

Abstract
Asthma is a chronic inflammatory disorder of the airways, the development of which is suppressed by regulatory T cells (Treg). Erythropoietin (EPO) is originally defined as a hematopoietic growth factor. Recently, the anti-inflammatory effects of EPO in asthma have been acknowledged. However, the underlying mechanisms remain ill-defined. Here, we showed that EPO treatment significantly reduced the severity of an ovalbumin (OVA)-induced asthma in mice, seemingly through promoting Foxp3-mediated activation of Treg cells in OVA-treated mouse lung. The activation of Treg cells resulted from increases in transforming growth factor β1 (TGFβ1), which were mainly produced by M2 macrophages (M2M). In vitro, Co-culture with M2M increased Foxp3 levels in Treg cells and the Treg cell number, in a TGFβ receptor signaling dependent manner. Moreover, elimination of macrophages abolished the therapeutic effects of EPO in vivo. Together, our data suggest that EPO may increase M2M, which activate Treg cells through TGFβ receptor signaling to mitigate the severity of asthma.
Keywords: OVA, asthma, erythropoietin (EPO), M2 macrophages (M2M), transforming growth factor β1 (TGFβ1), regulatory T cells (Treg), Foxp3
Introduction
Asthma is characterized by bronchial inflammation, hyperresponsive airways, and airflow obstruction. Most asthma cases are classified as allergic asthma, and characterized by activation of type-2 T helper cells, IgE production and eosinophilia, and is frequently related to an inappropriate T cell response to environmental allergens [1,2].
Regulatory T cells (Treg) are T cells that suppress potentially harmful immune responses. In animal models of asthma, Treg has been shown to suppress inflammatory responses and airway hyper-responsiveness to maintain peripheral immune tolerance [3-5]. Treg has been defined by markers including CD4, CD25 and a member of the forkhead box transcription factor -Foxp3- as a more specific marker and determinant for their development and function [5-7]. Foxp3 is crucial for naive T cell differentiation towards the Treg phenotype and its expansion [5-12]. However, the molecular basis underlying the activation of Treg through Foxp3 has not been clarified yet.
Erythropoietin (EPO) is a 30.4 kDa glycoprotein that regulates the rate of red blood cell production through binding to its specific cell surface receptors, has been widely used for the treatment of anemia [13]. Moreover, EPO has been shown to process antioxidant, anti-apoptotic, anti-inflammatory and angiogenic effects [14,15]. Recently, EPO has been reported as a treatment for experimental asthma [16]. However, the underlying mechanisms remain poorly defined.
Macrophages (marked by F4/80 expression) can regulate bronchial inflammation, hyperresponsive airways, and airflow obstruction in the lung diseases [17]. Besides the classically activated macrophages (also called M1 macrophages, M1M), which respond to inflammatory stimuli, and appear early on, there are also the alternatively activated macrophages (M2 macrophages, M2M), which appear later, to mediate humoral immunity and tissue repair [18,19]. M2M secrete a wide range of chemokines, enzymes and growth factors to promote neovascularization, fibrosis and tissue repair [20-22]. Of note, M2M have recently been shown to secret extremely high levels of transforming growth factor β1 (TGFβ1) in a pancreas injury model [20].
Here, we showed that EPO treatment significantly reduced the severity of an ovalbumin (OVA)-induced asthma in mice, seemingly through promoting Foxp3-mediated activation of Treg cells in OVA-treated mouse lung. The activation of Treg cells resulted from increases in TGFβ1, which were mainly produced by M2M. In vitro, Co-culture with M2M increased Foxp3 levels in Treg cells and the Treg cell number, in a TGFβ receptor signaling dependent manner. Moreover, elimination of macrophages abolished the therapeutic effects of EPO in vivo. Together, our data suggest that EPO may increase M2M, which activate Treg cells through TGFβ receptor signaling to mitigate the severity of asthma.
Materials and methods
Mouse handling
All mouse experiments were approved by the IACUC of General Hospital of Shenyang Military Area Command. Only 10-week-old male C57BL/6 mice (Jackson lab, Bar Harbor, ME, USA) were used for in vivo experiments. Mice were housed in a specific pathogen-free environment.
Ovalbumin (OVA)-induced allergic asthma model and erythropoietin (EPO) treatment
Male C57BL/6 mice of 10 weeks of age were sensitized with an intraperitoneal injection of 50 μg OVA (OVA, grade V; Sigma-Aldrich, St. Louis, MO, USA) with 2 mg aluminum hydroxide gel (Alum; Sigma-Aldrich) once per week for 3 times (week 0-2). Then the mice were challenged with 50 μg OVA by intranasal administration under light anesthesia every other day for another 7 weeks (week 3-9). Control mice received PBS. After another week, the mice were examined, sampled and analyzed (week 10). EPO (Sigma-Aldrich) was given after OCA challenge daily at a dose of 1000 IU/kg till the end of the experiment (week 9-10).
Macrophage depletion in vivo
For macrophage depletion, 200 μl clodronate-liposomes (Clodronateliposome, Netherlands) were intravenously injected every other day from the tail vein, from week 7 till week 10. Control mice were injected with liposomes containing PBS of same frequency and same dosage.
