

Abstract
Cardiac stem cells (CSCs) can differentiate into cardiac muscle-like cells upon stimulation by angiotensin II (Ang II). TNF receptor-associated factor 6 (TRAF6) has been shown to promote JNK- and p38-induced myogenic differentiation and mediate Smad-independent activation of TGF-β. However, the detailed mechanisms underlying the activation of these signaling pathways are not entirely known. Herein, we hypothesized that Ang II could promote the differentiation of CSCs into cardiac muscle-like cells by non-canonical TGF-β/TRAF6 signaling pathway, and sought to test the hypothesis. C-kit+ CSCs were isolated from neonatal Sprague Dawley (SD) rats, and their c-kit status was confirmed with immunofluorescence staining. A TGF-β type I receptor inhibitor (SB431542) was used to inhibit SMAD2/3 phosphorylation. The small interfering RNA (siRNA)-mediated knockdown of TRAF6 was used to investigate the role of TRAF6 in TGF-β signaling. Rescue of TRAF6 siRNA transfected cells with a 3’UTR-deleted siRNA insensitive construct was performed to rule out any off-target effects of the siRNA. TRAF6 dominant-negative (TRAF6DN) vector was constructed and used to infect c-kit+ CSCs. Our results showed that the increase in JNK and p38 activation by Ang-II was blocked by siRNA. After transfection by TRAF6-siRNA or Ad-TRAF6, the cardiac specific markers and Wnt signaling proteins were tested by Western blotting. Physical interactions between TRAF6 and TGF-β receptors were studied by co-immunoprecipitation. Forced expression of TRAF6 enhanced the expression of cTnT and Cx-43 but inhibited the expression of Wnt3a.Our data suggested that TRAF6 mediated Ang II-induced differential responses in c-kit+ CSCs via the non-canonical TGF-β signaling pathway.
Keywords: Cardiac stem cells, differentiation, TGF-β, TRAF6, ubiquitination
Introduction
Both the renin-angiotensin system (RAS) and transforming growth factor-β (TGF-β) are key mediators of cardiac differentiation. Recent studies indicate that angiotensin II (Ang II) and TGF-β do not act independently from one another, but rather act as part of a network that promotes cardiac differentiation. Cardiac stem cells (CSCs) have been used to regenerate heart muscle in patients who have suffered from heart attacks, and those with acute or chronic heart failure [1]. An important subpopulation of endogenous CSCs was identified in the adult heart: myogenic CSCs (mCSCs), which are characterized by their expression of c-kit [2]. Indeed, c-kit+ CSCs could differentiate into the myocyte lineage in vitro, and play a role in healing following myocardial injury [3]. Elevated levels of local Ang II and TGF-β1 are often observed under such conditions that cause myocardial loss or cardiac fibrosis after acute myocardial damage. Ang II upregulates TGF-β1 expression via activation of the AT1 receptor in cardiac myocytes and fibroblasts [4], and its induction is absolutely required for Ang II-induced cardiac hypertrophy in vivo. Recent studies indicate that the downstream mediators of cardiac Ang II/TGF-β networking may include Smad proteins, TGFβ-activated kinase-1 (TAK1), and induction of hypertrophic responsiveness to h-adrenergic stimulation in cardiac myocytes [5]. However, it remains unclear how Ang II might affect the differentiation of endogenous CSCs.
Canonical TGF-β signaling is initiated with ligand-induced oligomerization of serine/threonine receptor kinases and phosphorylation of the cytoplasmic signaling molecules Smad2 and Smad3, which transduce the TGF-β signal from the cell surface into the nucleus to regulate transcription. Recently, non-canonical TGF-β signaling pathway has been realized, and perhaps the best-characterized non-Smad pathway is the JNK and p38 MAPK signaling cascades. Non-Smad TGF-β signaling pathway plays important roles in proliferation, migration, differentiation, and apoptosis in a variety of cells [6]. It has also been well established that Ang II increases the expression of connective tissue growth factor (CTGF) via the TGF-β1/TRAF6/MAPK pathway in migration and contractility of heart, liver, and lung fibroblasts [7]. TGF-β receptors interact with TRAF6 and induce the formation of poly-ubiquitin chains on TRAF6. Poly-ubiquitinated TRAF6 recruits TGF-β activated kinase 1 (TAK1) to activate JNK and p38 MAPK signaling [8].
By activating TAK1 and subsequently p38/JNK (c-Jun NH2-terminalprotein kinase), TRAF6 significantly promoted myogenic differentiation and muscle regeneration in adult mice [9]. Furthermore, TAK1/JNK was required for the Smad-independent activation of the mitochondrial apoptotic pathway [10]. Betty [11] demonstrated the requirement for the TRAF6 RING domain and site-specific auto-ubiquitination (Ub) of TRAF6 for the activation of TAK1. Poly-ubiquitination of TRAF6 and subsequent activation of TAK1 were further confirmed by other groups [8,9].
Because of the critical roles of TRAF6/TAK1 in myocardial cells, we hypothesized that TRAF6 ubiquitination and its role in TAK1 activation may be essential for myogenic differentiation of c-kit+ CSCs. In this study, we used a combination of molecular and chemical approaches to analyze the roles of TRAF6 in the differentiation of rat c-kit+ CSCs. Our data demonstrates that TRAF6 poly-ubiquitination is a critical upstream mediator of MAPK activation and myogenic differentiation marker expression, revealing novel roles for TRAF6-TAK1 in the myogenic differentiation of rat c-kit+ CSCs.
