

Abstract
Numerous analyses including in vivo and in vitro experiments have demonstrated that inhalation exposure of NiONPs can result in pulmonary fibrosis. However, the potential mechanisms of this pathological process remain elusive. Here, we investigate the role of HIF-1α and TGF-ß1 in NiONPs-induced pulmonary fibrosis with a focus on the interplay of the above two proteins. In vivo, male Sprague&Dawley rats were exposed to NiONPs and pulmonary fibrosis was demonstrated using H&E staining and immunochemistry of αSMA. In vitro, NiONPs contributed to cell proliferation and increased expressions of collagen-1 and αSMA in human fetal lung fibroblasts. Both HIF-1α and TGF-ß1 were upregulated by NiONPs treatment. Inhibition of HIF-1α reduced TGF-ß1 expression and downregulation of TGF-ß1 reduced HIF-1α protein level. Mechanism investigation revealed that TGF-ß1 affects nuclear translocation activity of HIF-1α. Taken together, these finding provide evidence that HIF-1α and TGF-ß1 act in synergy to foster NiONPs-induced pulmonary fibrosis, and the cross talk between them is a pivotal mechanism of pulmonary fibrosis.
Keywords: Nanoparticles, transforming growth factor, hypoxia
Introduction
Pulmonary fibrosis is a progressive and lethal disease [1]. Exposure to chemicals, radiation, smoking and airborne pollutants have been reported to have a contribution to the progression of pulmonary fibrosis [2-5]. Recently, there is an increasing interest in the profibrotic risk of fine particles. Nanoparticles, at least one dimension of less than 100 nm, have been demonstrated to be correlated with oxidative stress, inflammatory response and fibrogenesis in vivo and in vitro [6-12]. A few evidence showed that inhalation exposure of NiONPs which were widely used industrial materials could result in pulmonary inflammation [13,14]. However, there is lacking of direct evidence to prove that NiONPs exposure can induce pulmonary fibrosis.
HIF-1α is a subunit of HIF-1 which is a transcriptional activator functioning as a global oxygen homeostasis [15]. HIF-1α can be recognized and ubiquitinated by von Hippel-Lindau tumor suppressor (pVHL) and ultimately degraded by proteasome under normixic conditions [16]. But hypoxia can prevent degradation of HIF-1α by inhibiting the activity of prolyl hydroxylase (PHD) by which HIF-1α could be recognized by pVHL [17-20]. Recently, HIF-1α has been identified as a key regulator of hypoxia-induced fibrosis in numerous organs, such as kidney, liver, derma and lung [21-25]. Furthermore, the HIF-1α pathway was demonstrated to be activated by NiONPs treatment in human lung epithelial cells [26].
Fibroblast-to-myofibroblast transition (FMT) is a critical mechanism to fibrogenesis [27]. Recent studies have confirmed the effect of EMT on pulmonary fibrosis [28,29]. TGF-ß1 can stimulate fibroblasts to produce collagen and promote fibroblasts to differentiate into myofibroblasts [30-32]. In addition, activation of TGF-ß1/Smad3 can contribute to collagen accumulation in kidney disease [33,34]. And TGF-ß/Smad3 signaling also plays a role in derma, breast, liver and lung fibrosis [35-38]. Accordingly, TGF-ß1 is definitely of great importance to organ fibrosis.
Some studies demonstrated that HIF-1α mediates the function of TGF-ß1 in pulmonary and liver fibrosis [39-41]. However, others reported that TGF-ß1 might contribute to tumor angiogenesis via HIF-2α signaling [42,43]. On the other hand, TGF-ß1 was reported to induce HIF-1α stabilization through selective inhibition of PHD2 expression [44,45]. And inhibition of TGF-ß1 could significantly counteract the TGF-β1-stimulated HIF-1α overexpression in renal epithelial cells [33]. Moreover, there are several identical target genes in renal fibrogenesis of HIF-1α and TGF-ß1 [45-47]. However, potential cross talk between HIF-1α and TGF-ß1 is still not well described.
