

Abstract
AMP-activated protein kinase (AMPK) is a critical regulator of cellular metabolism and plays an important role in diabetes, cancer and vascular disease. In the heart, AMPK activation is an essential component of the adaptive response to cardiomyocyte stress that occurs during myocardial ischemia. During ischemia-reperfusion, AMPK activation modulates glucose and fatty acid metabolism, mitochondrial function, endoplasmic reticulum stress, autophagy and apoptosis. Pharmacological activation of AMPK prevents myocardial necrosis and contractile dysfunction during ischemia-reperfusion and potentially represents a cardioprotective strategy for the treatment of myocardial infarction. This review will discuss novel mechanisms of AMPK activation in the ischemic heart, the role of endogenous AMPK activation during ischemia, and the potential therapeutic applications for AMPK directed therapy.
Introduction
AMP-activated protein kinase (AMPK) is a serine-threonine kinase, which functions as a fuel gauge and maintains energy homeostasis during cellular stress [1, 2]. When ATP consumption exceeds production, there is an ensuing increase in cellular ADP content. Conversion of two ADPs to AMP (and ATP) by adenylate kinase also increases the cytosolic concentration of AMP. The increase in both AMP and ADP activate AMPK by binding to the regulatory nucleotide binding domains of the AMPK gamma subunit. There is now increasing recognition that AMPK responds to extracellular cues, such as circulating hormones [3, 4] and local autocrine-paracrine factors that will be discussed subsequently in the context of myocardial ischemia.
The action of AMPK to regulate cellular metabolism is highly relevant to heart disease, where disturbances in energy balance can lead to cardiac contractile dysfunction and cell death. Normally, the heart maintains energy homeostasis by generating ATP, primarily from mitochondrial substrate oxidation. The energy demands of the heart are high because of the need for ATP to maintain both the membrane ionic gradients required for electrical excitability and the contractile function of myofibrillar proteins. AMPK activates critical steps responsible for the uptake and metabolism of glucose, when oxidative metabolism is diminished in the ischemic heart. After the onset of ischemia, down-regulation of regional myocardial contractility diminishes ATP requirements, but AMPK also inhibits protein synthesis, which otherwise would constitute a residual energy-consuming process that can also lead to ER stress. Thus, the endogenous AMPK pathway has both energy generating and energy conserving actions that preserve cellular ATP content during ischemia-reperfusion [5]. Specific molecular and cellular actions of AMPK in regulating metabolism during ischemia-reperfusion will be discussed.
The initial observations that AMPK modulated cardiac metabolism [5-8] stimulated interest in targeting the AMPK pathway as a novel strategy for the treatment of heart disease. Since that time, our understanding of the intrinsic physiological actions of AMPK activation has grown, while pre-clinical experiments in animal models have provided support for the hypothesis that AMPK activation might be cardioprotective [9, 10]. We will consider strategies to target AMPK in the setting of acute myocardial infarction that might have clinical benefit.
Molecular regulation of AMPK during ischemia
AMPK is a heterotrimeric complex composed of a catalytic α subunit and regulatory β and γ subunits. Each of the subunits has two or three isoforms, encoded by different genes. The heart expresses α1 and α2, β1 and β2, and γ1 and three distinct splice variants of γ2 isoforms [11, 12]. The pattern of expression changes during development and varies somewhat between rodent and human hearts [13]. AMPK expression is also altered under pathological conditions including heart failure [14].
AMPK functions as a cardiac energy sensor whose activation is highly regulated by changes in adenine nucleotide concentrations (Figure 1). AMP and ADP binding to site 3 in the γ subunit triggers a conformational change in the heterotrimeric complex, which has a very important action to facilitate the phosphorylation of the activating Thr172 site in the kinase activation domain [15-17]. This mechanism may predominate under physiological conditions that lead to mild energetic stress [18]. With greater increases in the intracellular AMP concentrations, such as those that occur during myocardial ischemia, AMP binds to site 1 and allosterically activates AMPK [15-17].
Figure 1. Mechanisms regulating AMPK activity.
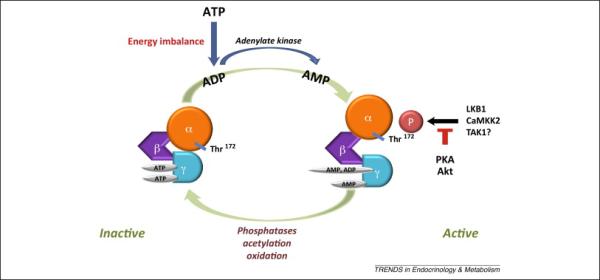
AMPK is a herotrimeric complex of α, β, and γ subunits. AMP binding to the γ subunit allosterically activates AMPK and facilitates the phosphorylation of the activating Thr172 site in the α subunit by upstream kinases including LKB1 and CaMKK2, and possibly TAK1. Akt phosphorylation of Ser485/491 in the α1/α2 subunits negatively modulates Thr172 phosphorylation. Deactivation of AMPK is mediated in part by protein phosphatase 2A and 2C, which dephosphorylate the Thr172 site. AMPK is also negatively regulated by acetylation and oxidation of cysteine residues in the AMPK α subunits.
Upstream kinases phosphorylate the critical Thr172 activating site and include the liver kinase B 1 (LKB1), calcium-calmodulin-dependent kinase kinase 2 (CaMKK2) and transforming growth factor-β-activated protein kinase-1 (TAK1) (Figure 1) [19-22]. In the heart, the major upstream kinase is LKB1 and phosphorylation of the AMPK complexes containing the α2 isoform during ischemia is completely abrogated in LKB1 KO mice [20]. The activity of LKB1 requires binding to its accessory proteins Ste20-related adaptor (STRAD) and mouse protein 25 (MO25) [23]. Although CaMKK2 is expressed at a relatively low level in cardiomyocytes [24], our recent findings indicated that CaMKK2 may play a role in mediating AMPK activation during ischemia in the heart [25]. Finally, TAK1 is known to phosphorylate SNF1, the yeast homolog of the mammalian AMPK α subunit [22]. In the mammalian heart, it appears to facilitate LKB1 activation, but whether it has direct effects on AMPK is uncertain [26].
