

Abstract
TCF3 (E2A) is a multifunctional basic helix loop helix (bHLH) transcription factor that is over-expressed in prostate cancer (PCa) as compared to normal prostate and that it acts as a tumor promoter in PCa. Given the diverse biological pathways regulated/influenced by TCF3, little is known about the mechanisms that regulate its expression. TCF3 expression in androgen sensitive LNCaP and insensitive C81 PCa cell lines was determined following treatments with androgen receptor (AR) agonist R1881 and antagonist Casodex. In silico analysis was used to discover putative Androgen Response Elements (ARE) in the TCF3 promoter/intron region. Chromatin Immunoprecipitation (ChIP) with AR antibody and luciferase reporter assays on the above mentioned cell lines was used to confirm AR biding and AR dependent transcriptional activity respectively. The results were confirmed by demonstrating TCF3 expression in LNCaP PCa xenograft models. The results suggested that TCF3 transcript increased in response to R1881 in LNCaP cells but was constitutively expressed in C-81 cell lines. The promoter/Intron region of the TCF3 gene was predicted to contain two putative ARE sites ARE1 and ARE2. ChIP after treatment of LNCaP and C81 cells with R1881 and Casodex showed that the ARE1 and ARE2 were bound by AR in LNCaP cells only in the presence of R1881, whereas C81 cells showed constitutive AR binding. Similar results were observed in luciferase reporter assays indicating that TCF3 is activated by AR in LNCaP cell lines whereas it is independent of androgens in C81 cell line. Luciferase reporter assays also confirmed that ARE1 alone drives androgen dependent transcription. TCF3 expression was only observed in castration resistant LNCaP xenografts in castrated mice. In conclusion, we demonstrate that in PCa androgen receptor regulates the expression of TCF3 which is mediated in part via a consensus androgen response element. The shift in TCF3 expression from androgen regulated to androgen independent during prostate cancer progression, together with lack of expression in normal prostate may provide mechanistic basis underlying the transition of androgen receptor from a tumor suppressor to an oncogene in prostate cancer.
Keywords: E2A, TCF3, bHLH, prostate cancer, androgen receptor, transcriptional regulation
Introduction
The transcription factor TCF3 belongs to the family of basic helix loop helix (bHLH) proteins [1]. TCF3 gene codes for two alternatively spliced variants E12 and E47 [2]. Both E12 and E47 are more than 99% similar overall except a 3 amino acid deletion in E47 immediately upstream of the bHLH domain due to alternative splicing as compared to E12. As a consequence, E12 only forms heterodimers with other bHLH proteins whereas E47 can homo as well as heterodimerize [3,4]. The bHLH interaction involves a C-terminal helix loop helix domain (HLH) which mediates protein-protein interactions and a basic region immediately upstream of the HLH domain which is responsible for the DNA binding. The N-terminal end of TCF3 also consists of two transcriptional activation domains AD1 and AD2 [5,6] which modulates the transcriptional activities of TCF3 target promoters by recruitment of CBP/p300 [7] or members of the ETO (eight twenty one encoded by RUNK1T1) family [8]. Recruitment of p300 leads to activation of gene expression, whereas association with members of the ETO family mediates transcriptional repression [7,8].
The heterodimerization of TCF3 with tissue specific bHLH proteins members results in multiple regulatory functions ranging from cellular differentiation to lineage commitment. For example, interaction of TCF3 with tissue specific bHLH proteins NeuroD and MyoD promotes neurogenesis and myogenesis, respectively [9,10].
The TCF3 proteins play particularly important roles during lymphoid development and hematopoietic stem cell development [11-13]. Evidence suggests that TCF3 proteins act as general negative regulator of cell proliferation in several normal cells and cancer cell lines [14-16]. Experimental evidence also demonstrated that expression of TCF3 is lower in CRC tissues than the normal mucosa and low TCF3 expression correlates with advanced TNM stage and larger tumor size, and predicted poor prognosis of CRC patients [17]. The growth inhibition by TCF3 occurs at multiple levels involving both bHLH dependent and independent mechanisms. Primary among these are the transcriptional up-regulation of multiple cyclin dependent kinase inhibitors CDKN1A (p21), p15INK4B and p16INK4B [15,16,18]. Ectopic expression of TCF3 also promotes apoptosis in TCF3 deficient lymphomas, independent of an arrest in cell cycle progression [14,16].
Contrary to its well established role as an inhibitor of proliferation, TCF3 expression is also observed in cells undergoing rapid proliferation in the rat embryo [19], in proliferating periventricular neuroepithelial cells in the developing brain and in centroblasts within germinal centers [20]. Ectopic expression of E47 also promoted proliferation of Pre-B cell line 697 in a cyclin D2/D3 dependent manner [21]. Furthermore, increased expression of E47 is also observed in breast cancer stem cells [22], breast cancer with basal like phenotype [23], gastric cancer [24], renal cell carcinoma [25] and hepatocellular carcinoma [26]. Over-expression of E47 in MDCK cells promotes angiogenesis and proliferation in tumor xenografts [27]. TCF3 also promotes epithelial to mesenchymal transition due to direct inhibition of E-cadherin expression at the promoter level [9,22,23,27,28], a mechanism central to cancer progression. A competition between CBP/p300 and ETO for binding to a region of AD1 domain referred to as the ‘p300/CBP and ETO target in E-proteins’ or PCET motif is considered as a potential mechanism for E-protein mediated transcriptional silencing. Thus the switch between the role of TCF3 as a tumor-suppressor/pro-differentiation to a tumor-/pro-proliferation likely involves the formation of selective transcriptional regulatory complexes.
We have shown in our previous study that TCF3 expression is significantly increased in prostate cancer in a stage dependent manner. At the molecular level, ablation of TCF3 leads to apoptosis and G1 arrest dependent proliferation block in PCa cell lines. Taken together, our results demonstrated that TCF3 acts as a potential tumor promoter [29] which is contrary to its role as a tumor suppressor. Collectively, the results suggest a complex transcription regulation of TCF3 which is contrary to earlier observations that TCF3 is ubiquitously expressed (largely based on in vitro cultured cell lines).
Despite the importance and function of TCF3, the TCF3 transcriptional regulatory mechanism remains unidentified. The major mechanism regulating TCF3 expression and function appears to be at the post-translational level [30] including dimerization with tissue specific bHLH and dominant negative HLH proteins of the inhibitor of differentiation (ID) family, ubiquitin and phosphorylation [31,32]. Apart from an earlier study characterizing the mouse TCF3 promoter [33], there is a lack of any study demonstrating the transcriptional regulation of the human TCF3 gene. The mouse TCF3 promoter has been shown to lack the conventional TATA and CAAT boxes. Our bioinformatics based analysis on human TCF3, using known transcriptional start sites for E12 and E47 suggested that the human TCF3 also lacks a conventional TATA box (unpublished observation). Potential binding sites for transcription factors GATA -1/-2, Sp1, CREB, AP-2 and E2F in the mouse promoter have been reported but lack experimental evidence [33]. The alternate transcription start sites for example putative initiator regions and GC rich regions could be involved in regulating TCF3 gene expression but remains to be identified. The lack of TATA boxes, a common feature of housekeeping genes is observed in more than 80% of all genes. Most of these TATA less genes are not housekeeping genes or constitutively expressed, but instead highly regulated suggesting that in these promoters, specific combinations between different core promoters and enhancers could determine restricted expression patterns in different tissues [20].
The present study was therefore designed to investigate the mechanism by which TCF3 expression is up-regulated specifically in prostate cancer. Here we show for the first time that TCF3 expression is regulated by androgen receptor in prostate cancer. Specifically we identify a putative androgen response element in the TCF3 promoter/intron that binds androgen receptor and promotes TCF3 transcription. The goal of this study was not to identify essential core/loose promoter or elements required to drive transcription but to determine putative enhancers, specifically Androgen receptor dependent elements as a potential cofactor required for androgen dependent TCF3 expression in prostate cancer.
Materials and methods
Cell culture and treatments
Human prostate cancer cell lines LNCaP and C81 were maintained at 37°C and 5% CO2 in RPMI-1640 supplemented with 10% (v/v) fetal bovine serum (FBS) and penicillin/streptomycin. Prior to the hormone treatments, the cells were grown in RPMI with 2% Charcoal-stripped fetal bovine serum (CSFBS). C81 cells were kindly provided by Prof. Ming-Fong Lin (University of Nebraska Medical Center) and were cultured as described earlier [34]. RWPE1 cells (Obtained from ATCC) were cultured and maintained as per supplier’s instructions. 10 nM R1881 (Metribolone) and 30 uM Casodex (Bicalutamide) (Selleck chemicals) was used for treatments as described previously [35].