In this study, we used male Sprague and Dawley rats and human fetal lung fibroblasts to evaluate the profibrotic effect of NiONPs. We first confirmed the lung toxicity of NiONPs treatment and demonstrated NiONPs-induced pulmonary fibrosis is HIF-1α and TGF-ß1 dependent. Then we showed that HIF-1α and TGF-ß1 potentiate each other to foster pulmonary fibrosis.
Materials and methods
Reagents
NiONPs were purchased from Sigma-Aldrich (St. Louis, MO, USA). 2-DG was purchased from Sangon biotech (China). SB431542 (ALK5 inhibitor) was obtained from Selleck Chemicals (Houston, TX, USA). Recombinant human TGF-ß1 was purchased from R&D systems (Minneapolis, MN, USA).
Preparation of NiONPs
NiONPs were prepared as we described previously [48]. Briefly, to ensure drying and sterility of NiONPs, NiONPs were baked in a drying oven for 24 h and then disposed by ultraviolet sterilization for 30 min. For cellular experiments, NiONPs suspensions were prepared by diluting them to 2 mg/mL stock solution in cell culture medium which was then sonicated for 1 h in ice bath. For optimal suspension, the stock solutions were re-sonicated for 10 min and diluted into required concentrations with fresh medium before administration to cells. For animal experiments, stock concentration of NiONPs suspensions was 2 mg/ml. 0.4 ml (800 μg) NiONPs suspension solution was administered to rats by a single intratracheal instillation. Rats in the control group underwent the same volume of physiological saline simultaneously.
Animals
Male Sprague and Dawley rats of 8 weeks old were purchased from and kept in the Laboratory Animal Center of the third military medical university (Chongqing, China). All handling and treatments were in line with the ethical standards set out by the Third Military Medical University Institutional Animal Care and Use Committee. To observe time-dependent change, at least 8 rats were treated per time point. On day 28 and day 60 after exposure, rats were anesthetized by pentobarbital sodium. Subsequently, the left lung was obtained for histopathological and immunohistochemistry analysis after the blood was perfused from the lung by right ventricle lavage with saline.
Cell culture and treatments
Human fetal lung fibroblasts were obtained from the Cell Bank of the Institute of Biochemistry and cell Biology (Shanghai, China). Cells were cultured in Ham’s F-12K (Kaighn’s) medium (Gibco, Invitrogen Corporation, NY, USA) supplemented with 10% fetal bovine serum (FBS, Gibco), 100 units/ml penicillin G and 100 μg/ml streptomycin (Beyotime, Haimen, Jiangsu, China) at 37°C under 5% CO2. Cells were passaged by trypsinization at 85% confluence. For cellular experiments, cells were seeded in plates or petri dishes (Corning, Costar, NY, USA) 24 hours before treatment. When appropriate, cells were treated with indicated concentrations of NiONPs, 2-DG, SB431542, and TGF-ß1 for indicated time.
Histopathological and immunohistochemistry analysis
The left lung was infused with 10% neutral paraformaldehyde, fixed for 24~48 h. The cross section through left principal bronchus of the left lung was embedded in paraffin wax and cut into 5-μm-thick slices. The slices were stained with H&E to observe the morphological changes. Immunohistochemical staining for αSMA was used to access the fibrosis. The slices were deparaffinized with xylene and endogenous peroxidase activity was quenched with 3% H2O2. After blocking with normal goat serum, the slices were incubated with αSMA antibody (1:100, ab5694) for 24 h and then with secondary antibody and peroxidase-labeled streptavidin incubation followed by diaminobenzidine and hematoxylin staining.
Western blot
After treatments, adherent cells were collected and whole-cell extracts were denatured and electrophoresed on SDS-PAGE and transferred onto a polyvinylidene fluoride membrane (PVDF, Bio-Rad, Hercules, CA, USA). After 1 hour incubation in blocking buffer [PBS (pH 7.4) supplemented with 5% low-fat milk powder], the membranes were incubated overnight at 4°C with primary antibodies: rabbit anti-HIF-1α (NB100-479) and rabbit anti-HIF-2α (NB100-122) were from Novus biologicals (USA) , rabbit anti-collagen-1 (ab34710), rabbit anti-αSMA (ab5694), rabbit anti-TGF-ß1 (ab92486) and rabbit anti-Smad3 (phospho S423+S425) (ab52903) antibodies were from Abcam (Cambridge, UK) and mouse anti-GAPDH was from Sigma-aldrich. Membranes then were incubated with anti-mouse or anti-rabbit secondary antibodies and processed for enhanced chemiluminescence (Thermo Fisher Scientific, Waltham MA, USA). Densitometric analysis of bands was performed using ChemiDoc XRS+System with Image Lab Software (Bio-Rad).