Thr172 phosphorylation is also modulated by phosphorylation of secondary serine residues in the AMPK α subunit (Figure 1). Phosphorylation of Ser485 in the α1 subunit and the corresponding Ser491 in the α2 subunit by Akt, and possibly α1 Ser173 by protein kinase A, attenuate Thr172 phosphorylation induced by LKB1 [27-29]. In the heart, insulin-stimulated Akt phosphorylation of Ser485 and Ser491 has been associated with decreased phosphorylation of the Thr172 site and blunted AMPK activation during ischemia [30]. Deactivation of AMPK is mediated by protein phosphatase 2A and 2C, which dephosphorylate the Thr172 site [31], although the specific role of these phosphatases is not well understood in the heart. Just as AMP and ADP binding to the γ subunit promotes α subunit Thr172 phosphorylation, it also inhibits the action of phosphatase 2C to inactivate AMPK [15-17]. Thus, changes in adenine nucleotides have multiple mechanisms that promote the activity of AMPK.
AMPK is also regulated by its acetylation state, which is determined by the actions of acetylase p300 and the histone deacetylase 1 (Figure 1) [32]. Acetylation is associated with low AMPK activity, while deacetylation facilitates AMPK binding with LKB1 and thereby promotes AMPK phosphorylation and activation [32]. In a parallel fashion, deacetylated LKB1 more readily forms an active complex with its partner protein, STRAD, in the cytosol [33]. Although this mechanism may be operative in the heart, the importance of deacetylation in cardiac AMPK activation during pathological states is still largely unknown.
Oxidative modification of AMPK is a third post-translational mechanism regulating its activity (Figure 1). During myocardial ischemia, the oxidation of Cys130 and Cys174 in the AMPK α2 catalytic subunit leads to alterations in AMPK structure through intra-molecular disulfide bond formation and interferes with the activation of AMPK by upstream kinases [34]. Oxidation potentially could contribute to the deactivation of AMPK that rapidly ensues during post-ischemic reperfusion. Interestingly, the ROS scavenger thioredoxin 1 cleaves disulfides in proteins, prevents AMPK oxidation and maintains its activity [34]. It might also be noted that cellular oxidative stress can also activate AMPK, via mechanisms that involve alterations in energy production and the AMP/ATP ratio [18].
AMPK regulation of cardiac metabolism in ischemia
During ischemia, activation of AMPK coordinates an increase in glucose utilization and glycolytic ATP production (Figure 2). The glycolytically-derived ATP is thought to have a critical function to maintain membrane ionic gradients that are important to preserve cell function and viability [35]. As an early adaptive mechanism, AMPK activation promotes cellular glucose uptake by triggering the trafficking of intracellular GLUT4-containing membrane vesicles to the sarcolemma membrane [5]. The translocation of GLUT4 vesicles stimulated by AMPK in skeletal muscle is mediated by phosphorylation of the Akt substrate protein AS160 [36], which is also expressed in the heart [37]. AS160 is a GTPase-activating protein (GAP), which functions to inhibit the small GTP-binding proteins that control many key steps of intracellular vesicle movement [38]. AMPK-induced phosphorylation of AS160 removes the GAP inhibition on Rab GTPases and allows translocation of GLUT4 vesicles to the cell surface [39, 40]. In addition, AMPK also inhibits the endocytotic cycling of GLUT4 from the sarcolemma, which further increases the content of GLUT4 in its active site [41]. AMPK also upregulates glycolysis through its phosphorylation of 6-phosphofructo-2-kinase (PFK 2) during myocardial ischemia [7]. The resulting increase in fructose 2,6-bisphosphate concentration allosterically activates PFK-1, which is the rate-limiting step in glycolysis [7]. AMPK may also inhibit glycogen synthesis, by suppressing the activity of glycogen synthase [42, 43], which would conserve ATP and indirectly promote the mobilization of glycogen during energy stress.
Figure 2. AMPK regulation of cardiac metabolism in ischemia-reperfusion.
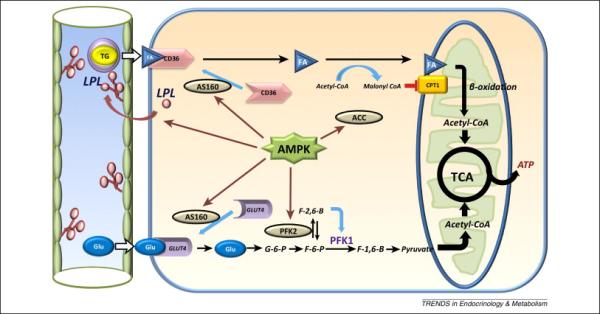
During ischemia, AMPK activation phosphorylates AS160, which promotes translocation of GLUT4-containing membrane vesicles to the sarcolemma and cellular glucose uptake. AMPK phosphorylates 6-phosphofructo-2-kinase (PFK 2), which increases the concentration of fructose 2,6-bisphosphate, an allosteric activator of PFK-1, which is the rate-limiting step in glycolysis. During reperfusion, the activation of AMPK facilitates both fatty acid uptake and oxidation. AMPK triggers lipoprotein lipase (LPL) translocation from cardiomyocytes to the vascular endothelial surface, where it catalyzes the release of fatty acid from circulating TG. AMPK phosphorylation of AS160 signaling also promotes translocation of the fatty acid transporter CD36 to the sarcolemma membrane. AMPK phosphorylates and inactivates acetyl-CoA carboxylase (ACC), decreasing the intracellular concentration of malonyl-CoA. Since malonyl- CoA is a negative regulator of carnitine palmitoyltransferase 1 (CPT–1), AMPK promotes fatty-acyl carnitine entry into the mitochondria.
Fatty acid metabolism accounts for the majority of ATP production in the normal aerobic heart and the activation of AMPK facilitates both fatty acid uptake and oxidation (Figure 2). AMPK increases LPL translocation from cardiomyocytes to its functional position on the vascular endothelial surface in the heart [44]. Active LPL catalyzes the release of fatty acid from circulating TG, which is required for their delivery to cardiomyocytes. In addition, AMPK triggered AS160 phosphorylation also recruits the fatty acid transporter CD36 to the sarcolemma membrane, thereby facilitating fatty acid uptake into cardiomyocytes [37]. Thus, AMPK has dual actions that promote cardiomyocyte uptake of fatty acid in the heart.