Materials and methods
Cell culture and the isolation of c-kit+ cells
One-to three-day-oldSprague-Dawley (SD) rats were anesthetized using methoxyflurane, and then perfused intracardially with 0.9% saline. The hearts of neonatal rats were then sliced, and explants were placed on poly-D-lysine-coated 10-cm plates containing complete explant medium (CEM) [12]. After 7 days, most primary cells from the explants had reached 50-70% confluency. To obtain c-kit+ CSCs, the cells were harvested and stained with FITC-conjugated monoclonal anti-c-kit antibodies (Cat. No. MAB1162F; Millipore) for 30 min. The c-kit+ cells were separated using fluorescence activated cell sorting (FACS), and then seeded in type I collagen from rat tail-coated plates (5-10 μg/cm2, C7661, Sigma) in cardiosphere-growing medium (CGM) (35% complete IMDM/65% DMEM/F-12 mix containing 2% B27, 0.1 mmol/L 2-mercaptoethanol, 10 ng/mL epidermal growth factor [EGF], 20 ng/mL basic fibroblast growth factor [bFGF], 40 nmol/L cardiotrophin-1, 40 nmol/L thrombin, and l-Glu, as in CEM) [13]. Cells (1 × 105) were seeded on coverslips, and then incubated with anti-c-kit primary antibodies (sc5535, Santa Cruz). Cells were then incubated with FITC-conjugated goat anti-rabbit antibodies (111-095-003, Jackson Laboratories) for 1 h, followed by 4’, 6-diamidino-2-phenylindole (DAPI; sc3598, Santa Cruz). An inverted fluorescence microscope was used to visualize the stained cells. Cells were used for the following experimentation before the fourth passage.
Flow Cytometry was used to analyze the expression patterns of CSCs markers. Cells were washed with PBS and incubated on ice for 30 min with fluorochrome-conjugated primary antibodies: anti CD34-PE; anti CD45-PE; anti CD133-PE (all from BD Bioscience).
The Medical Ethics Committee of the Xinhua Hospital affiliated with Shanghai Jiaotong University School of Medicine approved the study. The protocol conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication, 8th Edition, 2011).
Knockdown of TRAF6 using siRNA
Early-passage c-kit+ CSCs were seeded in 6-well plates at a density of 1 × 105 cells per well. Cells were transfected with TRAF6 siRNA (sc156004, Santa Cruz) or buffer (mock control) by using Oligofectamine™ Reagent (Catalog No. 12252-011, Invitrogen) according to the manufacturer’s protocol. Final concentration the siRNA duplex in culture medium was 0.1, 0.2, 0.5, or 1.0 pM. The transfection mixture was replaced with fresh CEM 24 h after transfection. A non-specific siRNA (sc36869, Santa Cruz) was used as the negative control for siRNA experiments. Knockdown efficiency was assessed by Western blot analysis of TRAF6.
Construction and expression of a dominant negative TRAF6 (TRAF6DN) from an adenoviral vector
The 2.7 kb protein coding region of TRAF6 was amplified with a SuperScript RT-PCR for long templates kit (Catalog No. 11922-010, Invitrogen). The sequencing primers were as follows: forward primer TGGAAGCACAGTGAAGAGGTTC, and reverse primer ATGTCATCAGCGAGGTCACATT. Amplified product was verified by DNA sequencing. Next, RING finger domain was removed to generate TRAF6DN (aa 289-522). TRAF6DN gene was cloned into the KpnI and BamHI sites of pShuttle-CMV-eGFP (enhanced green fluorescent protein) vector, resulting in pShuttle-eGFP-TRAF6DN plasmid. The shuttle plasmid was subsequently transformed into BJ5183 bacteria to recombine with the backbone vector pAdeno, and positive clones were selected for growth and propagation. Recombinant adenoviral vector pAd-eGFP-TRAF6DN was transfected into HEK293 cells for the packaging of Ad-eGFP-TRAF6DN. Virus was propagated through series infection of HEK293 cells and purified by CsCl equilibrium density centrifugation. Titration of the purified virus was carried out according to the instructions of the Adeno-X Rapid Titer Kit (Roche), and the titer of the virus stock used in this study was determined to be 2 × 109 TU/ml. Infection rate of CSC was > 95%. The TRAF6DN-eGFP fusion protein was stable in the infected cells, as confirmed by Western blotting.
Protein extraction and western blotting
To assess the phosphorylation of Smad2, p38, or JNK, c-kit+, cells were washed twice with PBS, and incubated in serum-free medium for 24 h followed by stimulation with SB431542 and/or Ang II. C-kit+ CSCs were treated with or without 0.5 μM SB431542 for 12 h. They were then treated with Ang II (200 ng/ml) for different time-periods (0, 15, or 30 min). Fresh serum-free medium was then added to the cells and incubated for 4 h. Immunoblotting was performed using phospho-specific antibodies to analyze various signaling pathways. JNK activity was assayed using substrate-specific phosphorylation and immunoprecipitation. In subsequent experiments, cell lysates were harvested at the time-point of maximal effect on phosphorylation of p38 and JNK. Western blotting was performed as described previously [14].
Statistical analysis
All analyses were performed with SPSS 17.0 software. All assays were performed independently three times and the quantitative data were shown as the mean ± SD or as indicated. Statistical significance was calculated with independent samples t test or one-way ANOVA, when appropriate. A significant difference was accepted when p < 0.05.
Results
Biological characteristics of c-kit+ CSCs
C-kit+ cell populations were purified from neonatal rat hearts using a stringent two-step strategy involving explant cultures followed by FACS. The heart explants from rats were grown as separate clones on plates (Figure 1A), and the cells migrated from the explants (Figure 1B-D). Newly isolated c-kit+ cells were seeded on to 100-mm tissue culture plates pre-coated with 0.2% gelatin (Figure 1E).
Figure 1.