Quantitative real-time PCR
Total RNA was isolated using Trizol (Takara Bio Inc., Shiga, Japan). cDNA was synthesized from total RNA by reverse transcription kit (Takara) according to the manufacturer’s protocol. qPCR analysis was then performed with gene-specific primers in the presence of SYBR green (Bio-Rad). Primers obtained from Invitrogen were as follows: HIF-1α, forward: 5’-TTT TGG CAG CAA CGA CAC AG-3’ and reverse: 5’-GCG GTG GGT AAT GGA GAC AT-3’; collagen-1, forward: 5’-GCC AAG ACG AAG ACA TCC-3’ and reverse: 5’-GTC ATC GCA CAA CAC CTT-3’; αSMA, forward: 5’-CTT GAG AAG AGT TAC GAG TTG-3’ and reverse: 5’-GAT GCT GTT GTA GGT GGT T-3’; TGF-ß1, forward: 5’-CTC GCC AGA GTG GTT ATC-3’ and reverse: 5’-GTT ATC CCT GCT GTC ACA-3’; GAPDH, forward: 5’-TAT GAC AAC AGC CTC AAG AT-3’ and reverse: 5’-AGT CCT TCC ACG ATA CCA-3’; LOX, forward: 5’-AGC CGA CCA AGA TAT TCC T-3’ and reverse: 5’-CTT CAG CCA CTC TCC TCT-3’; PAI-1, forward: 5’-CAG CAG CAG ATT CAA GCA-3’ and reverse: 5’-CTG ATC TCA TCC TTG TTC CA-3’; VEGF, forward: 5’-CTT GCC TTG CTG CTC TAC-3’ and reverse: 5’-ATC CAT GAA CTT CAC CAC TT-3’; Gene expression levels were determined by Cq method and normalized to GAPDH levels.
Cell proliferation assay
Cell proliferation rates were determined using EdU immunofluorescence assays (Guangzhou RiboBio, Guangzhou, China) according to the manufacturer’s instructions. Briefly, EdU was added to cells on slice in plate which were treated with NiONPs 24 h before and incubated 2 h. Cells were fixed with 4% paraformaldehyde and permeablized with TBS with 0.5% Triton X-100 and then stained with staining reactions for 30 min. Cell nuclei were counterstained with DAPI (Beyotime, Haimen, Jiangsu, China). Finally, the slices were analyzed under fluorescence microscopy (Leica, Germany).
Preparation of nuclear extracts and electrophoretic mobility shift assay (EMSA)
Nuclear extracts were obtained using NE-PER nuclear and cytoplasmic extraction reagents (Thermo scientific) following the instructions of the manufacturer. Briefly, treated cells were harvested with trypsin-EDTA and centrifuged at 500× g for 5 min and were washed with PBS. Then cells were pelleted by centrifugation at 500× g for 3 min. Cell pellet was incubated at 4°C for 10 min after adding ice-cold CER I. Afterward, ice-cold CER II was added. After vortex and centrifugation, the supernatant of cytoplasmic extracts was collected and pellet containing nuclei was suspended in ice-cold NER and placed on ice for 40 min. After centrifugation at 16000× g for 10 min, nuclear extracts were obtained from the supernatant.
