(5):1124–1137. doi: 10.1002/anie.201103110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.CDC 2013 Antibiotic Resistance Threats in the US. Available at www.cdc.gov/features/AntibioticResistanceThreats/index.html. Accessed November 21, 2015.
- 4.Taylor J, et al. 2014 Estimating the economic costs of antimicrobial resistance. Available at www.rand.org/pubs/research_reports/RR911.html. Accessed November 21, 2015.
- 5.East SP, Silver LL. Multitarget ligands in antibacterial research: Progress and opportunities. Expert Opin Drug Discov. 2013;8(2):143–156. doi: 10.1517/17460441.2013.743991. [DOI] [PubMed] [Google Scholar]
- 6.Oldfield E, Feng X. Resistance-resistant antibiotics. Trends Pharmacol Sci. 2014;35(12):664–674. doi: 10.1016/j.tips.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.WHO 2015 Global tuberculosis report 2015. Available at www.who.int/tb/publications/global_report/en/). Accessed November 21, 2015.
- 8.Zumla AI, et al. New antituberculosis drugs, regimens, and adjunct therapies: Needs, advances, and future prospects. Lancet Infect Dis. 2014;14(4):327–340. doi: 10.1016/S1473-3099(13)70328-1. [DOI] [PubMed] [Google Scholar]
- 9.Andries K, et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science. 2005;307(5707):223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 10.Protopopova M, et al. Identification of a new antitubercular drug candidate, SQ109, from a combinatorial library of 1,2-ethylenediamines. J Antimicrob Chemother. 2005;56(5):968–974. doi: 10.1093/jac/dki319. [DOI] [PubMed] [Google Scholar]
- 11.Zumla A, Nahid P, Cole ST. Advances in the development of new tuberculosis drugs and treatment regimens. Nat Rev Drug Discov. 2013;12(5):388–404. doi: 10.1038/nrd4001. [DOI] [PubMed] [Google Scholar]
- 12.Tahlan K, et al. SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2012;56(4):1797–1809. doi: 10.1128/AAC.05708-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li K, et al. Multitarget drug discovery for tuberculosis and other infectious diseases. J Med Chem. 2014;57(7):3126–3139. doi: 10.1021/jm500131s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Benaim G, et al. Amiodarone has intrinsic anti-Trypanosoma cruzi activity and acts synergistically with posaconazole. J Med Chem. 2006;49(3):892–899. doi: 10.1021/jm050691f. [DOI] [PubMed] [Google Scholar]
- 15.Benaim G, et al. Dronedarone, an amiodarone analog with improved anti-Leishmania mexicana efficacy. Antimicrob Agents Chemother. 2014;58(4):2295–2303. doi: 10.1128/AAC.01240-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mitchell P. Chemiosmotic coupling in oxidative and photosynthetic phosphorylation. Biol Rev Camb Philos Soc. 1966;41(3):445–502. doi: 10.1111/j.1469-185x.1966.tb01501.x. [DOI] [PubMed] [Google Scholar]
- 17.Mitchell P. Chemiosmotic coupling in energy transduction: A logical development of biochemical knowledge. J Bioenerg. 1972;3(1):5–24. doi: 10.1007/BF01515993. [DOI] [PubMed] [Google Scholar]
- 18.Benaim G, Paniz Mondolfi AE. The emerging role of amiodarone and dronedarone in Chagas disease. Nat Rev Cardiol. 2012;9(10):605–609. doi: 10.1038/nrcardio.2012.108. [DOI] [PubMed] [Google Scholar]
- 19.Veiga-Santos P, et al. SQ109, a new drug lead for Chagas disease. Antimicrob Agents Chemother. 2015;59(4):1950–1961. doi: 10.1128/AAC.03972-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li W, et al. Novel insights into the mechanism of inhibition of MmpL3, a target of multiple pharmacophores in Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2014;58(11):6413–6423. doi: 10.1128/AAC.03229-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thoma R, et al. Insight into steroid scaffold formation from the structure of human oxidosqualene cyclase. Nature. 2004;432(7013):118–122. doi: 10.1038/nature02993. [DOI] [PubMed] [Google Scholar]
- 22.Sacksteder KA, Protopopova M, Barry CE, 3rd, Andries K, Nacy CA. Discovery and development of SQ109: A new antitubercular drug with a novel mechanism of action. Future Microbiol. 2012;7(7):823–837. doi: 10.2217/fmb.12.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woods RA, Bard M, Jackson IE, Drutz DJ. Resistance to polyene antibiotics and correlated sterol changes in two isolates of Candida tropicalis from a patient with an amphotericin B-resistant funguria. J Infect Dis. 1974;129(1):53–58. doi: 10.1093/infdis/129.1.53. [DOI] [PubMed] [Google Scholar]
- 24.Ling LL, et al. A new antibiotic kills pathogens without detectable resistance. Nature. 2015;517(7535):455–459. doi: 10.1038/nature14098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldman RC. Why are membrane targets discovered by phenotypic screens and genome sequencing in Mycobacterium tuberculosis? Tuberculosis (Edinb) 2013;93(6):569–588. doi: 10.1016/j.tube.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 26.Stock U, et al. Measuring interference of drug-like molecules with the respiratory chain: Toward the early identification of mitochondrial uncouplers in lead finding. Assay Drug Dev Technol. 2013;11(7):408–422. doi: 10.1089/adt.2012.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.WHO 2014 Global network to support countries tackle rising tapeworm infection. Available at www.who.int/neglected_diseases/global_network_tackle_tapeworm/en/. Accessed November 21, 2015.