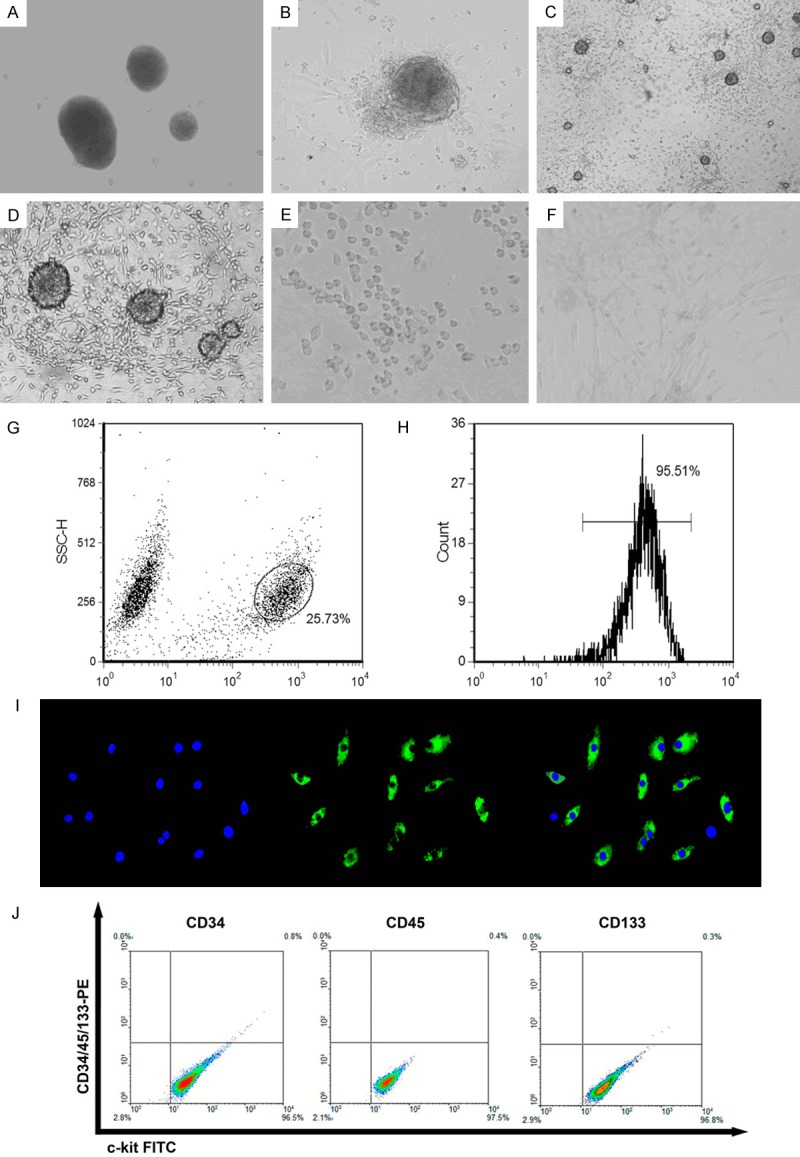
Isolation and flow cytometric sorting of c-kit+ CSCs. A. There was virtually no detectable outgrowth 24 h after plating. B. CEM stimulated the extensive outgrowth of cells, and only a small number of isolated cells were observed in the culture dish at 72 h. C and D. Most 7 day rat heart explants exhibited cellular outgrowth from the edge of the explants. E. FACS-isolated c-kit+ CSC cells from cardiac explants were replanted in CGM. F. Isolated cells were plated onto glass coverslips, which had been previously coated with type I collagen from rat tail. Angiotensin II could induce c-kit+ CSC cells into cardiomyocyte-like cells. G. Cells were isolated from cardiac explants and subjected to FACS as described in Methods section. Gating of c-kit+ CSCs. H. Representative post-sorting histogram identifying c-kit+ CSCs. After culture for 48 h, the c-kit-labeled cell population routinely constituted ~95.51% of the total gated cells. I. Immunocytochemistry of stained c-kit+ CSCs. These passage cells were incubated overnight with primary c-kit monoclonal antibodies (1:1500 dilution). The cell layers were then rinsed with PBS and incubated with FITC-conjugated anti-rabbit antibodies (1:200 dilution). Nuclei were counterstained with DAPI. J. These cells were positive for the expression of cell-surface marker c-kit and negative for expression of each one of cell-surface markers CD34, CD45, and CD133.
The cloned cardiosphere-derived cells were stained using anti-c-kit-FITC antibodies, and then sorted using FACS (Figure 1G and 1H).Cells were stained with anti-c-kit (green) antibodies, and counterstained with 1 µg/ml DAPI (blue) to visualize the nuclei (Figure 1I).
We investigated the expression of the stem cell markers CD117/c-kit, CD34, CD45, and CD133 in these cells. There was no detectable signals of CD34+, CD45+ and CD133+ cells in c-kit+ cells (Figure 1J).
The TGF-βR1 kinase activity is not required for Ang II-induced p38 and JNK activation
In the myocardium, both p38 and JNK transduction cascades have been implicated in regulating the hypertrophic response. We found that Ang II could induce c-kit+ CSC cells into cardiomyocyte-like cells (Figure 1F) and promote the expression of cardiac-specific proteins. To determine whether the activity of serine/threonine kinases was required for Ang II-induced p38 and JNK activation in c-kit+ CSCs, cells were treated with a specific TGF-βR1 kinase inhibitor (SB431542; cat. no. 616461, Merck). In Figure 2 (lane 1 vs. 2 and lane 1 vs. 3), we found that Ang II could induce Smad2, p38 and JNK activation in c-kit+ CSCs. Next, we sought to investigate whether the TGF-βR1 kinase activity was required for the activation of Smad2, p38 and JNK (lane 3 vs. 6). Treatment of c-kit+ CSCs with SB431542 decreased the phosphorylation of Smad2 regardless of Ang II stimulation, suggesting an important role of the TGF-βR1 kinase activity for activation of the canonical Smad-pathway (Figure 2C).