EMSA was carried out to assess activation of HIF-1α after indicated treatments. The sequences of the oligonucleotides used are 5’-TCT GTA CGT GAC CAC ACT CAC CTC-3’ for the biotin-end-labeled HIF-1α probe, and 5’-TCT GTA CGT GAC CAC ACT CAC CTC-3’ for the cold-competitive HIF-1α probe. Binding reactions were incubated at room temperature for 20 min in the presence of 2.5% glycerol, 5 mM MgCl2, 50 ng/μl poly (dI-dC), 0.05% Nonidet P-40 and 1×binding buffer (LightShiftTM chemiluminescent EMSA kit, Pierce) using 20 nM of biotin-end-labeled probe. The competition reactions were performed by adding 50-fold excess unlabeled probe to the reaction mixture. Following electrophoresis on a 6% pre-run Tris-borate-EDTA gel, the reactions were transferred to a nylon membrane. Then membrane was UV-crosslinked, blocked, and detected with the Chemiluminescent Nucleic Acid Detection Module (Pierce) in accordance with the manufacturer’s instructions.
Statistical analysis
All data are reported as the means ± SEM of at least three identical experiments. One-way analysis of variance (ANOVA) followed by LSD (L) comparison test was applied to analyze statistical significance. Significance was assumed when P-values <0.05 and all tests were performed using SPSS 16.0 software package (SPSS, Chicago, IL, USA).
Results
NiONPs increase fibrotic proliferation in vivo and in vitro
The pathological features of lungs at 28 days and 60 days after intratracheal instillation are shown in Figure 1A. In the NiONPs-administered groups, fibroblasts and collagen fibro proliferation and interstitial hyperplasia were obvious compared with control groups. There was no remarkable fibrotic pathology in control groups both 28 days and 60 days. And the severity of the symptoms of rats treated with NiONPs increased with time, the thickness of walls was significantly increased in 60 days treatment when compared to 28 days treatment. This phenomenon suggests pulmonary fibrosis induced by NiONPs exposure is persistent and aggressive. Pulmonary fibrosis is characterized by αSMA production, which is presented by immunochemistry assay in Figure 1B. We found that NiONPs significantly increased the production of αSMA at both 28 days and 60 days. Moreover, it appears that this effect progressed with time which corresponded to pathological changes. To study whether NiONPs causes the same effect in vitro as in vivo, human fetal lung fibroblasts