- 28.White CA., Jr Nitazoxanide: A new broad spectrum antiparasitic agent. Expert Rev Anti Infect Ther. 2004;2(1):43–49. doi: 10.1586/14787210.2.1.43. [DOI] [PubMed] [Google Scholar]
- 29.Makobongo MO, Einck L, Peek RM, Jr, Merrell DS. In vitro characterization of the anti-bacterial activity of SQ109 against Helicobacter pylori. PLoS One. 2013;8(7):e68917. doi: 10.1371/journal.pone.0068917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Carvalho LP, Lin G, Jiang X, Nathan C. Nitazoxanide kills replicating and nonreplicating Mycobacterium tuberculosis and evades resistance. J Med Chem. 2009;52(19):5789–5792. doi: 10.1021/jm9010719. [DOI] [PubMed] [Google Scholar]
- 31.de Carvalho LP, Darby CM, Rhee KY, Nathan C. Nitazoxanide disrupts membrane potential and intrabacterial pH homeostasis of Mycobacterium tuberculosis. ACS Med Chem Lett. 2011;2(11):849–854. doi: 10.1021/ml200157f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shigyo K, et al. Efficacy of nitazoxanide against clinical isolates of Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2013;57(6):2834–2837. doi: 10.1128/AAC.02542-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lam KK, et al. Nitazoxanide stimulates autophagy and inhibits mTORC1 signaling and intracellular proliferation of Mycobacterium tuberculosis. PLoS Pathog. 2012;8(5):e1002691. doi: 10.1371/journal.ppat.1002691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tao H, Zhang Y, Zeng X, Shulman GI, Jin S. Niclosamide ethanolamine-induced mild mitochondrial uncoupling improves diabetic symptoms in mice. Nat Med. 2014;20(11):1263–1269. doi: 10.1038/nm.3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang YC, et al. Drug screening identifies niclosamide as an inhibitor of breast cancer stem-like cells. PLoS One. 2013;8(9):e74538. doi: 10.1371/journal.pone.0074538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Imperi F, et al. New life for an old drug: The anthelmintic drug niclosamide inhibits Pseudomonas aeruginosa quorum sensing. Antimicrob Agents Chemother. 2013;57(2):996–1005. doi: 10.1128/AAC.01952-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perry RJ, et al. Reversal of hypertriglyceridemia, fatty liver disease, and insulin resistance by a liver-targeted mitochondrial uncoupler. Cell Metab. 2013;18(5):740–748. doi: 10.1016/j.cmet.2013.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perry RJ, Zhang D, Zhang XM, Boyer JL, Shulman GI. Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science. 2015;347(6227):1253–1256. doi: 10.1126/science.aaa0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Via LE, et al. Host-mediated bioactivation of pyrazinamide: Implications for efficacy, resistance, and therapeutic alternatives. ACS Infect Dis. 2015;1(5):203–214. doi: 10.1021/id500028m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Speirs RJ, Welch JT, Cynamon MH. Activity of n-propyl pyrazinoate against pyrazinamide-resistant Mycobacterium tuberculosis: Investigations into mechanism of action of and mechanism of resistance to pyrazinamide. Antimicrob Agents Chemother. 1995;39(6):1269–1271. doi: 10.1128/aac.39.6.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cholo MC, Steel HC, Fourie PB, Germishuizen WA, Anderson R. Clofazimine: Current status and future prospects. J Antimicrob Chemother. 2012;67(2):290–298. doi: 10.1093/jac/dkr444. [DOI] [PubMed] [Google Scholar]