Real time quantitative RT-PCR
RNA (2 µg) isolated from cultured cells was reverse transcribed in a final volume of 25 µl as per standard protocols. Reverse transcribed RNA was used for qRT-PCR using gene specific primers for TCF3: Forward Primer: 5’CGA GCT GGC CCT CAA CAG CC3’, Reverse Primer: 5’CCG GAC CTT CTT GGG CTG CG3’. Total RNA from xenografts was isolated by E.Z.N.A. DNA/RNA kit (Omega Biotech).
Immuno blot analysis
Cellular proteins were prepared from LNCaP and C81 cell lines using M-PER kit (Thermo Scientific). Immuno-blot analysis using TCF3 antibody (sc-349, Santa Cruz Biotechnology) followed by incubation with secondary antibody (SA1-9510, HRP- goat anti-rabbit, Thermo Scientific) as described earlier [29]. The LAS 3000 imager (Fuji) and image quant software was used to capture and quantify the images.
Chromatin immunoprecipitation (ChIP) assays
ChIP was performed using the ChIP kit from EMD Millipore (Billerica, MA) according to manufacturer’s instructions using Anti-Androgen Receptor N-terminal Rabbit polyclonal antibody (PG-21, Upstate Biotechnology). DNA samples from ChIP preparations were analyzed by PCR [36]. The primers used were designed flanking both the predicted ARE sites upstream of the TCF3 gene (417 bp fragment). TCF3-AR ChIP Forward Primer: 5’-CCC CCG GCA CTT TAA GTC TTG AAG-3’ and TCF3-AR ChIP Reverse Primer: 5’-GCA GGG AAG CTG GAA TTC CAG AGT-3’. The previously published primers used for PSA ChIP [37] were Forward Primer: 5’CAT GTT CAC ATT AGT ACA CCT TGC C-3’ and Reverse Primer: 5’TCTCAGATCCAGGCTTGCTTACTGTC-3’.
Site directed mutagenesis
Site-directed mutagenesis was used to mutate the putative ARE motifs in the TCF3 upstream region using a commercial QuickChange II Site-Directed Mutagenesis kit (Agilent Technologies, USA). The PCR primer used for mutagenesis was:
C48a_g54t ARE mutant Forward Primer3: 5’-GGG CGG GGT AGG Ata GGG CTt TTC CTC CC-3’ and c48a_g54t_asARE mutant Reverse Primer3: 5’-GGG AGG AAA AGC CCT ATC CTA CCC CGC CC-3’.
Corresponding to the ARE-1 (wild-type [WT] 5’-GGATcGGGCTgTTCC-3’) (Mutated nucleotides are shown as bold lowercase letters). The site-directed mutant on ARE motif (termed as ARE-1) was generated using the WT construct as template. Successful mutation was confirmed by direct sequencing, and the construct was used in luciferase reporter assays.
Transient transfection and luciferase assay
Luciferase reporter plasmids were created by cloning the PCR-generated fragments into minimal promoter luciferase reporter pGL4.26 vector (Promega Corp., Madison, WI). Four constructs were used for the assay: pGL 4.26 control vector backbone (CVB), pGL 4.26 with wtARE1+ARE2 (ARE1+ARE2), pGL4.26 with ARE1 (ARE1), pGL4.26 with ARE2 (ARE2) or pGL4.26 with mutantARE1+wild type ARE2 (mtARE1+ARE2). LNCaP and C81 cells (20,000 cells/well) were plated in 96-well plates in media containing 2% CSFBS. For transfection, the reporter luciferase construct and hRluc (pGL4.74, Renilla Luciferase, used as transfection control) plasmid DNA (Promega Corp., Madison, WI), in a 10:1 ratio were mixed with FuGENE HD transfection reagent (Promega Corp., Madison, WI) in a final volume of 100 ul of Opti-MEM and incubated for 15 min at room temperature. Twenty four hours after plating, the transfection mix was then added to the cells. After 24 h, the cells were treated with 10 nM R1881 or 10 nM R1881 plus 30 uM Casodex. The cells were assayed for Firefly and Renilla luciferase activities using the Dual-Glo Luciferase reporter assay system (Promega Corp., Madison, WI) 24 hrs after treatment. The results were normalized for the internal Renilla control and expressed as fold change of the mean relative light units.
Immuno-histochemistry (IHC)
IHC on paraffin embedded 5 um tissue sections was performed as described previously [29] using Anti-Human E12/E47 antibody (BD Pharmingen). Non-immune IgG, used as control for the immune localization studies resulted in lack of detection of respective antigens (data not shown).
Statistical analysis
Quantitative real time data was analyzed using the ΔΔCt method. The ChIP data was analyzed using % chromatin (1%) as input (Life Technologies). Within group Student’s t-test was used for evaluating the statistical differences between groups. All experiments were performed in triplicates.
Results
TCF3 expression in prostate cancer
Transcriptome wide analysis suggested that TCF3 expression is significantly higher in prostate adenocarcinoma as compared to normal prostate (Figure 1A, left panel [38]). TCF3 expression also increased significantly in hormone refractory metastatic prostate cancer as compared to prostate carcinoma (T-test: -6.268, P-value: 6.7E-5, Figure 1A, right panel) [39] (The data shown in Figure 1 was obtained from Oncomine v4.0). These results are consistent with increased TCF3 expression in PCa reported in our earlier studies using immunohistochemistry on prostate cancer tissue microarray [29].
Figure 1.
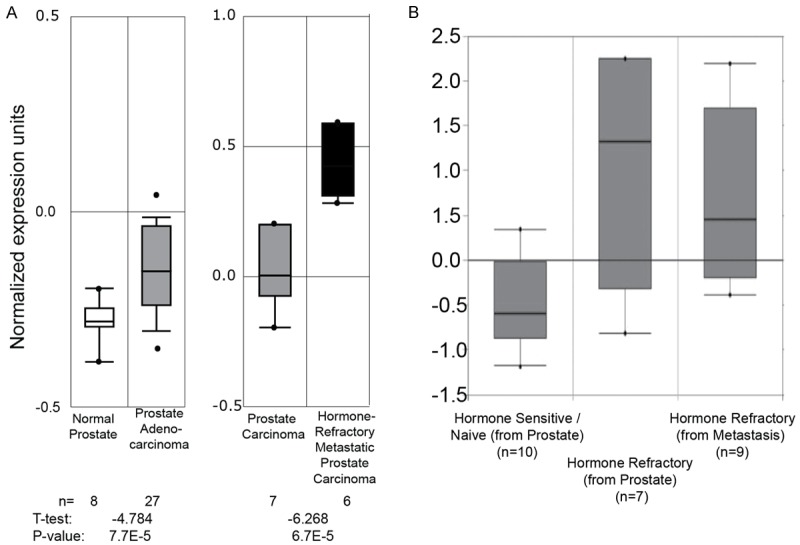
Box-whisker plots representing the normalized TCF3 expression in normal prostate and prostate cancer. A. The Oncomine V4.0 database was searched with “TCF3” and the prostate normal vs. normal data sets were analyzed. The study details and the microarray platform used in the analysis shown are described in. The Oncomine V4.0 database was searched with “TCF3” and the prostate normal vs. cancer (left panel) [38] and cancer vs. cancer (right panel) [39] data sets were analyzed. The number (n) of samples used in each analysis is shown. The statistical significance of the differences between each study in terms of T-test and P-value is shown at the bottom of each panel. B. A similar search (Oncomine v4.6) was used to assess TCF3 expression in hormone sensitive vs. hormone refractory (castration resistant) prostate cancer [40].
Interestingly, increased TCF3 expression in hormone refractory prostate adenocarcinoma as compared to hormone sensitive/Naïve prostate cancer was also observed in an independent dataset [40] (Figure 1B, Oncomine v4.5). These results suggested that TCF3 expression is low in the normal prostate, relatively high in hormone naïve prostate cancer and increases significantly in hormone refractory prostate cancer. Since AR activity and expression is central to normal prostate function and during all stages of prostate cancer [41-43] we therefore tested the hypothesis that AR could regulate TCF3 expression differentially in normal prostate and prostate cancer.