Airway hyper-responsiveness
Airway hyper-responsiveness (AHR) was measured by restrained invasive plethysmography 1 day after the last intranasal OVA challenge. Mice were anesthetized, after which a small incision was made to expose the trachea, and a cannula was inserted to connect to an inline nebulizer and ventilator. Mice were then challenged with aerosolized PBS followed by increasing doses of methacholine (Sigma-Aldrich). Airway resistance and dynamic compliance (Cdyn) were determined by analysis of pressure and flow waveforms.
Bronchoalveolar lavage, lung digestion and isolation of Treg or macrophages
Mice were euthanized by pentobarbitone overdose after AHR examination. Bronchoalveolar lavage fluid (BALF) was obtained by instilling three washes of 0.4 ml PBS with 0.1% BSA. BALF was centrifuged at 470 g for 5 min, and cells were enumerated and labeled for further analysis. For lung digestion, lungs were perfused with PBS, after which 0.8 ml 300 U/ml collagenase type I (Sigma-Aldrich), and 50 U/ml DNase I (Roche, Nutley, NJ, USA) in RPMI 1640 was injected into the trachea. Lungs were then removed, minced into small pieces, and digested at 37°C for 30 min. Lung pieces were disrupted with a syringe plunger, filtered through nylon mesh, and centrifuged. The cell pellet was resuspended in RBC lysis buffer, washed, enumerated, and labeled for flow cytometric analysis. Treg was analyzed and purified by fluorescence-activated cell sorting (FACS) with specific fluorescence-conjugated antibodies (CD4 and CD25, all from Becton-Dickinson Biosciences, San Jose, CA, USA) on a FACSAria cell sorter (Becton-Dickinson Biosciences), Macrophages were analyzed and purified by flow cytometry with specific fluorescence-conjugated antibodies (F4/80 and CD163, all from Becton-Dickinson Biosciences). Data were analyzed using Flowjo software (Flowjo LLC, Ashland, OR, USA).
Co-culture system
Purified Treg were co-cultured either with control media, or with M1M, or with M2M with/without 10 µmol/l SB431542, as has been described before [20]. Two days after co-culture, Treg was isolated for RNA and protein analyses.
RT-qPCR
RNA was extracted from purified macrophages or Treg after FACS, or from culture with RNeasy (Qiagen, Hilden, Germany). cDNA synthesis was performed by reserve transcription. Quantitative PCR (RT-qPCR) were performed in duplicates with QuantiTect SYBR Green PCR Kit (Qiagen). All primers were purchased from Qiagen. Data were collected and analyzed with the Rotorgene software accompanying the PCR machine, using 2-ΔΔCt method for quantification of the relative mRNA expression levels. Values of genes were first normalized against β-actin, and then compared to controls.
Western blot
The cells were lysed in RIPA lysis buffer (1% NP40, 0.1% SDS, 100 μg/ml phenylmethylsulfonyl fluoride, 0.5% sodium deoxycholate, in PBS) on ice. The supernatants were collected after centrifugation at 12000×g at 4°C for 20 min. Protein concentration was determined using a BCA protein assay kit (Bio-rad, China), and whole lysates were mixed with 4×SDS loading buffer (125 mmol/l Tris-HCl, 4% SDS, 20% glycerol, 100 mmol/l DTT, and 0.2% bromophenol blue) at a ratio of 1:3. Samples were heated at 100°C for 5 min and were separated on SDS-polyacrylamide gels. The separated proteins were then transferred to a PVDF membrane. The membrane blots were first probed with a primary antibody. After incubation with horseradish peroxidase-conjugated second antibody, autoradiograms were prepared using the enhanced chemiluminescent system to visualize the protein antigen. The signals were recorded using X-ray film. Primary antibodies for Western Blot are rabbit Foxp3 and β-actin (all purchased from Cell Signaling, San Jose, CA, USA). Secondary antibody is HRP-conjugated anti-rabbit (Jackson ImmunoResearch Labs, West Grove, PA, USA). Images shown in the figure were representative from 5 individuals.
ELISA assay
The TGFβ1 levels in the conditioned media (media in specific culture conditions) or in the cultured cells was determined by a TGFβ1 ELISA Kit (R&D Systems, Los Angeles, CA, USA). ELISAs were performed according to the instructions of the manufacturer. Briefly, the collected condition medium was added to a well coated with TGFβ1 polyclonal antibody, and then immunosorbented by biotinylated monoclonal anti-human TGFβ1 antibody at room temperature for 2 h. The color development catalyzed by horseradish peroxidase was terminated with 2.5 mol/l sulfuric acid and the absorption was measured at 450 nm. The protein concentration was determined by comparing the relative absorbance of the samples with the standards.
Statistics
Statistical analysis was performed with the unpaired two-tailed Student t test (for comparison between two groups), one-way ANOVA with the Tukey posttest (for comparison between three or more groups), or repeated-measures ANOVA with the Dunnett posttest (for AHR dose-response curves), using GraphPad Prism software (GraphPad Software, Inc. La Jolla, CA, USA). Data were represented as mean ± SD and were considered significant if p<0.05.