- 42.Karat ABA, Jeevaratnam A, Karat S, Rao PSS. Double-blind controlled clinical trial of clofazimine in reactive phases of lepromatous leprosy. BMJ. 1970;1(5690):198–200. doi: 10.1136/bmj.1.5690.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grant SS, Kaufmann BB, Chand NS, Haseley N, Hung DT. Eradication of bacterial persisters with antibiotic-generated hydroxyl radicals. Proc Natl Acad Sci USA. 2012;109(30):12147–12152. doi: 10.1073/pnas.1203735109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lechartier B, Cole ST. Mode of action of clofazimine and combination therapy with benzothiazinones against Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2015;59(8):4457–4463. doi: 10.1128/AAC.00395-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hards K, et al. Bactericidal mode of action of bedaquiline. J Antimicrob Chemother. 2015;70(7):2028–2037. doi: 10.1093/jac/dkv054. [DOI] [PubMed] [Google Scholar]
- 46.Rokitskaya TI, Ilyasova TM, Severina II, Antonenko YN, Skulachev VP. Electrogenic proton transport across lipid bilayer membranes mediated by cationic derivatives of rhodamine 19: Comparison with anionic protonophores. Eur Biophys J. 2013;42(6):477–485. doi: 10.1007/s00249-013-0898-9. [DOI] [PubMed] [Google Scholar]
- 47.Nagamune H, et al. The lipophilic weak base (Z)-5-methyl-2-[2-(1-naphthyl)ethenyl]-4-piperidinopyridine (AU-1421) is a potent protonophore type cationic uncoupler of oxidative phosphorylation in mitochondria. Biochim Biophys Acta. 1993;1141(2-3):231–237. doi: 10.1016/0005-2728(93)90047-j. [DOI] [PubMed] [Google Scholar]
- 48.Chen F-C, et al. Pros and cons of the tuberculosis drugome approach--an empirical analysis. PLoS One. 2014;9(6):e100829. doi: 10.1371/journal.pone.0100829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu W, et al. Antibacterial drug leads targeting isoprenoid biosynthesis. Proc Natl Acad Sci USA. 2013;110(1):123–128. doi: 10.1073/pnas.1219899110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Larsen SD, et al. Discovery and initial development of a novel class of antibacterials: Inhibitors of Staphylococcus aureus transcription/translation. Bioorg Med Chem Lett. 2006;16(24):6173–6177. doi: 10.1016/j.bmcl.2006.09.044. [DOI] [PubMed] [Google Scholar]
- 51.Peukert S, et al. Design and structure-activity relationships of potent and selective inhibitors of undecaprenyl pyrophosphate synthase (UPPS): Tetramic, tetronic acids and dihydropyridin-2-ones. Bioorg Med Chem Lett. 2008;18(6):1840–1844. doi: 10.1016/j.bmcl.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 52.Zhang Y, et al. HIV-1 integrase inhibitor-inspired antibacterials targeting isoprenoid biosynthesis. ACS Med Chem Lett. 2012;3(5):402–406. doi: 10.1021/ml300038t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang J, et al. Platensimycin is a selective FabF inhibitor with potent antibiotic properties. Nature. 2006;441(7091):358–361. doi: 10.1038/nature04784. [DOI] [PubMed] [Google Scholar]
- 54.Wu M, et al. Antidiabetic and antisteatotic effects of the selective fatty acid synthase (FAS) inhibitor platensimycin in mouse models of diabetes. Proc Natl Acad Sci USA. 2011;108(13):5378–5383. doi: 10.1073/pnas.1002588108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Davies Forsman L, et al. Intra- and extracellular activities of trimethoprim-sulfamethoxazole against susceptible and multidrug-resistant Mycobacterium tuberculosis. Antimicrob Agents Chemother. 2014;58(12):7557–7559. doi: 10.1128/AAC.02995-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Blein JP, Ducruet JM, Gauvrit C. Identification and biological-activity of an impurity of technical Diuron. Weed Res. 1979;19(2):117–121. [Google Scholar]
- 57.Routaboul JM, et al. Effects of N,N′-bis-(4-trifluoromethylphenyl)-urea on isolated plant-mitochondria and thylakoid membranes. Phytochemistry. 1991;30(3):733–738. [Google Scholar]