Activated AMPK is also critically important for the import of cytosolic fatty acid into the mitochondria (Figure 2). AMPK phosphorylates and inactivates acetyl-CoA carboxylase (ACC), leading to a decrease in the intracellular concentration of malonyl-CoA (Figure 2). Since malonyl- CoA is a negative regulator of carnitine palmitoyltransferase 1 (CPT–1), AMPK activation effectively reduces the inhibition of CPT-1 and promotes fatty acyl- carnitine entry into the mitochondria [6]. In the presence of oxygen, this mechanism accelerates fatty acid oxidation, although recent findings indicate that ACC activity and malonyl-CoA levels do not necessarily correlate with rates of heart fatty acid oxidation [45]. Although FA oxidation is limited by the lack of oxygen in the setting of ischemia, when oxygen delivery is restored during reperfusion, AMPK accelerates fatty acid oxidation [45]. The degree of residual AMPK activation following reperfusion appears to depend on the severity and duration of ischemia, so that the extent to which fatty acid oxidation is enhanced may be somewhat variable.
Concerns have been raised regarding whether persistent effects of AMPK activation on fatty acid oxidation might be detrimental during early reperfusion [46]. Excessive fatty acid oxidation may attenuate glucose oxidation, leading to increased lactate production in the face of increased glucose uptake and glycolysis. To the extent that uncoupled glycolysis perpetuates intracellular acidosis, it could lead to detrimental calcium overload, resulting from the combination of sodium-proton exchange and sodium-calcium exchange [46]. However, whether activated AMPK promotes acidosis or calcium overload in the reperfused heart is uncertain, and the integrated actions of activated AMPK appear to be protective in reducing myocardial necrosis during ischemia- reperfusion [5, 9, 10, 47-49].
Cellular effects of AMPK during ischemia-reperfusion
In addition to regulating metabolism and energy homeostasis, AMPK modulates several cellular processes that are important to cell survival during ischemia (Figure 3). In the ischemic heart, AMPK induces cardiomyocyte autophagy, which promotes cell survival [50, 51]. Autophagy removes damaged organelles, including malfunctioning mitochondria that otherwise lead to oxidative stress, and also generates substrates for cellular metabolism. AMPK induces autophagy by direct phosphorylation of the ULK1 (the mammalian homolog of yeast Atg1) kinase complex [52] and indirectly by inhibiting activation of the mTORC1 complex [53].
Figure 3. The actions of AMPK to mitigate cardiac injury during ischemia-reperfusion.
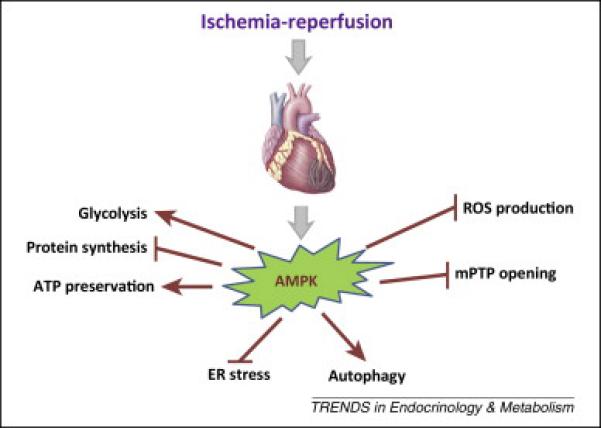
AMPK activation has metabolic actions to conserve energy during ischemia by promoting glycolytic ATP production and inhibiting protein synthesis. AMPK also prevents endoplasmic reticulum (ER) stress and induces autophagy. Durireperfusion, activated AMPK decreases ROS generation and mPTP opening. These actions prevent apoptosis and necrosis.
Ischemia results in the accumulation of unfolded or misfolded proteins, leading to endoplasmic reticulum (ER) stress [54]. AMPK attenuates ER stress by directly suppressing eEF2 regulated protein synthesis and indirectly attenuating oxidative stress in the heart during ischemia [55]. Intrinsic AMPK activation has also been shown to reduce apoptosis after ischemia-reperfusion in the mouse heart [5]. This might be due a reduction in ER stress-mediated apoptosis associated with the transcriptional induction of C/EBP homologous proteins (CHOP) [56] or activation of the capase 12 [57] and JNK pathways [58].
AMPK also inhibits opening of the mitochondrial permeability transition pore (mPTP) during ischemia-reperfusion [59]. Pore opening is a sentinel event in triggering cell death during the early post-ischemic reperfusion period. AMPK inactivation leads to increased markers of oxidative stress after ischemia-reperfusion in the mouse model [47]. The mechanism through which AMPK modulates pore opening is uncertain, but possibly could be due to reducing oxidative stress.
AMPK and cell survival in ischemia
Intrinsic AMPK activation has been shown to having a beneficial role to preventing cardiac injury during ischemia-reperfusion in most [5, 25, 47, 60], but not all [61], studies. The critical function of AMPK activation in the ischemic heart was elucidated in mouse models where AMPK activation was impaired [5, 10, 47, 62]. Early work from our laboratory showed that transgenic expression of an inactive K45R α2-subunit displaced native α1 and α2 subunits from the active AMPK heterotrimeric complex, resulting in reduced ischemic AMPK activation [5]. When these hearts were perfused ex vivo and coronary flow was decreased, they showed an exacerbation in LV contractile dysfunction, cardiac necrosis and apoptosis compared to wild type hearts during ischemia-reperfusion [5]. Impaired contractile dysfunction after ischemia has also been observed in some [63, 64], but not all [61], mouse models with cardiac AMPK α2 inactivation or deletion
Findings that intrinsic AMPK activation was a protective mechanism during ischemia-reperfusion generated interest in whether AMPK might be a potential target for pharmacologic treatment to reduce ischemic injury. Accumulating research now provides evidence to support this possibility [24, 49, 65, 66]. Metformin at high doses activates heart AMPK via inhibition of complex I activity and inhibition of mitochondrial ATP production. Metformin protects the heart from ischemic injury in both normal and diabetic mice [10]. This protective effect is seen not only when metformin was given prior to ischemia, but also after the start of ischemia at the onset of reperfusion [10]. Metformin also decreases acute ischemia-reperfusion injury and chronic rejection after murine cardiac transplantation [67]. A newer synthetic compound, A769662 that allosterically activates AMPK through interacting with the β subunit, preconditions against injury in both in vitro perfused hearts during ischemia-reperfusion and in vivo mice subjected to coronary ligation-reperfusion [9]. Pharmacological agents have potential additional off-target actions, which can confound the interpretation of their mechanisms of action. However, experiments combining pharmacologic and genetic approaches, demonstrate that the cardioprotective effects of both metformin and A769662 are AMPK-dependent based on their lack of efficacy in AMPK-inactivated mouse models [9, 10].