TCF3 is upregulated by androgen receptor in prostate cancer
In order to address the role of AR in regulating TCF3, we focused on investigating TCF3 expression in a panel of isogenic androgen receptor positive prostate cell lines LNCaP (AR+ve and hormone sensitive) and C81 (AR+ve but hormone insensitive) [34,44]. Additional prostate cancer cell lines such as PC3 and DU145 were not used in this study since these cell lines lack androgen receptor expression. Moreover, the immortalized normal prostate epithelial cell line RWPE1 expresses low to undetectable level of androgen receptor [45].
The steady state TCF3 transcript levels were significantly higher in androgen independent C81 cells as compared to androgen sensitive LNCaP cells cultured in media containing castrate levels of androgens (5% FBS). The TCF3 expression in both these cell lines was significantly higher that the RWPE1 cells (Figure 2A). The TCF3 expression in LNCaP cells increased significantly in the presence of 10 nm R1881 (4.6 fold ± 0.89, P < 0.001), a synthetic androgen analogue as compared to cells cultured in charcoal stripped FBS (CSFBS, Figure 2B). Casodex, an androgen receptor antagonist significantly attenuated TCF3 expression in the presence of R1881 suggesting an androgen dependent regulation of TCF3 in LNCaP cells (1.8 fold as compared to LNCaP+CSFBS, Figure 2B). No significant differences were observed in TCF3 expression in C81 cells cultured in the presence or absence of R1881 ± Casodex suggesting a loss of androgen dependent regulatory mechanism (Figure 2B). The changes in the protein levels of TCF3 in LNCaP and C81 cell lines in the presence or absence of R1881 also mimicked to those observed at the transcript level (Figure 2C), suggesting that the regulation of TCF3 by androgens is at the transcript and not at the protein level. Together with the clinical data (Figure 1) and isogenic cell line studies (Figure 2), the results suggested that TCF3 is regulated by androgens in hormone sensitive/naïve cells (e.g. LNCaP) but is constitutively expressed in hormone refractory (e.g. C-81) cells. These results led us to speculate that TCF3 promoter could be regulated by androgen receptor possibly through a putative androgen response element.
Figure 2.
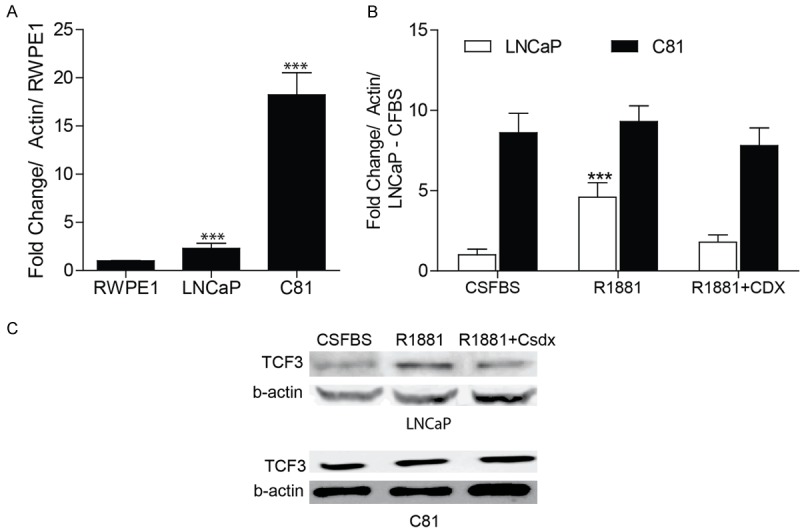
TCF3 expression in prostate cancer cell lines. A. Real time quantitative PCR (qPCR) based expression of TCF3 in immortalized normal prostate epithelial cell line (RWPE1), and isogenic prostate cancer cell lines LNCaP (androgen sensitive) and C81 (androgen insensitive). The results shown are fold change as compared to RWPE1 (normalized to actin). The data is mean + SEM of 3 experiments in triplicate (***P < 0.001). B. QPCR based TCF3 expression in LNCaP and C81 cells treated with synthetic androgen R1881 (10 nM, 24 hrs) alone or in the presence of anti-androgen Casodex (CDX, 30 uM). All treatments were on cell cultured in media containing charcoal stripped fetal bovine serum (CSFBS). The data expressed as fold change (mean + SEM, ***P < 0.001, 3 experiments in triplicate) was first normalized to actin and then to TCF3 expression in LNCaP cells cultured in CS+FBS alone. C. Immunoblot analysis of TCF3 expression in LNCaP and C81 cells in response to various treatments as indicated above. Beta actin (-actin) was used as loading control. The blot is representative of 3 experiments.
TCF3 gene contains consensus Androgen Response Elements (ARE)
The presence of a consensus ARE [46-49] in the TCF3 promoter/intron was discovered through the ConSite [50] (a web based tool that finds cis-regulatory elements in genomic sequences). Two putative ARE sequences were found to be present in close proximity of each other located in the first intron of the TCF3 gene (Figure 3A). These two consensus ARE regions were termed as ARE1 and ARE2. ARE1 and ARE2 are in the first intron of the E12 transcript but upstream of the first exon (and 5’UTR) in E47 due to alternative splicing (Figure 3B). The alternative splicing maintains the reading frame because translational initiation sites for both E12 and E47 are similar.
Figure 3.
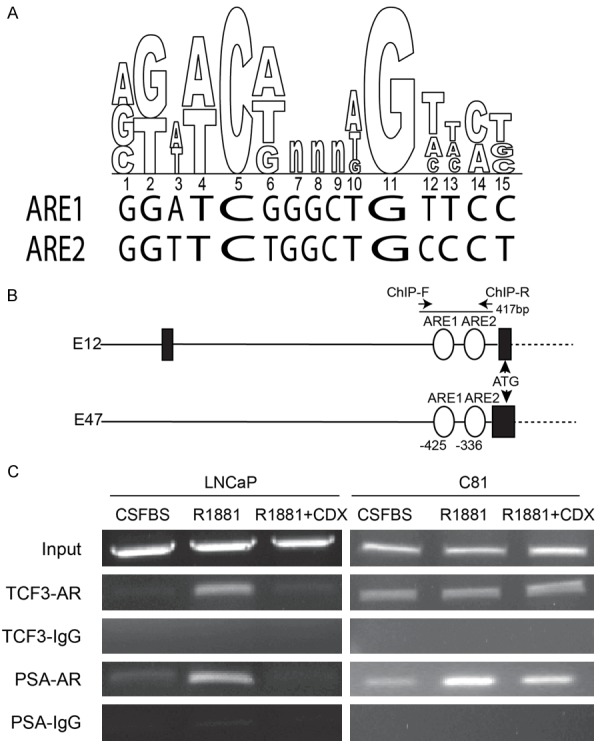
Consensus Androgen response element (ARE) on TCF3 binds androgen receptor. A. Two consensus androgen response elements ARE1 and ARE2 were found on the TCF3 promoter/intron (aligned with the consensus ARE sequence in upper panel). B. Schematic of the TCF3 gene showing the locations of ARE1 and ARE2, relative to the translational start site (ATG). The black bars and lines are exons and introns respectively in TCF3 splice variants E12 and E47. C. Chromatin immunoprecipitation (ChIP) demonstrating the binding of androgen receptor to the 417 bp fragment spanning both ARE1 and ARE2 on TCF3 promoter/intron and the known ARE on PSA promoter. The cells were treated as indicated (CSFBS: Charcoal stripped Fetal Bovine serum, R1881 (10 nM) CDX: Casodex (30 uM). ChIP was performed with androgen receptor (AR) antibody and non-immune rabbit IgG (IgG). The PCR is representative of 3 experiments.
The TCF3 ARE1 and ARE2 bind androgen receptor (AR)
Chromatin immuno-precipitation (ChIP) with androgen receptor (AR) antibody was performed to confirm whether AR is recruited on ARE1 and ARE2 sites in the TCF3 promoter/intron in presence or absence of R1881. Similar experiments were performed on the known ARE site on the PSA promoter [37]. PSA expression is androgen regulated in LNCaP cells that is dependent upon the binding of androgen receptor on the well-established ARE site on the PSA promoter, an effect that is reversed by the anti-androgen Casodex [51,52]. Analogous to TCF3, C81 cells also express PSA at levels significantly higher than LNCaP cells when cultured in media with castration level of androgens [53], possibly due to de novo androgen synthesis [54].
