

Abstract
Gastric cancer (GC) is one of the most common malignancies worldwide with poor prognosis for lack of early detection and effective treatment modalities. The significant influence of tumor microenvironment on malignant cells has been extensively investigated in this targeted-therapy era. Epithelial-mesenchymal transition (EMT) is a highly conserved and fundamental process that is critical for embryogenesis and some other pathophysiological processes, especially tumor genesis and progression. Aberrant gastric EMT activation could endow gastric epithelial cells with increased mesenchymal characteristics and less epithelial features, and promote cancer cell stemness, initiation, invasion, metastasis, and chemo-resistance with cellular adhesion molecules especially E-cadherin concomitantly repressed, which allows tumor cells to disseminate and spread throughout the body. Some pathogens, stress, and hypoxia could induce and aggravate GC via EMT, which is significantly correlated with prognosis. GC EMT is modulated by diverse micro-environmental, membrane, and intracellular cues, and could be triggered by various overexpressed transcription factors, which are downstream of several vital cross-talking signaling pathways including TGF-β, Wnt/β-catenin, Notch, etc. microRNAs also contribute significantly to GC EMT modulation. There are currently some agents which could suppress GC EMT, shedding light on novel anti-malignancy strategies. Investigating potential mechanisms modulating GC cell EMT and discovering novel EMT regulators will further elucidate GC biology, and may provide new biomarkers for early GC detection and potentially efficient targets for preventative and curative anti-GC intervention approaches to prevent local and distant invasions.
Keywords: Epithelial-mesenchymal transition, gastric cancer, microenvironment, transcription factor, microRNA, carcinogenesis, tumor progression, metastasis, prognosis, chemo-resistance
Introduction
Although the incidence and mortality have being declining in recent decades, gastric cancer (GC) remains one of the most common and lethal malignancies worldwide [1,2]. Surgical resection is the only possibly curative therapy, but most patients are diagnosed with an advanced disease for lack of specific early-stage symptoms and lose the opportunity of curative resection due to the aggressive GC nature, which results in poor prognosis, although chemoradiotherapy and targeted therapy have improved response rates [3-6]. Invasion and metastasis, the major causes of GC-related relapse and death, greatly impede the treatment efficiency [7]. A better understanding of the mechanism contributing to GC initiation and progression is warranting with the hope to improve early diagnosis and treatment efficacies.
Tumors are composed of malignant cells and the microenvironment, namely tumor stroma, and are regarded as wounds that do not heal [8]. Abundant evidences have revealed the importance of epithelial-mesenchymal transition (EMT), a process where epithelial cells are transformed into cells with mesenchymal phenotypes characterized by lost cellular polarity and adhesion and enhanced invasive and migratory properties, and an important mode responding to tumor microenvironment among cancer cells, in GC aggressiveness. The components of the complex tumor microenvironment, including various tumor stromal cells and cellular factors, modulate cancer cell growth and regulate their malignant behaviors via EMT induced by diverse intracellular signaling pathways which alter modes of transcription and translation, and which could not only regulate malignant cell behaviors directly, but also stimulate cancer tissues indirectly via modulating the microenvironment [9,10]. Diverse genes, proteins, and molecular pathways which are linked to transcriptional regulation, epigenetic modification, and cancer stem cells (CSCs), and which are potentially important during GC genesis and progression, have been extensively investigated during aberrant activation and regulation of GC EMT [11]. Detailed investigation into the role of EMT in GC could further our understanding of GC initiation, invasion, and metastasis. Herein GC EMT is systematically reviewed, with possibly involved regulators in the microenvironment and potential translational significance facilitating diagnosis and treatment discussed.
EMT
EMT is a multistage reprogramming process in which epithelial cells are closely arranged with weak deformability dedifferentiation. The epithelial cells may undergo phenotypic switch to acquire mesenchymal phenotype similar to fibroblasts that are loosely arranged with strong mobility, as well as stem cell properties. The common processes during EMT, which is important during embryogenesis and commonly found in various pathophysiological processes including wound healing, inflammation, and fibrosis [12], are epithelial marker repression, aberrant mesenchymal marker (e.g. vimentin, fibronectin, and α-smooth muscle actin [α-SMA]) up-regulation, loss of polarity and intercellular adhesion, cytoskeleton disorganization promoting motility, and surrounding microenvironment remodeling facilitating invasion [13]. It is believed that the leading cause of post-EMT biological behavior change is EMT-induced cytoskeleton-associated structure reconstruction [14]. During EMT, type I cadherin (epithelial-cadherin, E-cadherin, encoded by the CDH1 gene at human chromosome 16q22.1), which sustains key intracellular binding structures like desmosomes and claudins, is switched to neural cadherin (N-cadherin, encoded by the CDH2 gene), which is mostly expressed among mesenchymal cells. The reduction of E-cadherin with the immunoglobulin-like domain on cellular surface which is able to combine adjacent cells, and the intracellular region which is able to link α- and β-catenin to the actin cytoskeleton sustaining cell shape and polarity, is regarded as an important EMT feature which plays critical roles in EMT by changing the components of intercellular adhesion and regulating diverse signaling pathways [15]. E-cadherin down-regulation and initiation and execution of EMT are caused by several microRNAs (miRNAs) and various key EMT-inducing transcription factors (TFs) including Snail1 (Snail), Snail2 (Slug), Twist, Zeb1, Zeb2 (Sip1), forkhead box C2 (FoxC2), E47, Krüppel-like factor (KLF)4, KLF8, goosecoid, Sox9, et al., which interact with each other, and which are downstream in important signaling pathways activated by transforming growth factor-β (TGF-β), Wnt, Notch, integrin, interleukin (IL)-5, IL-6, fibroblast growth factor (FGF), signal transducer and activator of transcription-3 (STAT-3), epidermal growth factor (EGF), hepatic growth factor (HGF), nuclear factor-κB (NF-κB), et al. [16,17]. Other cellular surface proteins, extracellular matrix (ECM) proteins, and cytoskeletal markers including fibronectin, vimentin, fibroblast specific protein 1 (FSP1), β-catenin, and α-SMA might also characterize EMT, and are closely associated with tumor invasive ability [18]. TGF-β1-induced abnormal β-catenin nuclear translocation and its target gene transactivation are key processes activating EMT [19]. The complex mechanism of EMT regulated by various factors and signaling pathways makes it a hot topic.
EMT in tumor biology
The developmental reactivation in tumor cells generates EMT, leading to cell dissemination and playing a vital role in tumor progression [20]. In tumor cells, an essential EMT step is the down-regulation of E-cadherin, therefore disassembling the intercellular contact [21]. The E- to N-cadherin switch has critical functions in cancer progression for being essential for enhanced motility and migration [22]. Decreased expression of E-cadherin activates β-catenin which then translocates into the nucleus and transcriptionally modulates expression of various proteins including cyclin D1, CD44, c-Myc, and vascular endothelial growth factor (VEGF), promoting tumor initiation and progression [23]. Recently, EMT-associated reprogramming properties have been linked to improved cancer cell plasticity, as illustrated by its ability to induce a “CSC” phenotype [24]. The EMT-induced stemness endows cancer cells with the ability to self-renew, and to overexpress chemo-resistance-related genes, resulting in a challenging issue named multiple drug resistance (MDR) in cancer treatment [25].
In addition to tumorigenesis, aberrant EMT activation, which provides cancer cells with enhanced adaptability and migratory and invasive properties, and which prevents senescence and apoptosis and induces immunosuppression, also promotes tumor progression which is a multi-step process with EMT as one of the key links, gradually transforming early weakly-invasive tumor cells into cells with stronger metastatic abilities [17]. It is considered as the most important and common process during tumor local infiltration and distant metastasis [26]. When aberrant EMT is activated, N-cadherin expression could facilitate cell-stroma adhesion. Data from human solid cancers, xenograft models, and cell culture show that the EMT phenotype leads to poor outcome and enhances cell migration and invasiveness [27]. In both in vitro and in vivo studies, progression of cancer cells to metastatic ones frequently involves EMT-like epithelial plasticity changes towards a migratory fibroblastoid phenotype, particularly prominent at the invasive front of human tumors [12]. TGF-β pathway is a major promoter of tumor progression at later stages, which is attributed to its ability to activate EMT [28]. EMT could also generate tumor stromal cells, especially the cancer-associated fibroblasts (CAFs), which significantly facilitate tumor formation and progression possibly via EMT induction again [10,29]. Investigating possible mechanism modulating malignant cell EMT and discovering novel EMT regulators will further elucidate GC biology, and may provide novel biomarkers and efficient targets for preventative and curative anti-GC approaches.