were incubated with NiONPs (0~2.0 μg/cm2) for 24 h and then the incorporation of 5-ethynyl-2’-deoxyuridine (EdU) into cells was assayed. As illustrated in Figure 1C, incubation of human fetal lung fibroblasts with NiONPs induced dose-dependent increases in EdU incorporation, suggesting that NiONPs induced an increase in the proliferation of human fetal lung fibroblasts. Thus, NiONPs is a fibrotic activator in vivo and in vitro.
Figure 1.
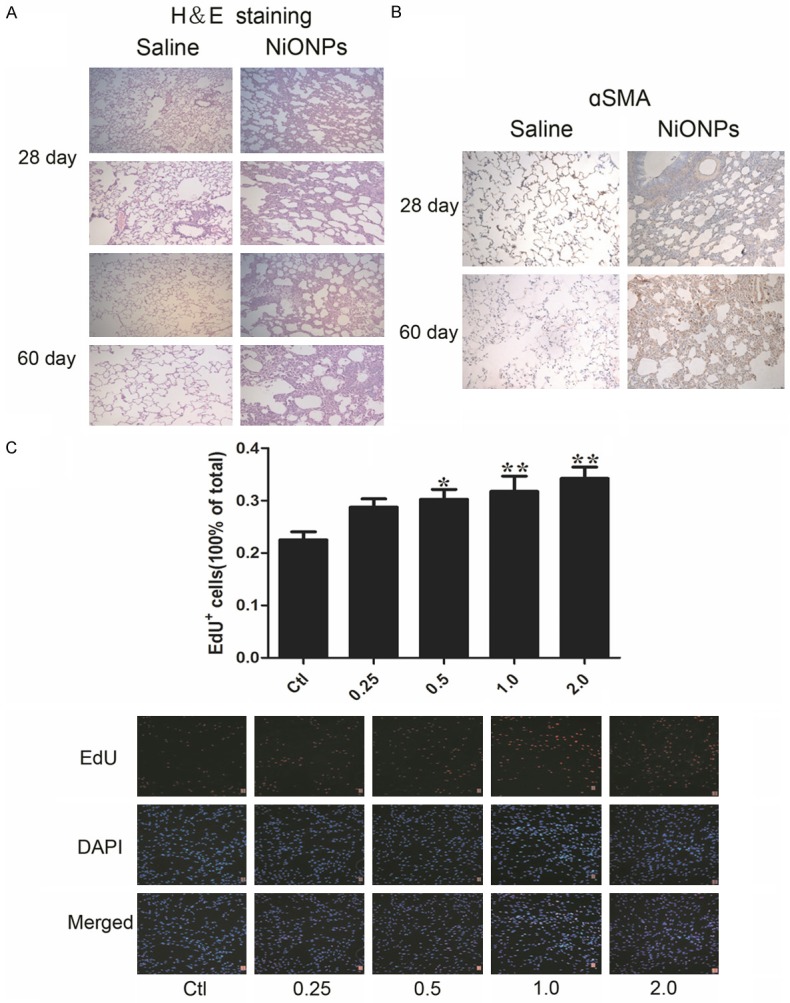
NiONPs treatment induces fibrosis in the lungs of Sprague&Dawley rats and the DNA synthesis of human fetal lung fibroblasts. Sprague&Dawley rats were treated with intratracheal instillation of 0.4 ml suspension containing 800 μg NiONPs for once time. The control group was given only 0.4 ml/rat of saline. The rats were sacrificed and lung tissues were obtained at day 28 and 60 after exposure. (A) Fibrotic damage was determined by H&E staining (magnification, ×100 and ×200) and (B) the expression of αSMA was detected by immunohistochemistry assay (magnification, ×200). (C) Effects of NiONPs (concentration: 0, 0.25, 0.5, 1.0 and 2.0 μg/cm2 ) on the DNA synthesis of human fetal lung fibroblasts were detected by EdU incorporation. *p<0.05, **p<0.01 compared with control. Results were represented as mean ± SEM at least three independent experiments.
NiONPs cause collagen-1 deposition and αSMA production
Accumulation of extracellular matrix (ECM) in lung is a characteristic marker in pulmonary fibrosis and deposition of collagen-1 from fibroblasts is an important constituent of ECM [49,50]. To investigate the mRNA expression level of collagen-1 in human fetal lung fibroblasts, quantitative real-time PCR (qPCR) was performed. As shown in Figure 2A, NiONPs exposure increased collagen-1 mRNA expression in a dose-dependent manner which corresponded to collagen-1 protein synthesis shown in Figure 2B FMT is a key event in pulmonary fibrosis and αSMA, a typical marker of myofibroblast, was detected by qPCR and western blot. And both mRNA and protein levels of αSMA were increased by NiONPs treatment (Figure 2C, 2D). Time-dependent experiments were conducted and mRNA expressions of both collagen-1 and αSMA were significantly increased compared with control, but the highest increments appeared at 24 h (Figure 2E, 2G). However, the protein levels of collagen-1 and αSMA were increased in a time-dependent manner.
Figure 2.