The compound AICAR has also been used extensively in experimental models as an AMPK activator. AICAR is mono-phosphorylated to the metabolite ZMP, which is an AMP mimetic and activates AMPK through binding to the gamma subunit nucleotide binding domains. AICAR administration before ischemia improves the ability of the heart to recover from ischemia-reperfusion in the rat [68]. In a heart transplantation model, AICAR administration during cardioplegia and reperfusion led to functional protection against cardiac injury [68]. However, it should be noted that AICAR has additional effects, including blockade of adenosine reuptake that effectively increases extracellular adenosine action [68]. AICAR was used in clinical studies to reduce complications related to cardiac bypass with some benefit [69, 70], although it is unclear whether its effect was related to AMPK activation, since the doses used were much lower than those that typically activate AMPK in vivo.
Autocrine-paracrine activation of AMPK during ischemia
Although AMPK is a well-recognized cellular energy sensor that is activated by changes in adenine nucleotide concentrations, it is also responsive to extracellular cues, including hormones, cytokines, adipokines and autocrine/paracrine factors (Figure 4). During ischemia-reperfusion, the heart releases proteins that appear to protect against injury and have been termed “cardiokines” [71]. Our interest in this field started with the observation that cardiac AMPK was regulated during ischemia by macrophage migration inhibitory factor (MIF) [60], a cytokine with an important role in inflammatory disease. Curiously, MIF is also highly expressed in cardiomyocytes [60] and its expression is regulated by HIF-1alpha [72, 73]. MIF is secreted during ischemia, so that tissue hypoxia leads to both release and increased synthesis of MIF. When released from the cardiomyocyte, MIF has an autocrine-paracrine effect to activate the cell surface MIF-receptor CD74, which has a physiological action to limit cardiac injury by enhancing AMPK activity [60]. In addition, secreted MIF blunts the post-ischemic activation of the stress kinase JNK, which is known to contribute to reperfusion injury [74]. Because MIF expression and consequent AMPK activation is diminished in patients with a common polymorphism in the MIF promoter [60], as well as with aging in animals [73], individuals with these conditions may prove susceptible to cardiac ischemic injury. These observations hold potential clinical therapeutic implications in the era of personalized medicine to the extent that individuals with impairment in the MIF-AMPK axis might derive enhanced benefit from AMPK directed therapy at the time of myocardial infarction.
Figure 4. Autocrine-paracrine activation of AMPK.
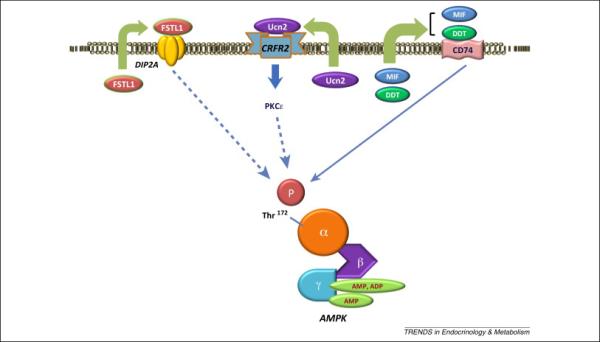
During ischemia-reperfusion, the heart releases proteins that have autocrine-paracrine actions that amplify AMPK activation and protect the heart from injury. These include the cytokines macrophage migration inhibitory factor (MIF) and its homolog D-dopachrome tautomerase (DDT), the corticotropin-releasing factor (CRF) family peptide urocortin 2 (Ucn2), and the glycoprotein follistatin-like 1 (FSTL1).
Although endogenous MIF appears to have an important role in the response to ischemia [60], pharmacological treatment with MIF may have limited therapeutic potential because of its narrow therapeutic window [25, 74]. MIF reduces cardiac contractility via activation of the chemokine receptor CXCR4 [75] and might increase inflammation after severe ischemic injury [75]. In this regard, a more selective CD74 agonist potentially would potentially have greater therapeutic potential. In studying the role of CD74 in ischemia-reperfusion, we observed more dramatic detrimental effects of CD74 deletion compared to MIF deletion in mouse models [25]. This finding led to the discovery that CD74 had a second ligand, D-dopachrome tautomerase (DDT), which shares partial sequence and structural homology with MIF [76]. DDT lacks a known physiological substrate in mammalian species and might be considered a vestigial enzyme. However, DDT is highly expressed in cardiomyocytes and is secreted by the heart during ischemic stress [25]. DDT increases AMPK activation through binding to the MIF receptor CD74 and triggers CaMKK2-mediated phosphorylation of the Thr172 site on AMPK [25]. However, DDT appears to be a more selective than MIF as a CD74 agonist and lacks the adverse effects on cardiac contractility that are seen with MIF treatment [25]. DDT treatment protects both isolated mouse hearts in vitro and intact hearts in vivo against injury and contractile dysfunction after ischemia-reperfusion [25]. Thus, it will be of interest to determine whether DDT has similar therapeutic effects in pre-clinical large animal models of coronary artery ligation and reperfusion.
Additional secreted factors activate AMPK in an autocrine-paracrine manner in the heart, including urocortin 2 (Ucn2), a peptide of the corticotropin-releasing factor (CRF) family [77]. Pharmacological administration of Ucn2 treatment also increases AMPK activation, downstream acetyl-CoA carboxylase phosphorylation and glucose uptake in isolated heart muscles [77]. These actions of Ucn2 are mediated by the cell surface CRFR2 receptor and require PKCε translocation, although the mechanism linking PKCε to AMPK activation remains uncertain [77]. Follistatin-like 1 is a secreted glycoprotein that also activates AMPK and inhibits apoptosis and inflammation [78]. Follistatin-like 1 interacts with transforming growth factor-β signaling [79] and plasma levels of follistatin-like 1 are elevated in heart failure, acute coronary syndromes and ischemia [80, 81]. In both rodent and large animal models, follistatin-like 1 prevents injury during experimental myocardial infarction [78].
