

Abstract
Tumors require a vascular supply to grow and can achieve this via the expression of pro-angiogenic growth factors. Many potential oncogenic mutations have been identified in tumor angiogenesis. Somatic mutations in the small GTPase KRAS are the most common activating lesions found in human cancer, and are generally associated with poor response to standard therapies. Biguanides, such as the diabetes therapeutics metformin and phenformin, have demonstrated anti-tumor activity both in vitro and in vivo. The extracellular regulated protein kinases (ERK) signaling is known to be a major cellular target of biguanides. Based on KRAS activates several down-stream effectors leading to the stimulation of the RAF/mitogen-activated protein kinase/extracellular signal-regulated kinase (RAF/MEK/ERK) and phosphatidylinositol-3-kinase (PI3K) pathways, we investigated the anti-tumor effects of biguanides on the proliferation of KRAS-mutated tumor cells in vitro and on KRAS-driven tumor growth in vivo. In cancer cells harboring oncogenic KRAS, phenformin switches off the ERK pathway and inhibit the expression of pro-angiogenic molecules. In tumor xenografts harboring the KRAS mutation, phenformin extensively modifies the tumor growth causing abrogation of angiogenesis. These results strongly suggest that significant therapeutic advantage may be achieved by phenformin anti-angiogenesis for the treatment of tumor.
Keywords: KRAS, phenformin, NSCLC, angiogenesis
Introduction
Angiogenesis has been understood to be an important therapeutic target, and drugs targeting vascular endothelial growth factor (VEGF) such as a bevacizumab has been developed and approved for clinical use, however, few other angiogenic factors switch on during cancer progression [1,2]. To date, many other potential “driver mutations” occurring in genes encoding cellular signaling proteins have also been identified in tumor angiogenesis and are causally associated with the neoplastic process [3]. It has been suggested that alterations in oncogenes involved in the intracellular RAS signaling cascade play a central role in regulating tumor angiogenesis [4]. Nearly 30% of human cancers possess activating RAS mutations, 85% of which are KRAS mutations [5]. KRAS is a membrane-bound GTPase that cycle between an active GTP-bound form and an inactive GDP-bound form due to the hydrolysis of the bound GTP. The most common KRAS mutation is a G12C, which results in constitutive activation of the kinase activity [6]. Among numerous downstream effectors of KRAS, the best characterized include RAF and phosphoinositide-3 kinase (PI3K). The major axes of RAS signaling through the RAF/MEK/ERK and PI3K/AKT cascades ultimately control processes such as cell growth and survival [7]. This is accomplished in part by ERK-regulated activation of transcription factors that promote cell cycle progression, and by AKT-mediated inactivation of pro-apoptotic proteins for apoptosis suppression. In addition, a number of alternate effectors of KRAS have been described in an extensive body of literature, which regulates processes such as cell migration, endocytosis, changes in cytoskeleton, and calcium signaling [8].
The role of KRAS oncogenes in promoting cellular transformation is well established. In addition, KRASG12C modulates tumor-stroma interaction and supports cancer invasiveness by influencing the expression of metalloproteinases and cytokines that involved in angiogenesis [9]. KRAS mutations lead to constitutive activation of downstream pathways and the mutations of KRAS is associated with tumor angiogenesis, indicating that this mutation and pathologic mechanism may be a suitable target for anticancer agents. Biguanides, such as metformin and phenformin, are common therapeutics for type 2 diabetics. Emerging evidence from retrospective population-based studies and preclinical studies using cultured cancer cells and mouse models have demonstrated that biguanides also possess antitumor activity [10]. The attenuation of ERK signaling is known to contribute to the anti-tumor effects of metformin. Furthermore, phenformin inhibited the growth of breast cancer cells by de-activating MAPK. KRAS regulates several pathways that synergistically induce cellular transformation, including the well-characterized ERK cascade [11]. With respect to these findings, we hypothesized that treatment of KRAS-mutant lung cancer with biguanides could offer therapeutic advantages in cancer therapy. In this report, we show that phenformin results in anti-cancer efficacy in both in vitro and in vivo models of KRAS mutant tumors. We show that this effect is, in part, explained by the ability of phenformin to suppress the growth of tumor cells and angiogenesis. The effected pathway and regulatory proteins were also identified.
Materials and methods
Cells and transfection
hTERT-HME1, H1792, H358, H1299, A549 and HUVEC were purchased from American Type Culture Collection, and were cultured in DMEM, 1640 or M199 with 10% FBS, and 1% Penicilin/Streptomycin mix and maintained at 37°C in a humidified atmosphere containing 5% CO2. The short small interfering RNA (siRNA) was constructed by Nanjing genscript biotechnology co., LTD with sequence specifically targeted to KRASG12C gene: (#1: 5’-GAAGUGCAUACACCGAGAC-3’ or #2: 5’-GUGCAAUGAAGGGACCAGUA-3’). A constitutively active mutant KRAS (G12C) plasmid was a gift from Channing Der (Addgene plasmid #58901). Transient transfection was performed using the Lipofectamine RNAi MAX reagent (Invitrogen) and following the manufacturer’s instructions.