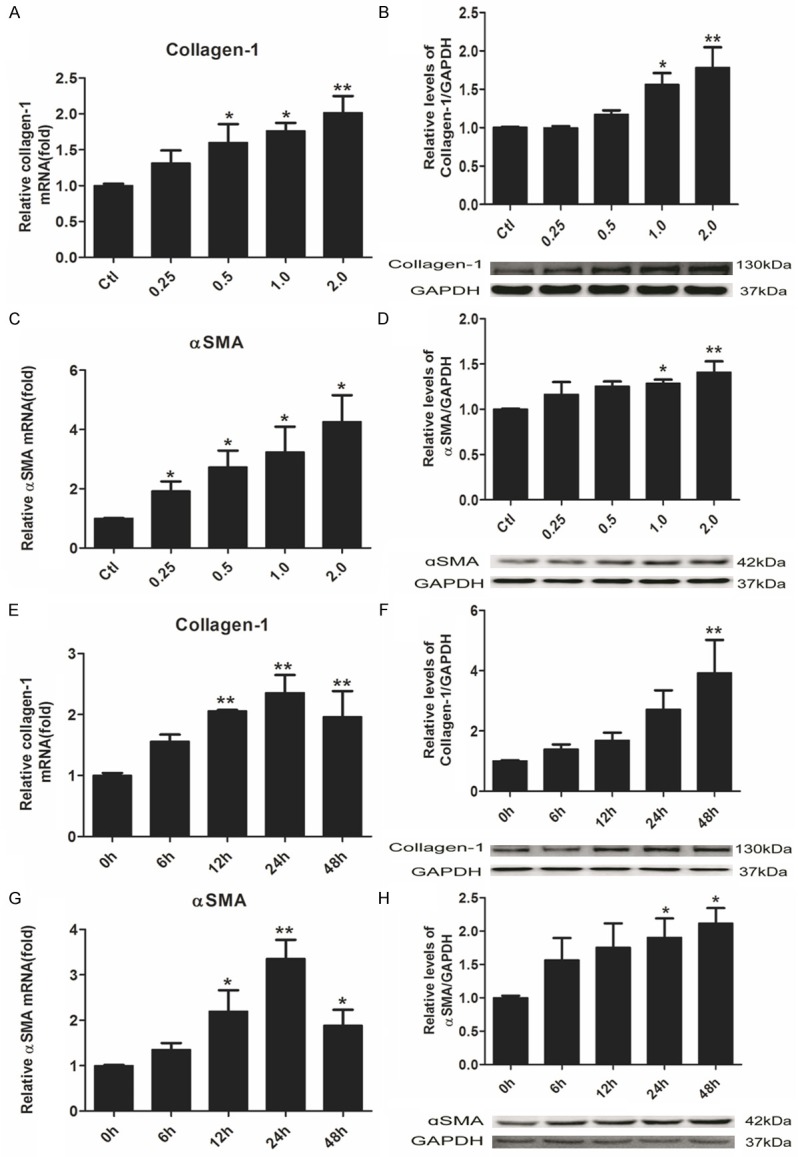
NiONPs cause collagen-1 deposition and production of αSMA by upregulating collagen-1 and αSMA mRNA transcriptions in human fetal lung fibroblasts. (A-D) Human fetal lung fibroblasts were exposed to NiONPs suspensions with concentrations of 0, 0.25, 0.5, 1.0 and 2.0 μg/cm2 for 24 h. (E-H) Time-dependent experiments were conducted with 1 μg/cm2 NiONPs for various time points. (A, C, E, G) mRNA transcriptions of collagen-1 and αSMA were detected by qPCR, normalized to GAPDH and (B, D, F, G) Protein levels of collagen-1 and αSMA were determined by western blot and normalized to GAPDH. The value was expressed as fold changes relative to controls. *p<0.05, **p<0.01 compared with control. Results were represented as mean ± SEM at least three independent experiments.
NiONPs-induced fibrosis is HIF-1α dependent but not HIF-2α
It has been previously reported that protein accumulation of HIFs, including HIF-1α and HIF-2α is common in the pulmonary fibrosis [25]. To determine whether HIF-1α or HIF-2α is the critical factor in NiONPs-induced fibrosis, western blot was used to detect the protein expression of the above molecules. NiONPs exposure strongly induced HIF-1α expression (Figure 3A). There was no such change of HIF-2α in human fetal lung fibroblasts exposed to NiONPs (Figure 3B). To further confirm the changes of HIF-1α, HIF-1α-controlled profibrotic genes, including LOX, PAI-1 and VEGF were detected by qPCR. As shown in Figure 3C-E, the expressions of LOX, PAI-1 and VEGF were stimulated in accordance with HIF-1α. Furthermore, incorporation of 2-Deoxy-D-glucose (2-DG) could significant prevent NiONPs-induced expressions of collagen-1 and αSMA at the level of mRNA and protein (Figure 3G-J). 2-DG has been proved to be a valid inhibitor of HIF-1α [51,52] and the above results showed that NiONPs-induced fibrosis is HIF-1α dependent.
Figure 3.