Figure 2.
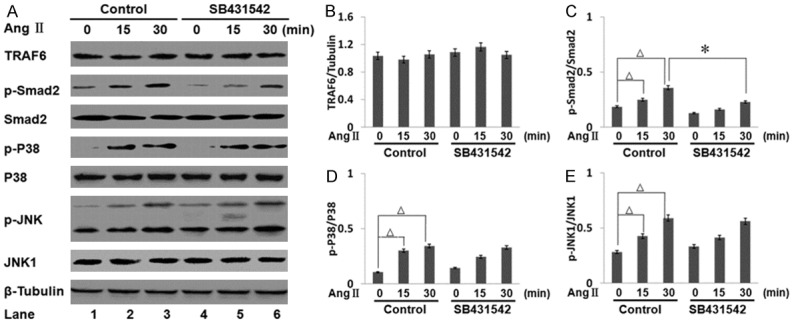
Effect of TGF-βR1 inhibitor SB431542 on the activation of Smad2, JNK, and p38 induced by Ang II. A. c-kit+ CSCs were treated with or without 0.5 μM SB431542 for 12 h, then treated with Ang II for 30 min or mock treated. The levels of phospho- or total Smad2, JNK1, and p38 were analyzed by Western blotting. All bar graphs are displayed as mean ± standard deviation (n = 3 per group; *p < 0.05). B. The expression of TRAF6 was unchanged by Ang II or SB431542. C. SB431542 inhibited Smad2 phosphorylation. D and E. JNK1 and p38 phosphorylation were unchanged by SB431542 treatment. Ang II could induce Smad2, p38 and JNK phosphorylation in c-kit+ CSCs (Δp < 0.05, lane 1 vs. 2, lane 1 vs. 3).
However, the expression of TRAF6 was affected by neither Ang II nor SB431542 treatment (Figure 2B). The Ang II-stimulated phosphorylation of p38 and JNK was elevated in c-kit+ CSCs in a time-dependent manner, and was not suppressed by SB431542 (Figures 2D and 3E). These results suggested that the TGF-βR1 kinase activity might be crucial for the activation of the canonical Smad-pathway, but dispensable for the Ang II-induced phosphorylation of p38 and JNK.
Figure 3.
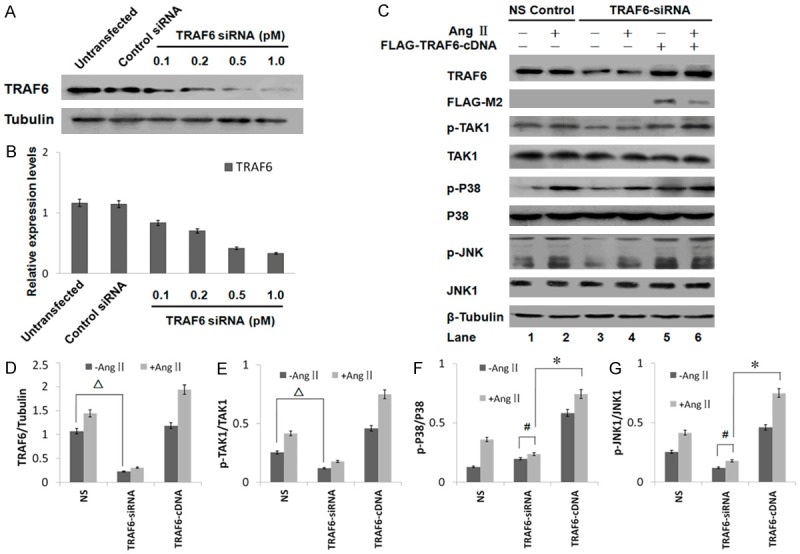
The expression of TRAF6 in c-kit+ CSCs transfected with TRAF6 siRNA. A. C-kit+ CSCs were transfected with increasing amounts of TRAF6 siRNA. Western blot showing TRAF6 expression in untransfected c-kit+ CSCs (Lane 1), and cells transfected with control siRNA (Lane 2), 0.1 pM TRAF6 siRNA (Lane 3), 0.2 pM TRAF6 siRNA (Lane 4), 0.5 pM TRAF6 siRNA (Lane 5), and 1.0 pM TRAF6 siRNA (Lane 6). The efficiency of RNAi was assessed by immunoblotting with anti-TRAF6 antibodies. B. Quantitative analysis of the Western blots. TRAF6 expression was regulated by the transfection of TRAF6 siRNA in a dose-dependent manner. All bar graphs are presented as mean ± standard deviation (n = 3). C. The expression of TRAF6 in c-kit+ CSCs treated with TRAF6 siRNA. C-kit+ CSCs were treated with or without 1.0 pmol of the indicated siRNA, then treated with Ang II for 30 min or mock treated. The levels of phospho- or total TAK1, JNK1, and p38 were analyzed by Western blotting. All bar graphs are presented as mean ± standard deviation (n = 3 per group). D. TRAF6 protein expression was decreased in cells transfected with TRAF6-targeting siRNA compared with control siRNA (∆p < 0.05, lane 1 vs. lane 3). E. TRAF6 siRNA decreased the stimulatory effect of Ang II on TAK1 phosphorylation (∆p < 0.05, lane 1 vs. lane 3). F and G. The levels of phospho- or total JNK and p38 were analyzed by Western blotting. Ang II-induced JNK and p38 phosphorylation was attenuated in TRAF6 siRNA-transfected cells (#p < 0.05, lane 3 vs. lane 4). JNK and p38 activation was rescued in TRAF6-siRNA transfected CSCs after transfection with FLAG-TRAF6, a siRNA insensitive TRAF6 construct. (*p < 0.05, lane 4 vs. lane 6).