Chorioallantoic membrane (CAM) assay
Fertilized white leghorn chicken embryos were incubated for 3 days at 37°C and 70% humidity. A small hole was made over the air sac at the end of the egg, and a second hole was made directly over the embryonic CAM. After 10 d, 1 × 106 hTERT-HME1 WT or KRASG12C knock-in cells were mixed with 50 μL of serum-free DMEM plus 50 μL of Matrigel and dropped onto the CAM to form a plug. After 48 h, CAMs were fixed with PBS solution/3.7% paraformaldehyde for 10 min at room temperature, and images were taken with a Nikon digital color camera [12].
In vivo Matrigel plug assay
A Matrigel plug assay was performed in BALB/c mice, as described previously with some modifications. Matrigel (500 µL) containing H1792 or H358 cells was inoculated subcutaneously into the right flank of Balb/c mice. All treatment groups contained six mice. The mice were treated with phenformin or 30% PEG400/0.5% Tween80/5% propylene glycol (vehicle) daily by inject into Matrigel plugs. After 10 days, the Matrigel plugs were removed and hemoglobin content was determined according to Drabkin’s method [13].
Cell viability assay
Briefly, cells (3 × 104 cells per well) were seeded in 96-well plates, and exposed to various concentrations of phenformin or metformin for 24 hours. Cell viability was measured by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay and three independent experiments with triplicate were carried out.
Wound healing assay
We examined the migration of human umbilical vein endothelial cells (HUVEC) using a wound-healing assay. Briefly, cells were each grown on 3.5-cm plates with M199. After the growing cell layers had reached confluence, we inflicted a uniform wound in each plate using a pipette tip, and washed the wounded layers with PBS to remove all cell debris. Then, cells were cultured with FBS or VEGFA in the presence of phenformin. We evaluated the closure at 24 h using bright-field microscopy [14].
Invasion assay
To determine the effect of KRASG12C on HUVEC invasion in vitro, we conducted cell assay by Matrigel-coated Boyden inserts (8 μm; BD Biosciences). Cells were then seeded on the upper chamber of Boyden and allowed to invasive to the lower chamber with 500 μL supernatant from hTERT-HME1 WT, KRASG12C knock-in cells or cells treated by phenformin. After 7 hours incubation, noninvasive cells were removed with cotton swabs, and invasive cells were fixed with cold 4% paraformaldehyde and stained with 1% crystal violet. Images were taken with an inverted microscope, and migrated cells in random 5 fields were quantified by manual counting [15]. Three independent experiments with triplicate were carried out.
ELISA
Cells (7 × 105) were plated in six-well dishes and treated for 24 h with phenformin, or vehicle. Supernatants were collected and ELISA for VEGFA was performed with a Quantikine immunoassay kit (DVE00; R&D Systems) following the manufacturer’s instructions.
Real-time PCR
Total RNA was isolated using TRIzol according to the manufacturer’s instructions (Invitrogen, USA) and the concentration of total RNA was detected by spectrophotometry at OD260. Reverse transcription (RT) was carried out using superscript III reverse transcriptase (Invitrogen, USA) as described in the manufacturer’s manual. The real-time PCR was performed on ABI Prism 7500 Sequence detection system (Applied Biosystems, CA) with the KAPA SYBR® qPCR Kit (KAPA Biosystems, USA) according to the manufacturer’s instructions. The primers used were as follow in Supplementary Table 1. The target mRNA level of control cells normalized to the level of β-actin mRNA, was defined as 1. Results were obtained from three independent experiments.
Western blot analysis
The whole-cell extracts were prepared by lysis buffer supplement with different kinds of protein inhibitors. Equal protein aliquot of each lysate was subjected to SDS-PAGE (8%), blotted onto polyvinylidene difluoride (PVDF) membrane (Bio-Rad), probed with specific antibodies and subsequently detected by chemiluminescence. Protein concentration was determined by Micro BCA Protein Assay Kit (Pierce Biotechnology).
Mouse xenografts
All animal procedures were approved by the ethical commission of the Shanxi Baoji People’s Hospital. H1792 (5 × 106 cells per mouse) or H358 (8 × 106 cells per mouse) were injected s.c. into the right posterior flanks of 7-wk-old immunodeficient NOD/SCID female mice (6 mice per group). On the seventh day, mice with appropriate size (250 mm3) of tumors were divided randomly into six groups including vehicle-treated group and phenformin dosage groups. The mice were treated with phenformin or 30% PEG400/0.5% Tween80/5% propylene glycol (vehicle) daily by intragastric administration. Tumor volume and mice body weight were measured every 3 days. Tumor volume was calculated as mm3 = 0.5 × length (mm)3 width (mm)2. After sacrificing mice on day 25, deparaffinized tumor sections were stained with specific antibodies including CD31 (Abbiotec) and Ki-67. Detection was done with avidin-biotin-HRP complex (Thermo scientific) and diaminobenzidine as chromogen [17]. Nuclei were counterstained with hematoxylin. All animal experiments were carried out in compliance with the Guidelines for the Capital University of Medical Sciences.