The AR ChIP was performed on LNCaP and C81 cells treated with R1881 as well as R1881 plus Casodex. A single set of ChIP Primers were designed flanking both the ARE1 and ARE2 sites (Figure 3B). In LNCaP cells, binding of AR to these ARE sites was found only in the presence of R1881 whereas no enrichment was observed in presence of AR antagonist Casodex, suggesting specificity of AR binding (Figure 3C). Interestingly, constitutive AR binding was observed irrespective of the treatments in C81 cell lines (Figure 3C). Similar results were observed when PSA, a known AR regulated gene was used as a positive control. Thus the increased AR binding to the ARE sites (ARE1 and ARE2) in TCF3 promoter/intron corresponds with expression in LNCaP and C81 cells (Figure 2).
ARE1 but not ARE2 regulates AR dependent basal expression
The role of ARE1 and ARE2 in regulating transcription of the TCF3 promoter/intron was further investigated in a luciferase reporter assay system. ARE1 and ARE2 either alone (ARE1 or ARE2) or together (ARE1+ARE2) were cloned upstream of a minimum promoter in a luciferase reporter plasmid (Figure 4A). The cloned reporter luciferase constructs were transiently co-transfected with Renilla luciferase in LNCaP and C81 cells followed by treatment of cells with R1881, R1881+CDX or left untreated (CSFBS only, control). The luciferase activity was measured after 24 hrs of treatment and results were normalized to Renilla Luciferase. In untreated cells (CSFBS) ARE1+ARE2 and ARE1 alone resulted in at least 2 fold increases in luciferase activity as compared to the ARE2 alone in LNCaP cells (Figure 4C). The magnitude of expression of luciferase in ARE1+ARE2 and ARE1 alone was at least 10 fold higher in C81 cells as compared to LNCaP cells cultured in CSFBS alone (Figure 4C). These results suggested that 1) ARE1 but not ARE2 is the primary androgen response element in the TCF3 promoter/intron and 2) ARE1 alone promotes constitutive luciferase activity in C81 cells which is consistent with higher basal expression (in CSFBS) of TCF3 in these cells (Figure 2). In response to R1881, a significant increase in luciferase activity was observed in LNCaP cells transfected with ARE1+ARE2 or ARE1 alone (< 6 fold, P < 0.001) as compared to ARE2 Figure 4C). Furthermore, the R1881 dependent increase in ARE1+ARE2 or ARE1 luciferase activity in LNCaP cells was significantly attenuated in the presence of the anti-androgen Casodex (Figure 4C). Together, these results suggested that that ARE1 (or ARE1+ARE2) luciferase activity in LNCaP cells is strictly dependent upon ligand dependent activation and binding of androgen response element to ARE1. These results are consistent with the AR ChIP assay shown in Figure 3C that demonstrated ligand dependent AR binding on ARE1+ARE2 that was sensitive to Casodex in LNCaP cells. No significant difference between ARE1 (and ARE1+ARE2) luciferase activity in untreated or R1881 treated C81 cells suggested a constitutively active androgen receptor (Figure 4D). Interestingly, no difference was observed between the ARE1 (and ARE1+ARE2) luciferase activity in R1881 versus R1881+CDX treated C81 cells suggesting resistance to anti-androgen CDX. The resistance to antiandrogen in androgen insensitive cells such as C4-2 was also demonstrated in earlier studies [55]. Similar results, demonstrating ligand independent and anti-androgen insensitive binding of AR to ARE1+ARE2 was also observed in ChIP assays (Figure 3C).
Figure 4.
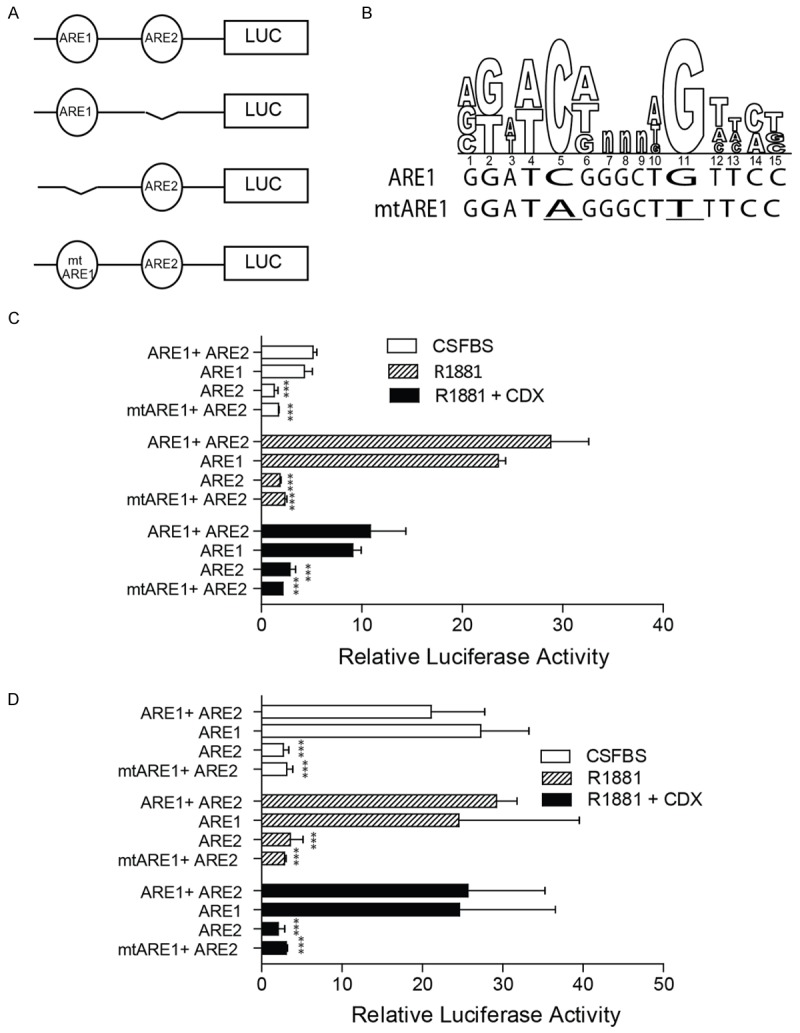
Transcriptional activity of ARE1 and ARE2 measured in terms of luciferase reporter activity in LNCaP and C81 cells. (A) ARE1 and ARE2 either together (ARE1+ARE2), alone (ARE1 or ARE2) or mutant ARE1 (mtARE1) was cloned upstream of a minimal promoter luciferase reporter plasmid. (B) The mutant ARE1 was created by mutating the conserved “C” and G” in the ARE to “A” and “T” respectively (underlined). (C and D) The reporter plasmids were transiently transfected in LNCaP (C) or C81 (D) cells cultured in media containing CFBS and treated as indicated. The cells were also co-transfected with Renilla luciferase used as a transfection control. The luciferase activity measured 24 hrs after transfection was normalized to Renilla luciferase and expressed as relative luciferase activity. The data is expressed as mean ± SEM of 3 experiments performed in quadruplicates. The t-test significance (***P < 0.001) is measured against the activity of ARE1+ARE2 within respective treatment groups.
Mutations of conserved sites within ARE1 were used to further confirm that ARE1 contributes to the majority of AR dependent luciferase activity (Figure 4B). The mutant ARE1 in ARE1+ARE2 (mtARE1+ARE2) significantly attenuated Ligand dependent (LNCaP, Figure 4C) and ligand independent (C81) luciferase activity (Figure 4D). These results indicated that it is only the ARE1 which is functional and not the ARE2. Collectively, the results demonstrated that in LNCaP the activation of ARE1 was ligand dependent whereas in case of C81 the ARE1 was activity was ligand independent possibly due to constitutive binding of AR to ARE1 (Figure 3C).
TCF3 expression is increased in mouse model of Prostatic Intra-epithelial neoplasia (PIN)
The Id4-/-mouse was shown earlier to develop PIN lesions as early as 6 weeks of age post-partum [56]. The increase in the number of PIN lesions in Id4-/-mice was associated with persistent AR expression, increased expression of c-Myc, Id1, Ki-67 and p-Akt, decreased expression NKX3.1 and Pten, generally associated with prostate cancer progression, as compared to wild type counterparts [56]. We used this well established mouse PIN model to investigate TCF3 expression. Interestingly, TCF3 expression was essentially un-detectable in wild type prostates (Figure 5A1) except some cells (black arrowheads) whereas high TCF3 expression was observed in the prostates of Id4-/-mice (Figure 5A2). The TCF3 expression in Id4-/-mice prostate was essentially nuclear (Figure 5A2) that is consistent with primarily nuclear TCF3 expression in early stages of prostate cancer [29]. These results clearly establish that increased TCF3 expression is an early event in prostate cancer initiation.