Results
EPO attenuates OVA-induced hallmarks of the asthma
We used a well-established mouse allergic asthma model in our study [23-25]. In this model, mice were first sensitized to alum-adsorbed OVA for 2 weeks, and then exposed to repeated airway provocation for 7 weeks to develop AHR. EPO was given after OVA challenge for one week, and then the mice were analyzed (Figure 1A). The establishment of asthma model was demonstrated by a dose-dependent increase in lung resistance (Rl) and decrease in Cdyn in response to a cholinergic stimulus (methacholine), which were significantly attenuated by EPO treatment (Figure 1B, 1C). Another hallmark feature of OVA-induced allergic asthma -eosinophilic accumulation in the pulmonary airways- was also evaluated. We found that the influx of inflammatory eosinophils increased to about 50% of total BALF cells by OVA-treatment, which was also significantly attenuated by EPO treatment (Figure 1D). These data suggest that EPO attenuates OVA-induced hallmarks of the asthma in mice.
Figure 1.
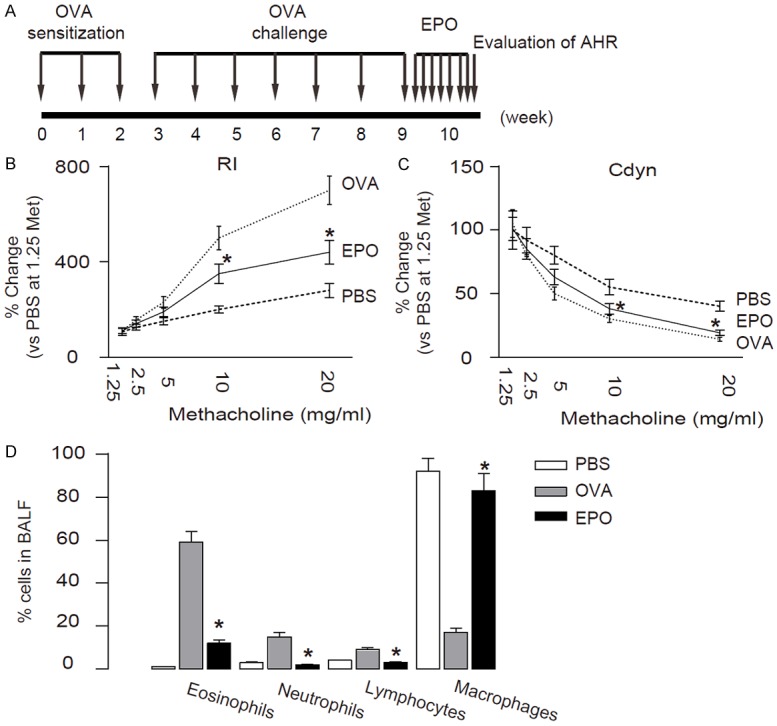
EPO attenuates OVA-induced hallmarks of the asthma. (A) Experimental schematic for OVA sensitization, inhalation challenge, and EPO treatment. (B, C) RI (B) and Cdyn (C) in response to increasing doses of methacholine. (D) Percentage of eosinophils in BALF. *p<0.05. N=10. Statistics: one-way ANOVA with a Bonferroni Correction.
EPO increases Treg cells in OVA-treated mouse lung
Then we aimed to find out the molecular mechanisms underlying the therapeutic effects of EPO. We hypothesized that Treg cells may be regulated by EPO, since Treg cells are potent inflammation suppressor in asthma. We used CD4 and CD25 to purify Treg from the lung digests of the mice with different treatments by flow cytometry (Figure 2A). We detected a significant increase in Treg cell number in the EPO-treated mouse lung (Figure 2B). Moreover, we did not find a difference of Foxp3 levels in CD4+CD25+ Treg cells from PBS, OVA and OVA+EPO treated mice (Figure 2C). Thus, EPO increases Treg in OVA-treated mouse lung but did not alter the features of Treg cells.
Figure 2.
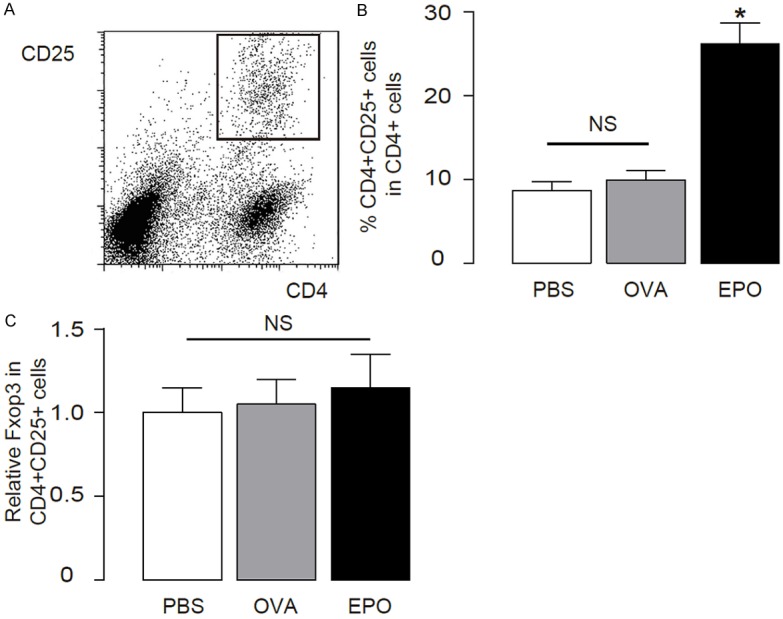
EPO increases Treg cells in OVA-treated mouse lung. (A, B) Treg cells were purified from the lung digests of OVA/EPO-treated mice based on CD4 and CD25 positivity by flow cytometry, shown by a representative flow chart (A), and quantification (B). (C) Foxp3 levels in Treg cells from different sources. *p<0.05. N=10. Statistics: one-way ANOVA with a Bonferroni Correction.