Statistical analysis
Numerical results were analyzed using independent mean T-test and expressed in mean ± standard deviation (SD). Statistical analysis was performed using post hoc testing using Bonferroni’s method. Differences were considered statistically significant at P < 0.05.
Results
Phenformin inhibits KRAS mutated NSCLC cell lines growth
To examine the effects of metformin and phenformin on cell viability as single agents, we performed MTT cell assays by using KRAS mutated NSCLC cell lines H1792 and H358. Metformin had limited effect on reducing cell viability in both two cell lines, when used at concentrations of up to 5 mM (Figure 1A). In contrast, phenformin was much more potent, with an estimated IC50 in the range of 2.1 mM for H1792 cells and 2.4 mM for H358 (Figure 1B). Furthermore, the effect of phenformin depended, at least partially, on KRASG12C, because siRNA knockdown of KRASG12C in cells (Figure S1) attenuated the reduction in cell viability in response to phenformin (Figure 1C). As H1972 and H358 cells harbor KRASG12C mutation, we tested whether the killing effects of phenformin is pendent on KRAS status. NSCLC cancer cells carrying wild type (WT) KRAS A549 and H1299 were used as control and proved insensitive to phenformin treatment (Figure 1D).
Figure 1.
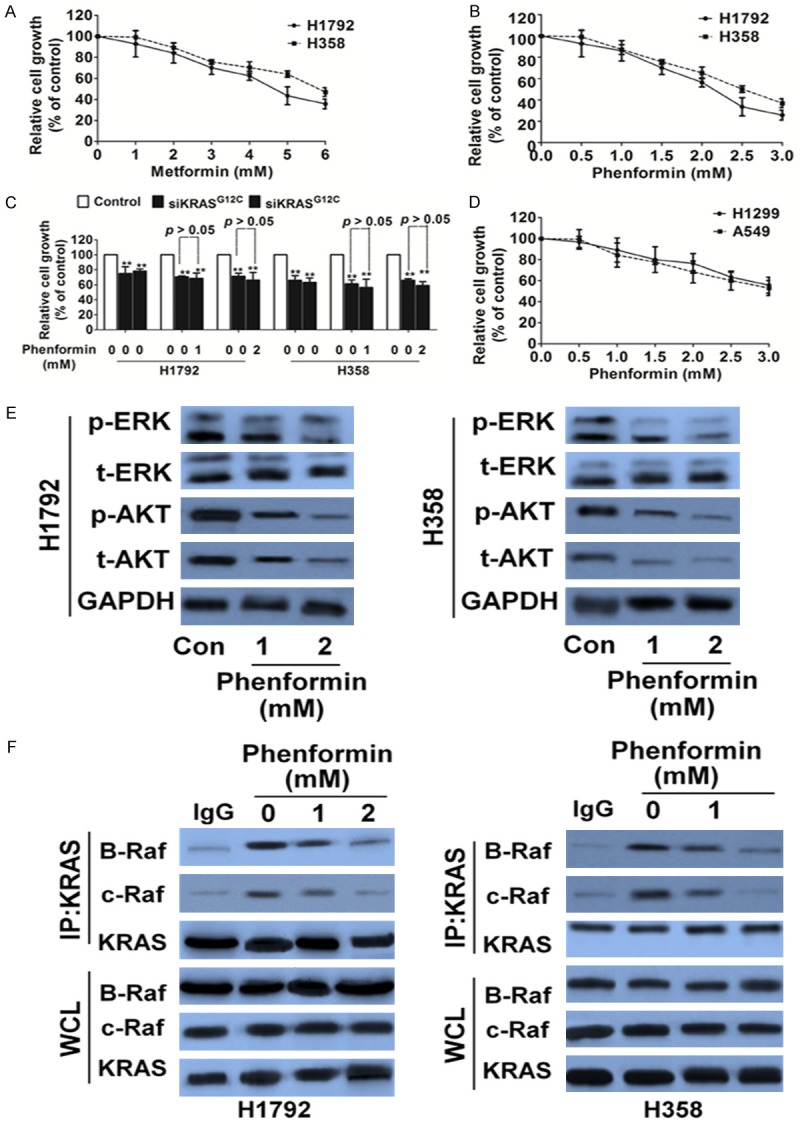
Phenformin inhibits proliferation and turns off ERK signaling in tumor cells carrying KRASG12C. A. Proliferation of H1792 and H358 cells were assessed with metformin. Data are from three independent experiments and are mean ± SD. N = 3. B. H1792 and H358 cells were assessed with phenformin and the growth was assayed by MTT. Data are from three independent experiments and are mean ± SD. N = 3. C. The proliferation inhibitory effects of phenformin on H1792 and H358 cells were abolished by KRASG12C siRNA. Data are from three independent experiments and are mean ± SD. N = 3, **P < 0.01 versus control. D. Proliferation of H1299 and A549 cells were treated with indicated phenformin and was assessed by MTT. Data are from three independent experiments and are mean ± SD. N = 3. E. Biochemical analysis of phospho-ERK and phospho-AKT (Ser473 and Thr308) in H1792 and H358. GAPDH was used as loading control. F. Co-immunoprecipitation (IP) of B-Raf and C-Raf with KRAS from KRASG12C cell lines after treatment with phenformin. Data were from three independent experiments. WCL, whole cell lysate.