Figure 5.
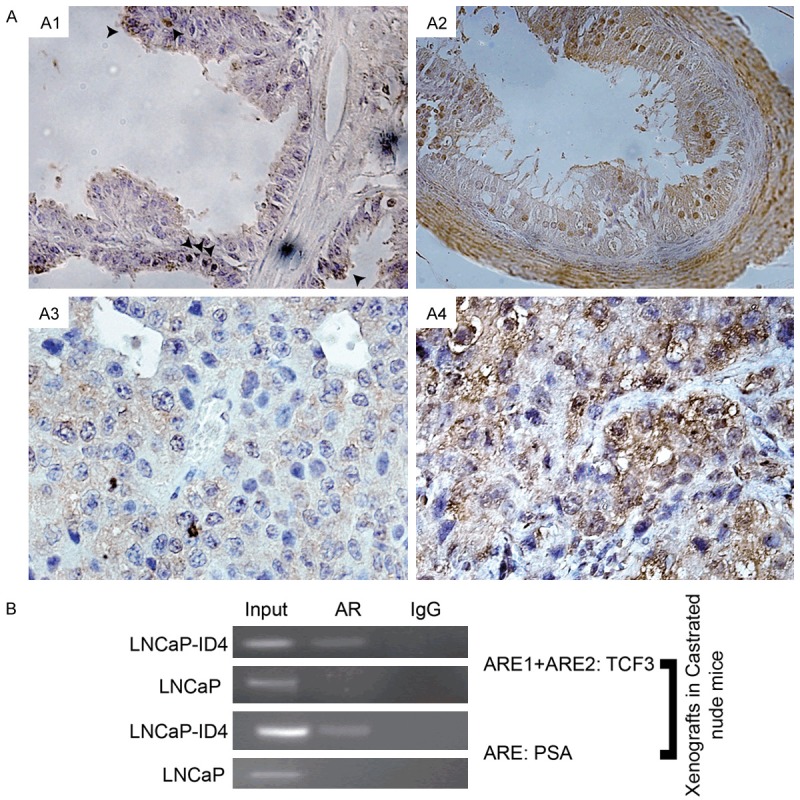
Increased TCF3 expression in Id4 null mice model and LNCaP xenografts. A. tcf3 expression (brown staining) is rarely observed in the prostate epithelial cells in wild type mice (A1: Arrow heads indicate tcf3 expressing cells) as compared to widespread expression in Id4-/-null mice (A2). Small and rare LNCaP xenografts from castrated mice (A3) have undetectable levels of TCF3 whereas xenografts from LNCaP cells lacking ID4 (A4, LNCaP-ID4) which form large tumors express high levels of TCF3. Representative image from 3 different xenografts is shown. B. ChIP assay demonstrating increased binding of AR on the 417 bp region of TCF3 promoter/intron spanning ARE1 and ARE2 in LNCaP-ID4 xenografts as compared to LNCaP xenografts (Top panel). The bottom panel shows the binding of AR to the ARE on PSA promoter in LNCaP and LNCaP-ID4 promoter. Note that AR binding was not observed on TCF3 ARE1+ARE2 region and PSA ARE in LNCaP cells possibly due to lack of circulating androgens. Representative data from 3 experiments is shown.
TCF3 expression is increased in castration resistant LNCaP xenograft model
We recently reported a new castration resistant LNCaP cell model [35]. The LNCaP cells lacking Id4 (LNCaP(-)ID4) forms tumors in castrated mice as compared to the parental LNCaP cells, which rarely forms tumors in castrated mice [35]. The LNCaP(-)ID4 tumor xenografts from castrated mice were used to investigate whether TCF3 expression is constitutive in this castration resistance model. Interestingly, TCF3 expression was absent in the tumors derived from parental LNCaP cells in castrated mice (Figure 5A3) suggesting that circulating androgens are required for TCF3 expression in LNCaP cells. Surprisingly, a robust TCF3 expression was observed in LNCaP(-)ID4 tumors in castrated mice (Figure 5A4). These studies essentially confirmed our results described above that TCF3 expression is androgen dependent in LNCaP cells but transitions and androgen -independent in castration resistant LNCaP(-)ID4 cells in vivo.
The ChIP using AR antibody on LNCaP and LNCaP(-)ID4 xenograft tissue demonstrated significant binding of AR to ARE1+ARE2 fragment as compared to LNCaP xenografts (Figure 5B). Similar results were obtained with the ARE site on the PSA promoter. One of the mechanisms to support these observations could the gain of de novo steroidogenesis as shown earlier in these xenografts [35] resulting in a constitutively active AR. Nevertheless these results suggest that increased TCF3 expression continues to be AR dependent even in castration resistance prostate cancer.
Discussion
TCF3 spliced products E12 and E47 are multifunctional transcriptional factors that can act as tumor suppressors and tumor promoters. Meta-analysis (Oncomine, TCF3) and experimental studies demonstrated increased TCF3 expression in many cancers [25,26] including prostate [29]. Promoting EMT via direct transcriptional repression of E-cadherin is perhaps the most well-established mechanism of action of TCF3 in tumorigenesis [22,27,28].
In this study we present compelling evidence that TCF3 is regulated by androgen receptor in prostate cancer. Based on meta-analysis, prostate cancer isogenic cell line transition (LNCaP and C-81), and in vivo castration resistant (LNCaP(-)ID4) models, a strong correlation between TCF3 expression during transition of prostate cancer from hormone naïve to castration independent disease was observed. This expression profile was further explored to investigate whether TCF3 could be regulated by androgen receptor in transition of prostate cancer to castration resistance. The up-regulation of TCF3 transcript by androgens in hormone sensitive LNCaP cells and subsequent down regulation by the anti-androgen Casodex suggests that TCF3 regulation is indeed at the transcriptional level.
A functional androgen receptor response element, ARE1 appears to direct androgen dependent transcription of TCF3 in LNCaP cells. The ARE1 is 1459 base pairs downstream of the transcriptional start site in intron 1 of E12. In E47, ARE1 is -425 bp upstream of the transcriptional start site. ARE1 in a minimal promoter luciferase reporter construct was sufficient to drive luciferase expression. Mutations and/or loss of ARE1 but not ARE2 were sufficient to attenuate luciferase reporter activity. Chromatin immunoprecipitation studies also revealed androgen dependent binding of Androgen receptor on the fragment containing both ARE1 and ARE2. However, given the close proximity of ARE1 and ARE2, we were unable to identify the specific response element to which AR was bound. It is anticipated that AR bounds specifically to ARE1 but not ARE2, based on the results demonstrating the activity of ARE1 in luciferase reporter assays, however, the binding of AR to ARE2 cannot be ruled out.
The regulation of TATA less TCF3 gene by androgen receptor is not surprising. Many other TATA less genes such as Sterol Regulatory Element-binding Protein Cleavage-activating Protein (SREBP-SCAP) (ARE in intron 8 [57]), GNMT (ARE in exon 1 [58]), murine Pem homeobox gene [59] and androgen receptor itself [60] are also regulated by androgen receptor in an androgen dependent manner.
Transition of prostate cancer from hormone naïve to castration resistance is a complex process that involves multiple mechanisms including altered growth factor signaling and protein phosphorylation [61]. However, central to this process of transition is androgen receptor [41,43,62,63]. Increased stability [64] and mutations [65] that broaden AR ligand specificity promotes AR activity in castration resistant prostate cancer. Recent studies also suggest that de novo steroidogenesis by prostate cancer cells is sufficient to drive androgen dependent gene expression in castration resistant prostate cancer [66]. Therefore it is not surprising that in androgen dependent LNCaP cells ARE1/ARE2 was occupied only after activation of AR by the ligand R1881. The C81 cells which are ligand independent in part due to gain in de novo steroidogenesis [54], ARE1/2 was constitutively bound by AR. Thus the binding of AR to ARE1/2 site on the TCF3 promoter/intron is consistent with TCF3 expression in the isogenic LNCaP and C81 cell line prostate cancer transition model. Our results therefore strongly suggest that the androgen regulated TCF3 is a prostate cancer associated gene which is constitutively expressed in castration resistant prostate cancer. These results are consistent with other studies that demonstrated constitutive expression of androgen regulated genes in castration resistant prostate cancer [67].