EPO increases TGFβ1 that activates Foxp3 in Treg cells
TGFβ1 has been shown to activate Foxp3 in Treg cells to promote cell differentiation and expansion [6]. Thus, we tested whether the increases in Treg cells may result from increases in TGFβ1 by EPO. Indeed, we detected higher levels of TGFβ1 in the mouse lung from the OVA-mice treated with EPO (Figure 3A). The purified CD4+CD25+ Treg cells were treated with 10 µmol/l TGFβ1, with or without 10 µmol/l SB431542, a specific inhibitor of TGFβ receptor I [20]. We found that TGFβ1 significantly increased Foxp3 in Treg, by RT-qPCR (Figure 3B), and by Western blot (Figure 3C). However, the increase in Foxp3 in Treg by TGFβ1 was completely abolished by SB431542 (Figure 3B, 3C). These data suggest that TGFβ1 may activate Foxp3 through augmenting TGFβ receptor signaling in Treg cells.
Figure 3.
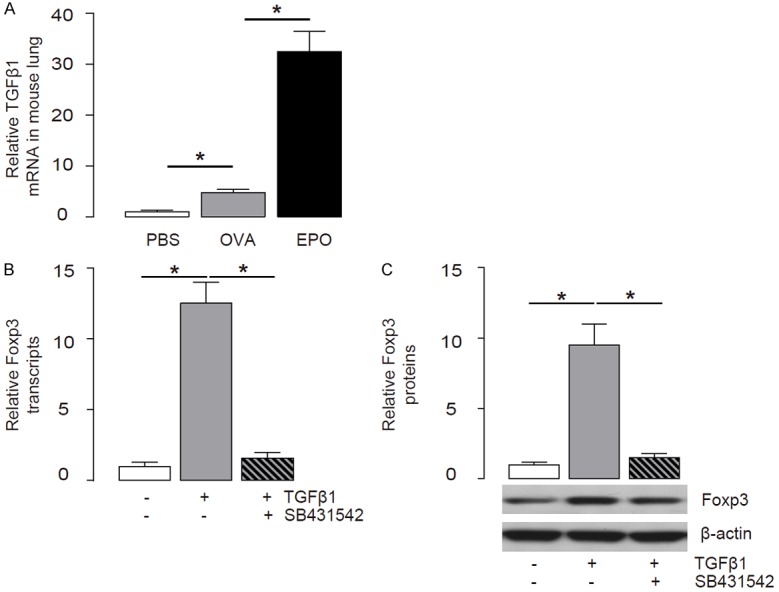
EPO increases TGFβ1 that activates Foxp3 in Treg cells. (A) TGFβ1 levels in the mouse lung from the OVA/EPO-treated mice by RT-qPCR. (B, C) The purified CD4+CD25+ Treg cells were treated with 10 µmol/l TGFβ1, with or without 10 µmol/l SB431542, a specific inhibitor of TGFβ receptor I. Foxp3 levels in Treg cells were examined by RT-qPCR (B), and by Western blot (C). *p<0.05. N=10. Statistics: one-way ANOVA with a Bonferroni Correction.
M2M produce high levels of TGFβ1
Since M2M have recently been shown to secret high levels of TGFβ1 in a pancreas injury model [20], we aimed to figure out whether M2M may also secret high levels of TGFβ1 to activate Treg in asthma. We thus analyzed the macrophages in OVA/EPO-treated mice by flow cytometry based on a pan-macrophage marker F4/80. We also sorted out M1 and M2 macrophage subtype based on a specific M2M marker, CD163 [20,25] (Figure 4A, 4B). We found that F4/80+CD163+ M2M contained more than 50-fold greater TGFβ1 mRNA compared to those in total BALF cells, or more than 30-fold greater TGFβ1 mRNA compared to those in F4/80+CD163- M1M (Figure 4C). Moreover, the high TGFβ1 mRNA in M2M resulted in comparably greater cellular protein (Figure 4D) and secreted protein in the media (Figure 4E). These data suggest that M2M are the substantial source of TGFβ1 in OVA/EPO-treated mouse lung.
Figure 4.