Heart AMPK is also activated by circulating plasma adipokines released from tat tissue. Adiponectin is the prototype adipokine and adiponectin knockout mice demonstrate impaired AMPK activation during ischemia and increased susceptibility to ischemic injury [82]. Conversely, mice with adenoviral-mediated adiponectin expression have increased AMPK activation and are protected against ischemic injury [82]. In isolated cells, the anti-apoptotic effects of adiponectin are partially mediated by AMPK [82]. More recently, additional adipokines, including C1q/TNF-related protein 9 [83] and omentin [84], have also been found to activate heart AMPK, contributing to their cardioprotective effects.
Intracellular mechanisms also appear to regulate AMPK activation during ischemia. AMPK activation is modulated in the heart by its association with intracellular scaffold proteins, including the TAK1-binding protein TAB1 [85] and Sestrin 2 [86]. Whether these protein scaffolds simply facilitate AMPK activation or functionally compartmentalize AMPK to specific subcellular locations is of interest, but is not yet well understood.
AMPK, ischemia-reperfusion and diabetes
The AMPK pathway may be highly relevant to ischemia in the setting of diabetes. Although it remains controversial as to whether the diabetic heart is more sensitive to ischemic injury, diabetic patients are particularly susceptible to developing heart failure and have an increased mortality after myocardial infarction.
Substantial evidence suggests that AMPK activity is downregulated in striated muscle, liver and adipose tissue from animal models of insulin resistance and type 2 diabetes [87, 88]. High fat diets suppress cardiac AMPK activation and glucose metabolism in the mouse heart [88]. After streptozotocin treatment, insulin-deficient mice also have reduced heart AMPK activation [89]. Treatment of in insulin-deficient mice with metformin or resveratrol activates AMPK, decreases autophagy [89] and improves cardiac microvascular function [90].
Despite the down-regulation of intrinsic AMPK activation, pharmacological agents that activate AMPK appear to have beneficial actions to limit cardiac injury induced by ischemia-reperfusion in diabetic models. A single dose of metformin, administered either before ischemia or at the time of reperfusion, has cardioprotective effects in db/db diabetic mice [10]. The beneficial effect of metformin pre-treatment was AMPK-dependent and associated with augmented eNOS activation during early reperfusion [10]. Augmentation of AMPK activation by A769662 during in vivo local ischemia also reduced infarct size [59]. This protective action was associated with increased phosphorylation of GSK-3β and reduced mPTP opening [59]. Thus, initial evidence suggests that AMPK activation might be beneficial in the setting of myocardial infarction in the setting of diabetes, although further work is needed to support this hypothesis.
Clinical implications
Acute myocardial infarction affects over 1.5 million individuals annually in the US alone. Thus, it is critical to address whether AMPK activating agents might protect against injury in patients during acute myocardial infarction. The cardioprotective actions of AMPK would be most directly applicable to the setting of acute myocardial infarction. Since acute coronary syndromes such as ST-segment elevation myocardial infarction cannot be anticipated, AMPK activators would need to be effective when administered after the onset of ischemia or during early reperfusion. As such they could be used in the cardiac catheterization laboratory in conjunction with percutaneous coronary revascularization and stent placement during ST-segment elevation myocardial infarction. To the extent that these drugs might lower blood glucose in non-diabetic subjects, glucose monitoring and glucose infusion would be required to maintain euglycemia.
If AMPK activating drugs prove to be suitable for long-term administration, it is possible that they might have utility as a chronic preconditioning strategy. This might have particular utility in patients with type 2 diabetes, since AMPK activators improve glycemic control and may have utility in the treatment of long-term metabolic disease [91]. The beneficial effects of AMPK activation during ischemia may also extend beyond the heart to include other solid organs, including the kidneys [92].
Concluding remarks
There has been significant progress in our knowledge about the biological actions of AMPK during the last several years. AMPK activation appears to be instrumental in the cardiomyocyte response to ischemia-reperfusion. Whether this strategy will prove beneficial in the clinical setting of acute myocardial infarction remains to be determined. However, with the development of more specific and potent AMPK activators, there is an opportunity to harness the discoveries of the basic science investigation to test this hypothesis.
Intrinsic AMPK activation protects the heart from injury during ischemia-reperfusion.
AMPK orchestrates metabolic and cellular responses to ischemia.
Secreted cardiokines have autocrine-paracrine effects to modulate AMPK activation.