KRAS protein plays a central role in controlling the activity of several crucial downstream signaling pathways such as Raf and AKT/ERK that regulate tumor cellular proliferation, differentiation and survival. As expected, phenformin inactivated ERK and AKT phosphorylation in the cells harboring KRASG12C (Figure 1E). We then measured RAS-Raf association in two KRASG12C-mutant lung cancer cell lines treated by phenformin, using co-immunoprecipitation. As predicted, treatment with phenformin decreased the association of B-Raf and C-Raf with KRAS (Figure 1F).
Phenformin treatment inhibits tumor growth and angiogenesis in xenograft mice
We next measured the effect of phenformin in vivo by growing H1972 and H358 subcutaneously in immunocompromised mice. Phenformin induced a prolonged cytostatic effect and shown evident shrinkage at the end of the experiments in both xenograft models (Figure 2A). None of the treatment groups demonstrated a weight loss of more than 10%, indicating no significant signs of toxicity (Figure 2B). This was further supported by the analysis of proliferation and apoptosis assessed through Ki-67 staining (Figure 2C) and inactivation of caspase-3 (Figure 2D), respectively. We found that phenformin markedly inhibited tumor cell proliferation, whereas it was ineffective in inducing apoptosis (Figure 2D). Similar to what was observed in vitro, phenformin treatment of H1972 and H358 tumors decreased the phosphorylation of ERK and AKT (Figure 2D).
Figure 2.

Phenformin treatment inhibits growth in H1792 and H358 xenograft models. A. Tumor growth curve of H1792 and H358 xenografts. H1792 and H358 xenografts were treated with phenformin 50 mg/kg or vehicle for 25 days after tumor volume reached an average of 200 to 300 mm3. Data are presented as means ± SD, n = 6, **P < 0.01 versus vehicle group. B. Body weight changes in phenformin and vehicle treated mice. Data are presented as means ± SD, n = 6. C. Representative images of Ki-67 staining in H1792 and H358 xenografts. Scale bar represents 50 μm. Quantification of proliferating cells by Ki-67 staining in H1792 and H358 xenografts. Data are presented as means ± SD, n = 6, **P < 0.01 versus vehicle group. D. Biochemical analysis of ERK, AKT and Caspase 3 in protein extract of H1792 and H358 xenografts. Protein loading was normalized by GAPDH. Three independent samples were evaluated.
To further examine whether phenformin suppress angiogenesis, tumor tissues were stained with specific antibody against CD31. Cluster of differentiation (CD31) is a widely used endothelial marker for quantifying angiogenesis by calculating microvessel density (MVD). Tumor sections stained with anti-CD31 antibody revealed that phenformin inhibited MVD (Figure 3A). This supports that phenformin is not only effective in vitro but also acts as an effective anti-cancer regimen in vivo as well. VEGF is one of the most potent proangiogenic peptides known, and modulation of this peptide will likely have significant consequence on angiogenesis. Western blot analysis further verified these findings. The phenformin treatment group had the low levels of CD31 and VEGFA than control group (Figure 3B). We had shown that phenformin was able to decrease the intreaction of B-Raf and C-Raf with KRAS by targeting the KRAS pathway in NSCLC cells. A similar trend was also seen in vivo. In vivo, there were significantly lower levels of CR-af and BR-af in the phenformin group compared to control (Figure 3C).
Figure 3.