TRAF6 is critical for Ang II-induced activation of p38 and JNK
Cardiac stem cells have been shown to spontaneously differentiate into a cardiomyocyte phenotype under standard culturing conditions, which can be enhanced by Ang II stimulation. Thus far, our results suggested that TGF-βR1 kinase activity might not be required for the Ang II-induced p38 and JNK phosphorylation. Therefore, using RNAi, we next investigated whether TRAF6 was involved in this signaling cascade, as TRAF6 had previously been reported to interact with TGF-βR1 to induce p38 and JNK phosphorylation through the Lys 63-linked ubiquitination of TAK1 [6]. TRAF6 siRNA transfected in c-kit+ CSCs inhibited the expression of TRAF6 in a dose-dependent manner (Figure 3A and 3B); 1 pM TRAF6 siRNA transfection resulted in an 80% decrease of TRAF6 expression as compared to control siRNA. As a result, the phosphorylation of TAK1, a downstream target of TRAF6, was also inhibited by TRAF6 siRNA. Ang II-induced phosphorylation of JNK and p38 in TRAF6 wild type c-kit+ CSC cells showed negligible effect in TRAF6 siRNA-transfected cells (Figure 3C, lane 2 vs. 4).
Next, rescue experiments were performed to exclude any off-target effect of TRAF6 knockdown. The re-introduction of a FLAG-tagged siRNA-insensitive TRAF6 construct lacking its 3’UTR into c-kit+ CSCs reversed the effect of TRAF6-specific siRNA on the ability of Ang II to activate TAK1, JNK, and p38 (Figure 3C, lane 4 vs. 6). Therefore, TRAF6 was an important upstream component of the MAPKs pathway which mediated Ang II-induced p38 and JNK activation in c-kit+ CSC cells.
The RING domain is required for the Lys63-linked poly-ubiquitination of TRAF6
It was previously reported that the interaction between TGF-βR1 and TRAF6 was required for TGF-β-induced auto-ubiquitination of TRAF6 and the subsequent activation of the TAK1-p38/JNK pathway [15]. Therefore, we assessed whether TGF-β receptors engaged TRAF6 to activate TAK1 in a TβR-dependent manner. As shown in Figure 4A, TRAF6 could bind to TβRI and TβRII after 30 min stimulation with Ang II. TRAF6 was co-immunoprecipitated with both TβRI and TβRII. In addition, Ang II promoted the association of endogenous TRAF6 with TGF-β receptors (Figure 4A). To assess the functional significance of this interaction, we examined the ubiquitination of TRAF6 in cultured c-kit+ CSCs. As depicted in Figure 4B, a ladder of high molecular weight TRAF6 bands were observed in the anti-TRAF6 immunocomplex, suggesting that 30 min of Ang II treatment promoted TRAF6 ubiquitination (Figure 4B). It has been proposed that TRAF6 is a RING-dependent Ub ligase that, in conjunction with Ubc13/Uev1A, catalyzes its own auto-ubiquitination via Lys63-linked poly-Ub chains [10].
Figure 4.
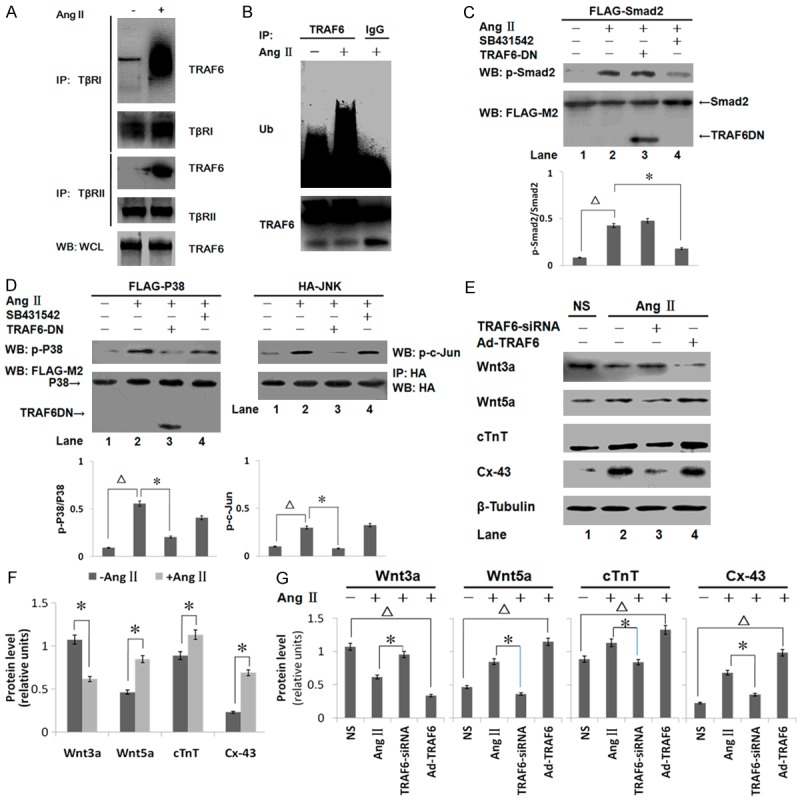
Ang II induces the interaction between TRAF6 and TGF-β receptors and promotes the poly-ubiquitination of TRAF6. A. Ang II stimulates the interaction between endogenous TRAF6 and TβRI or TβRII. C-kit+ CSCs were cross-linked using DSS, and TRAF6 levels were analyzed by Western blotting after immunoprecipitation with either TGF-βR1 or TGF-βR2 antibodies. WCL, whole cell lysates. B. Ang II-induced ubiquitination of endogenous TRAF6 in c-kit+ CSCs. Cells were lysed in RIPA buffer after treatment with Ang II. The amount of ubiquitinated TRAF6 was visualized using anti-Ub and anti-TRAF6 antibodies after immunoprecipitation. C. Smad2 phosphorylation was significantly elevated by Ang II (∆p < 0.05, lane 1 vs. lane 2). The TRAF6 deletion mutant (TRAF6DN) did not affect Ang II-induced Smad2 phosphorylation (lane 2 vs. lane 3). SB431542 reduced Smad2 phosphorylation (*p < 0.05, lane 2 vs. lane 4). D. The levels of phosphorylated and total p38 were analyzed by Western blotting. In addition, HA-JNK was immunopurified from 2 × 106 cells and tested for kinase activity using purified GST-tagged c-Jun. The phosphorylation of p38 and c-Jun were increased markedly by Ang II (∆p < 0.05, lane 1 vs. lane 2). The TRAF6 mutant inhibited the activation of both p38 and JNK (*p < 0.05, lane 2 vs. lane 3). SB431542 had no effect on the phosphorylation of p38 or c-Jun (lane 2 vs. lane 4). E. The effect of transfecting TRAF6-siRNA or Ad-TRAF6 into c-kit+ CSCs was assessed after treatment with or without Ang II. The levels of Wnt3a, Wnt5a, and cTnT in cell lysates were analyzed by Western blotting. F. Expression levels were normalized against β-Tubulin. Wnt3a protein expression was decreased after Ang II treatment, whereas Wnt5a, Cx-43 and cTnT were increased in c-kit+ CSCs (*p < 0.05 lane 1 vs. lane 2). G. RNAi-mediated silencing of TRAF6 altered the expression of Wnt and cardiac-specific proteins. The overexpression of TRAF6 inhibited Wnt3a expression. The levels of Wnt5a, cTnT, and Cx-43 were increased significantly in the Ad-TRAF6 group compared with the NS group. All bar graphs are displayed as mean ± standard deviation (n = 3 per group; *p < 0.05 lane 2 vs. lane 3; ∆p < 0.05 lane 1 vs. lane 4).