- 58.Brown JR, et al. The structure-activity relationship of urea derivatives as anti-tuberculosis agents. Bioorg Med Chem. 2011;19(18):5585–5595. doi: 10.1016/j.bmc.2011.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kenwood BM, et al. Identification of a novel mitochondrial uncoupler that does not depolarize the plasma membrane. Mol Metab. 2014;3(2):114–123. doi: 10.1016/j.molmet.2013.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Farha MA, Verschoor CP, Bowdish D, Brown ED. Collapsing the proton motive force to identify synergistic combinations against Staphylococcus aureus. Chem Biol. 2013;20(9):1168–1178. doi: 10.1016/j.chembiol.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 61.Singh AP, Nicholls P. Cyanine and safranine dyes as membrane potential probes in cytochrome c oxidase reconstituted proteoliposomes. J Biochem Biophys Methods. 1985;11(2-3):95–108. doi: 10.1016/0165-022x(85)90045-4. [DOI] [PubMed] [Google Scholar]
- 62.Gange DM, Donovan S, Lopata RJ, Henegar K. 1995. The QSAR of insecticidal uncouplers. Classical and Three-Dimensional QSAR in Agrochemistry, ACS Symposium Series, eds Hansch C, Fujita T, Vol 606, pp 199–212.
- 63.Brennan PJ, Nakaido H. The envelope of mycobacteria. Annu Rev Biochem. 1995;64:29–63. doi: 10.1146/annurev.bi.64.070195.000333. [DOI] [PubMed] [Google Scholar]
- 64.Oldfield E, Chapman D. Effects of cholesterol and cholesterol derivatives on hydrocarbon chain mobility in lipids. Biochem Biophys Res Commun. 1971;43(3):610–616. doi: 10.1016/0006-291x(71)90658-9. [DOI] [PubMed] [Google Scholar]
- 65.Kelley LA, Sternberg MJ. Protein structure prediction on the Web: A case study using the Phyre server. Nat Protoc. 2009;4(3):363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 66.Cheng W, Li W. Structural insights into ubiquinone biosynthesis in membranes. Science. 2014;343(6173):878–881. doi: 10.1126/science.1246774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lin F-Y, et al. Head-to-head prenyl tranferases: Anti-infective drug targets. J Med Chem. 2012;55(9):4367–4372. doi: 10.1021/jm300208p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guo R-T, et al. Bisphosphonates target multiple sites in both cis- and trans-prenyltransferases. Proc Natl Acad Sci USA. 2007;104(24):10022–10027. doi: 10.1073/pnas.0702254104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang W, et al. The structural basis of chain length control in Rv1086. J Mol Biol. 2008;381(1):129–140. doi: 10.1016/j.jmb.2008.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chan HC, et al. Structure and inhibition of tuberculosinol synthase and decaprenyl diphosphate synthase from Mycobacterium tuberculosis. J Am Chem Soc. 2014;136(7):2892–2896. doi: 10.1021/ja413127v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Farha MA, et al. Antagonism screen for inhibitors of bacterial cell wall biogenesis uncovers an inhibitor of undecaprenyl diphosphate synthase. Proc Natl Acad Sci USA. 2015;112(35):11048–11053. doi: 10.1073/pnas.1511751112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jacobs AC, et al. Adenylate kinase release as a high-throughput-screening-compatible reporter of bacterial lysis for identification of antibacterial agents. Antimicrob Agents Chemother. 2013;57(1):26–36. doi: 10.1128/AAC.01640-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Derbyshire ER, Prudêncio M, Mota MM, Clardy J. Liver-stage malaria parasites vulnerable to diverse chemical scaffolds. Proc Natl Acad Sci USA. 2012;109(22):8511–8516. doi: 10.1073/pnas.1118370109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Layre E, et al. Molecular profiling of Mycobacterium tuberculosis identifies tuberculosinyl nucleoside products of the virulence-associated enzyme Rv3378c. Proc Natl Acad Sci USA. 2014;111(8):2978–2983. doi: 10.1073/pnas.1315883111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Young DC, et al. In vivo biosynthesis of terpene nucleosides provides unique chemical markers of Mycobacterium tuberculosis infection. Chem Biol. 2015;22(4):516–526. doi: 10.1016/j.chembiol.2015.