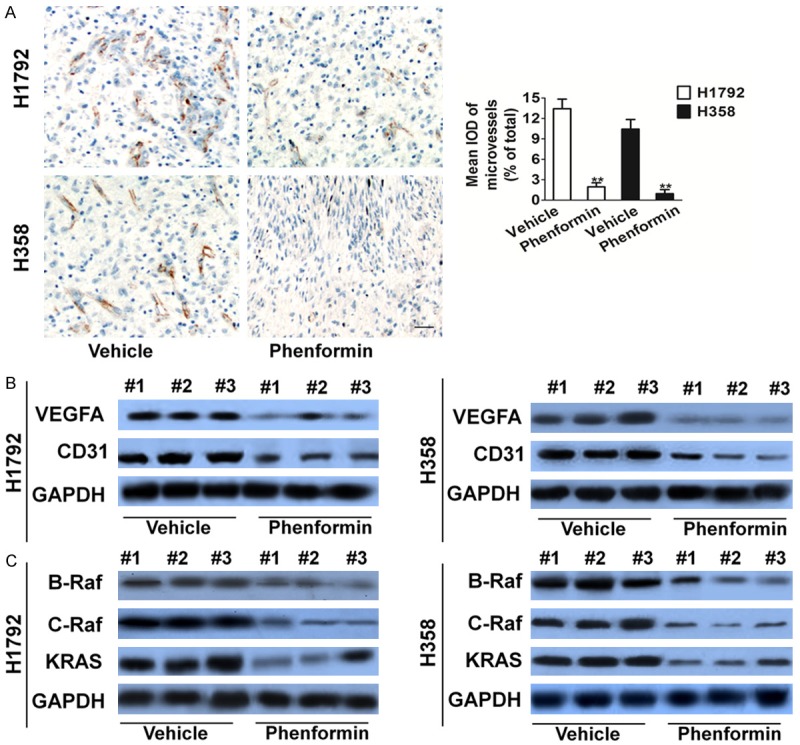
Phenformin treatment inhibits angiogenesis in H1792 and H358 xenograft models. A. Tumor tissues were prepared for immunohistochemistry detection with antibody against CD31 (Scale bar represents 50 μm). Data are presented as means ± SD, n = 6, **P < 0.01 versus vehicle group. B. VEGFA and CD31 levels from tumor samples were analyzed through Western blot analysis. Lower CD31 and VEGFA levels were measured in the phenformin treatment group. C. Similarly, through Western blot, phenformin treatment had the most profound decrease in CRAF, BRAF and NRAS protein levels in tumor samples.
Confirming phenformin treatment inhibits tumor angiogenesis dependent on KRASG12C
To study the specific influence of phenformin on tumor angiogenesis depedent on KRASG12C, we used an isogenic model in which KRASG12C was knocked into the genome of the nontumorigenic human mammary epithelial cell line (hTERT-HME1) and the expression of KRASG12C was confirmed by western blot with anti-HA-tag antibody (Figure 4A). We used the chicken chorioallantoic membrane (CAM) assay to assess whether phenformin modulate angiogenesis dependent on KRASG12C (Figure 4B). We next compared the transcriptional profile of genes involved in angiogenesis between KRASG12C knock-in hTERT-HME1 cell and phenformin treated cells. This assay revealed that oncogenic KRASG12C enhanced the expression of several pro-angiogenic molecules, including VEGF-A and VEGF-C, PDGFA, and chemokines, such as CCL-2, IL6, and IL8, while phenformin decrease the expression of proangiogenic factors (Figure 4C). Among the factors that were pinpointed by the transcriptional profile, we focused on VEGF-A because this molecule is angiogenic program. KRASG12C knock in cells released higher amounts of VEGF-A in the supernatant that enhanced HUVECs invasion compared with the WT counterpart. While, phenformin treatment significantly inhibited HUVECs invasion stimulated by KRASG12C (Figure 4C).
Figure 4.

Phenformin inhibits tumor angiogenesis dependent on KRASG12C. A. Western blot analysis to assay the KRASG12C in empty vector transfected cells and KRASG12C transfected cells. GAPDH was used as a loading control. B. Representative images of hTERT-HME1 cells plated on the CAM. Scale bar: 0.5 cm. Qualitative assessment of angiogenesis in the CAM assay. Data are from three independent experiments and are mean ± SD. N = 6, **P < 0.01 compared with wild type cells, ##P < 0.01 compared with KRASG12C knock-in cells. C. KRASG12C up-regulates the expression of pro-angiogenic factors in KRASG12C knock-in hTERT-HME1 cells. Gene expression analysis was performed by real-time PCR comparing parental hTERT-HME1 with KRASG12C clone. Data are from three independent experiments and are mean ± SD. N = 6, *P < 0.05, **P < 0.01 compared with wild type cells and #P < 0.05, ##P < 0.01 compared with KRASG12C knock-in cells. D. Quantification of secreted VEGFA in hTERT-HME1 cells by ELISA. Data are from three independent experiments and are mean ± SD. N = 3, **P < 0.01 compared with wild type cells, ##P < 0.01 compared with KRASG12C knock-in cells. E. Invasion of HUVECs was enhanced by the supernatant of hTERT-HME1 KRASG12C knock-in cells compared with the wild type counterpart. Scale bar represents 50 μm. Data are from three independent experiments and are mean ± SD. N = 3, **P < 0.01 compared with supernatant from wild type cells, ##P < 0.01 compared with supernatant from KRASG12C knock-in cells.