Microenvironment, membrane, and intracellular regulators in GC EMT
EMT is induced in epithelia by diverse extracellular stimuli including TGF-β, HGF, TNF-α, hypoxia inducible factor 1α (HIF1α), and inflammatory signals from surrounding microenvironment, which activate growth factor (GF) and chemokine receptors, the downstream signaling cascades, and several TFs [30]. In GC stroma, there are various cues which can influence EMT (Figure 1). Transduction of exogenous WNT5A into GC cells upregulates EMT-related genes, indicating that WNT5A regulates GC EMT [31]. Down-regulation of Wnt5a by EGF, which increases Arf6 and ERK activity, is necessary for EGF-induced EMT in GC cells [32]. Gastric epithelia are especially sensitized for TGF-β-induced EMT, and the resultant Lgr5 induction is enhanced by the compounding effect of aberrantly activated and dysregulated TGF-β and Wnt pathways [33]. Tipα could induce vimentin expression, and cause EMT phenotype changes in human GC cells [34]. Besides its ability to induce apoptosis, Fas signaling can also induce non-apoptotic events in tumor cells, including EMT, which promotes motility and metastasis in GC [35]. Fas ligand (FasL)-treated GC cells have increased EMT TF expression in the nucleus and obtain an EMT-related morphology. FasL treatment inhibits E-cadherin transcription by up-regulation of Snail, inducing EMT in GC cells. The nuclear expression and transcriptional activity of Snail and β-catenin are increased by inhibitory phosphorylation of glycogen synthase kinase-3β (GSK-3β) at Ser9 by FasL-induced extracellular signal-regulated kinase (ERK)/mitogen-activated protein kinase (MAPK) signaling. Snail associates with β-catenin in the nucleus and thus increases β-catenin transcriptional activity [36-38]. Amplification of ERBB2, MET, FGFR2, PIK3CA, AKT1, WNT2, WNT2B and WNT8B, and down-regulation of SFRP1 and PAR3/PAR6/aPKC complex could lead to GC EMT also through GSK3β inhibition [39]. Some TFs with partially overlapping downstream target genes can activate EMT. These key EMT TFs are under control of several upstream mechanisms, and some are directly induced at the transcriptional level by the activated form of Notch (NICD), which directly regulates major EMT TFs consequently triggerring EMT in cancers [40]. Notch signal can be activated among endothelia in a paracrine manner [41]. In NICD/p53-/- mice, Notch and p53 are altered in every carcinoma cell, but only a small proportion of these cells undergo EMT [42]. EMT is associated with an invasion phenotype in NICD/p53-/- GC [43]. Notch activation and p53 deletion have a synergistic effect in activating EMT [43]. In the Notch gain-of-function mutants, transcriptional activation of EMT inducers may be counterbalanced by p53-activated miRNAs at the translational level, possibly causing lack of invasive tumors in some NICD-activating cases [44]. p53 inactivation could cause evasion from senescence, facilitating EMT induction by TFs including Twist1, Twist2 and Zeb1, as well as GFs like EGF and TGF-β [45]. Reduced Twist expression is associated with increased E-cadherin, supporting that Twist may be associated with GC EMT [46]. Zeb-1 expression is significantly correlated with the mesenchymal phenotype in GC [47]. Erythropoietin-producing hepatocellular A2 (EphA2) expression is positively correlated with EMT markers in human GC [48]. EphA2 overexpression results in up-regulation of mesenchymal markers N-cadherin and Snail, as well as the Wnt/β-catenin signaling targets TCF4, Cyclin-D1 and c-Myc, and correlates with the loss of epithelial proteins, while silencing EphA2 has the opposite effect. Furthermore, Wnt/β-catenin pathway inhibition negates the EphA2 overexpression effect, whereas activating the pathway impairs the EphA2 knockdown effect on EMT [48,49]. These observations suggest that EphA2 promotes GC cell EMT through Wnt/β-catenin signaling activation. Periostin could induce GC EMT, which is facilitated by nicotine. The expression of EMT-related proteins is closely related to each other in GC. The known GC stem cell maker, CD44, is significantly associated with the expression of Snail-1, ZEB-1, and E-cadherin proteins [50]. Through crosstalk with TGF-β, Notch and Wnt signaling pathways, KLF4 negatively regulates GC EMT [51]. TGF-β1 could induce EMT via down-regulating E-cadherin and up-regulating vimentin expression in GC cells. KLF8, a downstream TF of TGF-β1, contributes to EMT induction. Small interfering RNA (siRNA)-mediated KLF8 silence blocks TGF-β1-induced EMT-like transformation and subsequently reverses the loss of E-cadherin and the gain of vimentin [52]. Overexpression of Ets variant gene 1 (ETV1; also known as ER81) in normal gastric epithelia induces EMT. ETV1 transcriptionally upregulates Snail expression, and its expression is significantly correlated with Snail expression in human GC samples. It promotes Snail expression to induce EMT-like metastatic progression in GC [53]. The high mobility group protein A2 (HMGA2) is able to elicit GC EMT and the Wnt/β-catenin pathway activated by HMGA2 might be the underlying mechanism [54]. Aquaporin 3 (AQP3) up-regulation represses E-cadherin, thus promoting EMT in human GC. It also up-regulates the expression of vimentin and fibronectin in vitro. The PI3K/AKT/SNAIL signaling pathway is likely involved in the EMT induction by AQP3 in GC [55]. Accumulation of pSTAT3 in the nucleus results in the decreased expression of E-cadherin and increased expression of mesenchymal markers (Slug, α-SMA, N-cadherin and vimentin). Crosstalk of EGFR and TGF-β signaling with Wnt signaling upregulates EMT. Caveolin-1 (Cav-1) is modulated by HSP90 and functions as a crucial regulator of GC EMT [56]. Knockdown of RUNX3 increases the expression of vimentin in human GC cells, and overexpression of RUNX3 decreases vimentin expression and inhibits GC cell colonization in nude mice [57]. In excised human GCs, expression of epidermal growth factor-like domain-containing protein 7 (EGFL7) is positively correlated with expression of vimentin and Snail, and negatively correlated with E-cadherin expression. In GC cell lines, EGFL7 knockdown reverses morphological signs of EMT and decreases both vimentin and Snail expression [58]. Down-regulation of FoxM1 significantly inhibits vimentin and N-cadherin expression. Cells transfected with FoxM1 siRNA displays an elongated/irregular fibroblastoid morphology and reduction of the vimentin expression [59]. CEACAM6 is negatively correlated with E-cadherin expression in GC tissues. Overexpression of CEACAM6 induces GC EMT, characterized by increases in the EMT markers N-cadherin, vimentin and Slug, while E-cadherin expression is decreased [60]. Paired-related homeobox 1 (PRRX1) has been identified as a new EMT inducer. PRRX1 is upregulated and positively correlated with EMT markers in human GC specimens. It promotes EMT in GC cells through the activation of Wnt/β-catenin signaling [61]. Insulin-like growth factor I (IGF-I) induces EMT by upregulating the levels of ZEB2, which is dependent on the phosphoinositide 3-kinase (PI3K)/Akt signaling pathway in GC cells. In addition, GSK-3β, an intracellular downstream effector of PI3K/Akt, sustains the epithelial phenotype by repressing ZEB2 expression and the subsequent inhibition of EMT induced by IGF-I, suggesting the involvement of a potential activation of the PI3K/Akt-GSK-3β-ZEB2 signaling pathway in IGF-I-induced EMT in GC cells [62]. Zipper-interacting Protein Kinase (ZIPK) induces and promotes EMT with increased expression of β-catenin, mesenchymal markers, Snail and Slug, and with decreased expression of E-cadherin, via activating the AKT/IκB/NF-κB pathway [63]. Ectopic activated Cdc42-associated kinase 1 (ACK1) expression induces and promotes EMT by activating the AKT-POU2F1-ECD signaling in GC cells. Overexpression of cell cycle-related protein ecdysoneless homologue (ECD) also promotes EMT, similar to the effects of ACK1 overexpression. Silencing of ECD completely blocks the augmentation of ACK1 overexpression-induced EMT [64]. Nuclear protein C23 and bone morphogenetic protein-2 (BMP2) have been linked into EMT. BMP2 increases the expression of p-Erk1/2, p-Akt, vimentin, N-cadherin, and matrix metalloproteinase (MMP) 2 in GC cells, which is in a dose-dependent manner. si-C23 treatment attenuates the BMP2-stimulated expression of the molecules. These suggests that C23 mediates BMP2-induced EMT via the up-regulation of Erk1/2 and Akt signaling pathway in GC [65]. CCL19 treatment could increase p-ERK, p-AKT, Snail and MMP-9 expression, and decrease E-cadherin in GC cells in a dose-dependent manner. The blockade of Snail abrogates the up-regulation of MMP-9 and the down-regulation of E-cadherin [66]. Positive expression of CD146 is strongly associated with loss of E-cadherin and acquisition of the mesenchymal markers nuclear β-catenin and vimentin, suggesting that CD146 might promote EMT [67]. GC cells acquires an “activated” carcinoma-associated fibroblast (CAF) phenotype after close contact with mesenchymal stem cells (MSCs) with enhanced tumor metastasis and growth in vivo. The paracrine signals could induces EMT and promotes transwell and trans-endothelial migration, and the changes are dependent on β-catenin, MMP-16, snail and twist [68]. Cell fusion between gastric epithelial cells and MSCs may also result in EMT [69].
Figure 1.
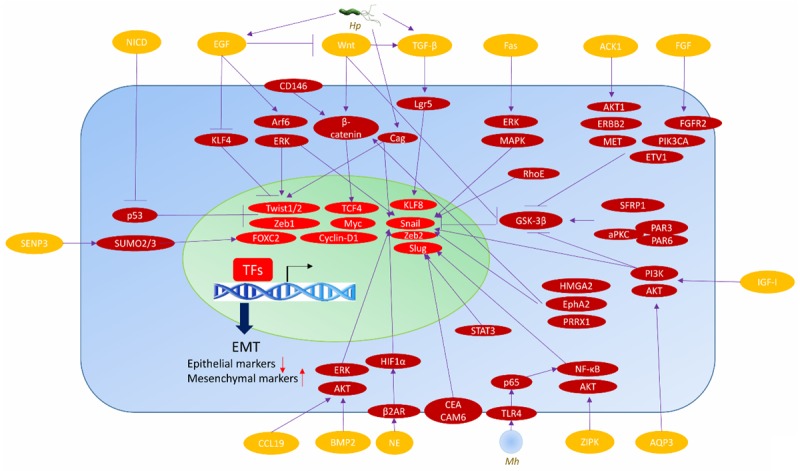
Key inducers and regulators of EMT in gastric cancer. ACK1, activated Cdc42-associated kinase 1; AQP3, Aquaporin 3; BMP2, bone morphogenetic protein-2; Cag, Cytotoxin-associated gene; EGF, epidermal growth factor; EMT, epithelial-mesenchymal transition; EphA2, Erythropoietin-producing hepatocellular A2; ERK, extracellular signal-regulated kinase; ETV1, Ets variant gene 1; FGF, fibroblast growth factor; FoxC2, forkhead box C2; GSK-3β, glycogen synthase kinase-3β; HIF1α, hypoxia inducible factor 1α; HMGA2, high mobility group protein A2; Hp, Helicobacter pylori; IGF-I, Insulin-like growth factor I; KLF, Krüppel-like factor; MAPK, mitogen-activated protein kinase; Mh, Mycoplasma hyorhinis; NF-κB, nuclear factor-κB; NICD, activated form of Notch; PI3K, phosphoinositide 3-kinase; PRRX1, Paired-related homeobox 1; STAT3, signal transducer and activator of transcription-3; TF, transcriptional factor; TGF-β, transforming growth factor-β; ZIPK, Zipper-interacting Protein Kinase.
Pathogens and GC EMT
Chronic Helicobacter pylori (Hp) infection provokes an inflammation of the gastric mucosa, a high risk for cancer development. The most virulent strains harbor the cag pathogenicity island encoding a type IV secretion system, which allows delivery of bacterial effectors into gastric epithelial cells, inducing phenotypic alterations reminiscent of an EMT [70]. Pathogenic Hp up-regulates soluble HB-EGF shedding, a factor implicated in EMT, in gastric epithelia [71]. This process is partially dependent on and facilitated by both gastrin and MMP-7 expression in Hp-infected cells. siRNA against MMP-7 and MMP-7 neutralizing antibody significantly reduces EMT gene expression. The effects of Hp on EMT are also reversed by gastrin siRNA. Neutralization of gastrin reduces expression of key EMT proteins. Thus, Hp could induce EMT via up-regulating MMP-7, which is partially dependent on gastrin, by indirectly increasing soluble HB-EGF level [72]. GC EMT could be induced by Hp Cytotoxin-associated gene A (CagA), which promotes TWIST1 and vimentin expression, and inhibits E-cadherin expression by down-regulating PDCD4, revealing a new signaling pathway of EMT in GC cell lines [73]. Eradication of Hp reduces the expression of TGF-β1, Twist, Snail, Slug, and vimentin mRNAs, whereas it enhances the expression of E-cadherin, suggesting that Hp infection may trigger the TGF-β1-induced EMT pathway, and that its eradication may prevent the GC genesis by inhibiting these 2 pathways [74]. Mycoplasma hyorhinis (Mh) could also induce EMT, which could be counteracted by inhibition of NF-κB signaling or p65 knockdown in GC cells. Knock-down or inhibition of TLR4 could also antagonize Mh-induced EMT [75].
Stress, hypoxia and GC EMT
GC is one of the typical oxidative stress-related malignances [76]. Some redox-sensitive factors including TGF-β, HIF-1, NF-κB and p53, are important EMT modulators in various tumors [77]. SENP3, a redox-sensitive SUMO2/3-specific protease, is correlated with, induces and promotes GC cell EMT, which could be possibly explained by the fact that it could de-conjugate SUMO2/3 during EMT, activating an EMT-inducing TF, FOXC2 [78]. This finding adds novel clues to the role of the SENP family in EMT for only SENP7 has been previously shown to be contributive during oxidative stress originating from the microenvironment [79]. Norepinephrine (NE) not only obviously induces EMT-related morphological alterations in GC cells, but also up-regulates EMT markers including vimentin, and decreases E-cadherin expression through the regulation of β2-AR-HIF-1α-Snail activity, further enhancing cell motility and invasiveness [80].