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pethe K, et al. Isolation of Mycobacterium tuberculosis mutants defective in the arrest of phagosome maturation. Proc Natl Acad Sci USA. 2004;101(37):13642–13647. doi: 10.1073/pnas.0401657101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim MO, et al. A Molecular Dynamics Investigation of Mycobacterium tuberculosis Prenyl Synthases: Conformational Flexibility and Implications for Computer-aided Drug Discovery. Chem Biol Drug Des. 2015;85(6):756–769. doi: 10.1111/cbdd.12463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53(7):2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 79.Yang X, et al. Prevention of multidrug resistance (MDR) in osteosarcoma by NSC23925. Br J Cancer. 2014;110(12):2896–2904. doi: 10.1038/bjc.2014.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kitambi SS, et al. Vulnerability of glioblastoma cells to catastrophic vacuolization and death induced by a small molecule. Cell. 2014;157(2):313–328. doi: 10.1016/j.cell.2014.02.021. [DOI] [PubMed] [Google Scholar]
- 81.Brown RF, et al. alpha-(2-Piperidyl)-2-aryl-4-quinolinemethanols. J Am Chem Soc. 1946;68(12):2705–2708. doi: 10.1021/ja01216a087. [DOI] [PubMed] [Google Scholar]
- 82.Mead JF, Senear AE, Koepfli JB. The synthesis of potential antimalarials; 2-alkyl-alpha-(2-piperidyl)-4-quinolinemethanols. J Am Chem Soc. 1946;68(12):2708–2710. doi: 10.1021/ja01216a088. [DOI] [PubMed] [Google Scholar]
- 83.Duan Z, et al. Synthesis and evaluation of (2-(4-methoxyphenyl)-4-quinolinyl)(2-piperidinyl)methanol (NSC23925) isomers to reverse multidrug resistance in cancer. J Med Chem. 2012;55(7):3113–3121. doi: 10.1021/jm300117u. [DOI] [PubMed] [Google Scholar]
- 84.Duan Z, Choy E, Hornicek FJ. NSC23925, identified in a high-throughput cell-based screen, reverses multidrug resistance. PLoS One. 2009;4(10):e7415. doi: 10.1371/journal.pone.0007415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rybniker J, et al. Lansoprazole is an antituberculous prodrug targeting cytochrome bc1. Nat Commun. 2015;6:7659. doi: 10.1038/ncomms8659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sjøstrøm JE, Kühler T, Larsson H. Basis for the selective antibacterial activity in vitro of proton pump inhibitors against Helicobacter spp. Antimicrob Agents Chemother. 1997;41(8):1797–1801. doi: 10.1128/aac.41.8.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tsutsui N, et al. A novel action of the proton pump inhibitor rabeprazole and its thioether derivative against the motility of Helicobacter pylori. Antimicrob Agents Chemother. 2000;44(11):3069–3073. doi: 10.1128/aac.44.11.3069-3073.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ohara T, et al. Inhibitory action of a novel proton pump inhibitor, rabeprazole, and its thioether derivative against the growth and motility of clarithromycin-resistant Helicobacter pylori. Helicobacter. 2001;6(2):125–129. doi: 10.1046/j.1523-5378.2001.00018.x. [DOI] [PubMed] [Google Scholar]
- 89.Fryklund J, Wallmark B. Sulfide and sulfoxide derivatives of substituted benzimidazoles inhibit acid formation in isolated gastric glands by different mechanisms. J Pharmacol Exp Ther. 1986;236(1):248–253. [PubMed] [Google Scholar]
- 90.Lu P, et al. Pyrazinoic acid decreases the proton motive force, respiratory ATP synthesis activity, and cellular ATP levels. Antimicrob Agents Chemother. 2011;55(11):5354–5357. doi: 10.1128/AAC.00507-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Li K, et al. Oxa, thia, heterocycle, and carborane analogues of SQ109: Bacterial and protozoal cell growth inhibitors. ACS Infect Dis. 2015;1(5):215–221. doi: 10.1021/acsinfecdis.5b00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gostimskaya I, Galkin A. Preparation of highly coupled rat heart mitochondria. J Vis Exp 2010 doi: 10.3791/2202. , 10.3791/220. [DOI] [PMC free article] [PubMed] [Google Scholar]