Pharmacological AMPK activation has a cardioprotective effect during ischemia-reperfusion.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zaha VG, Young LH. AMP-activated protein kinase regulation and biological actions in the heart. Circ Res. 2012;111:800–814. doi: 10.1161/CIRCRESAHA.111.255505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hardie DG, Carling D. The AMP-activated protein kinase--fuel gauge of the mammalian cell? Eur J Biochem. 1997;246:259–273. doi: 10.1111/j.1432-1033.1997.00259.x. [DOI] [PubMed] [Google Scholar]
- 3.Minokoshi Y, et al. Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature. 2002;415:339–343. doi: 10.1038/415339a. [DOI] [PubMed] [Google Scholar]
- 4.Yamauchi T, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8:1288–1295. doi: 10.1038/nm788. [DOI] [PubMed] [Google Scholar]
- 5.Russell RR, 3rd, et al. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest. 2004;114:495–503. doi: 10.1172/JCI19297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kudo N, et al. High rates of fatty acid oxidation during reperfusion of ischemic hearts are associated with a decrease in malonyl-CoA levels due to an increase in 5′-AMP-activated protein kinase inhibition of acetyl-CoA carboxylase. J Biol Chem. 1995;270:17513–17520. doi: 10.1074/jbc.270.29.17513. [DOI] [PubMed] [Google Scholar]
- 7.Marsin AS, et al. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol. 2000;10:1247–1255. doi: 10.1016/s0960-9822(00)00742-9. [DOI] [PubMed] [Google Scholar]
- 8.Russell RR, 3rd, et al. Translocation of myocardial GLUT-4 and increased glucose uptake through activation of AMPK by AICAR. Am J Physiol. 1999;277:H643–649. doi: 10.1152/ajpheart.1999.277.2.H643. [DOI] [PubMed] [Google Scholar]
- 9.Kim AS, et al. A small molecule AMPK activator protects the heart against ischemiareperfusion injury. J Mol Cell Cardiol. 2011;51:24–32. doi: 10.1016/j.yjmcc.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Calvert JW, et al. Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNOS-mediated signaling. Diabetes. 2008;57:696–705. doi: 10.2337/db07-1098. [DOI] [PubMed] [Google Scholar]
- 11.Kim AS, et al. AMP-activated protein kinase: a core signalling pathway in the heart. Acta physiologica. 2009;196:37–53. doi: 10.1111/j.1748-1716.2009.01978.x. [DOI] [PubMed] [Google Scholar]
- 12.Li J, et al. Activation of AMPK alpha- and gamma-isoform complexes in the intact ischemic rat heart. Am J Physiol Heart Circ Physiol. 2006;291:H1927–1934. doi: 10.1152/ajpheart.00251.2006. [DOI] [PubMed] [Google Scholar]
- 13.Pinter K, et al. Embryonic expression of AMPK gamma subunits and the identification of a novel gamma2 transcript variant in adult heart. Journal of molecular and cellular cardiology. 2012;53:342–349. doi: 10.1016/j.yjmcc.2012.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim M, et al. AMPK isoform expression in the normal and failing hearts. J Mol Cell Cardiol. 2012;52:1066–1073. doi: 10.1016/j.yjmcc.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiao B, et al. Structure of mammalian AMPK and its regulation by ADP. Nature. 2011;472:230–233. doi: 10.1038/nature09932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gowans GJ, et al. AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab. 2013;18:556–566. doi: 10.1016/j.cmet.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oakhill JS, et al. AMPK is a direct adenylate charge-regulated protein kinase. Science. 2011;332:1433–1435. doi: 10.1126/science.1200094. [DOI] [PubMed] [Google Scholar]
- 18.Hardie DG, et al. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hawley SA, et al. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine. 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem. 1996;271:27879–27887. doi: 10.1074/jbc.271.44.27879. [DOI] [PubMed] [Google Scholar]
- 20.Sakamoto K, et al. Deficiency of LKB1 in heart prevents ischemia-mediated activation of AMPKalpha2 but not AMPKalpha1. Am J Physiol Endocrinol Metab. 2006;290:E780–788. doi: 10.1152/ajpendo.00443.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Woods A, et al. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell metab. 2005;2:21–33. doi: 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 22.Momcilovic M, et al. Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro. J Biol Chem. 2006;281:25336–25343. doi: 10.1074/jbc.M604399200. [DOI] [PubMed] [Google Scholar]
- 23.Hawley SA, et al. Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol. 2003;2:28. doi: 10.1186/1475-4924-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang J, et al. Activated protein C protects against myocardial ischemic/reperfusion injury through AMP-activated protein kinase signaling. J Thromb Haemost. 2011;9:1308–1317. doi: 10.1111/j.1538-7836.2011.04331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qi D, et al. The vestigial enzyme D-dopachrome tautomerase protects the heart against ischemic injury. J Clin Invest. 2014;124:3540–3550. doi: 10.1172/JCI73061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie M, et al. A pivotal role for endogenous TGF-beta-activated kinase-1 in the LKB1/AMP-activated protein kinase energy-sensor pathway. Proc Natl Acad Sci U S A. 2006;103:17378–17383. doi: 10.1073/pnas.0604708103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pulinilkunnil T, et al. Adrenergic regulation of AMP-activated protein kinase in brown adipose tissue in vivo. J Biol Chem. 2011;286:8798–8809. doi: 10.1074/jbc.M111.218719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kovacic S, et al. Akt activity negatively regulates phosphorylation of AMP-activated protein kinase in the heart. J Biol Chem. 2003;278:39422–39427. doi: 10.1074/jbc.M305371200. [DOI] [PubMed] [Google Scholar]
- 29.Djouder N, et al. PKA phosphorylates and inactivates AMPKalpha to promote efficient lipolysis. EMBO J. 2010;29:469–481. doi: 10.1038/emboj.2009.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Horman S, et al. Insulin antagonizes ischemia-induced Thr172 phosphorylation of AMP-activated protein kinase alpha-subunits in heart via hierarchical phosphorylation of Ser485/491. J Biol Chem. 2006;281:5335–5340. doi: 10.1074/jbc.M506850200. [DOI] [PubMed] [Google Scholar]
- 31.Davies SP, et al. 5′-AMP inhibits dephosphorylation, as well as promoting phosphorylation, of the AMP-activated protein kinase. Studies using bacterially expressed human protein phosphatase-2C alpha and native bovine protein phosphatase-2AC. FEBS Lett. 1995;377:421–425. doi: 10.1016/0014-5793(95)01368-7. [DOI] [PubMed] [Google Scholar]