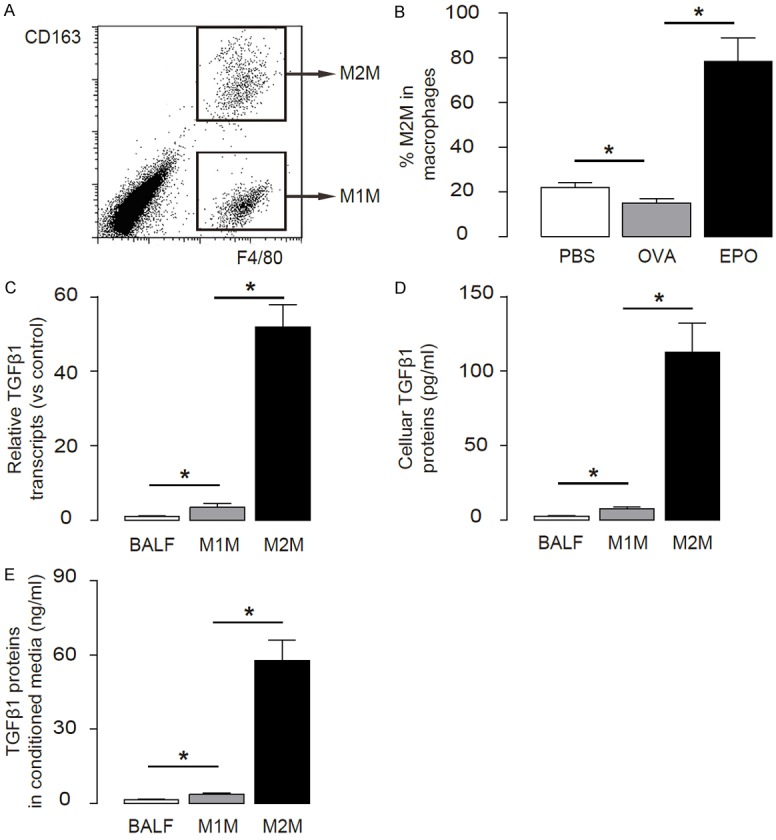
M2M produce high levels of TGFβ1. (A, B) M1M and M2M were purified from the lung digests of OVA/EPO-treated mice based on F4/80 and CD163 positivity by flow cytometry, shown by a flow chart (A), and quantification (B). (C, D) TGFβ1 levels in total BALF cells, M1AM and M2AM were examined by RT-qPCR (C), and by ELISA (D). (E) TGFβ1 levels in conditioned media were examined by ELISA. *p<0.05. N=10. Statistics: one-way ANOVA with a Bonferroni Correction.
Co-culture with M2M increases Foxp3 in Treg cells
We then used a co-culture system to examine whether M2M specifically induce Foxp3 in Treg cells. Purified Treg cells from OVA/EPO-lung were cultured either with control media, or with M1M, or with M2M, with/without 10 µmol/l SB431542, as has been described in a previous study [20]. Two days after co-culture, Treg cells were isolated for RNA and protein analyses. We found that only co-culture with M2M significantly upregulated Foxp3 in Treg cells, which could be completely abolished by SB431542, by mRNA (Figure 5A), and by protein (Figure 5B). Moreover, co-culture with M2M significantly increased the Treg cell number in an MTT assay (Figure 5C). These data suggest that M2M may secrete TGFβ1 to activate Foxp3 in Treg cells.
Figure 5.
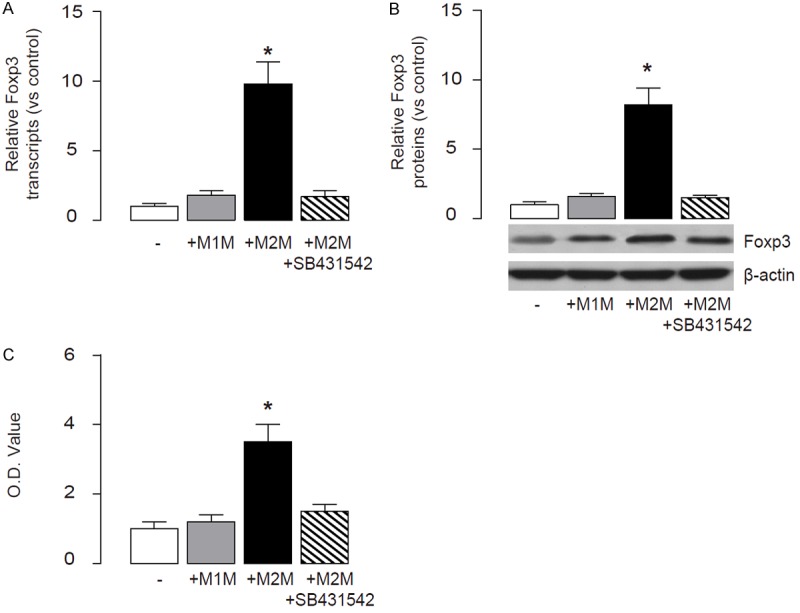
Co-cultured with M2M increases Foxp3 in Treg cells. (A, B) Purified Treg cells were co-cultured either with control media, or with M1AM, or with M2AM with/without 10 µmol/l SB431542. Two days after co-culture, Foxp3 levels in Treg cells were examined by RT-qPCR (A), and by Western blot (B). (C) MTT assay for Treg cells. *p<0.05. N=10. Statistics: one-way ANOVA with a Bonferroni Correction.
Elimination of macrophages abolishes the therapeutic effects of EPO in vivo
To figure out whether M2M are indispensable for the EPO effects on OVA-asthma, we depleted macrophages by injection of clodronate-liposomes, as has been described before [20,26,27]. Control mice were injected with liposomes containing PBS (Figure 6A). The depletion of macrophages was assured by flow cytometry quantification (Figure 6B, 6C). We found that depletion of macrophages resulted in a significant decrease in Treg cells in the lung after OVA-treatment (Figure 6D). These data suggest that elimination of macrophages may reduce the EPO-induced increases in Treg cells in vivo. Moreover, macrophage depletion abolished the therapeutic effects of EPO in vivo (Figure 6E). Taken together, our study suggests that EPO-induced M2M may activate Treg cells through TGFβ receptor signaling and Foxp3 to mitigate the severity of asthma.