NiONPs-induced collagen-1 and αSMA expressions are HIF-1α dependent. (A-E) Human fetal lung fibroblasts were exposed to NiONPs suspensions with concentrations of 0, 0.25, 0.5, 1.0 and 2.0 μg/cm2 for 24 h. (A) HIF-1α and (B) HIF-2α expressions were analyzed by western blot and normalized to GAPDH. (C-E) Relative levels of HIF-1α-controlled genes (LOX, PAI-1 and VEGF) were determined by qPCR and normalized to GAPDH. (G-J) Human fetal lung fibroblasts were pretreated with 10 mM (+) and 20 mM (++) 2-DG (HIF-1α inhibitor) for 2 h and then exposed to 2 μg/cm2 NiONPs for 24 h. Collagen-1 and αSMA expressions were analyzed by qPCR and western blot and normalized to GAPDH. The value was expressed as fold changes relative to controls. *p<0.05, **p<0.01 compared with control. Results were represented as mean ± SEM at least three independent experiments.
NiONPs-induced fibrosis is correlated with TGF-ß1/Samd3 pathway
TGF-ß1 is a primary mediator of FMT during fibrogenesis [30-32]. Previous studies have reported that TGF-ß1 secreted from macrophages in lungs plays an important role in pulmonary fibrosis [53,54]. There was only a few works pointed out that fibroblasts could be stimulated to secrete TGF-ß1 [53]. Our study examined the effect of NiONPs on stimulation of TGF-ß1/Smad3 pathway in fibroblasts. qPCR results showed that TGF-ß1 mRNA expression can be induced by NiONPs in a dose-dependent manner (Figure 4A). Western blot results showed that both TGF-ß1 and phosphorylated Smad3 (P-Smad3) were upregulated by NiONPs in fibroblasts (Figure 4B and 4C). It has been reported exogenous TGF-ß1 could promote collagen deposition and αSMA production [34,55-57]. Thus, we applied SB431542, which can selectively inhibit TGF-ß receptor I kinase (ALK5), to fibroblasts treated with NiONPs. As shown in Figure 4D-G, SB431542 could counteract the effects of NiONPs on the expressions of collagen-1 and αSMA which proves that NiONPs-induced production of collagen-1 and αSMA is in a TGF-ß1 dependent manner in fibroblasts.
Figure 4.

NiONPs-induced collagen-1 and αSMA expressions are TGF-ß1 dependent. (A-C) Human fetal lung fibroblasts were exposed to NiONPs suspensions with concentrations of 0, 0.25, 0.5, 1.0 and 2.0 μg/cm2 for 24 h. TGF-ß1 expression was detected by qPCR (A) and western blot (B) and normalized to GAPDH. (C) The levels of P-Smad3 was determined by western blot and normalized to GAPDH. (D-G) Human fetal lung fibroblasts were treated with 10 nM SB431542 (TGF-β receptor I kinase (ALK5) inhibitor) and 2 μg/cm2 NiONPs simultaneously for 24 h. Collagen-1 and αSMA expressions were analyzed by qPCR and western blot and normalized to GAPDH. The value was expressed as fold changes relative to controls. *p<0.05, **p<0.01 compared with control. Results were represented as mean ± SEM at least three independent experiments.