We next assessed whether the ubiquitination of TRAF6 was associated with the activation of downstream protein kinases. We co-expressed FLAG-tagged Smad2, FLAG-tagged p38, or HA-tagged JNK with a dominant-negative TRAF6 mutant lacking the RING domain in c-kit+ CSCs. Smad2 phosphorylation in response to Ang II was not affected by the TRAF6 mutant (TRAF6DN) (Figure 4C). However, Ang II-induced p38 activation was inhibited significantly by TRAF6DN, but not by SB431542, suggesting a specific requirement for the TRAF6 RING domain for p38 phosphorylation (Figure 4D). A similar result was observed when the activation of JNK was assessed. C-Jun is activated via JNK-stimulated double phosphorylation. In this study, recombinant GST-c-Jun was used to study the activation of JNK in vitro following the immunoprecipitation of HA-tagged JNK in transfected c-kit+ CSCs (Figure 4D). TRAF6 RING domain deletion inhibited JNK and p38 activities, suggesting that the Lys63-linked ubiquitination of TRAF6 was required for Ang II-induced p38 and JNK activation.
TRAF6 is a key signal transducer during Ang II-mediated myogenic differentiation
We investigated whether TRAF6 played a role in Ang II-mediated myogenic differentiation responses using TRAF6-specific siRNA. Treating c-kit+ CSCs with Ang II promoted the expression of the cardiac-specific proteins cTnT and Cx-43. However, Ang II-stimulated upregulation of Wnt5a, cTnT, and Cx-43 was inhibited by TRAF6 knockdown (p < 0.05, Figure 4E and 4F, Ang II vs. TRAF6-siRNA), indicating that Ang II-induced myogenic differentiation might be mediated by TRAF6. To further confirm this, we overexpressed a TRAF6 siRNA-insensitive TRAF6 transcript from an adenoviral vector and rescued the decrease of the expression of Wnt5a, Cx-43, and cTnT. Overexpression of TRAF6 promoted the expression of cardiac-specific proteins. These data suggest that TRAF6 participates in Ang II-induced cardiac-specific protein expression by regulating non-canonical TGF-β signaling.
Discussion
Cardiovascular diseases, including heart failure, are the second leading cause of death in China. The recent heart failure epidemic has stimulated interest in understanding cardiac regeneration. Methods of transplantation to replace weakened or lost cardiomyocytes would be extremely useful for the treatment of heart failure [16]. However, it is difficult to obtain sufficient transplantable cardiomyocytes. CSCs are necessary and sufficient for the regeneration and repair of myocardial damage [17,18]. C-kit positive CSCs are critical for cardiomyogenesis in the developing heart.
However, endogenous c-kit+ cells have been controversial, with some critics arguing whether they contribute to differentiated cardiomyocytes in the heart during development [19]. The study found that endogenous c-kit+ cells could generate cardiomyocytes at a functionally insignificant level in vivo. Therefore, there is a need for more in-depth studies of the mechanism of myogenic differentiation of c-kit+ CSCs.
Ang II plays a critical role in cardiac remodeling and promotes cardiac myocyte hypertrophy. Ang II induced cardiac-specific protein expression in cultured rat neonatal cardiomyocytes in a dose-dependent manner [20]. However, the direct effect of Ang II on myogenic differentiation has not been previously investigated in resident CSCs.
Adult stem cells are insufficient to repair the ischemic myocardium after injury. We demonstrated previously that bone marrow mesenchymal stem cells (BMMSCs) could promote the differentiation of c-kit+ CSCs via the Smad-dependent TGF-β pathway [14]. However, TGF-β-induced Smad activation was necessary, but not sufficient, for the induction of myocardial differentiation in c-kit+ CSCs. Besides canonical Smad pathways, TGF-β also signals via non-Smad pathways such as the p38 and JNK pathways, which also participate in myogenesis [21].