Figure 6.
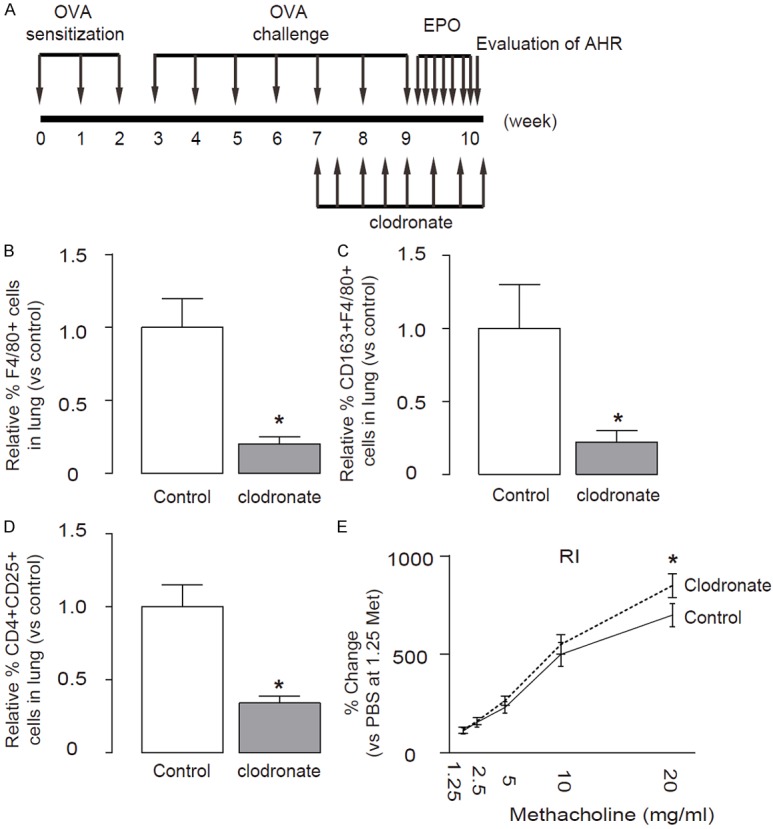
Elimination of macrophages abolishes the therapeutic effects of EPO in vivo. (A) Macrophages were depleted in vivo by injection of clodronate-liposomes, while control mice were injected with liposomes containing PBS. (B, C) The depletion of macrophages (B), and M2M (C) in the lung were confirmed by flow cytometry. (D) The depletion of macrophages resulted in a significant decrease in the percentage of Treg cells in the lung after OVA/EPO-treatment. (E) Rl. *p<0.05. N=10. Statistics: one-way ANOVA with a Bonferroni Correction.
Discussion
In tissues, macrophages are activated in a dynamic response to stimuli to acquire specialized functional phenotypes. A dichotomy has been proposed for macrophage activation: classic vs. alternative, also M1 and M2, respectively. Besides the classically activated macrophages (M1M), there are also the alternatively activated macrophages (M2M), which mediates humoral immunity and repair after injury [18,19]. Although M2M are generally thought to be anti-inflammatory and promote tissue repair, their role in asthma is contentious. There is evidence that M2M, induced by the Th2 cytokine environment during asthma, actually promote allergic inflammation [28,29]. However, the polarization of macrophages during lung injury and their specific effects on Foxp3 activation in Treg have not been systematically investigated.
Here, we sought to determine the critical cellular and molecular signaling that mediate the activation of Treg cells by EPO to alleviate allergic asthma. We used a well-established mouse allergic asthma model [23-25], and purified CD4+CD25+ Treg, F4/80+CD163- M1M and F4/80+CD163+ M2M. By analyzing gene profiling and protein levels in these purified cells, we found that EPO increased M2M, which specifically expressed high levels of TGFβ1. Moreover, after compared to total cells in BALF and to M1M, we conclude that M2M are the substantial source of TGFβ1 in OVA/EPO-treated lung. Although there may be other sources for TGFβ1 in the injured lung, e.g. myofibroblasts, our data suggest that in this model, TGFβ1 is predominantly produced by M2M. Since TGFβ1 has been shown by others [6], and by us here, to be a major trigger for Foxp3 activation in Treg cells to promote cell differentiation and expansion, we thus conclude that M2M may be responsible for activation of Treg cells after OVA/EPO-treatment. Besides TGFβ1, we also checked expression of other TGFβ receptor ligands, for example, TGFβ2 and TGFβ3. However, their expression levels were very low.
In a co-culture system, Foxp3 in Treg was specifically activated by M2M, but not by M1M. Moreover, in all abovementioned experiments, the effect of TGFβ1 was efficiently inhibited by SB431542, a specific inhibitor of TGFβ receptor I. SB431542 inhibits the phosphorylation of TGFβ receptor I, which is induced by binding of TGFβ1 to TGFβ receptor II. The phosphorylated TGFβ receptor I is required for phosphorylation of downstream SMADs for activation of Foxp3 in Treg [20,30-33]. These data suggest that the M2M may secrete TGFβ1, which binds to the receptor in Treg, and subsequently activates Foxp3 through TGFβ receptor signaling. Of note, this activation of Foxp3 has been shown to be at transcriptional level in our study.