Hypoxia is also a significant GC EMT inducer. Under hypoxia, E-cadherin decreases and N-cadherin, Vimentin, Snail, Sox2, Oct4, and Bmi1 increase, indicating that the hypoxic microenvironment induces EMT, accompanied by cytoskeleton remodeling [81]. Hypoxia or overexpression of RhoE in normoxia up-regulates Vimentin, and down-regulates E-cadherin in GC, and silencing of HIF-1α or RhoE by specific siRNAs could reverse these hypoxia-induced effects, suggesting that RhoE up-regulation represents a pivotal cellular adaptive response to hypoxia with implications in GC EMT [82]. Hypoxia could also induce EMT in some GC cell lines via autocrine TGF-β/TGF-βR signaling, and TGF-βR inhibition significantly suppresses the process [83]. Hypoxia is able to induce KLF8-mediated EMT in GC cells. KLF8 and HIF-1 siRNAs strongly reverse EMT in cells undergoing hypoxia [84]. UPR protein potentiates the EMT of GC cells under conditions of severe hypoxia, and Knockdown of PERK, ATF4 or ATF6 impedes EMT of GC cells induced by severe hypoxia [85]. The specific anti-EMT drugs in combination with anti-hypoxia regimens are new promising anti-GC therapies [84].
EMT and Lauren classification
Lauren classified GCs into intestinal and diffuse types based on morphology. The different phenotypes of the cells indicate the presence of different molecular mechanisms, which can be approached with the help of current techniques. The best described are the germline/somatic mutations and the hyper-methylations of the CDH1 gene promotor. In diffuse-type GC, up-regulation of Snail, a direct repressor of E-cadherin, is associated with down-regulation of E-cadherin [86]. Mesenchymal-related genes (WNT5A, CDH2, PDGFRB, EDNRA, ROBO1, ROR2, and MEF2C) are activated by an EMT regulator, SIP1/ZFHX1B/ZEB2, which is a target of a primary transcriptional regulator GLI1 in the hedgehog (Hh) signaling. The Hh-EMT pathway is preferentially activated in diffuse-type GCs compared with intestinal-type GCs [87]. However, the mesenchymal features are weaker in diffuse-type GCs than in MSCs. Diffuse-type GC which has undergone extensive EMT, and which has a poor prognosis, can be identified by quantitative PCR analysis of the mRNA ratio of CDH2 to CDH1 [88]. In intestinal-type GC, higher expression of β-catenin and MMP-16 is correlated with tumor invasion and metastasis [68]. Targeting the key molecules involved may interfere with EMT in different types of GC.
EMT and GC tumorigenesis
GC genesis is closely related to EMT. The levels of TGF-β1, Twist, Snail, Slug, and vimentin, in addition to the level of CD44, a CSC marker, are all up-regulated in patients with dysplasia or early GC, while the level of E-cadherin is decreased in these patients [74]. E-cadherin and Vimentin expressions in GC tissues are significantly higher than those in adjacent normal tissues, while the E-cadherin level in GC tissues is significantly lower than that in the adjacent normal tissues [46]. Increased expression of the EMT-related regulatory protein TWIST1 and the mesenchymal marker vimentin in cancer tissues, and decreased expression of programmed cell death factor 4 (PDCD4) and E-cadherin expression in cancer specimens are associated with tissue malignant degree [73]. The existence of gastric CSCs has been demonstrated. EMT-induced CSC phenotype may contribute largely to GC genesis [89]. In gastric epithelia, the stem cells at the base of the pyloric gastric glands are dependent on an active and dynamically regulated Wnt pathway [90], which is characterized by the exclusive expression of Lgr5, an exclusive gastric stem cell marker, and which could amplify the Wnt signaling, an important activated pathway during EMT [91]. EMT activation in Runx3-/-p53-/- gastric epithelial cells is accompanied by the Lgr5 induction [92]. EMT induced by TF Runx3 loss in gastric epithelial cells produces a tumorigenic stem cell-like subpopulation, which remarkably expresses Lgr5. Runx3 plays a vital protective role in protecting gastric epithelial cells against aberrant GF signaling and the resultant cellular plasticity and stemness. E-cadherin down-regulation is associated with an undifferentiated and invasive GC phenotype [33]. These suggest that EMT is associated with CSCs and is able to induce stemness and tumorigenesis. GC is closely associated with chronic inflammation, and Hp infection is a significant risk factor [93]. MMP-7 up-regulation by Hp may play a role in GC formation, potentially through EMT, by indirectly increasing the soluble heparin-binding (HB)-EGF level [72]. Mh induces EMT and promotes cell migration via TLR4-NF-κB signaling, which provides a clue to the GC-genesis role of Mh [75]. In diffuse type GC, there exist a decreased expression of adhesion molecules including E-cadherin, α-catenin and β-catenin, and relevant morphological changes, suggesting that EMT may be involved not only in GC genesis, but also in treatment resistance [94].
EMT and GC progression and metastasis
Recent evidence indicates that EMT is a key GC progression driver, and plays a fundamental role during early steps of GC invasion, metastasis and relapse. EMT-induced cell migration and reach their metastatic niche through both the lymphatic system and blood. The EMT phenotype is correlated with an advanced GC stage [95]. Among GC patients, non-EMT phenotype is mainly distributed in those at early stage, while patients with Fas signaling-induced EMT, which promotes GC cell motility and metastasis, are mostly at an advanced stage [36]. MSCs within the tumor niche significantly facilitates GC growth and metastasis by paracrine cues and close physical connection, which occurs partly through snail, twist and its downstream targets, specifically β-catenin/MMP-16 [68]. The connective tissue growth factor (CTGF)-induced EMT of human peritoneal mesothelial cells is associated with an increased adhesion of GC cells to mesothelial cells, suggesting that CTGF promotes GC cell adhesion to peritoneum, which facilitates malignant cell dissemination [96]. Malignant cell entering vasculature is an essential procedure during tumor metastasis. In gastric circulating tumor cells (CTCs), epithelial markers pan-CK and E-cadherin are decreased, and mesenchymal markers N-cadherin and vimentin are overexpressed, suggesting that gastric CTCs exhibits a remarkable EMT process [97]. Sonic hedgehog (Shh) signaling activation could induce GC EMT through activating the PI3K/Akt pathway, which induces lymphangiogenesis and improves tumor invasiveness and metastasis, indicating that Shh might be a candidate therapeutic target for treating metastatic GC [98,99]. Histone demethylase Jumonji domain-containing protein 2B (JMJD2B) promotes EMT and GC invasion and metastasis via β-catenin-induced H3K9 demethylation, which causes vimentin up-regulation, implicating JMJD2B as a potential target for reversing EMT and intervening GC progression [11]. EMT induced by Tipα, a TNF-α-inducing protein and a carcinogenic factor in the gastric epithelia which is produced by Hp, is also probably associated with GC progression [34]. Rho GDP dissociation inhibitor 2 (RhoGDI2) plays a critical role in GC progression through EMT induction with E-cadherin repression via up-regulating Snail [100]. Targeting RhoGDI2 may thus be a useful strategy to inhibit GC cell invasion and metastasis. Highly metastatic GC cell lines have decreased expression of E-cadherin, together with enhanced expression of vimentin and Snail. The decreased expression of E-cadherin could be restored by Snail knockdown. NDRG1 knockdown in the highly metastatic cell line shows enhanced expression of E-cadherin and decreased expression of vimentin and Snail, suggesting that NDRG1 plays a pivotal role in the malignant progression of GC through EMT [101]. Fas signaling has been shown to induce EMT to promote GC metastasis. The expressions of FasL, phospho-GSK-3β, Snail, and β-catenin increase during GC progression [38]. The EMT process can be triggered by the BMP2/BMPR axis in GC cells and is then involved in the tumor cell migration, invasion and metastasis via the activation of PI3K/AKT and MEK/ERK pathways [102]. KLF8 is a novel EMT regulating TF that is involved in the progression of GC [84]. Effective therapy targeting the RUNX3 pathway may help control GC cell invasion and metastasis by inhibiting EMT [57]. EGFL7 promotes metastasis by activating EMT through an EGFR-AKT-Snail signaling pathway [58]. FoxM1 signaling plays important roles in tumor cell aggressiveness in GC cells. The over-expression of FoxM1 promotes cell migration, invasion and proliferation through the acquisition of an EMT phenotype by up-regulating the mesenchymal cell markers ZEB1, ZEB2 and vimentin and by down-regulating E-cadherin in gastric epithelial cells [59]. shRNA-mediated knockdown of ZEB2 results in reduced invasion and migration of GC cells, along with the up-regulation of E-cadherin and down-regulation of fibronectin and vimentin, further suggesting that ZEB2 promotes GC migration and invasion partly via the regulation of EMT [103]. WAVE3, an actin cytoskeleton remodeling protein, could promote cancer invasion and metastasis by participating in EMT. Elevated WAVE3 expression could induce and promote EMT in GC cells by dampening the expression of E-cadherin while increasing the expression of vimentin via up-regulation of Snail. Down-regulating Snail could particularly weaken EMT and tumor metastasis, invasion and proliferation promoted by overexpression of WAVE3 in GC cells [104]. CEACAM6 enhances invasion and metastasis in GC by promoting EMT via the PI3K/AKT signaling pathway [60]. CCR7 promotes Snail expression to induce EMT, resulting in progression, migration and invasion in GC [66].
miRNAs and GC EMT
Small non-protein coding RNAs (miRNAs) may act as powerful regulators of GC EMT (Figure 2). miRNA-506 could suppress SNAI2 expression by binding to its 3’ untranslated region (UTR), resulting in increased expression of E-cadherin [105]. Fibronectin, vimentin, N-cadherin and SNAIL protein levels are decreased, and E-cadherin expression is increased in GC cells transfected with miRNA-503, indicating that miRNA-503 can also inhibit GC EMT. Up-regulation of miRNA-1228* decreases the expression of mesenchymal markers and increases the epithelial marker E-cadherin, also suggesting its potential role in suppressing EMT [106]. miRNA-148a overexpression also down-regulates vimentin expression and upregulates E-cadherin expression, inhibiting GC EMT, likely via SMAD2 [107]. Exogenous expression of miRNA-194 inhibits the acquisition of the EMT phenotype in GC cells by down-regulating FoxM1, thereby inhibiting cell migration and invasion during cancer progression [108]. Expression of EMT-related TFs, snail1, zeb1 and twist1, are negatively correlated with miRNA-200s, and are positively correlated with phospho-Akt. Up-regulation of miRNA-200s down-regulates twist1 and zeb1, resulting in the suppression of EMT, which impairs migration and invasion in gastric CTCs [97]. Upregulated miRNA-200a expression increases E-cadherin and suppresses the Wnt/β-catenin pathway by interacting with β-catenin and targeting ZEB1 and ZEB2 in GC, thus inhibiting tumor migration, invasion and proliferation in vivo, which may provide a therapeutic target against EMT. When miRNA-200a is elevated, the expression of N-cadherin, β-catenin, Twist1 and Snail2 decreases [109]. Up-regulation of miRNA-204 could increase E-cadherin expression and decrease vimentin expression. The regulation of EMT by miRNA-204 involves an cooperation with LKB1 [110]. miRNA-137 increases expression of E-cadherin and cytokeratin, and suppresses expression of N-cadherin and vimentin. In vitro experiments have shown that miRNA-137 enhances the epithelial cell morphology in GIST [111]. Over-expression of RUNX3 increases the expression of miRNA-30a, which directly targets the 3’ UTR of vimentin and decreases its protein level. miRNA-30a inhibitor abrogates RUNX3-mediated inhibition of GC cell invasion and down-regulation of vimentin. In GC patients, RUNX3 level is positively correlated with miRNA-30a and negatively associated with the vimentin level [57].
Figure 2.