The strict androgen dependent regulation of TCF3 is also reflected in the mouse xenograft models. The androgen dependent parental LNCaP cells rarely formed tumors in castrated mice but the ones that do form small tumors did not express any detectable TCF3 suggesting that TCF3 expression is indeed androgen dependent. The LNCaP cells lacking ID4 (LNCaP(-)ID4) formed large tumors in castrated mice and expressed significantly higher levels of TCF3, possibly due to gain of de novo steroidogenic capacity [35]. The androgen insensitive LNCaP(-)ID4 cells also retain PSA expression both in vitro and in vivo (androgen deprived conditions) suggesting a constitutively active androgen receptor [35]. The strong correlation between TCF3 expression and binding of AR to ARE1/ARE2 in LNCaP-ID4 but not on LNCaP cells clearly establishes the role of AR in regulating TCF3 in prostate cancer.
The ID4 knockout mice which results in PIN lesions as reported earlier also expresses TCF3 at significantly higher levels than the wild type mice. The increase in TCF3 in Id4-/-mice is also consistent with the expression of the tumor promoter Id1 which is increased in Id4-/-mice [56] and in castration resistant prostate cancer [67]. These results suggest that the TCF3 promoter/intron shifts from being transcriptionally silent to androgen responsive and transcriptionally active as normal prostate epithelial cells acquire cancerous phenotype.
In silico analysis suggests that the TCF3 promoter/intron is not enriched in CpG islands (UCSC genome browser) hence cancer specific promoter hypomethylation does not appear to be the mechanism involved in the regulation of TCF3 in prostate cancer. The lack of this mechanism is reflected in the TCGA prostate adenocarcinoma DNA methylation (TCGA PRAD methylation 450K, n = 549) profile which is similar between adjacent normal and prostate adenocarcinoma samples (data not shown). These observations suggest that the transcriptional silencing of TCF3 in the normal prostate without promoter DNA hyper-methylation could involve histone modifications only (hypo-acetylation and hypermethylation). For example, hypo-acetylation only without DNA methylation is involved in transcriptional silencing of CDKN1A in HT-29 [68] and T24 cells [69]. Similarly ID1 is transcriptionally inactive without DNA methylation in AML cells [70]. An alternate mechanism may involve the recruitment of transcriptional repressor complex on the TCF3 promoter that may lead its transcriptional silencing, similar to the down-regulation of ID1 in non-transformed mammary epithelial cells. In these cells, ID1 is down-regulated in part due to recruitment of NF-1/Rb/HDAC-1 repressor complexes [71]. Loss of this repressor complex results in high and constitutive expression of ID1 in poorly differentiated and highly metastatic breast cancer cells. Both these mechanisms could be inter-dependent because recruitment of HDAC1 as part of the repressor complex could result in hypo-acetylation, eventually leading to transcriptional silencing. Whether such a mechanism regulates TCF3 expression in normal prostate epithelial cells vs. prostate cancer cells remains to be investigated but the net outcome at least in the prostate cancer cells is the gain of TCF3 transcriptional activity.
Conclusions
In conclusion, we provide compelling evidence that increased TCF3 expression is associated with progression of prostate cancer to hormone insensitive/castration resistance phenotype. At the mechanistic level, increase in TCF3 is in part due to constitutive binding of androgen receptor to the newly identified androgen response element in the TCF3 promoter/intron. We believe that this is the first study to demonstrate the regulation of TCF3 gene by androgens at the transcriptional level. The regulatory pathways involved in transcriptional silencing of TCF3 in the normal prostate and subsequent activation in prostate cancer could play a key role in transition/initiation of prostate cancer.
Acknowledgements
The work was supported by NIH/NCI CA128914 (JC) and in part by NIH/NCRR/RCMI G12MD007590 (CCRTD).
Disclosure of conflict of interest
None.
Authors’ contribution
DP: performed the cell based assays site directed mutagenesis, discovered ARE1/2, cloning, transfections and IHC and wrote the first draft; SC: performed ChIP analysis on xenograft tissues; JC: conceived the study and edited the final draft.
References
- 1.Murre C, McCaw PS, Baltimore D. A new DNA binding and dimerization motif in immunoglobulin enhancer binding, daughterless, MyoD, and myc proteins. Cell. 1989;56:777–783. doi: 10.1016/0092-8674(89)90682-x. [DOI] [PubMed] [Google Scholar]
- 2.Henthorn P, Kiledjian M, Kadesch T. Two distinct transcription factors that bind the immunoglobulin enhancer microE5/kappa 2 motif. Science. 1990;247:467–470. doi: 10.1126/science.2105528. [DOI] [PubMed] [Google Scholar]
- 3.Sun XH, Baltimore D. An inhibitory domain of E12 transcription factor prevents DNA binding in E12 homodimers but not in E12 heterodimers. Cell. 1991;64:459–470. doi: 10.1016/0092-8674(91)90653-g. [DOI] [PubMed] [Google Scholar]
- 4.Shirakata M, Paterson BM. The E12 inhibitory domain prevents homodimer formation and facilitates selective heterodimerization with the MyoD family of gene regulatory factors. Embo J. 1995;14:1766–1772. doi: 10.1002/j.1460-2075.1995.tb07165.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Massari ME, Jennings PA, Murre C. The AD1 transactivation domain of E2A contains a highly conserved helix which is required for its activity in both Saccharomyces cerevisiae and mammalian cells. Mol Cell Biol. 1996;16:121–129. doi: 10.1128/mcb.16.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Massari ME, Murre C. Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol Cell Biol. 2000;20:429–440. doi: 10.1128/mcb.20.2.429-440.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Denis CM, Langelaan DN, Kirlin AC, Chitayat S, Munro K, Spencer HL, LeBrun DP, Smith SP. Functional redundancy between the transcriptional activation domains of E2A is mediated by binding to the KIX domain of CBP/p300. Nucleic Acids Res. 2014;42:7370–7382. doi: 10.1093/nar/gku206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo C, Hu Q, Yan C, Zhang J. Multivalent binding of the ETO corepressor to E proteins facilitates dual repression controls targeting chromatin and the basal transcription machinery. Mol Cell Biol. 2009;29:2644–2657. doi: 10.1128/MCB.00073-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Slattery C, Ryan MP, McMorrow T. E2A proteins: regulators of cell phenotype in normal physiology and disease. Int J Biochem Cell Biol. 2008;40:1431–1436. doi: 10.1016/j.biocel.2007.05.014. [DOI] [PubMed] [Google Scholar]
- 10.Zhuang Y, Kim CG, Bartelmez S, Cheng P, Groudine M, Weintraub H. Helix-loop-helix transcription factors E12 and E47 are not essential for skeletal or cardiac myogenesis, erythropoiesis, chondrogenesis, or neurogenesis. Proc Natl Acad Sci U S A. 1992;89:12132–12136. doi: 10.1073/pnas.89.24.12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kee BL, Quong MW, Murre C. E2A proteins: essential regulators at multiple stages of B-cell development. Immunol Rev. 2000;175:138–149. [PubMed] [Google Scholar]
- 12.Bain G, Maandag EC, Izon DJ, Amsen D, Kruisbeek AM, Weintraub BC, Krop I, Schlissel MS, Feeney AJ, van Roon M, et al. E2A proteins are required for proper B cell development and initiation of immunoglobulin gene rearrangements. Cell. 1994;79:885–892. doi: 10.1016/0092-8674(94)90077-9. [DOI] [PubMed] [Google Scholar]