- 32.Lin YY, et al. Functional dissection of lysine deacetylases reveals that HDAC1 and p300 regulate AMPK. Nature. 2012;482:251–255. doi: 10.1038/nature10804. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Lan F, et al. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J Biol Chem. 2008;283:27628–27635. doi: 10.1074/jbc.M805711200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shao D, et al. A redox-dependent mechanism for regulation of AMPK activation by Thioredoxin1 during energy starvation. Cell Metab. 2014;19:232–245. doi: 10.1016/j.cmet.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weiss J, Hiltbrand B. Functional compartmentation of glycolytic versus oxidative metabolism in isolated rabbit heart. J Clin Invest. 1985;75:436–447. doi: 10.1172/JCI111718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kramer HF, et al. Distinct signals regulate AS160 phosphorylation in response to insulin, AICAR, and contraction in mouse skeletal muscle. Diabetes. 2006;55:2067–2076. doi: 10.2337/db06-0150. [DOI] [PubMed] [Google Scholar]
- 37.Samovski D, et al. Insulin and AMPK regulate FA translocase/CD36 plasma membrane recruitment in cardiomyocytes via Rab GAP AS160 and Rab8a Rab GTPase. J Lipid Res. 2012;53:709–717. doi: 10.1194/jlr.M023424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fukuda M. TBC proteins: GAPs for mammalian small GTPase Rab? Biosci Rep. 2011;31:159–168. doi: 10.1042/BSR20100112. [DOI] [PubMed] [Google Scholar]
- 39.Ishikura S, et al. Rabs 8A and. 14 are targets of the insulin-regulated Rab-GAP AS160 regulating GLUT4 traffic in muscle cells. Biochem Biophys Res Commun. 2007;353:1074–1079. doi: 10.1016/j.bbrc.2006.12.140. [DOI] [PubMed] [Google Scholar]
- 40.Sano H, et al. Insulin-stimulated GLUT4 protein translocation in adipocytes requires the Rab10 guanine nucleotide exchange factor Dennd4C. J Biol Chem. 2011;286:16541–16545. doi: 10.1074/jbc.C111.228908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang J, Holman GD. Insulin and contraction stimulate exocytosis, but increased AMP-activated protein kinase activity resulting from oxidative metabolism stress slows endocytosis of GLUT4 in cardiomyocytes. J Biol Chem. 2005;280:4070–4078. doi: 10.1074/jbc.M410213200. [DOI] [PubMed] [Google Scholar]
- 42.Nielsen JN, et al. Role of 5′AMP-activated protein kinase in glycogen synthase activity and glucose utilization: insights from patients with McArdle's disease. The Journal of physiology. 2002;541:979–989. doi: 10.1113/jphysiol.2002.018044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Halse R, et al. Regulation of glycogen synthase by glucose and glycogen: a possible role for AMP-activated protein kinase. Diabetes. 2003;52:9–15. doi: 10.2337/diabetes.52.1.9. [DOI] [PubMed] [Google Scholar]
- 44.An D, et al. The metabolic “switch” AMPK regulates cardiac heparin-releasable lipoprotein lipase. Am J Physiol Endocrinol Metab. 2005;288:E246–253. doi: 10.1152/ajpendo.00211.2004. [DOI] [PubMed] [Google Scholar]
- 45.Zordoky BN, et al. AMPK-dependent inhibitory phosphorylation of ACC is not essential for maintaining myocardial fatty acid oxidation. Circ Res. 2014;115:518–524. doi: 10.1161/CIRCRESAHA.115.304538. [DOI] [PubMed] [Google Scholar]
- 46.Dyck JR, Lopaschuk GD. AMPK alterations in cardiac physiology and pathology: enemy or ally? J Physiol. 2006;574:95–112. doi: 10.1113/jphysiol.2006.109389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y, et al. AMP-activated protein kinase deficiency enhances myocardial ischemia/reperfusion injury but has minimal effect on the antioxidant/antinitrative protection of adiponectin. Circulation. 2009;119:835–844. doi: 10.1161/CIRCULATIONAHA.108.815043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Costa R, et al. Activated protein C modulates cardiac metabolism and augments autophagy in the ischemic heart. Journal of thrombosis and haemostasis : JTH. 2012;10:1736–1744. doi: 10.1111/j.1538-7836.2012.04833.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ma Y, et al. Antithrombin up-regulates AMP-activated protein kinase signalling during myocardial ischaemia/reperfusion injury. Thromb Haemost. 2015;113:338–349. doi: 10.1160/TH14-04-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Matsui Y, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin. 1 in mediating autophagy. Circ Res. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 51.Takagi H, et al. AMPK mediates autophagy during myocardial ischemia in vivo. Autophagy. 2007;3:405–407. doi: 10.4161/auto.4281. [DOI] [PubMed] [Google Scholar]
- 52.Kim J, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Herrero-Martin G, et al. TAK1 activates AMPK-dependent cytoprotective autophagy in TRAIL-treated epithelial cells. EMBO J. 2009;28:677–685. doi: 10.1038/emboj.2009.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110:1389–1398. doi: 10.1172/JCI16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Terai K, et al. AMP-activated protein kinase protects cardiomyocytes against hypoxic injury through attenuation of endoplasmic reticulum stress. Mol Cell Biol. 2005;25:9554–9575. doi: 10.1128/MCB.25.21.9554-9575.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- 57.Nakagawa T, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 58.Urano F, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- 59.Paiva MA, et al. Enhancing AMPK activation during ischemia protects the diabetic heart against reperfusion injury. Am J Physiol Heart Circ Physiol. 2011;300:H2123–2134. doi: 10.1152/ajpheart.00707.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miller EJ, et al. Macrophage migration inhibitory factor stimulates AMP-activated protein kinase in the ischaemic heart. Nature. 2008;451:578–582. doi: 10.1038/nature06504. [DOI] [PubMed] [Google Scholar]
- 61.Folmes CD, et al. Suppression of 5′-AMP-activated protein kinase activity does not impair recovery of contractile function during reperfusion of ischemic hearts. Am J Physiol Heart Circ Physiol. 2009;297:H313–321. doi: 10.1152/ajpheart.01298.2008. [DOI] [PubMed] [Google Scholar]
- 62.Xing Y, et al. Glucose metabolism and energy homeostasis in mouse hearts overexpressing dominant negative alpha2 subunit of AMP-activated protein kinase. J Biol Chem. 2003;278:28372–28377. doi: 10.1074/jbc.M303521200. [DOI] [PubMed] [Google Scholar]
- 63.Zarrinpashneh E, et al. Role of the alpha2-isoform of AMP-activated protein kinase in the metabolic response of the heart to no-flow ischemia. Am J Physiol Heart Circ Physiol. 2006;291:H2875–2883. doi: 10.1152/ajpheart.01032.2005. [DOI] [PubMed] [Google Scholar]