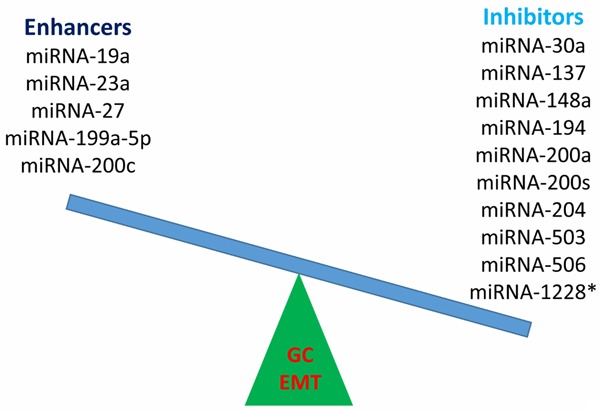
miRNAs either enhance or inhibit EMT in GC. EMT, epithelial-mesenchymal transition; GC, gastric cancer; miRNA, microRNA.
Some miRNAs act as EMT enhancers. Increased serum response factor (SRF)/miRNA-199a-5p/E-cadherin pathway activity possibly promotes GC cell metastasis though EMT, also indicating the relevant regulators as potential therapeutic targets or biomarkers for GC progression [112]. Increased expression of miRNA-27 up-regulates the EMT-associated genes ZEB1, ZEB2, Slug and Vimentin, and decreases E-cadherin levels in GC. It promotes EMT by activating the Wnt pathway [113]. Overexpression of miRNA-19a can promote EMT through activating the PI3K/AKT pathway [114]. miRNA-23a might participate in the mechanism of FasL-induced EMT and serve as a potential therapeutic target for GC metastasis [95]. IGF-I-induced GC cell EMT is accompanied by Zeb2 up-regulation, and both Akt/ERK inhibitors and Akt/ERK gene knockdown could reverse IGF-I-induced EMT through miRNA-200c up-regulation, suggesting the involvement of an Akt/ERK-miRNA-200c-ZEB2 axis in the IGF-I-induced GC EMT. The ubiquitin ligase Cbl-b ubiquitinates and degrades IGF-IR and inhibits the Akt/ERK-miRNA-200c-ZEB2 axis, causing IGF-I-induced EMT repression in GC cells [115]. Suppression of long non-coding RNA (lncRNA) HOTAIR could also reverse EMT process [116]. LEIGC lncRNA acts as a tumor suppressor in GC by inhibiting EMT [117].
EMT and prognosis of GC patients
Currently the important EMT activators and drivers have been reported to be closely correlated with cancer prognosis. The EMT status (vimentin/E-cadherin ration) is a critical independent prognosticator for GC [47]. The EMT phenotype correlates with an advanced GC stage, and is significantly correlated with a worse GC prognosis [36,38,95]. In GC, EMT is associated with a diffuse type, a poorly differentiated histology, an advanced TNM stage, and a poor prognosis, suggesting that inhibition of EMT could be promising in prevention of metastatic progression and invasion [118]. There exist significant negative correlations between overall survival (OS) and the expression of some EMT-related molecules including TGF-α, cyclooxygenase-2 (COX-2), MMP-7, MMP-9, and C-X-C chemokine receptor (CXCR4) in GC [119]. The expression of EphA2 and vimentin is significantly higher in GC tissues than that in normal gastric mucosa tissues, and similar results are for negative E-cadherin expression and ectopic β-catenin expression. Down-regulated expression of E-cadherin, overexpression of vimentin and ectopic expression of β-catenin are associated with inferior depth of tumor invasion, tumor differentiation, TNM stage, and lymph node metastasis in GC tissues [60]. The expression of EphA2 is negatively correlated with E-cadherin expression and is positively correlated with ectopic β-catenin and vimentin expression. The overexpression of EphA2 and vimentin, ectopic expression of β-catenin and down-regulation of E-cadherin indicate a poor outcome, and are independent prognostic factors for postoperative GC [49]. In high-risk GC patients, Snail-1, vimentin, E-cadherin and CD44 significantly associate with an advanced pT stage, lymph node metastasis, vascular invasion and undifferentiated histologic type, and predict disease-free survival (DFS) independent of pTNM stage and histologic differentiation. The acquired mesenchymal phenotype of GC cells at the primary site is restored to an epithelial phenotype in lymph node metastases. Loss of E-cadherin expression and aberrant expression of vimentin and the known gastric CSC maker CD44 are significantly associated with aggressive clinicopathologic features, a combination of EMT and CSC-like phenotypes is an important predictor of aggressive biologic behavior and has an independent prognostic value in predicting outcomes of primary GC [50]. The 5-year survival rate is also reduced for gastric neuroendocrine tumor (NET) patients showing high levels of Snail1, a cytoplasmic E-cadherin pattern, aberrant N-cadherin expression and loss of E-cadherin/β-catenin adhesion complex integrity on the cell membrane. Interestingly, high β-catenin expression is useful in identifying a grade 1 NET subgroup with a favorable clinical course [120]. Positive staining of S100A4, an EMT-related mesenchymal protein, in tumor body (TB) and of Snail1 at invasive edge (IE) is associated with involvement of circumferential resection margins. Positive staining of S100A4 in the TB and luminal surface (LS) is associated with a poor 5-year OS. The acquisition of S100A4 is associated with a poor prognosis in patients with gastroesophageal junction tumors who undergo potentially curative surgery [121]. Stage III GC patients with an early recurrence have larger tumors and more lymph node metastases, coupled with aberrant expression of EMT and CSC markers, compared with patients with a late recurrence. A combination of EMT and CSC-like markers is also a predictor of recurrence after radical resection for GC [122]. Mesenchymal epithelial transition (MET) has also been recognized as an important poor prognostic marker in some GCs [123].
EMT and GC chemo-resistance
Besides their therapeutic effects, accumulating evidences suggest that chemotherapeutic agents also induce EMT, which might be one mechanism of cancer chemo-resistance, significantly reducing treatment responsiveness and worsening prognosis. Lapatinib-resistant GC cell line (SNU216 LR) presents an EMT phenotype and maintains activation of Met, Her3, Stat3, Akt and MAPK signaling with lapatinib presence. Up-regulation of various EMT-related genes and ECM molecules, especially Testican-1, is identified in SNU216 LR cells. Testican-1 inhibition by siRNA decreases Testican-1-induced and Met-dependent downstream signaling, and restores cellular sensitivity to lapatinib. Furthermore, selective inhibition of β-catenin-mediated transcription and Testican-1-induced EMT signaling lead to G1 arrest. These results suggest the potential role of EMT in acquired resistance to HER2-directed treatment in HER2-positive GC, and shed lights on strategies for preventing this resistance among patients [124]. Moreover, oxaliplatin, a common chemotherapeutic agent, induces EMT partly through Fas signaling, which possibly contributes to chemo-resistance [36]. The cell lines resistant to SNU-16, a selective FGFR inhibitor, demonstrates changes characteristic of EMT. For GC patients with EMT-mediated resistance following treatment with FGFR inhibitors, mubritinib or AUY922 may be an alternative therapeutic strategy [125]. Doxorubicin (Dox) treatment could also induce EMT in human GC cells, with β-catenin signaling activated. Inhibition of β-catenin by indomethacin or siRNA suppresses Dox-induced EMT [126]. Inhibition of p300 by siRNA suppresses the expression of vimentin, and an epigenetic mechanism is involved in the EMT of the survival cells after long term Dox culture [127].
Anti-GC EMT strategies
Anti-EMT treatment modalities could inhibit GC progression. The non-cytostatic concentrations of Celastrus orbiculatus (COE) effectively inhibits TGF-β1-induced EMT in GC cells by suppressing the expression of HSP27, which is correlated with inhibition of the NF-κB/Snail signaling pathway in GC cells and which is characterized by prevented morphological changes, increased E-cadherin expression, and decreased vimentin and N-cadherin expression. Treatment with COE dose-dependently and effectively inhibits the proliferation, adhesion, invasion, migration and metastasis of GC cells in vitro, which is mediated by its anti-EMT activity. Overexpression of HSP27 significantly decreases the inhibitory effect of COE on EMT and the NF-κB/Snail pathway [128]. COE effectively suppresses tumor growth and metastasis in the nude mouse model based on reduced expression of N-cadherin, vimentin, NF-κB, p65, and Snail, and increased expression of E-cadherin in the tumor tissues [129]. VGLL4 is associated with the location change of nuclear β-catenin, suggesting β-catenin as a downstream factor of VGLL4. It suppresses EMT partly via negative regulation of the Wnt/β-catenin signaling pathway [130]. Danusertib inhibits EMT with involvement of signaling pathways mediated by PI3K/Akt/mTOR, p38 MAPK, and 5’ AMP-activated protein kinase [131]. Pantoprazole, a PPI, can effectively reverse the aggressiveness and EMT marker expression of adriamycin-resistant (ADR) GC cells, the aggressive phenotype of which is mediated by induction of EMT. It suppresses the invasiveness of the ADR cells by targeting the Akt/GSK-3β/β-catenin signaling involved in EMT [132]. Gastrokine 1 (GKN1), which plays an important role in the gastric mucosal defense mechanism and which also acts as a functional GC suppressor, could inhibit EMT in sporadic GCs [133]. The γ-secretase inhibitor DAPT could inhibit GC EMT with decreased expression of mesenchymal markers, vimentin, N-cadherin and Snail, and increased levels of E-cadherin in GC cell lines [134]. Wnt5a, a ligand activating the non-canonical Wnt signaling pathway, is commonly associated with EMT in cancer cell metastasis, and is a potential suppressor of EMT. Blockade of Arf6 represses ERK activity, up-regulates Wnt5a expression and represses EMT in response to EGF. Inhibition of ERK phosphorylation decreases movement of ERK from the cytoplasm to the nucleus following rescued Wnt5a expression, and favors an epithelial phenotype of GC cells [32]. LY294002, a PI3K inhibitor, could reverse CEACAM6-induced EMT via MET [60]. However, MET might also promote tumor progression. Dormant GC cells could be activated through EMT before migration, and MET is actually a key step facilitating tumor cell localization in the metastatic niche. The HGF/MET pathway is one of the most promising pathways among important signaling pathways regulating tumor proliferation and aggressiveness. The clinical evaluation of the antitumor therapy with several MET-targeting agents in GC is important, and is eagerly awaited as they may improve our understanding of the precise role of these agents in the treatment of advanced GC [123].