Secretion of pro-angiogenic factors in KRASG12C mutant cancer cells
To further assess whether the anti-angiogenic effect of phenformin was direct (on the tumor vasculature) or indirect (via epithelial cells), we investigated if phenformin affected HUVECs in vitro. We found that proliferation and migration of HUVECs were unaffected by phenformin treatment (Figure 5A-C), thus ruling out a possible direct effect of phenformin inhibition on the endothelial compartment. We next considered if phenformin treatment was capable of modulating the production of angiogenic molecules by cancer cells, which in turn might influence the tumor environment. To assess this, we analyzed the effect of phenformin on the expression of angiogenic factors in NSCLC cells carrying mutant KRAS. We found that, upon phenformin treatment, multiple mediators of angiogenesis (e.g., IL-6, IL-8 and VEGF-A) were down-regulated in H1792 and H358 cells (Figure 5D). On the contrary, the same genes were not modulated in the KRAS WT cancer cells (Figure S2). In vivo Matrigel plug angiogenesis assays were performed to test the effects of phenformin treatment on tumor angiogenesis. The hemoglobin contents in Matrigels containing drug was significantly inhibited (Figure 5E). Western blot analysis for CD31 and VEGFA levels further verified phenformin had inhibition potential to tumor angiogenesis (Figure 5F).
Figure 5.

Phenformin does not affect HUVECs proliferation and mobility. A~C. HUVECs were used to evaluate proliferation, migration, and invasion. HUVECs were stimulated by FBS or 50 ng/mL VEGFA and were treated with phenformin or vehicle. Cells proliferation is shown as mean percentage of cells viability compared with serum-Free untreated samples (SF) ± SD in quadruplicate. HUVECs migration is represented as mean number of migrated cells ± SD in triplicate. In both cases, representative results of three independent experiments are shown. D. Expression of proangiogenic factors was evaluated by real-time PCR. Cells carrying KRASG12C was treated for 24 h with phenformin. Data are expressed as relative quantity (RQ) of phenformin compared with vehicle-treated samples. Bars show mean ± SD of triplicate measurements. E. Quantitative analysis of hemoglobin levels in Matrigels plugs. Data are from three independent experiments and are mean ± SD. N = 6, **P < 0.01 compared with wild type cells. F. VEGFA and CD31 levels from plugs tissues were analyzed through Western blot analysis.
Discussion
Angiogenesis has been recognized as an important event as it plays an essential role for tumor growth and survival. In fact, in NSCLC, there is a direct correlation between decreased survival with high levels of angiogenesis and controlled by a variety of growth factors, and the most important proangiogenic peptide is VEGF [18]. For this reason, there continues to be active research and development of anti-angiogenic agents such as VEGF receptor TKIs and a monoclonal antibody that targets VEGF. Unfortunately, the clinical benefit conferred by these therapies is variable and other angiogenic factors switch on during cancer progression, which facilitates tumor initiation and induce resistance to RTK inhibitors [19]. Among these, RAS family and related downstream pathways play a critical role in cancer development and over recent years has become a validated target in NSCLC. Activating KRAS mutations are present in more than 80% of all NSCLCs and most often are due to G12C point mutation [20]. This mutation lead to constant phosphorylation of the receptor and activation of downstream cascade pathways (such as the RAS, Phosphoinositide 3-kinase, and AKT signaling pathways) that are important in regulating cell proliferation and growth. It has been shown that activating mutations of KRAS or NRAS lead to a consecutive activation of the RAS-RAF pathway. Subtle changes in the molecular nature of KRAS oncogene activating mutations occurring in tumor cells have a major impact on the vascular strategy devised providing with new insights on the role of KRAS mutations on angiogenesis [21].
Phenformin, is widely used as a first-line therapy for type 2 diabetes. Recent epidemiological stidues have found that patients with type 2 diabetes who were treated with phenformin had lower cancer risk and lower cancer-related mortality rates compared with patients treated with other therapeutic [22]. Moreover, phenformin has antitumor activities in various xenograft, carcinogen-induced, and genetically modified mouse models, raising strong interest in repurposing these drugs for cancer therapy. Phenformin lowers elevated insulin levels associated with type 2 diabetes by inhibiting hepatic gluconeogenesis via AMP-activated protein kinase (AMPK) activation. It increases insulin sensitivity and glucose utilization by skeletal muscle and adipose tissue resulting in reduced blood glucose and insulin levels [23]. Phenformin can have a direct anti-tumoral effect, but also can act indirectly to improve insulin sensitivity, decrease hyperinsulinaemia and consequently decrease tumor proliferation. The decrease in insulin levels caused by phenformin can reduce the activation of insulin pathways such as PI3K/Akt/mTOR and MEK/ERK1/2 and lead to a decrease in tumor growth [24]. Therefore, in this study, we wanted to test whether we can augment phenformin treatment to achieve even better outcomes in NSCLC harboring KRAS mutations.