In order to figure out whether this model occurs in vivo, we depleted macrophages by injection of clodronate-liposomes [20,26,27]. Clodronate has been widely used in numerous studies, and has been shown of its specificity on macrophage targeting and elimination, whereas being lack of selectivity on M1M and M2M. Indeed, we detected significant decrease in lung macrophages after clodronate treatment, and most importantly, this decrease in macrophages resulted in a significant decrease in the number of Treg. Thus, our proposed model was validated. The ultimate outcome of clodronate-mediated macrophage depletion on asthma may result from the combined effects of M2M on Treg activation (to inhibit asthma), a direct effect of TGFβ1 by M2M (to promote asthma), and M1M on asthma.
Collectively, our data demonstrate a previously unrecognized cross-talk between M2M and Treg, through TGFβ receptor signaling and Foxp3 activation, to regulate the severity of asthma, specifically after EPO treatment. Thus, our studies demonstrate a cellular inter-regulation network that controls the pathogenesis of asthma, suggesting that modulation of the cross-talk among different cell types may shed light on novel therapeutic strategies for allergic asthma.
Disclosure of conflict of interest
None.
References
- 1.Holgate ST. Pathogenesis of asthma. Clin Exp Allergy. 2008;38:872–897. doi: 10.1111/j.1365-2222.2008.02971.x. [DOI] [PubMed] [Google Scholar]
- 2.Holgate ST. The airway epithelium is central to the pathogenesis of asthma. Allergol Int. 2008;57:1–10. doi: 10.2332/allergolint.R-07-154. [DOI] [PubMed] [Google Scholar]
- 3.Schreiber TH, Wolf D, Tsai MS, Chirinos J, Deyev VV, Gonzalez L, Malek TR, Levy RB, Podack ER. Therapeutic Treg expansion in mice by TNFRSF25 prevents allergic lung inflammation. J Clin Invest. 2010;120:3629–3640. doi: 10.1172/JCI42933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doganci A, Eigenbrod T, Krug N, De Sanctis GT, Hausding M, Erpenbeck VJ, Haddad el-B, Lehr HA, Schmitt E, Bopp T, Kallen KJ, Herz U, Schmitt S, Luft C, Hecht O, Hohlfeld JM, Ito H, Nishimoto N, Yoshizaki K, Kishimoto T, Rose-John S, Renz H, Neurath MF, Galle PR, Finotto S. The IL-6R alpha chain controls lung CD4+CD25+ Treg development and function during allergic airway inflammation in vivo. J Clin Invest. 2005;115:313–325. doi: 10.1172/JCI22433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trzonkowski P, Szmit E, Mysliwska J, Mysliwski A. CD4+CD25+ T regulatory cells inhibit cytotoxic activity of CTL and NK cells in humans-impact of immunosenescence. Clin Immunol. 2006;119:307–316. doi: 10.1016/j.clim.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 6.Pyzik M, Piccirillo CA. TGF-beta1 modulates Foxp3 expression and regulatory activity in distinct CD4+ T cell subsets. J Leukoc Biol. 2007;82:335–346. doi: 10.1189/jlb.1006644. [DOI] [PubMed] [Google Scholar]
- 7.Palomares O, Ruckert B, Jartti T, Kucuksezer UC, Puhakka T, Gomez E, Fahrner HB, Speiser A, Jung A, Kwok WW, Kalogjera L, Akdis M, Akdis CA. Induction and maintenance of allergen-specific FOXP3+ Treg cells in human tonsils as potential first-line organs of oral tolerance. J Allergy Clin Immunol. 2012;129:510–520. 520.e1–9. doi: 10.1016/j.jaci.2011.09.031. [DOI] [PubMed] [Google Scholar]
- 8.Soroosh P, Doherty TA, Duan W, Mehta AK, Choi H, Adams YF, Mikulski Z, Khorram N, Rosenthal P, Broide DH, Croft M. Lungresident tissue macrophages generate Foxp3+ regulatory T cells and promote airway tolerance. J Exp Med. 2013;210:775–788. doi: 10.1084/jem.20121849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aggarwal NR, Tsushima K, Eto Y, Tripathi A, Mandke P, Mock JR, Garibaldi BT, Singer BD, Sidhaye VK, Horton MR, King LS, D’Alessio FR. Immunological priming requires regulatory T cells and IL-10-producing macrophages to accelerate resolution from severe lung inflammation. J Immunol. 2014;192:4453–4464. doi: 10.4049/jimmunol.1400146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coleman MM, Ruane D, Moran B, Dunne PJ, Keane J, Mills KH. Alveolar macrophages contribute to respiratory tolerance by inducing FoxP3 expression in naive T cells. Am J Respir Cell Mol Biol. 2013;48:773–780. doi: 10.1165/rcmb.2012-0263OC. [DOI] [PubMed] [Google Scholar]
- 11.Zaslona Z, Przybranowski S, Wilke C, van Rooijen N, Teitz-Tennenbaum S, Osterholzer JJ, Wilkinson JE, Moore BB, Peters-Golden M. Resident alveolar macrophages suppress, whereas recruited monocytes promote, allergic lung inflammation in murine models of asthma. J Immunol. 2014;193:4245–4253. doi: 10.4049/jimmunol.1400580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tran DQ, Ramsey H, Shevach EM. Induction of FOXP3 expression in naive human CD4+FOXP3 T cells by T-cell receptor stimulation is transforming growth factor-beta dependent but does not confer a regulatory phenotype. Blood. 2007;110:2983–2990. doi: 10.1182/blood-2007-06-094656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kelada O, Marignol L. Erythropoietinstimulating agents and clinical outcomes in metastatic breast cancer patients with chemotherapy-induced anemia: a closed debate? Tumour Biol. 2014;35:5095–5100. doi: 10.1007/s13277-014-1730-6. [DOI] [PubMed] [Google Scholar]