Conclusions and outlook
The investigation of EMT as a key contributor to malignant progression is a current trend in cancer research. EMT, which is regulated by the tumor microenvironment, is associated with many important processes of GC initiation and progression, including tumor cell stemness, proliferation, invasion, migration and chemo-resistance. The mechanism of transition highlights the integration of nuclear regulation and network signaling with alterations in the microenvironment to create a moving cell. GC EMT could be triggered by various TFs which are downstream of several vital cross-talking signaling pathways which play critical roles in EMT regulation. Recognition of the active role of EMT in GC not only adds a new level of complexity to cancer biology, but also brings an opportunity for new therapeutic strategies and early detection and precise prognostic markers. Loss of E-cadherin is a key step during EMT. Transcriptional repressors of E-cadherin including Snail, Zeb and Twist may be useful therapeutic targets for the prevention of GC invasion and metastasis. Inhibition of the key signaling proteins may allow early intervention avoiding an inoperable cancer. Other modulators participating in EMT regulation especially miRNAs could also be potentially developed as novel important therapeutic targets in GC. The relevant novel therapeutic antibodies, RNA interference compounds and small molecular inhibitors of EMT targeting the extra- and intra-cellular mediators should be developed against GC. Regulators of EMT may provide novel clinical targets to detect GC early. However, it is unclear which signaling pathway should be inhibited in order to most effectively block tumor progression with minimal toxicity caused in normal tissues simultaneously. A better understanding of the effect of targeting these pathways is required. It is notable that the transcriptional repressors, or EMT regulators, might also have other cellular functions in the complex intra- and inter-cellular networks. It is also probable that the same factors also have vital effects on other stromal cells, including fibroblasts and inflammatory cells. GC differentiation, stage, grade and location are also needed to be considered before conducting chemotherapy targeting EMT regulators. Personalized medicine with new anti-EMT agents would dramatically improve GC patients’ prognosis. Besides, further understanding of the relevant epigenetics will be potentially vital to develop EMT as a patient-specific therapeutic target. Notably, the results discussed may not be applicable to the real clinical situation due to the fact that not all key micro-environmental factors and cells are considered in basic researches. It is warranting to further explore the interactions between various EMT-activating signaling molecules, and their signaling pathways in GC, which could contribute to developing novel combination regimens to improve clinical outcome. Moreover, EMT investigation may provide new insights into the CSC theory. Some underlying mechanisms especially in GC initiation, and CSC reprogramming and maintaining remain obscure, and more in-depth researches into GC EMT are required to better elucidate its role and significance in GC genesis and progression. Future efforts should also further focus on how the events of cell attachment, matrix proteolysis and cell migration are controlled and integrated in GC, which requires a better understanding of the transcriptional regulations and the cell signaling mechanism involved. Animal models and clinical specimens should be carefully investigated to allow early identification and to clarify the role of EMT and the reverse process MET in primary GC lesion, pre-metastatic niche and metastasis pre-, during and post-treatment. Besides, single nucleotide polymorphisms (SNPs) and copy number variations (CNVs) of EMT signaling molecule-encoding genes could be identified as novel potential GC risk factors. Partial EMT and mechanisms alternative to EMT during GC invasion and metastasis should also be of note.
Acknowledgements
The authors would sincerely thank the reviewers and editors for critically reviewing this paper and for the constructive and thoughtful comments and suggestions.
Disclosure of conflict of interest
None.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Xu AM, Huang L, Liu W, Gao S, Han WX, Wei ZJ. Neoadjuvant chemotherapy followed by surgery versus surgery alone for gastric carcinoma: systematic review and meta-analysis of randomized controlled trials. PLoS One. 2014;9:e86941. doi: 10.1371/journal.pone.0086941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Memon MA, Subramanya MS, Khan S, Hossain MB, Osland E, Memon B. Meta-analysis of D1 versus D2 gastrectomy for gastric adenocarcinoma. Ann Surg. 2011;253:900–911. doi: 10.1097/SLA.0b013e318212bff6. [DOI] [PubMed] [Google Scholar]
- 4.Xu AM, Huang L, Zhu L, Wei ZJ. Significance of peripheral neutrophil-lymphocyte ratio among gastric cancer patients and construction of a treatment-predictive model: a study based on 1131 cases. Am J Cancer Res. 2014;4:189–195. [PMC free article] [PubMed] [Google Scholar]
- 5.Huang L, Xu A, Li T, Han W, Wu S, Wang Y. Detection of perioperative cancer antigen 72-4 in gastric juice pre- and post-distal gastrectomy and its significances. Med Oncol. 2013;30:651. doi: 10.1007/s12032-013-0651-3. [DOI] [PubMed] [Google Scholar]
- 6.Xu AM, Huang L, Han WX, Wei ZJ. Monitoring of peri-distal gastrectomy carbohydrate antigen 19-9 level in gastric juice and its significance. Int J Clin Exp Med. 2014;7:230–238. [PMC free article] [PubMed] [Google Scholar]
- 7.Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331:1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- 8.Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–1659. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 9.Sleeman JP, Thiery JP. SnapShot: The epithelial-mesenchymal transition. Cell. 2011;145:162.e1. doi: 10.1016/j.cell.2011.03.029. [DOI] [PubMed] [Google Scholar]
- 10.Huang L, Xu AM, Liu S, Liu W, Li TJ. Cancer-associated fibroblasts in digestive tumors. World J Gastroenterol. 2014;20:17804–17818. doi: 10.3748/wjg.v20.i47.17804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao L, Li W, Zang W, Liu Z, Xu X, Yu H, Yang Q, Jia J. JMJD2B promotes epithelial-mesenchymal transition by cooperating with betacatenin and enhances gastric cancer metastasis. Clin Cancer Res. 2013;19:6419–6429. doi: 10.1158/1078-0432.CCR-13-0254. [DOI] [PubMed] [Google Scholar]
- 12.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–142. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 13.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 14.Li J, Yang B, Zhou Q, Wu Y, Shang D, Guo Y, Song Z, Zheng Q, Xiong J. Autophagy promotes hepatocellular carcinoma cell invasion through activation of epithelial-mesenchymal transition. Carcinogenesis. 2013;34:1343–1351. doi: 10.1093/carcin/bgt063. [DOI] [PubMed] [Google Scholar]
- 15.Qiao Y, Jiang X, Lee ST, Karuturi RK, Hooi SC, Yu Q. FOXQ1 regulates epithelial-mesenchymal transition in human cancers. Cancer Res. 2011;71:3076–3086. doi: 10.1158/0008-5472.CAN-10-2787. [DOI] [PubMed] [Google Scholar]
- 16.Li R, Liang J, Ni S, Zhou T, Qing X, Li H, He W, Chen J, Li F, Zhuang Q, Qin B, Xu J, Li W, Yang J, Gan Y, Qin D, Feng S, Song H, Yang D, Zhang B, Zeng L, Lai L, Esteban MA, Pei D. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell Stem Cell. 2010;7:51–63. doi: 10.1016/j.stem.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 17.De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13:97–110. doi: 10.1038/nrc3447. [DOI] [PubMed] [Google Scholar]
- 18.Zeisberg M, Neilson EG. Biomarkers for epithelial-mesenchymal transitions. J Clin Invest. 2009;119:1429–1437. doi: 10.1172/JCI36183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Z, Li Q, Li K, Chen L, Li W, Hou M, Liu T, Yang J, Lindvall C, Bjorkholm M, Jia J, Xu D. Telomerase reverse transcriptase promotes epithelial-mesenchymal transition and stem cell-like traits in cancer cells. Oncogene. 2013;32:4203–4213. doi: 10.1038/onc.2012.441. [DOI] [PubMed] [Google Scholar]
- 20.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–1428. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harris TJ, Tepass U. Adherens junctions: from molecules to morphogenesis. Nat Rev Mol Cell Biol. 2010;11:502–514. doi: 10.1038/nrm2927. [DOI] [PubMed] [Google Scholar]
- 22.Christofori G. New signals from the invasive front. Nature. 2006;441:444–450. doi: 10.1038/nature04872. [DOI] [PubMed] [Google Scholar]
- 23.Yang J, Weinberg RA. Epithelial-mesenchymal transition: at the crossroads of development and tumor metastasis. Dev Cell. 2008;14:818–829. doi: 10.1016/j.devcel.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 24.Scheel C, Eaton EN, Li SH, Chaffer CL, Reinhardt F, Kah KJ, Bell G, Guo W, Rubin J, Richardson AL, Weinberg RA. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell. 2011;145:926–940. doi: 10.1016/j.cell.2011.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu J, Lamouille S, Derynck R. TGF-betainduced epithelial to mesenchymal transition. Cell Res. 2009;19:156–172. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hur K, Toiyama Y, Takahashi M, Balaguer F, Nagasaka T, Koike J, Hemmi H, Koi M, Boland CR, Goel A. MicroRNA-200c modulates epithelial-to-mesenchymal transition (EMT) in human colorectal cancer metastasis. Gut. 2013;62:1315–1326. doi: 10.1136/gutjnl-2011-301846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nieto MA. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu Rev Cell Dev Biol. 2011;27:347–376. doi: 10.1146/annurev-cellbio-092910-154036. [DOI] [PubMed] [Google Scholar]
- 28.Ikushima H, Miyazono K. TGFbeta signalling: a complex web in cancer progression. Nat Rev Cancer. 2010;10:415–424. doi: 10.1038/nrc2853. [DOI] [PubMed] [Google Scholar]