- 13.Ramirez J, Lukin K, Hagman J. From hematopoietic progenitors to B cells: mechanisms of lineage restriction and commitment. Curr Opin Immunol. 2010;22:177–184. doi: 10.1016/j.coi.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Engel I, Murre C. Ectopic expression of E47 or E12 promotes the death of E2A-deficient lymphomas. Proc Natl Acad Sci U S A. 1999;96:996–1001. doi: 10.1073/pnas.96.3.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peverali FA, Ramqvist T, Saffrich R, Pepperkok R, Barone MV, Philipson L. Regulation of G1 progression by E2A and Id helix-loop-helix proteins. Embo J. 1994;13:4291–4301. doi: 10.1002/j.1460-2075.1994.tb06749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Prabhu S, Ignatova A, Park ST, Sun XH. Regulation of the expression of cyclin-dependent kinase inhibitor p21 by E2A and Id proteins. Mol Cell Biol. 1997;17:5888–5896. doi: 10.1128/mcb.17.10.5888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang A, Zhao H, Quan Y, Jin R, Feng B, Zheng M. E2A predicts prognosis of colorectal cancer patients and regulates cancer cell growth by targeting miR-320a. PLoS One. 2014;9:e85201. doi: 10.1371/journal.pone.0085201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pagliuca A, Gallo P, De Luca P, Lania L. Class A helix-loop-helix proteins are positive regulators of several cyclin-dependent kinase inhibitors’ promoter activity and negatively affect cell growth. Cancer Res. 2000;60:1376–1382. [PubMed] [Google Scholar]
- 19.Roberts VJ, Steenbergen R, Murre C. Localization of E2A mRNA expression in developing and adult rat tissues. Proc Natl Acad Sci U S A. 1993;90:7583–7587. doi: 10.1073/pnas.90.16.7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rutherford MN, LeBrun DP. Restricted expression of E2A protein in primary human tissues correlates with proliferation and differentiation. Am J Pathol. 1998;153:165–173. doi: 10.1016/S0002-9440(10)65557-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao F, Vilardi A, Neely RJ, Choi JK. Promotion of cell cycle progression by basic helix-loop-helix E2A. Mol Cell Biol. 2001;21:6346–6357. doi: 10.1128/MCB.21.18.6346-6357.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hwang-Verslues WW, Chang PH, Wei PC, Yang CY, Huang CK, Kuo WH, Shew JY, Chang KJ, Lee EY, Lee WH. miR-495 is upregulated by E12/E47 in breast cancer stem cells, and promotes oncogenesis and hypoxia resistance via downregulation of E-cadherin and REDD1. Oncogene. 2011;30:2463–2474. doi: 10.1038/onc.2010.618. [DOI] [PubMed] [Google Scholar]
- 23.Cubillo E, Diaz-Lopez A, Cuevas EP, Moreno-Bueno G, Peinado H, Montes A, Santos V, Portillo F, Cano A. E47 and Id1 interplay in epithelial-mesenchymal transition. PLoS One. 2013;8:e59948. doi: 10.1371/journal.pone.0059948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Castro Alves C, Rosivatz E, Schott C, Hollweck R, Becker I, Sarbia M, Carneiro F, Becker KF. Slug is overexpressed in gastric carcinomas and may act synergistically with SIP1 and Snail in the down-regulation of E-cadherin. J Pathol. 2007;211:507–515. doi: 10.1002/path.2138. [DOI] [PubMed] [Google Scholar]
- 25.Krishnamachary B, Zagzag D, Nagasawa H, Rainey K, Okuyama H, Baek JH, Semenza GL. Hypoxia-inducible factor-1-dependent repression of E-cadherin in von Hippel-Lindau tumor suppressor-null renal cell carcinoma mediated by TCF3, ZFHX1A, and ZFHX1B. Cancer Res. 2006;66:2725–2731. doi: 10.1158/0008-5472.CAN-05-3719. [DOI] [PubMed] [Google Scholar]
- 26.Kim J, Hong SJ, Park JY, Park JH, Yu YS, Park SY, Lim EK, Choi KY, Lee EK, Paik SS, Lee KG, Wang HJ, Do IG, Joh JW, Kim DS. Epithelialmesenchymal transition gene signature to predict clinical outcome of hepatocellular carcinoma. Cancer Sci. 2010;101:1521–1528. doi: 10.1111/j.1349-7006.2010.01536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peinado H, Marin F, Cubillo E, Stark HJ, Fusenig N, Nieto MA, Cano A. Snail and E47 repressors of E-cadherin induce distinct invasive and angiogenic properties in vivo. J Cell Sci. 2004;117:2827–2839. doi: 10.1242/jcs.01145. [DOI] [PubMed] [Google Scholar]
- 28.Perez-Moreno MA, Locascio A, Rodrigo I, Dhondt G, Portillo F, Nieto MA, Cano A. A new role for E12/E47 in the repression of E-cadherin expression and epithelial-mesenchymal transitions. J Biol Chem. 2001;276:27424–27431. doi: 10.1074/jbc.M100827200. [DOI] [PubMed] [Google Scholar]
- 29.Patel D, Chaudhary J. Increased expression of bHLH transcription factor E2A (TCF3) in prostate cancer promotes proliferation and confers resistance to doxorubicin induced apoptosis. Biochem Biophys Res Commun. 2012;422:146–151. doi: 10.1016/j.bbrc.2012.04.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun L, Trausch-Azar JS, Ciechanover A, Schwartz AL. E2A protein degradation by the ubiquitin-proteasome system is stage-dependent during muscle differentiation. Oncogene. 2007;26:441–448. doi: 10.1038/sj.onc.1209793. [DOI] [PubMed] [Google Scholar]
- 31.Huggins GS, Chin MT, Sibinga NE, Lee SL, Haber E, Lee ME. Characterization of the mUBC9-binding sites required for E2A protein degradation. J Biol Chem. 1999;274:28690–28696. doi: 10.1074/jbc.274.40.28690. [DOI] [PubMed] [Google Scholar]
- 32.Norton JD. ID helix-loop-helix proteins in cell growth, differentiation and tumorigenesis. J Cell Sci. 2000;113:3897–3905. doi: 10.1242/jcs.113.22.3897. [DOI] [PubMed] [Google Scholar]
- 33.Hata K, Mizuguchi J. Genomic organization and characterization of the promoter for the E2A gene. Gene. 2004;325:53–61. doi: 10.1016/j.gene.2003.09.040. [DOI] [PubMed] [Google Scholar]
- 34.Lin MF, Meng TC, Rao PS, Chang C, Schonthal AH, Lin FF. Expression of human prostatic acid phosphatase correlates with androgenstimulated cell proliferation in prostate cancer cell lines. J Biol Chem. 1998;273:5939–5947. doi: 10.1074/jbc.273.10.5939. [DOI] [PubMed] [Google Scholar]
- 35.Patel D, Knowell AE, Korang-Yeboah M, Sharma P, Joshi J, Glymph S, Chinaranagari S, Nagappan P, Palaniappan R, Bowen NJ, Chaudhary J. Inhibitor of differentiation 4 (ID4) inactivation promotes de novo steroidogenesis and castration resistant prostate cancer. Mol Endocrinol. 2014;28:1239–53. doi: 10.1210/me.2014-1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knowell A, Patel D, Morton D, Sharma P, Glymph S, Chaudhary J. Id4 dependent acetylation restores mutant-p53 transcriptional activity. Molecular Cancer. 2013;12:161. doi: 10.1186/1476-4598-12-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Louie MC, Yang HQ, Ma AH, Xu W, Zou JX, Kung HJ, Chen HW. Androgen-induced recruitment of RNA polymerase II to a nuclear receptor-p160 coactivator complex. Proc Natl Acad Sci U S A. 2003;100:2226–2230. doi: 10.1073/pnas.0437824100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vanaja DK, Cheville JC, Iturria SJ, Young CY. Transcriptional silencing of zinc finger protein 185 identified by expression profiling is associated with prostate cancer progression. Cancer Res. 2003;63:3877–3882. [PubMed] [Google Scholar]
- 39.Varambally S, Yu J, Laxman B, Rhodes DR, Mehra R, Tomlins SA, Shah RB, Chandran U, Monzon FA, Becich MJ, Wei JT, Pienta KJ, Ghosh D, Rubin MA, Chinnaiyan AM. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell. 2005;8:393–406. doi: 10.1016/j.ccr.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 40.Tamura K, Furihata M, Tsunoda T, Ashida S, Takata R, Obara W, Yoshioka H, Daigo Y, Nasu Y, Kumon H, Konaka H, Namiki M, Tozawa K, Kohri K, Tanji N, Yokoyama M, Shimazui T, Akaza H, Mizutani Y, Miki T, Fujioka T, Shuin T, Nakamura Y, Nakagawa H. Molecular Features of Hormone-Refractory Prostate Cancer Cells by Genome-Wide Gene Expression Profiles. Cancer Res. 2007;67:5117–5125. doi: 10.1158/0008-5472.CAN-06-4040. [DOI] [PubMed] [Google Scholar]
- 41.Culig Z, Bartsch G. Androgen axis in prostate cancer. J Cell Biochem. 2006;99:373–81. doi: 10.1002/jcb.20898. [DOI] [PubMed] [Google Scholar]