- 14.Wang L, Di L, Noguchi CT. Erythropoietin, a novel versatile player regulating energy metabolism beyond the erythroid system. Int J Biol Sci. 2014;10:921–939. doi: 10.7150/ijbs.9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanchis-Gomar F, Garcia-Gimenez JL, Pareja-Galeano H, Romagnoli M, Perez-Quilis C, Lippi G. Erythropoietin and the heart: physiological effects and the therapeutic perspective. Int J Cardiol. 2014;171:116–125. doi: 10.1016/j.ijcard.2013.12.011. [DOI] [PubMed] [Google Scholar]
- 16.Karaman M, Firinci F, Kiray M, Tuncel T, Bagriyanik A, Yilmaz O, Uzuner N, Karaman O. Beneficial effects of erythropoietin on airway histology in a murine model of chronic asthma. Allergol Immunopathol (Madr) 2012;40:75–80. doi: 10.1016/j.aller.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 17.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–483. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- 19.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 20.Xiao X, Gaffar I, Guo P, Wiersch J, Fischbach S, Peirish L, Song Z, El-Gohary Y, Prasadan K, Shiota C, Gittes GK. M2 macrophages promote beta-cell proliferation by up-regulation of SMAD7. Proc Natl Acad Sci U S A. 2014;111:E1211–1220. doi: 10.1073/pnas.1321347111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ricardo SD, van Goor H, Eddy AA. Macrophage diversity in renal injury and repair. J Clin Invest. 2008;118:3522–3530. doi: 10.1172/JCI36150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pull SL, Doherty JM, Mills JC, Gordon JI, Stappenbeck TS. Activated macrophages are an adaptive element of the colonic epithelial progenitor niche necessary for regenerative responses to injury. Proc Natl Acad Sci U S A. 2005;102:99–104. doi: 10.1073/pnas.0405979102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee HH, Meyer EH, Goya S, Pichavant M, Kim HY, Bu X, Umetsu SE, Jones JC, Savage PB, Iwakura Y, Casasnovas JM, Kaplan G, Freeman GJ, DeKruyff RH, Umetsu DT. Apoptotic cells activate NKT cells through T cell Ig-like mucin-like-1 resulting in airway hyperreactivity. J Immunol. 2010;185:5225–5235. doi: 10.4049/jimmunol.1001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pichavant M, Goya S, Hamelmann E, Gelfand EW, Umetsu DT. Animal models of airway sensitization. Curr Protoc Immunol. 2007 doi: 10.1002/0471142735.im1518s79. Chapter 15: Unit 15 18. [DOI] [PubMed] [Google Scholar]
- 25.Song X, Xie S, Lu K, Wang C. Mesenchymal Stem Cells Alleviate Experimental Asthma by Inducing Polarization of Alveolar Macrophages. Inflammation. 2015;38:485–92. doi: 10.1007/s10753-014-9954-6. [DOI] [PubMed] [Google Scholar]
- 26.Cao X, Han ZB, Zhao H, Liu Q. Transplantation of mesenchymal stem cells recruits trophic macrophages to induce pancreatic beta cell regeneration in diabetic mice. Int J Biochem Cell Biol. 2014;53:372–379. doi: 10.1016/j.biocel.2014.06.003. [DOI] [PubMed] [Google Scholar]
- 27.Shen B, Liu X, Fan Y, Qiu J. Macrophages Regulate Renal Fibrosis Through Modulating TGFbeta Superfamily Signaling. Inflammation. 2014;37:2076–84. doi: 10.1007/s10753-014-9941-y. [DOI] [PubMed] [Google Scholar]
- 28.Moreira AP, Hogaboam CM. Macrophages in allergic asthma: fine-tuning their pro- and anti-inflammatory actions for disease resolution. J Interferon Cytokine Res. 2011;31:485–491. doi: 10.1089/jir.2011.0027. [DOI] [PubMed] [Google Scholar]
- 29.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Inman GJ, Nicolas FJ, Callahan JF, Harling JD, Gaster LM, Reith AD, Laping NJ, Hill CS. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol. 2002;62:65–74. doi: 10.1124/mol.62.1.65. [DOI] [PubMed] [Google Scholar]
- 31.Derynck R, Feng XH. TGF-beta receptor signaling. Biochim Biophys Acta. 1997;1333:F105–150. doi: 10.1016/s0304-419x(97)00017-6. [DOI] [PubMed] [Google Scholar]
- 32.Xiao X, Wiersch J, El-Gohary Y, Guo P, Prasadan K, Paredes J, Welsh C, Shiota C, Gittes GK. TGFbeta Receptor Signaling Is Essential for Inflammation-Induced but Not beta-Cell Workload-Induced beta-Cell Proliferation. Diabetes. 2013;62:1217–1226. doi: 10.2337/db12-1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Massague J. TGFbeta signalling in context. Nat Rev Mol Cell Biol. 2012;13:616–630. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]