- 29.Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu CC, Simpson TR, Laklai H, Sugimoto H, Kahlert C, Novitskiy SV, De Jesus-Acosta A, Sharma P, Heidari P, Mahmood U, Chin L, Moses HL, Weaver VM, Maitra A, Allison JP, LeBleu VS, Kalluri R. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25:719–734. doi: 10.1016/j.ccr.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 31.Kanzawa M, Semba S, Hara S, Itoh T, Yokozaki H. WNT5A is a key regulator of the epithelial-mesenchymal transition and cancer stem cell properties in human gastric carcinoma cells. Pathobiology. 2013;80:235–244. doi: 10.1159/000346843. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Y, Du J, Zheng J, Liu J, Xu R, Shen T, Zhu Y, Chang J, Wang H, Zhang Z, Meng F, Wang Y, Chen Y, Xu Y, Gu L. EGF-reduced Wnt5a transcription induces epithelial-mesenchymal transition via Arf6-ERK signaling in gastric cancer cells. Oncotarget. 2015;6:7244–7261. doi: 10.18632/oncotarget.3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Voon DC, Wang H, Koo JK, Nguyen TA, Hor YT, Chu YS, Ito K, Fukamachi H, Chan SL, Thiery JP, Ito Y. Runx3 protects gastric epithelial cells against epithelial-mesenchymal transition-induced cellular plasticity and tumorigenicity. Stem Cells. 2012;30:2088–2099. doi: 10.1002/stem.1183. [DOI] [PubMed] [Google Scholar]
- 34.Watanabe T, Takahashi A, Suzuki K, Kurusu-Kanno M, Yamaguchi K, Fujiki H, Suganuma M. Epithelial-mesenchymal transition in human gastric cancer cell lines induced by TNFalpha-inducing protein of Helicobacter pylori. Int J Cancer. 2014;134:2373–2382. doi: 10.1002/ijc.28582. [DOI] [PubMed] [Google Scholar]
- 35.Chen L, Park SM, Tumanov AV, Hau A, Sawada K, Feig C, Turner JR, Fu YX, Romero IL, Lengyel E, Peter ME. CD95 promotes tumour growth. Nature. 2010;465:492–496. doi: 10.1038/nature09075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng HX, Cai YD, Wang YD, Cui XB, Xie TT, Li WJ, Peng L, Zhang Y, Wang ZQ, Wang J, Jiang B. Fas signaling promotes motility and metastasis through epithelial-mesenchymal transition in gastrointestinal cancer. Oncogene. 2013;32:1183–1192. doi: 10.1038/onc.2012.126. [DOI] [PubMed] [Google Scholar]
- 37.Zheng HX, Cai YD, Wang YD, Cui XB, Xie TT, Li WJ, Peng L, Zhang Y, Wang ZQ, Wang J, Jiang B. Fas signaling promotes motility and metastasis through epithelial-mesenchymal transition in gastrointestinal cancer. Oncogene. 2013;32:1183–1192. doi: 10.1038/onc.2012.126. [DOI] [PubMed] [Google Scholar]
- 38.Zheng H, Li W, Wang Y, Liu Z, Cai Y, Xie T, Shi M, Wang Z, Jiang B. Glycogen synthase kinase-3 beta regulates Snail and beta-catenin expression during Fas-induced epithelial-mesenchymal transition in gastrointestinal cancer. Eur J Cancer. 2013;49:2734–2746. doi: 10.1016/j.ejca.2013.03.014. [DOI] [PubMed] [Google Scholar]
- 39.Katoh M. Epithelial-mesenchymal transition in gastric cancer (Review) Int J Oncol. 2005;27:1677–1683. [PubMed] [Google Scholar]
- 40.Maier HJ, Schmidt-Strassburger U, Huber MA, Wiedemann EM, Beug H, Wirth T. NFkappaB promotes epithelial-mesenchymal transition, migration and invasion of pancreatic carcinoma cells. Cancer Lett. 2010;295:214–228. doi: 10.1016/j.canlet.2010.03.003. [DOI] [PubMed] [Google Scholar]
- 41.Lu J, Ye X, Fan F, Xia L, Bhattacharya R, Bellister S, Tozzi F, Sceusi E, Zhou Y, Tachibana I, Maru DM, Hawke DH, Rak J, Mani SA, Zweidler-McKay P, Ellis LM. Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of Jagged-1. Cancer Cell. 2013;23:171–185. doi: 10.1016/j.ccr.2012.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tsai JH, Donaher JL, Murphy DA, Chau S, Yang J. Spatiotemporal regulation of epithelialmesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell. 2012;22:725–736. doi: 10.1016/j.ccr.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chanrion M, Kuperstein I, Barriere C, El Marjou F, Cohen D, Vignjevic D, Stimmer L, Paul-Gilloteaux P, Bieche I, Tavares Sdos R, Boccia GF, Cacheux W, Meseure D, Fre S, Martignetti L, Legoix-Ne P, Girard E, Fetler L, Barillot E, Louvard D, Zinovyev A, Robine S. Concomitant Notch activation and p53 deletion trigger epithelial-to-mesenchymal transition and metastasis in mouse gut. Nat Commun. 2014;5:5005. doi: 10.1038/ncomms6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fre S, Huyghe M, Mourikis P, Robine S, Louvard D, Artavanis-Tsakonas S. Notch signals control the fate of immature progenitor cells in the intestine. Nature. 2005;435:964–968. doi: 10.1038/nature03589. [DOI] [PubMed] [Google Scholar]
- 45.Ansieau S, Bastid J, Doreau A, Morel AP, Bouchet BP, Thomas C, Fauvet F, Puisieux I, Doglioni C, Piccinin S, Maestro R, Voeltzel T, Selmi A, Valsesia-Wittmann S, Caron de Fromentel C, Puisieux A. Induction of EMT by twist proteins as a collateral effect of tumorpromoting inactivation of premature senescence. Cancer Cell. 2008;14:79–89. doi: 10.1016/j.ccr.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 46.Liu AN, Zhu ZH, Chang SJ, Hang XS. Twist expression associated with the epithelial-mesenchymal transition in gastric cancer. Mol Cell Biochem. 2012;367:195–203. doi: 10.1007/s11010-012-1333-8. [DOI] [PubMed] [Google Scholar]
- 47.Murai T, Yamada S, Fuchs BC, Fujii T, Nakayama G, Sugimoto H, Koike M, Fujiwara M, Tanabe KK, Kodera Y. Epithelial-to-mesenchymal transition predicts prognosis in clinical gastric cancer. J Surg Oncol. 2014;109:684–689. doi: 10.1002/jso.23564. [DOI] [PubMed] [Google Scholar]
- 48.Huang J, Xiao D, Li G, Ma J, Chen P, Yuan W, Hou F, Ge J, Zhong M, Tang Y, Xia X, Chen Z. EphA2 promotes epithelial-mesenchymal transition through the Wnt/beta-catenin pathway in gastric cancer cells. Oncogene. 2014;33:2737–2747. doi: 10.1038/onc.2013.238. [DOI] [PubMed] [Google Scholar]
- 49.Hou F, Yuan W, Huang J, Qian L, Chen Z, Ge J, Wu S, Chen J, Wang J, Chen Z. Overexpression of EphA2 correlates with epithelialmesenchymal transition-related proteins in gastric cancer and their prognostic importance for postoperative patients. Med Oncol. 2012;29:2691–2700. doi: 10.1007/s12032-011-0127-2. [DOI] [PubMed] [Google Scholar]
- 50.Ryu HS, Park do J, Kim HH, Kim WH, Lee HS. Combination of epithelial-mesenchymal transition and cancer stem cell-like phenotypes has independent prognostic value in gastric cancer. Hum Pathol. 2012;43:520–528. doi: 10.1016/j.humpath.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 51.Cui J, Shi M, Quan M, Xie K. Regulation of EMT by KLF4 in gastrointestinal cancer. Curr Cancer Drug Targets. 2013;13:986–995. doi: 10.2174/15680096113136660104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang H, Liu L, Wang Y, Zhao G, Xie R, Liu C, Xiao X, Wu K, Nie Y, Zhang H, Fan D. KLF8 involves in TGF-beta-induced EMT and promotes invasion and migration in gastric cancer cells. J Cancer Res Clin Oncol. 2013;139:1033–1042. doi: 10.1007/s00432-012-1363-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li Z, Zhang L, Ma Z, Yang M, Tang J, Fu Y, Mao Y, Hong X, Zhang Y. ETV1 induces epithelial to mesenchymal transition in human gastric cancer cells through the upregulation of Snail expression. Oncol Rep. 2013;30:2859–2863. doi: 10.3892/or.2013.2776. [DOI] [PubMed] [Google Scholar]
- 54.Zha L, Zhang J, Tang W, Zhang N, He M, Guo Y, Wang Z. HMGA2 elicits EMT by activating the Wnt/beta-catenin pathway in gastric cancer. Dig Dis Sci. 2013;58:724–733. doi: 10.1007/s10620-012-2399-6. [DOI] [PubMed] [Google Scholar]
- 55.Chen J, Wang T, Zhou YC, Gao F, Zhang ZH, Xu H, Wang SL, Shen LZ. Aquaporin 3 promotes epithelial-mesenchymal transition in gastric cancer. J Exp Clin Cancer Res. 2014;33:38. doi: 10.1186/1756-9966-33-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kannan A, Krishnan A, Ali M, Subramaniam S, Halagowder D, Sivasithamparam ND. Caveolin-1 promotes gastric cancer progression by up-regulating epithelial to mesenchymal transition by crosstalk of signalling mechanisms under hypoxic condition. Eur J Cancer. 2014;50:204–215. doi: 10.1016/j.ejca.2013.08.016. [DOI] [PubMed] [Google Scholar]
- 57.Liu Z, Chen L, Zhang X, Xu X, Xing H, Zhang Y, Li W, Yu H, Zeng J, Jia J. RUNX3 regulates vimentin expression via miR-30a during epithelial-mesenchymal transition in gastric cancer cells. J Cell Mol Med. 2014;18:610–623. doi: 10.1111/jcmm.12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Luo BH, Xiong F, Wang JP, Li JH, Zhong M, Liu QL, Luo GQ, Yang XJ, Xiao N, Xie B, Xiao H, Liu RJ, Dong CS, Wang KS, Wen JF. Epidermal growth factor-like domain-containing protein 7 (EGFL7) enhances EGF receptor-AKT signaling, epithelial-mesenchymal transition, and metastasis of gastric cancer cells. PLoS One. 2014;9:e99922. doi: 10.1371/journal.pone.0099922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miao L, Xiong X, Lin Y, Cheng Y, Lu J, Zhang J, Cheng N. Down-regulation of FoxM1 leads to the inhibition of the epithelial-mesenchymal transition in gastric cancer cells. Cancer Genet. 2014;207:75–82. doi: 10.1016/j.cancergen.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 60.Zang M, Zhang B, Zhang Y, Li J, Su L, Zhu Z, Gu Q, Liu B, Yan M. CEACAM6 promotes gastric cancer invasion and metastasis by inducing epithelial-mesenchymal transition via PI3K/AKT signaling pathway. PLoS One. 2014;9:e112908. doi: 10.1371/journal.pone.0112908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guo J, Fu Z, Wei J, Lu W, Feng J, Zhang S. PRRX1 promotes epithelial-mesenchymal transition through the Wnt/beta-catenin pathway in gastric cancer. Med Oncol. 2015;32:393. doi: 10.1007/s12032-014-0393-x. [DOI] [PubMed] [Google Scholar]
- 62.Li H, Xu L, Zhao L, Ma Y, Zhu Z, Liu Y, Qu X. Insulin-like growth factor-I induces epithelial to mesenchymal transition via GSK-3beta and ZEB2 in the BGC-823 gastric cancer cell line. Oncol Lett. 2015;9:143–148. doi: 10.3892/ol.2014.2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li J, Deng Z, Wang Z, Wang D, Zhang L, Su Q, Lai Y, Li B, Luo Z, Chen X, Chen Y, Huang X, Ma J, Wang W, Bi J, Guan X. Zipper-interacting protein kinase promotes epithelial-mesenchymal transition, invasion and metastasis through AKT and NF-kB signaling and is associated with metastasis and poor prognosis in gastric cancer patients. Oncotarget. 2015;6:8323–8338. doi: 10.18632/oncotarget.3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xu SH, Huang JZ, Xu ML, Yu G, Yin XF, Chen D, Yan GR. ACK1 promotes gastric cancer epithelial-mesenchymal transition and metastasis through AKT-POU2F1-ECD signalling. J Pathol. 2015;236:175–185. doi: 10.1002/path.4515. [DOI] [PubMed] [Google Scholar]
- 65.Yang Y, Yang C, Zhang J. C23 protein meditates bone morphogenetic protein-2-mediated EMT via up-regulation of Erk1/2 and Akt in gastric cancer. Med Oncol. 2015;32:76. doi: 10.1007/s12032-015-0547-5. [DOI] [PubMed] [Google Scholar]
- 66.Zhang J, Zhou Y, Yang Y. CCR7 pathway induces epithelial-mesenchymal transition through up-regulation of Snail signaling in gastric cancer. Med Oncol. 2015;32:467. doi: 10.1007/s12032-014-0467-9. [DOI] [PubMed] [Google Scholar]