Studies with experimental models of myocardial infarction (MI) and with pressure overload have shown increased myocardial TGF-β1, TβRI, and TAK1 expression, suggesting the involvement of TGF-β1 in fibrosis and hypertrophic growth of cardiomyocytes [8]. More studies have confirmed that non-canonical TGF-β signaling interacts with the TGF-β receptor complex, and is required for the Smad-independent activation of JNK and p38. Although TRAF6 was initially identified as a kinase that regulates skeletal muscle differentiation and the regeneration of mouse myoblasts [20], these effects have not been described previously in cardiac progenitor cells. Therefore, we examined whether TGF-β activated TRAF6/TAK1 in rat c-kit+ CSCs, a specific cell population that improves post-infarction left ventricular dysfunction.
A recent study suggested that Ang II-induced cardiomyocyte hypertrophy in vitro occurs in a TAK1-dependent, but Smad-independent, manner [22]. In our studies, we confirmed that TAK1 was involved in the Ang II-induced MAPK activation through TRAF6 activation (Figure 3C). Ang II promoted the phosphorylation of Smad2, p38, and JNK. The inhibition of TGF-βRI serine/threonine kinase activity by SB431542 led to downregulated Smad2 phosphorylation in rat c-kit+ CSCs, but spared the phosphorylation of p38 and JNK.
TRAF6 might play a critical role in the induction of cardiogenesis through the activation of p38 and JNK. RNA interference (RNAi) was used to silence TRAF6 expression, which inhibited Ang II-induced TAK1 phosphorylation. In addition, transfection with TRAF6-siRNA inhibited p38 and JNK phosphorylation. These data suggested that TRAF6 mediates the stimulatory effect of Ang II on p38 and JNK in c-kit+ CSCs.
We then determined whether TRAF6 interacted with TGF-βR after Ang II-stimulation. C-kit+ CSCs were treated with DSS to fix protein-protein interactions, and the protein complexes were then immunoprecipitated using anti-TβRI or anti-TβRII antibodies followed by immunoblotting for TRAF6. TRAF6 has a tumor necrosis factor receptor (TNFR)-associated factor (TRAF) domain in its carboxyl terminus, as well as a RING finger domain (a cluster of zinc fingers) and a coiled-coil domain, consistent with other TRAF family proteins [23]. Through the employment of a RING domain deletion mutant, we established that the E3 ligase activity of TRAF6 was essential for its function as a mediator for Ang II-induced MAPK and JNK activation.
Wnt signaling plays important roles in the regulation of early cardiomyogenesis and the differentiation of cardiac progenitor cells into myocardial cells [23]. For example, the treatment of cardiac progenitor cells with Wnt inhibitors stimulated cardiomyocyte formation, whereas Wnt-3a inhibited this process [24]. TAK1, a member of the mitogen-activated protein kinase kinase kinase (MAPKKK) family, was originally identified as an effector of TGF-β-induced p38 activation, which counteracts the Wnt pathway [25]. Recent findings also demonstrated that TAK1 functions as a MAPKKK in the p38 pathway to promote myogenic differentiation [27]. Specifically, the TAK1 pathway provides a negative feedback mechanism for Wnt signaling. We observed a much lower expression of Wnt3a after Ad -TRAF6 transfection in CSCs. At the same time, the expression of cardiac-specific proteins increased significantly. The modulatory effect of Ang II was attenuated by TRAF6-siRNA. Therefore, we proposed that TRAF6/TAK1 played an important role in the induction of cardiogenesis by regulating the Wnt signaling cascade in rat c-kit+ CSCs. This study provided experimental evidence demonstrating that TRAF6 has the capacity to promote the differentiation of rat c-kit+ CSCs induced by Ang II.
In conclusion, this study demonstrated that TRAF6 functions as an important mediator for Ang II-induced p38 and JNK activation, and cardiac-specific proteins cTnT and Cx-43 expression in c-kit+ CSCs. To achieve this function, an intact RING domain of TRAF6 as well as poly-ubiquitination and activation of TAK1 are required.
Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant No. 81270205), Shanghai Jiao Tong University School of MedicineScience and Technology Fund (Grant No. 14XJ10065) and Xinhua Hospital (Grant No. 13YJ01).
Disclosure of conflict of interest
None.