- 64.Carvajal K, et al. Dual cardiac contractile effects of the alpha2-AMPK deletion in low-flow ischemia and reperfusion. Am J Physiol Heart Circ Physiol. 2007;292:H3136–3147. doi: 10.1152/ajpheart.00683.2006. [DOI] [PubMed] [Google Scholar]
- 65.Wang J, et al. Limiting cardiac ischemic injury by pharmacological augmentation of macrophage migration inhibitory factor-AMP-activated protein kinase signal transduction. Circulation. 2013;128:225–236. doi: 10.1161/CIRCULATIONAHA.112.000862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Morrison A, et al. Acute rosiglitazone treatment is cardioprotective against ischemiareperfusion injury by modulating AMPK, Akt, and JNK signaling in nondiabetic mice. Am J Physiol Heart Circ Physiol. 2011;301:H895–902. doi: 10.1152/ajpheart.00137.2011. [DOI] [PubMed] [Google Scholar]
- 67.Chin JT, et al. A novel cardioprotective agent in cardiac transplantation: metformin activation of AMP-activated protein kinase decreases acute ischemia-reperfusion injury and chronic rejection. Yale J Biol Med. 2011;84:423–432. [PMC free article] [PubMed] [Google Scholar]
- 68.Galinanes M, et al. Acadesine and myocardial protection. Studies of time of administration and dose-response relations in the rat. Circulation. 1992;86:598–608. doi: 10.1161/01.cir.86.2.598. [DOI] [PubMed] [Google Scholar]
- 69.Rosenkranz ER, et al. Biochemical studies: failure of tissue adenosine triphosphate levels to predict recovery of contractile function after controlled reperfusion. J Thorac Cardiovasc Surg. 1986;92:488–501. [PubMed] [Google Scholar]
- 70.Mangano DT. Effects of acadesine on myocardial infarction, stroke, and death following surgery. A meta-analysis of the 5 international randomized trials. The Multicenter Study of Perioperative Ischemia (McSPI) Research Group. Jama. 1997;277:325–332. doi: 10.1001/jama.277.4.325. [DOI] [PubMed] [Google Scholar]
- 71.Shimano M, et al. Cardiokines: recent progress in elucidating the cardiac secretome. Circulation. 2012;126:e327–332. doi: 10.1161/CIRCULATIONAHA.112.150656. [DOI] [PubMed] [Google Scholar]
- 72.Welford SM, et al. HIF1alpha delays premature senescence through the activation of MIF. Genes Dev. 2006;20:3366–3371. doi: 10.1101/gad.1471106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ma H, et al. Impaired macrophage migration inhibitory factor-AMP-activated protein kinase activation and ischemic recovery in the senescent heart. Circulation. 2010;122:282–292. doi: 10.1161/CIRCULATIONAHA.110.953208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Qi D, et al. Cardiac macrophage migration inhibitory factor inhibits JNK pathway activation and injury during ischemia/reperfusion. J Clin Invest. 2009;119:3807–3816. doi: 10.1172/JCI39738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liehn EA, et al. Compartmentalized protective and detrimental effects of endogenous macrophage migration-inhibitory factor mediated by CXCR2 in a mouse model of myocardial ischemia/reperfusion. Arterioscler Thromb Vasc Biol. 2013;33:2180–2186. doi: 10.1161/ATVBAHA.113.301633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Merk M, et al. D-dopachrome tautomerase (D-DT or MIF-2): doubling the MIF cytokine family. Cytokine. 2012;59:10–17. doi: 10.1016/j.cyto.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li J, et al. Urocortin 2 autocrine/paracrine and pharmacologic effects to activate AMP-activated protein kinase in the heart. Proc Natl Acad Sci U S A. 2013;110:16133–16138. doi: 10.1073/pnas.1312775110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ogura Y, et al. Therapeutic impact of follistatin-like 1 on myocardial ischemic injury in preclinical models. Circulation. 2012;126:1728–1738. doi: 10.1161/CIRCULATIONAHA.112.115089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 80.Lara-Pezzi E, et al. Expression of follistatin-related genes is altered in heart failure. Endocrinology. 2008;149:5822–5827. doi: 10.1210/en.2008-0151. [DOI] [PubMed] [Google Scholar]
- 81.Oshima Y, et al. Follistatin-like 1 is an Akt-regulated cardioprotective factor that is secreted by the heart. Circulation. 2008;117:3099–3108. doi: 10.1161/CIRCULATIONAHA.108.767673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Shibata R, et al. Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat Med. 2005;11:1096–1103. doi: 10.1038/nm1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kambara T, et al. C1q/TNF-related protein 9 protects against acute myocardial injury through an AdipoR1-AMPK dependent mechanism. Mol Cell Biol. 2015;35:2173–2185. doi: 10.1128/MCB.01518-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kataoka Y, et al. Omentin prevents myocardial ischemic injury through AMP-activated protein kinase- and Akt-dependent mechanisms. J Am Coll Cardiol. 2014;63:2722–2733. doi: 10.1016/j.jacc.2014.03.032. [DOI] [PubMed] [Google Scholar]
- 85.Li J, et al. AMP-activated protein kinase activates p38 mitogen-activated protein kinase by increasing recruitment of p38 MAPK to TAB1 in the ischemic heart. Circ Res. 2005;97:872–879. doi: 10.1161/01.RES.0000187458.77026.10. [DOI] [PubMed] [Google Scholar]
- 86.Morrison A, et al. Sestrin2 promotes LKB1-mediated AMPK activation in the ischemic heart. FASEB J. 2015;29:408–417. doi: 10.1096/fj.14-258814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sriwijitkamol A, et al. Effect of acute exercise on AMPK signaling in skeletal muscle of subjects with type 2 diabetes: a time-course and dose-response study. Diabetes. 2007;56:836–848. doi: 10.2337/db06-1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ko HJ, et al. Nutrient stress activates inflammation and reduces glucose metabolism by suppressing AMP-activated protein kinase in the heart. Diabetes. 2009;58:2536–2546. doi: 10.2337/db08-1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Xie Z, et al. Improvement of cardiac functions by chronic metformin treatment is associated with enhanced cardiac autophagy in diabetic OVE26 mice. Diabetes. 2011;60:1770–1778. doi: 10.2337/db10-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kusmic C, et al. Improved myocardial perfusion in chronic diabetic mice by the up-regulation of pLKB1 and AMPK signaling. J Cell Biochem. 2010;109:1033–1044. doi: 10.1002/jcb.22486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ruderman NB, et al. AMPK, insulin resistance, and the metabolic syndrome. J Clin Invest. 2013;123:2764–2772. doi: 10.1172/JCI67227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Seo-Mayer PW, et al. Preactivation of AMPK by metformin may ameliorate the epithelial cell damage caused by renal ischemia. Am J Physiol Renal Physiol. 2011;301:F1346–1357. doi: 10.1152/ajprenal.00420.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]