- 67.Liu WF, Ji SR, Sun JJ, Zhang Y, Liu ZY, Liang AB, Zeng HZ. CD146 Expression Correlates with Epithelial-Mesenchymal Transition Markers and a Poor Prognosis in Gastric Cancer. Int J Mol Sci. 2012;13:6399–6406. doi: 10.3390/ijms13056399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xue Z, Wu X, Chen X, Liu Y, Wang X, Wu K, Nie Y, Fan D. Mesenchymal stem cells promote epithelial to mesenchymal transition and metastasis in gastric cancer though paracrine cues and close physical contact. J Cell Biochem. 2015;116:618–627. doi: 10.1002/jcb.25013. [DOI] [PubMed] [Google Scholar]
- 69.He X, Li B, Shao Y, Zhao N, Hsu Y, Zhang Z, Zhu L. Cell fusion between gastric epithelial cells and mesenchymal stem cells results in epithelial-to-mesenchymal transition and malignant transformation. BMC Cancer. 2015;15:24. doi: 10.1186/s12885-015-1027-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Watson SA, Grabowska AM, El-Zaatari M, Takhar A. Gastrin-active participant or bystander in gastric carcinogenesis? Nat Rev Cancer. 2006;6:936–946. doi: 10.1038/nrc2014. [DOI] [PubMed] [Google Scholar]
- 71.McCaig C, Duval C, Hemers E, Steele I, Pritchard DM, Przemeck S, Dimaline R, Ahmed S, Bodger K, Kerrigan DD, Wang TC, Dockray GJ, Varro A. The role of matrix metalloproteinase-7 in redefining the gastric microenvironment in response to Helicobacter pylori. Gastroenterology. 2006;130:1754–1763. doi: 10.1053/j.gastro.2006.02.031. [DOI] [PubMed] [Google Scholar]
- 72.Yin Y, Grabowska AM, Clarke PA, Whelband E, Robinson K, Argent RH, Tobias A, Kumari R, Atherton JC, Watson SA. Helicobacter pylori potentiates epithelial:mesenchymal transition in gastric cancer: links to soluble HB-EGF, gastrin and matrix metalloproteinase-7. Gut. 2010;59:1037–1045. doi: 10.1136/gut.2009.199794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yu H, Zeng J, Liang X, Wang W, Zhou Y, Sun Y, Liu S, Li W, Chen C, Jia J. Helicobacter pylori promotes epithelial-mesenchymal transition in gastric cancer by downregulating programmed cell death protein 4 (PDCD4) PLoS One. 2014;9:e105306. doi: 10.1371/journal.pone.0105306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Choi YJ, Kim N, Chang H, Lee HS, Park SM, Park JH, Shin CM, Kim JM, Kim JS, Lee DH, Jung HC. Helicobacter pylori-induced epithelial-mesenchymal transition, a potential role of gastric cancer initiation and an emergence of stem cells. Carcinogenesis. 2015;36:553–563. doi: 10.1093/carcin/bgv022. [DOI] [PubMed] [Google Scholar]
- 75.Duan H, Qu L, Shou C. Mycoplasma hyorhinis induces epithelial-mesenchymal transition in gastric cancer cell MGC803 via TLR4-NF-kappaB signaling. Cancer Lett. 2014;354:447–454. doi: 10.1016/j.canlet.2014.08.018. [DOI] [PubMed] [Google Scholar]
- 76.Chiba T, Marusawa H, Ushijima T. Inflammation-associated cancer development in digestive organs: mechanisms and roles for genetic and epigenetic modulation. Gastroenterology. 2012;143:550–563. doi: 10.1053/j.gastro.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 77.Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, Saito Y, Johnson RS, Kretzler M, Cohen CD, Eckardt KU, Iwano M, Haase VH. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. 2007;117:3810–3820. doi: 10.1172/JCI30487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ren YH, Liu KJ, Wang M, Yu YN, Yang K, Chen Q, Yu B, Wang W, Li QW, Wang J, Hou ZY, Fang JY, Yeh ET, Yang J, Yi J. De-SUMOylation of FOXC2 by SENP3 promotes the epithelial-mesenchymal transition in gastric cancer cells. Oncotarget. 2014;5:7093–7104. doi: 10.18632/oncotarget.2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bawa-Khalfe T, Lu LS, Zuo Y, Huang C, Dere R, Lin FM, Yeh ET. Differential expression of SUMO-specific protease 7 variants regulates epithelial-mesenchymal transition. Proc Natl Acad Sci U S A. 2012;109:17466–17471. doi: 10.1073/pnas.1209378109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shan T, Cui X, Li W, Lin W, Li Y, Chen X, Wu T. Novel regulatory program for norepinephrine-induced epithelial-mesenchymal transition in gastric adenocarcinoma cell lines. Cancer Sci. 2014;105:847–856. doi: 10.1111/cas.12438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Guo J, Wang B, Fu Z, Wei J, Lu W. Hypoxic Microenvironment Induces EMT and Upgrades Stem-Like Properties of Gastric Cancer Cells. Technol Cancer Res Treat. 2015 doi: 10.1177/1533034614566413. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 82.Zhou J, Li K, Gu Y, Feng B, Ren G, Zhang L, Wang Y, Nie Y, Fan D. Transcriptional upregulation of RhoE by hypoxia-inducible factor (HIF)-1 promotes epithelial to mesenchymal transition of gastric cancer cells during hypoxia. Biochem Biophys Res Commun. 2011;415:348–354. doi: 10.1016/j.bbrc.2011.10.065. [DOI] [PubMed] [Google Scholar]
- 83.Matsuoka J, Yashiro M, Doi Y, Fuyuhiro Y, Kato Y, Shinto O, Noda S, Kashiwagi S, Aomatsu N, Hirakawa T, Hasegawa T, Shimizu K, Shimizu T, Miwa A, Yamada N, Sawada T, Hirakawa K. Hypoxia stimulates the EMT of gastric cancer cells through autocrine TGFbeta signaling. PLoS One. 2013;8:e62310. doi: 10.1371/journal.pone.0062310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liu N, Wang Y, Zhou Y, Pang H, Zhou J, Qian P, Liu L, Zhang H. Kruppel-like factor 8 involved in hypoxia promotes the invasion and metastasis of gastric cancer via epithelial to mesenchymal transition. Oncol Rep. 2014;32:2397–2404. doi: 10.3892/or.2014.3495. [DOI] [PubMed] [Google Scholar]
- 85.Shen X, Xue Y, Si Y, Wang Q, Wang Z, Yuan J, Zhang X. The unfolded protein response potentiates epithelial-to-mesenchymal transition (EMT) of gastric cancer cells under severe hypoxic conditions. Med Oncol. 2015;32:447. doi: 10.1007/s12032-014-0447-0. [DOI] [PubMed] [Google Scholar]
- 86.Rosivatz E, Becker I, Specht K, Fricke E, Luber B, Busch R, Hofler H, Becker KF. Differential expression of the epithelial-mesenchymal transition regulators snail, SIP1, and twist in gastric cancer. Am J Pathol. 2002;161:1881–1891. doi: 10.1016/S0002-9440(10)64464-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ohta H, Aoyagi K, Fukaya M, Danjoh I, Ohta A, Isohata N, Saeki N, Taniguchi H, Sakamoto H, Shimoda T, Tani T, Yoshida T, Sasaki H. Cross talk between hedgehog and epithelialmesenchymal transition pathways in gastric pit cells and in diffuse-type gastric cancers. Br J Cancer. 2009;100:389–398. doi: 10.1038/sj.bjc.6604846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tanabe S, Aoyagi K, Yokozaki H, Sasaki H. Gene expression signatures for identifying diffuse-type gastric cancer associated with epithelial-mesenchymal transition. Int J Oncol. 2014;44:1955–1970. doi: 10.3892/ijo.2014.2387. [DOI] [PubMed] [Google Scholar]
- 89.Greaves M. Cancer stem cells: back to Darwin? Semin Cancer Biol. 2010;20:65–70. doi: 10.1016/j.semcancer.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 90.Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- 91.de Lau W, Barker N, Low TY, Koo BK, Li VS, Teunissen H, Kujala P, Haegebarth A, Peters PJ, van de Wetering M, Stange DE, van Es JE, Guardavaccaro D, Schasfoort RB, Mohri Y, Nishimori K, Mohammed S, Heck AJ, Clevers H. Lgr5 homologues associate with Wnt receptors and mediate R-spondin signalling. Nature. 2011;476:293–297. doi: 10.1038/nature10337. [DOI] [PubMed] [Google Scholar]
- 92.Barker N, Huch M, Kujala P, van de Wetering M, Snippert HJ, van Es JH, Sato T, Stange DE, Begthel H, van den Born M, Danenberg E, van den Brink S, Korving J, Abo A, Peters PJ, Wright N, Poulsom R, Clevers H. Lgr5(+ve) stem cells drive self-renewal in the stomach and build long-lived gastric units in vitro. Cell Stem Cell. 2010;6:25–36. doi: 10.1016/j.stem.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 93.Ferlay J, Shin HR, Bray F, Forman D, Mathers C, Parkin DM. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer. 2010;127:2893–2917. doi: 10.1002/ijc.25516. [DOI] [PubMed] [Google Scholar]
- 94.Susman S, Barnoud R, Bibeau F, Borini F, Pocard M, Tomuleasa C, Sabourin JC. The Lauren classification highlights the role of epithelial-to-mesenchymal transition in gastric carcinogenesis: an immunohistochemistry study of the STAT3 and adhesion molecules expression. J Gastrointestin Liver Dis. 2015;24:77–83. doi: 10.15403/jgld.2014.1121.sus. [DOI] [PubMed] [Google Scholar]
- 95.Zheng H, Li W, Wang Y, Xie T, Cai Y, Wang Z, Jiang B. miR-23a inhibits E-cadherin expression and is regulated by AP-1 and NFAT4 complex during Fas-induced EMT in gastrointestinal cancer. Carcinogenesis. 2014;35:173–183. doi: 10.1093/carcin/bgt274. [DOI] [PubMed] [Google Scholar]
- 96.Jiang CG, Lv L, Liu FR, Wang ZN, Na D, Li F, Li JB, Sun Z, Xu HM. Connective tissue growth factor is a positive regulator of epithelial-mesenchymal transition and promotes the adhesion with gastric cancer cells in human peritoneal mesothelial cells. Cytokine. 2013;61:173–180. doi: 10.1016/j.cyto.2012.09.013. [DOI] [PubMed] [Google Scholar]
- 97.Yuan D, Xia H, Zhang Y, Chen L, Leng W, Chen T, Chen Q, Tang Q, Mo X, Liu M, Bi F. P-Akt/miR200 signaling regulates epithelial-mesenchymal transition, migration and invasion in circulating gastric tumor cells. Int J Oncol. 2014;45:2430–2438. doi: 10.3892/ijo.2014.2644. [DOI] [PubMed] [Google Scholar]
- 98.Yoo YA, Kang MH, Lee HJ, Kim BH, Park JK, Kim HK, Kim JS, Oh SC. Sonic hedgehog pathway promotes metastasis and lymphangiogenesis via activation of Akt, EMT, and MMP-9 pathway in gastric cancer. Cancer Res. 2011;71:7061–7070. doi: 10.1158/0008-5472.CAN-11-1338. [DOI] [PubMed] [Google Scholar]
- 99.Hooper JE, Scott MP. Communicating with Hedgehogs. Nat Rev Mol Cell Biol. 2005;6:306–317. doi: 10.1038/nrm1622. [DOI] [PubMed] [Google Scholar]
- 100.Cho HJ, Park SM, Kim IK, Nam IK, Baek KE, Im MJ, Yoo JM, Park SH, Ryu KJ, Han HT, Kim HJ, Hong SC, Kim KD, Pak Y, Kim JW, Lee CW, Yoo J. RhoGDI2 promotes epithelial-mesenchymal transition via induction of Snail in gastric cancer cells. Oncotarget. 2014;5:1554–1564. doi: 10.18632/oncotarget.1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Ureshino H, Murakami Y, Watari K, Izumi H, Kawahara A, Kage M, Arao T, Nishio K, Yanagihara K, Kinoshita H, Kuwano M, Ono M. N-myc downstream regulated gene 1 (NDRG1) promotes metastasis of human scirrhous gastric cancer cells through epithelial mesenchymal transition. PLoS One. 2012;7:e41312. doi: 10.1371/journal.pone.0041312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liao A, Wang W, Sun D, Jiang Y, Tian S, Li J, Yang X, Shi R. Bone morphogenetic protein 2 mediates epithelial-mesenchymal transition via AKT and ERK signaling pathways in gastric cancer. Tumour Biol. 2015;36:2773–8. doi: 10.1007/s13277-014-2901-1. [DOI] [PubMed] [Google Scholar]