References
- 1.Tateishi K, Takehara N, Matsubara H, Oh H. Stemming heart failure with cardiac- or reprogrammed-stem cells. J Cell Mol Med. 2008;12:2217–2232. doi: 10.1111/j.1582-4934.2008.00487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hosoda T. C-kit-positive cardiac stem cells and myocardial regeneration. Am J Cardiovasc Dis. 2012;2:58–67. [PMC free article] [PubMed] [Google Scholar]
- 3.Cimini M, Fazel S, Zhuo S, Xaymardan M, Fujii H, Weisel RD, Li RK. c-kit dysfunction impairs myocardial healing after infarction. Circulation. 2007;116:I77–82. doi: 10.1161/CIRCULATIONAHA.107.708107. [DOI] [PubMed] [Google Scholar]
- 4.Akhurst RJ. The paradoxical TGF-beta vasculopathies. Nat Genet. 2012;44:838–839. doi: 10.1038/ng.2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferreira JP, Santos M. Heart failure and atrial fibrillation: from basic science to clinical practice. Int J Mol Sci. 2015;16:3133–3147. doi: 10.3390/ijms16023133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gu J, Liu X, Wang QX, Tan HW, Guo M, Jiang WF, Zhou L. Angiotensin II increases CTGF expression via MAPKs/TGF-beta1/TRAF6 pathway in atrial fibroblasts. Exp Cell Res. 2012;318:2105–2115. doi: 10.1016/j.yexcr.2012.06.015. [DOI] [PubMed] [Google Scholar]
- 7.Ehanire T, Ren L, Bond J, Medina M, Li G, Bashirov L, Chen L, Kokosis G, Ibrahim M, Selim A, Blobe GC, Levinson H. Angiotensin II stimulates canonical TGF-beta signaling pathway through angiotensin type 1 receptor to induce granulation tissue contraction. J Mol Med (Berl) 2015;93:303. doi: 10.1007/s00109-014-1211-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matsumoto-Ida M, Takimoto Y, Aoyama T, Akao M, Takeda T, Kita T. Activation of TGF-beta1-TAK1-p38 MAPK pathway in spared cardiomyocytes is involved in left ventricular remodeling after myocardial infarction in rats. Am J Physiol Heart Circ Physiol. 2006;290:H709–715. doi: 10.1152/ajpheart.00186.2005. [DOI] [PubMed] [Google Scholar]
- 9.Landstrom M. The TAK1-TRAF6 signalling pathway. Int J Biochem Cell Biol. 2010;42:585–589. doi: 10.1016/j.biocel.2009.12.023. [DOI] [PubMed] [Google Scholar]
- 10.Lamothe B, Webster WK, Gopinathan A, Besse A, Campos AD, Darnay BG. TRAF6 ubiquitin ligase is essential for RANKL signaling and osteoclast differentiation. Biochem Biophys Res Commun. 2007;359:1044–1049. doi: 10.1016/j.bbrc.2007.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lamothe B, Campos AD, Webster WK, Gopinathan A, Hur L, Darnay BG. The RING domain and first zinc finger of TRAF6 coordinate signaling by interleukin-1, lipopolysaccharide, and RANKL. J Biol Chem. 2008;283:24871–80. doi: 10.1074/jbc.M802749200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith AJ, Lewis FC, Aquila I, Waring CD, Nocera A, Agosti V, Nadal-Ginard B, Torella D, Ellison GM. Isolation and characterization of resident endogenous c-Kit+ cardiac stem cells from the adult mouse and rat heart. Nat Protoc. 2014;9:1662–1681. doi: 10.1038/nprot.2014.113. [DOI] [PubMed] [Google Scholar]
- 13.Messina E, De Angelis L, Frati G, Morrone S, Chimenti S, Fiordaliso F, Salio M, Battaglia M, Latronico MV, Coletta M, Vivarelli E, Frati L, Cossu G, Giacomello A. Isolation and expansion of adult cardiac stem cells from human and murine heart. Circ Res. 2004;95:911–921. doi: 10.1161/01.RES.0000147315.71699.51. [DOI] [PubMed] [Google Scholar]
- 14.Cao Q, Wang F, Lin J, Xu Q, Chen S. Mesenchymal stem cells enhance the differentiation of c-kit+ cardiac stem cells. Front Biosci (Landmark Ed) 2012;17:1323–8. doi: 10.2741/3989. [DOI] [PubMed] [Google Scholar]
- 15.Mu Y, Gudey SK, Landstrom M. Non-Smad signaling pathways. Cell Tissue Res. 2012;347:11–20. doi: 10.1007/s00441-011-1201-y. [DOI] [PubMed] [Google Scholar]
- 16.Torella D, Ellison GM, Nadal-Ginard B. Adult c-kitpos Cardiac Stem Cells Fulfill Koch’s Postulates as Causal Agents for Cardiac Regeneration. Circ Res. 2014;114:e24–26. doi: 10.1161/CIRCRESAHA.113.303313. [DOI] [PubMed] [Google Scholar]
- 17.Martinez EC, Kofidis T. Adult stem cells for cardiac tissue engineering. J Mol Cell Cardiol. 2011;50:312–319. doi: 10.1016/j.yjmcc.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 18.Circulation Research Thematic Synopsis: stem cells & cardiac progenitor cells. Circ Res. 2013;113:e10–29. doi: 10.1161/CIRCRESAHA.113.301919. [DOI] [PubMed] [Google Scholar]
- 19.van Berlo JH, Kanisicak O, Maillet M, Vagnozzi RJ, Karch J, Lin SC, Middleton RC, Marban E, Molkentin JD. c-kit+ cells minimally contribute cardiomyocytes to the heart. Nature. 2014;509:337–341. doi: 10.1038/nature13309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sadoshima J, Izumo S. Molecular characterization of angiotensin II--induced hypertrophy of cardiac myocytes and hyperplasia of cardiac fibroblasts. Critical role of the AT1 receptor subtype. Circ Res. 1993;73:413–423. doi: 10.1161/01.res.73.3.413. [DOI] [PubMed] [Google Scholar]
- 21.Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009;19:128–139. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Watkins SJ, Borthwick GM, Oakenfull R, Robson A, Arthur HM. Angiotensin II-induced cardiomyocyte hypertrophy in vitro is TAK1-dependent and Smad2/3-independent. Hypertens Res. 2012;35:393–398. doi: 10.1038/hr.2011.196. [DOI] [PubMed] [Google Scholar]
- 23.Lanier M, Schade D, Willems E, Tsuda M, Spiering S, Kalisiak J, Mercola M, Cashman JR. Wnt inhibition correlates with human embryonic stem cell cardiomyogenesis: a structure-activity relationship study based on inhibitors for the Wnt response. J Med Chem. 2012;55:697–708. doi: 10.1021/jm2010223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brade T, Manner J, Kuhl M. The role of Wnt signalling in cardiac development and tissue remodelling in the mature heart. Cardiovasc Res. 2006;72:198–209. doi: 10.1016/j.cardiores.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 25.Chon E, Thompson V, Schmid S, Stein TJ. Activation of the canonical Wnt/beta-catenin signalling pathway is rare in canine malignant melanoma tissue and cell lines. J Comp Pathol. 2013;148:178–187. doi: 10.1016/j.jcpa.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]