In this study, we have demonstrated that the phenformin offers a therapeutic advantage against KRAS mutant NSCLC in both cell culture and animal models. In vitro treatment with the phenformin resulted in cell growth inhibition and ERK, AKT inactivation in both H1792 and H358 cell lines. As in vitro, phenformin treatment was also effective in vivo. Xenograft mice with implanted H1792 or H358 cells demonstrated significantly greater inhibition of tumor growth and shrinkage when treated with phenformin. Other than causing cell death, there are likely additional mechanisms in which phenformin treatment is able to inhibit tumor growth; yet these mechanisms have to be elucidated or reported. Intuitively, previous studies have shown that the treatment of an anticancer agent with an anti-angiogenesis potential results in enhanced inhibition of tumor growth. This is somewhat expected considering one of the drugs specifically targets angiogenesis. In this study, we show for the first time that low dose phenformin treatment is able to profoundly inhibit angiogenesis through down-regulation of VEGF in vitro and even more effectively in vivo. To explore the possibility that phenformin affects the angiogenic potential of cancer cells harboring mutational oncogene. As a model, we chose KRASG12C, one of the most aggressive oncogenes frequently detected in colorectal tumors and melanomas. We took advantage of a knock-in model, in which KRAS mutation has been introduced into the genome of non-transformed epithelial cells, hence closely recapitulating the situation observed in human neoplasms. We report that KRAS mutant cells display up-regulation of proangiogenic factors. In tumor cells, blockage of KRASG12C with phenformin not only exerted a cytostatic activity, but influenced the tumor vasculature. Notably, phenformin treatment did not affect ECs directly; rather, it down-regulated the expression of angiogenic factors in tumor cells.
In this regard, our work suggests that the pharmacological inhibition of an oncogenic mutation, which causally contributes to decrease pro-angiogenic mediators, may represent another valuable strategy to target tumor angiogenesis.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Kubota Y. Tumor angiogenesis and anti-angiogenic therapy. Keio J Med. 2012;61:47–56. doi: 10.2302/kjm.61.47. [DOI] [PubMed] [Google Scholar]
- 2.Bertolini F, Marighetti P, Martin-Padura I, Mancuso P, Hu-Lowe DD, Shaked Y, D’Onofrio A. Anti-VEGF and beyond: shaping a new generation of anti-angiogenic therapies for cancer. Drug Discov Today. 2011;16:1052–1060. doi: 10.1016/j.drudis.2011.08.007. [DOI] [PubMed] [Google Scholar]
- 3.Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3:401–10. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- 4.Desroches-Castan A, Quélard D, Demeunynck M, Constant JF, Dong C, Keramidas M, Coll JL, Barette C, Lafanechère L, Feige JJ. A new chemical inhibitor of angiogenesis and tumorigenesis that targets the VEGF signaling pathway upstream of Ras. Oncotarget. 2015;6:5382–411. doi: 10.18632/oncotarget.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–551. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guin S, Theodorescu D. The RAS-RAL axis in cancer: evidence for mutation-specific selectivity in non-small cell lung cancer. Acta Pharmacol Sin. 2015;36:291–297. doi: 10.1038/aps.2014.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hofmann I, Weiss A, Elain G, Schwaederle M, Sterker D, Romanet V, Schmelzle T, Lai A, Brachmann SM, Bentires-Alj M, Roberts TM, Sellers WR, Hofmann F, Maira SM. K-RAS Mutant Pancreatic Tumors Show Higher Sensitivity to MEK than to PI3K Inhibition In Vivo. PLoS One. 2012;7:e44146. doi: 10.1371/journal.pone.0044146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Konstantinidou G, Bey EA, Rabellino A, Schuster K, Maira MS, Gazdar AF, Amici A, Boothman DA, Scaglioni PP. Dual PI3K/mTOR blockade is an effective radiosensitizing strategy for the treatment of non-small cell lung cancer harboring K-RAS mutations. Cancer Res. 2009;69:7644–7652. doi: 10.1158/0008-5472.CAN-09-0823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sparmann A, Bar-Sagi D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell. 2004;6:447–458. doi: 10.1016/j.ccr.2004.09.028. [DOI] [PubMed] [Google Scholar]