- 103.Dai YH, Tang YP, Zhu HY, Lv L, Chu Y, Zhou YQ, Huo JR. ZEB2 promotes the metastasis of gastric cancer and modulates epithelial mesenchymal transition of gastric cancer cells. Dig Dis Sci. 2012;57:1253–1260. doi: 10.1007/s10620-012-2042-6. [DOI] [PubMed] [Google Scholar]
- 104.Yue Z, Feng W, Xiangke L, Liuxing W, Qingxia F, Jianbo G. WAVE3 promotes epithelial-mesenchymal transition of gastric cancer through upregulation of Snail. Cancer Gene Ther. 2014;21:499–506. doi: 10.1038/cgt.2014.52. [DOI] [PubMed] [Google Scholar]
- 105.Sakimura S, Sugimachi K, Kurashige J, Ueda M, Hirata H, Nambara S, Komatsu H, Saito T, Takano Y, Uchi R, Sakimura E, Shinden Y, Iguchi T, Eguchi H, Oba Y, Hoka S, Mimori K. The miR-506-Induced Epithelial-Mesenchymal Transition is Involved in Poor Prognosis for Patients with Gastric Cancer. Ann Surg Oncol. 2015;22(Suppl 3):1436–43. doi: 10.1245/s10434-015-4418-2. [DOI] [PubMed] [Google Scholar]
- 106.Jia L, Wu J, Zhang L, Chen J, Zhong D, Xu S, Xie C, Cai J. Restoration of miR-1228* expression suppresses epithelial-mesenchymal transition in gastric cancer. PLoS One. 2013;8:e58637. doi: 10.1371/journal.pone.0058637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang SH, Li X, Zhou LS, Cao ZW, Shi C, Zhou CZ, Wen YG, Shen Y, Li JK. microRNA-148a suppresses human gastric cancer cell metastasis by reversing epithelial-to-mesenchymal transition. Tumour Biol. 2013;34:3705–3712. doi: 10.1007/s13277-013-0954-1. [DOI] [PubMed] [Google Scholar]
- 108.Li Z, Ying X, Chen H, Ye P, Shen Y, Pan W, Zhang L. MicroRNA-194 inhibits the epithelialmesenchymal transition in gastric cancer cells by targeting FoxM1. Dig Dis Sci. 2014;59:2145–2152. doi: 10.1007/s10620-014-3159-6. [DOI] [PubMed] [Google Scholar]
- 109.Cong N, Du P, Zhang A, Shen F, Su J, Pu P, Wang T, Zjang J, Kang C, Zhang Q. Downregulated microRNA-200a promotes EMT and tumor growth through the wnt/betacatenin pathway by targeting the E-cadherin repressors ZEB1/ZEB2 in gastric adenocarcinoma. Oncol Rep. 2013;29:1579–1587. doi: 10.3892/or.2013.2267. [DOI] [PubMed] [Google Scholar]
- 110.Zhang L, Wang X, Chen P. MiR-204 down regulates SIRT1 and reverts SIRT1-induced epithelial-mesenchymal transition, anoikis resistance and invasion in gastric cancer cells. BMC Cancer. 2013;13:290. doi: 10.1186/1471-2407-13-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liu S, Cui J, Liao G, Zhang Y, Ye K, Lu T, Qi J, Wan G. MiR-137 regulates epithelial-mesenchymal transition in gastrointestinal stromal tumor. Tumour Biol. 2014;35:9131–9138. doi: 10.1007/s13277-014-2177-5. [DOI] [PubMed] [Google Scholar]
- 112.Zhao X, He L, Li T, Lu Y, Miao Y, Liang S, Guo H, Bai M, Xie H, Luo G, Zhou L, Shen G, Guo C, Bai F, Sun S, Wu K, Nie Y, Fan D. SRF expedites metastasis and modulates the epithelial to mesenchymal transition by regulating miR-199a-5p expression in human gastric cancer. Cell Death Differ. 2014;21:1900–13. doi: 10.1038/cdd.2014.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang Z, Liu S, Shi R, Zhao G. miR-27 promotes human gastric cancer cell metastasis by inducing epithelial-to-mesenchymal transition. Cancer Genet. 2011;204:486–491. doi: 10.1016/j.cancergen.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 114.Lu W, Xu Z, Zhang M, Zuo Y. MiR-19a promotes epithelial-mesenchymal transition through PI3K/AKT pathway in gastric cancer. Int J Clin Exp Pathol. 2014;7:7286–7296. [PMC free article] [PubMed] [Google Scholar]
- 115.Li H, Xu L, Li C, Zhao L, Ma Y, Zheng H, Li Z, Zhang Y, Wang R, Liu Y, Qu X. Ubiquitin ligase Cbl-b represses IGF-I-induced epithelial mesenchymal transition via ZEB2 and microRNA-200c regulation in gastric cancer cells. Mol Cancer. 2014;13:136. doi: 10.1186/1476-4598-13-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Xu ZY, Yu QM, Du YA, Yang LT, Dong RZ, Huang L, Yu PF, Cheng XD. Knockdown of long non-coding RNA HOTAIR suppresses tumor invasion and reverses epithelial-mesenchymal transition in gastric cancer. Int J Biol Sci. 2013;9:587–597. doi: 10.7150/ijbs.6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Han Y, Ye J, Wu D, Wu P, Chen Z, Chen J, Gao S, Huang J. LEIGC long non-coding RNA acts as a tumor suppressor in gastric carcinoma by inhibiting the epithelial-to-mesenchymal transition. BMC Cancer. 2014;14:932. doi: 10.1186/1471-2407-14-932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kim MA, Lee HS, Lee HE, Kim JH, Yang HK, Kim WH. Prognostic importance of epithelialmesenchymal transition-related protein expression in gastric carcinoma. Histopathology. 2009;54:442–451. doi: 10.1111/j.1365-2559.2009.03247.x. [DOI] [PubMed] [Google Scholar]
- 119.Fanelli MF, Chinen LT, Begnami MD, Costa WL Jr, Fregnami JH, Soares FA, Montagnini AL. The influence of transforming growth factor-alpha, cyclooxygenase-2, matrix metalloproteinase (MMP)-7, MMP-9 and CXCR4 proteins involved in epithelial-mesenchymal transition on overall survival of patients with gastric cancer. Histopathology. 2012;61:153–161. doi: 10.1111/j.1365-2559.2011.04139.x. [DOI] [PubMed] [Google Scholar]
- 120.Galvan JA, Astudillo A, Vallina A, Fonseca PJ, Gomez-Izquierdo L, Garcia-Carbonero R, Gonzalez MV. Epithelial-mesenchymal transition markers in the differential diagnosis of gastroenteropancreatic neuroendocrine tumors. Am J Clin Pathol. 2013;140:61–72. doi: 10.1309/AJCPIV40ISTBXRAX. [DOI] [PubMed] [Google Scholar]
- 121.Mirza A, Foster L, Valentine H, Welch I, West CM, Pritchard S. Investigation of the epithelial to mesenchymal transition markers S100A4, vimentin and Snail1 in gastroesophageal junction tumors. Dis Esophagus. 2014;27:485–492. doi: 10.1111/j.1442-2050.2012.01435.x. [DOI] [PubMed] [Google Scholar]
- 122.Xu GF, Zhang WJ, Sun Q, Xu X, Zou X, Guan W. Combined epithelial-mesenchymal transition with cancer stem cell-like marker as predictors of recurrence after radical resection for gastric cancer. World J Surg Oncol. 2014;12:368. doi: 10.1186/1477-7819-12-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Abdel-Rahman O. Targeting the hepatocyte growth factor/mesenchymal epithelial transition pathway in gastric cancer: biological rationale and clinical applications. Expert Rev Anticancer Ther. 2015;15:235–245. doi: 10.1586/14737140.2014.974564. [DOI] [PubMed] [Google Scholar]
- 124.Kim HP, Han SW, Song SH, Jeong EG, Lee MY, Hwang D, Im SA, Bang YJ, Kim TY. Testican-1-mediated epithelial-mesenchymal transition signaling confers acquired resistance to lapatinib in HER2-positive gastric cancer. Oncogene. 2014;33:3334–3341. doi: 10.1038/onc.2013.285. [DOI] [PubMed] [Google Scholar]
- 125.Grygielewicz P, Dymek B, Bujak A, Gunerka P, Stanczak A, Lamparska-Przybysz M, Wieczorek M, Dzwonek K, Zdzalik D. Epithelialmesenchymal transition confers resistance to selective FGFR inhibitors in SNU-16 gastric cancer cells. Gastric Cancer. 2014 doi: 10.1007/s10120-014-0444-1. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Han R, Xiong J, Xiao R, Altaf E, Wang J, Liu Y, Xu H, Ding Q, Zhang Q. Activation of betacatenin signaling is critical for doxorubicin-induced epithelial-mesenchymal transition in BGC-823 gastric cancer cell line. Tumour Biol. 2013;34:277–284. doi: 10.1007/s13277-012-0548-3. [DOI] [PubMed] [Google Scholar]
- 127.Han RF, Ji X, Dong XG, Xiao RJ, Liu YP, Xiong J, Zhang QP. An epigenetic mechanism underlying doxorubicin induced EMT in the human BGC-823 gastric cancer cell. Asian Pac J Cancer Prev. 2014;15:4271–4274. doi: 10.7314/apjcp.2014.15.10.4271. [DOI] [PubMed] [Google Scholar]
- 128.Zhu Y, Liu Y, Qian Y, Dai X, Yang L, Chen J, Guo S, Hisamitsu T. Research on the efficacy of Celastrus Orbiculatus in suppressing TGFbeta1-induced epithelial-mesenchymal transition by inhibiting HSP27 and TNF-alpha-induced NF-kappa B/Snail signaling pathway in human gastric adenocarcinoma. BMC Complement Altern Med. 2014;14:433. doi: 10.1186/1472-6882-14-433. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 129.Zhu Y, Liu Y, Qian Y, Dai X, Yang L, Chen J, Guo S, Hisamitsu T. Antimetastatic Effects of Celastrus orbiculatus on Human Gastric Adenocarcinoma by Inhibiting Epithelial-Mesenchymal Transition and NF-kappaB/Snail Signaling Pathway. Integr Cancer Ther. 2015;14:271–281. doi: 10.1177/1534735415572880. [DOI] [PubMed] [Google Scholar]
- 130.Li H, Wang Z, Zhang W, Qian K, Liao G, Xu W, Zhang S. VGLL4 inhibits EMT in part through suppressing Wnt/beta-catenin signaling pathway in gastric cancer. Med Oncol. 2015;32:83. doi: 10.1007/s12032-015-0539-5. [DOI] [PubMed] [Google Scholar]
- 131.Yuan CX, Zhou ZW, Yang YX, He ZX, Zhang X, Wang D, Yang T, Pan SY, Chen XW, Zhou SF. Danusertib, a potent pan-Aurora kinase and ABL kinase inhibitor, induces cell cycle arrest and programmed cell death and inhibits epithelial to mesenchymal transition involving the PI3K/Akt/mTOR-mediated signaling pathway in human gastric cancer AGS and NCI-N78 cells. Drug Des Devel Ther. 2015;9:1293–1318. doi: 10.2147/DDDT.S74964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhang B, Yang Y, Shi X, Liao W, Chen M, Cheng AS, Yan H, Fang C, Zhang S, Xu G, Shen S, Huang S, Chen G, Lv Y, Ling T, Zhang X, Wang L, Zhuge Y, Zou X. Proton pump inhibitor pantoprazole abrogates adriamycin-resistant gastric cancer cell invasiveness via suppression of Akt/GSK-beta/beta-catenin signaling and epithelial-mesenchymal transition. Cancer Lett. 2015;356:704–712. doi: 10.1016/j.canlet.2014.10.016. [DOI] [PubMed] [Google Scholar]
- 133.Yoon JH, Kang YH, Choi YJ, Park IS, Nam SW, Lee JY, Lee YS, Park WS. Gastrokine 1 functions as a tumor suppressor by inhibition of epithelial-mesenchymal transition in gastric cancers. J Cancer Res Clin Oncol. 2011;137:1697–1704. doi: 10.1007/s00432-011-1051-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Li LC, Peng Y, Liu YM, Wang LL, Wu XL. Gastric cancer cell growth and epithelial-mesenchymal transition are inhibited by gammasecretase inhibitor DAPT. Oncol Lett. 2014;7:2160–2164. doi: 10.3892/ol.2014.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]