- 42.Culig Z, Klocker H, Bartsch G, Hobisch A. Androgen receptors in prostate cancer. Endocr Relat Cancer. 2002;9:155–170. doi: 10.1677/erc.0.0090155. [DOI] [PubMed] [Google Scholar]
- 43.Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev. 2004;25:276–308. doi: 10.1210/er.2002-0032. [DOI] [PubMed] [Google Scholar]
- 44.Karan D, Kelly DL, Rizzino A, Lin MF, Batra SK. Expression profile of differentially-regulated genes during progression of androgen-independent growth in human prostate cancer cells. Carcinogenesis. 2002;23:967–975. doi: 10.1093/carcin/23.6.967. [DOI] [PubMed] [Google Scholar]
- 45.Bennett NC, Hooper JD, Johnson DW, Gobe GC. Expression profiles and functional associations of endogenous androgen receptor and caveolin-1 in prostate cancer cell lines. Prostate. 2014;74:478–487. doi: 10.1002/pros.22767. [DOI] [PubMed] [Google Scholar]
- 46.Lin B, Ferguson C, White JT, Wang S, Vessella R, True LD, Hood L, Nelson PS. Prostatelocalized and androgen-regulated expression of the membrane-bound serine protease TMPRSS2. Cancer Res. 1999;59:4180–4184. [PubMed] [Google Scholar]
- 47.Roche PJ, Hoare SA, Parker MG. A consensus DNA-binding site for the androgen receptor. Mol Endocrinol. 1992;6:2229–2235. doi: 10.1210/mend.6.12.1491700. [DOI] [PubMed] [Google Scholar]
- 48.Nelson PS, Clegg N, Arnold H, Ferguson C, Bonham M, White J, Hood L, Lin B. The program of androgen-responsive genes in neoplastic prostate epithelium. Proc Natl Acad Sci U S A. 2002;99:11890–11895. doi: 10.1073/pnas.182376299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Asirvatham AJ, Schmidt M, Gao B, Chaudhary J. Androgens regulate the immune/inflammatory response and cell survival pathways in rat ventral prostate epithelial cells. Endocrinology. 2006;147:257–271. doi: 10.1210/en.2005-0942. [DOI] [PubMed] [Google Scholar]
- 50.Sandelin A, Wasserman WW, Lenhard B. ConSite: web-based prediction of regulatory elements using cross-species comparison. Nucleic Acids Res. 2004;32:W249–252. doi: 10.1093/nar/gkh372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fujii Y, Kawakami S, Okada Y, Kageyama Y, Kihara K. Regulation of prostate-specific antigen by activin A in prostate cancer LNCaP cells. Am J Physiol Endocrinol Metab. 2004;286:E927–31. doi: 10.1152/ajpendo.00443.2003. [DOI] [PubMed] [Google Scholar]
- 52.Arnold JT, Liu X, Allen JD, Le H, McFann KK, Blackman MR. Androgen receptor or estrogen receptor-beta blockade alters DHEA-, DHT-, and E(2)-induced proliferation and PSA production in human prostate cancer cells. Prostate. 2007;67:1152–1162. doi: 10.1002/pros.20585. [DOI] [PubMed] [Google Scholar]
- 53.Igawa T, Lin FF, Lee MS, Karan D, Batra SK, Lin MF. Establishment and characterization of androgen-independent human prostate cancer LNCaP cell model. Prostate. 2002;50:222–235. doi: 10.1002/pros.10054. [DOI] [PubMed] [Google Scholar]
- 54.Dillard PR, Lin MF, Khan SA. Androgenindependent prostate cancer cells acquire the complete steroidogenic potential of synthesizing testosterone from cholesterol. Mol Cell Endocrinol. 2008;295:115–120. doi: 10.1016/j.mce.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dehm SM, Tindall DJ. Ligand-independent androgen receptor activity is activation function-2-independent and resistant to antiandrogens in androgen refractory prostate cancer cells. J Biol Chem. 2006;281:27882–27893. doi: 10.1074/jbc.M605002200. [DOI] [PubMed] [Google Scholar]
- 56.Sharma P, Knowell AE, Chinaranagari S, Komaragiri S, Nagappan P, Patel D, Havrda MC, Chaudhary J. Id4 deficiency attenuates prostate development and promotes PIN-like lesions by regulating androgen receptor activity and expression of NKX3.1 and PTEN. Mol Cancer. 2013;12:67. doi: 10.1186/1476-4598-12-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Heemers H, Verrijdt G, Organe S, Claessens F, Heyns W, Verhoeven G, Swinnen JV. Identification of an androgen response element in intron 8 of the sterol regulatory element-binding protein cleavage-activating protein gene allowing direct regulation by the androgen receptor. J Biol Chem. 2004;279:30880–30887. doi: 10.1074/jbc.M401615200. [DOI] [PubMed] [Google Scholar]
- 58.Lee CM, Yen CH, Tzeng TY, Huang YZ, Chou KH, Chang TJ, Arthur Chen YM. Androgen response element of the glycine N-methyltransferase gene is located in the coding region of its first exon. Biosci Rep. 2013:33. doi: 10.1042/BSR20130030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Barbulescu K, Geserick C, Schüttke I, Schleuning WD, Haendler B. New Androgen Response Elements in the Murine Pem Promoter Mediate Selective Transactivation. Mol Endocrinol. 2001;15:1803–1816. doi: 10.1210/mend.15.10.0708. [DOI] [PubMed] [Google Scholar]
- 60.Le Dai J, Maiorino CA, Gkonos PJ, Burnstein KL. Androgenic up-regulation of androgen receptor cDNA expression in androgen-independent prostate cancer cells. Steroids. 1996;61:531–539. doi: 10.1016/s0039-128x(96)00086-4. [DOI] [PubMed] [Google Scholar]
- 61.Nadiminty N, Gao A. Mechanisms of persistent activation of the androgen receptor in CRPC: recent advances and future perspectives. World J Urol. 2012;30:287–295. doi: 10.1007/s00345-011-0771-3. [DOI] [PubMed] [Google Scholar]
- 62.Hodgson MC, Bowden WA, Agoulnik IU. Androgen receptor footprint on the way to prostate cancer progression. World J Urol. 2012;30:279–285. doi: 10.1007/s00345-011-0743-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mohler JL, Gregory CW, Ford OH 3rd, Kim D, Weaver CM, Petrusz P, Wilson EM, French FS. The androgen axis in recurrent prostate cancer. Clin Cancer Res. 2004;10:440–448. doi: 10.1158/1078-0432.ccr-1146-03. [DOI] [PubMed] [Google Scholar]
- 64.Gregory CW, Johnson RT Jr, Mohler JL, French FS, Wilson EM. Androgen receptor stabilization in recurrent prostate cancer is associated with hypersensitivity to low androgen. Cancer Res. 2001;61:2892–2898. [PubMed] [Google Scholar]
- 65.Seton-Rogers S. Therapeutic resistance: Two steps ahead. Nat Rev Cancer. 2013;13:382–383. doi: 10.1038/nrc3532. [DOI] [PubMed] [Google Scholar]
- 66.Mitsiades N, Sung CC, Schultz N, Danila DC, He B, Eedunuri VK, Fleisher M, Sander C, Sawyers CL, Scher HI. Distinct Patterns of Dysregulated Expression of Enzymes Involved in Androgen Synthesis and Metabolism in Metastatic Prostate Cancer Tumors. Cancer Res. 2012;72:6142–6152. doi: 10.1158/0008-5472.CAN-12-1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Decker KF, Zheng D, He Y, Bowman T, Edwards JR, Jia L. Persistent androgen receptormediated transcription in castration-resistant prostate cancer under androgen-deprived conditions. Nucleic Acids Res. 2012;40:10765–10779. doi: 10.1093/nar/gks888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Archer SY, Meng S, Shei A, Hodin RA. p21 (WAF1) is required for butyrate-mediated growth inhibition of human colon cancer cells. Proc Natl Acad Sci U S A. 1998;95:6791–6796. doi: 10.1073/pnas.95.12.6791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Histone deacetylase inhibitor selectively induces p21WAF1 expression and geneassociated histone acetylation. Proc Natl Acad Sci U S A. 2000;97:10014–10019. doi: 10.1073/pnas.180316197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yu WP, Scott SA, Dong WF. Induction of ID1 expression and apoptosis by the histone deacetylase inhibitor (trichostatin A) in human acute myeloid leukaemic cells. Cell Prolif. 2008;41:86–97. doi: 10.1111/j.1365-2184.2007.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Singh J, Murata K, Itahana Y, Desprez PY. Constitutive expression of the Id-1 promoter in human metastatic breast cancer cells is linked with the loss of NF-1/Rb/HDAC-1 transcription repressor complex. Oncogene. 2002;21:1812–1822. doi: 10.1038/sj.onc.1205252. [DOI] [PubMed] [Google Scholar]