- 10.Fasih A, Elbaz HA, Hüttemann M, Konski AA, Zielske SP. Radiosensitization of Pancreatic Cancer Cells by Metformin through the AMPK Pathway. Radiat Res. 2014;182:50–59. doi: 10.1667/RR13568.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soares HP, Ni Y, Kisfalvi K, Sinnett-Smith J, Rozengurt E. Different Patterns of Akt and ERK Feedback Activation in Response to Rapamycin, Active-Site mTOR Inhibitors and Metformin in Pancreatic Cancer Cells. PLoS One. 2013;8:e57289. doi: 10.1371/journal.pone.0057289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lokman NA, Elder AS, Ricciardelli C, Oehler MK. Chick Chorioallantoic Membrane (CAM) Assay as an In Vivo Model to Study the Effect of Newly Identified Molecules on Ovarian Cancer Invasion and Metastasis. Int J Mol Sci. 2012;13:9959–9970. doi: 10.3390/ijms13089959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murakami M, Nguyen LT, Hatanaka K, Schachterle W, Chen PY, Zhuang ZW, Black BL, Simons M. FGF-dependent regulation of VEGF receptor 2 expression in mice. J Clin Invest. 2011;121:2668–2678. doi: 10.1172/JCI44762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen L, Zhang JJ, Huang XY. cAMP Inhibits Cell Migration by Interfering with Rac-induced Lamellipodium Formation. J Biol Chem. 2008;283:13799–805. doi: 10.1074/jbc.M800555200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Banyard J, Chung I, Migliozzi M, Phan DT, Wilson AM, Zetter BR, Bielenberg DR. Identification of genes regulating migration and invasion using a new model of metastatic prostate cancer. BMC Cancer. 2014;14:387. doi: 10.1186/1471-2407-14-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee C, Liu A, Miranda-Ribera A, Hyun SW, Lillehoj EP, Cross AS, Passaniti A, Grimm PR, Kim BY, Welling PA, Madri JA, DeLisser HM, Goldblum SE. NEU1 Sialidase Regulates the Sialylation State of CD31 and Disrupts CD31-driven Capillary-like Tube Formation in Human Lung Microvascular Endothelia. J Biol Chem. 2014;289:9121–9135. doi: 10.1074/jbc.M114.555888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li J, Ouyang Y, Zhang X, Zhou W, Wang F, Huang Z, Wang X, Chen Y, Zhang H, Fu L. Effect of HM910, a novel camptothecin derivative, on the inhibition of multiple myeloma cell growth in vitro and in vivo. Am J Cancer Res. 2015;5:1000–1016. [PMC free article] [PubMed] [Google Scholar]
- 18.Yu Y, Cai W, Pei CG, Shao Y. Rhamnazin, a novel inhibitor of VEGFR2 signaling with potent antiangiogenic activity and antitumor efficacy. Biochem Biophys Res Commun. 2015;458:913–919. doi: 10.1016/j.bbrc.2015.02.059. [DOI] [PubMed] [Google Scholar]
- 19.Leali D, Bianchi R, Bugatti A, Nicoli S, Mitola S, Ragona L, Tomaselli S, Gallo G, Catello S, Rivieccio V, Zetta L, Presta M. Fibroblast growth factor 2-antagonist activity of a long-pentraxin 3-derived anti-angiogenic pentapeptide. J Cell Mol Med. 2010;14:2109–2121. doi: 10.1111/j.1582-4934.2009.00855.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shimamura T, Chen Z, Soucheray M, Carretero J, Kikuchi E, Tchaicha JH, Gao Y, Cheng KA, Cohoon TJ, Qi J, Akbay E, Kimmelman AC, Kung AL, Bradner JE, Wong KK. Efficacy of BET bromodomain inhibition in Kras-mutant non-small cell lung cancer. Clin Cancer Res. 2013;19:6183–6192. doi: 10.1158/1078-0432.CCR-12-3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kelly-Spratt KS, Philipp-Staheli J, Gurley KE, Hoon-Kim K, Knoblaugh S, Kemp CJ. Inhibition of PI-3K restores nuclear p27Kip1 expression in a mouse model of K-ras driven lung cancer. Oncogene. 2009;28:3652–3662. doi: 10.1038/onc.2009.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Appleyard MV, Murray KE, Coates PJ, Wullschleger S, Bray SE, Kernohan NM, Fleming S, Alessi DR, Thompson AM. Phenformin as prophylaxis and therapy in breast cancer xenografts. Br J Cancer. 2012;106:1117–1122. doi: 10.1038/bjc.2012.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Z, Ren L, Liu C, Xia T, Zha X, Wang S. Phenformin Induces Cell Cycle Change, Apoptosis, and Mesenchymal-Epithelial Transition and Regulates the AMPK/mTOR/p70s6k and MAPK/ERK Pathways in Breast Cancer Cells. PLoS One. 2015;10:e0131207. doi: 10.1371/journal.pone.0131207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang L, Sha H, Davisson RL, Qi L. Phenformin Activates the Unfolded Protein Response in an AMP-activated Protein Kinase (AMPK)-dependent Manner. J Biol Chem. 2013;288:13631–13638. doi: 10.1074/jbc.M113.462762. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.