HIF-1α and TGF-ß1 interact to promote NiONPs-induced fibrosis
Our above results have shown that both HIF-1α and TGF-ß1 were involved in the NiONPs-induced fibrosis. Some studies reported that TGF-ß1 could regulate HIF-1α expression [44,45]. Also it has been previously reported that HIF-1α could affect the production of TGF-ß1 [39-41]. Thus, we wondered that whether the mutual regulation of HIF-1α and TGF-ß1 could be existed in human fetal lung fibroblasts. To elucidate the interplay of HIF-1α and TGF-ß1, TGF-ß1 and P-Smad3 were detected by qPCR and western blot in fibroblasts which were exposed to 2-DG and NiONPs, it was obvious that overexpression of TGF-ß1 and P-Smad3 resulted from NiONPs treatment could be downregulated by 2-DG which was an inhibitor of HIF-1α (Figure 5A-C). Conversely, significant decrease of NiONPs-induced HIF-1α protein expression was observed in our studies which was caused by SB431542 (Figure 5E). We tested the downstream genes of HIF-1α and effects on these genes mediated by SB431542 validated the conclusion that TGF-ß1 is a regulator of HIF-1α (Figure 5F-H). Furthermore, to clarify the mechanism of how TGF-ß1 regulates HIF-1α, we evaluated the nuclear translocation of HIF-1α using electrophoretic mobility shift assays (EMSA) analyses. As shown in Figure 5I, there was no band in the negative control (lane 1) and a slight shadow was shown in the competitor control (lane 8) which should be compared with lane 7 as they had the equal nuclear extracts. The binding of HIF-1α in the NiONPs treatment group (lane 5) was higher than ordinary control group (lane 2). And this impact could be improved by exogenous TGF-ß1 (lane 6) and reduced by SB431542 (lane 7). Together with the result of Figure 5D, we concluded that TGF-ß1 could affect protein accumulation and nuclear translocation of HIF-1α without regulating transcription activity of HIF-1α.
Figure 5.
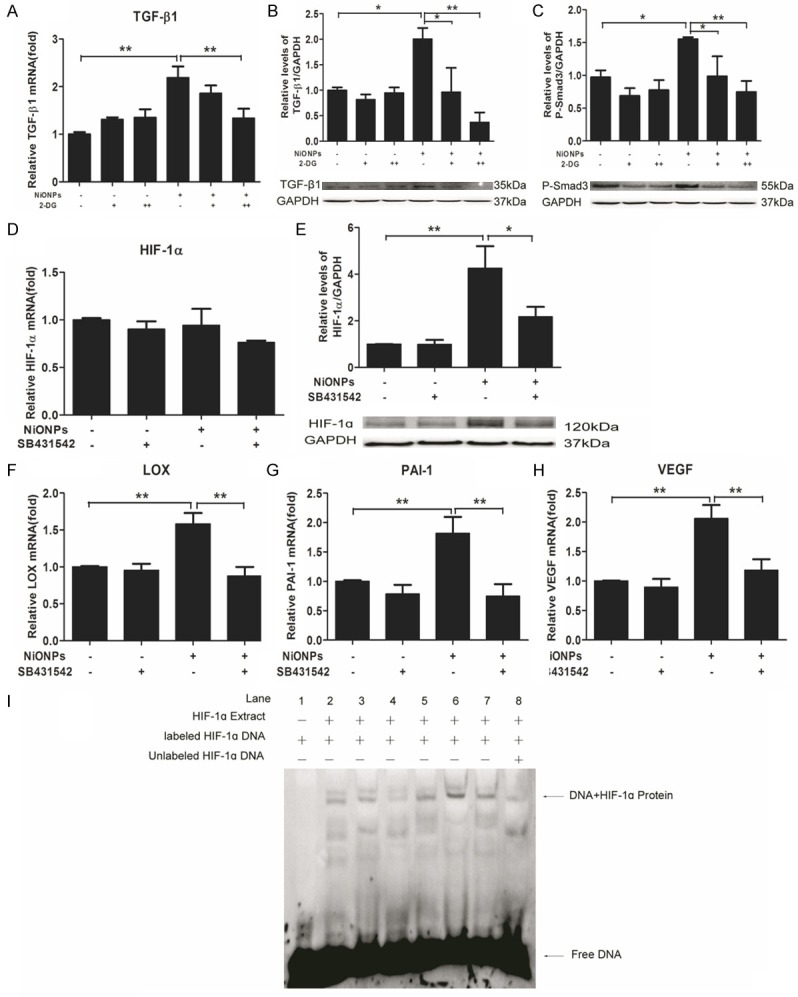
Reciprocal regulation between HIF-1α and TGF-ß1. (A-C) Human fetal lung fibroblasts were pretreated with 10 mM (+) and 20 mM (++) 2-DG (HIF-1α inhibitor) for 2 h and then exposed to 2 μg/cm2 NiONPs for 24 h. (D-H) Human fetal lung fibroblasts were treated with 10 nM SB431542 and 2 μg/cm2 NiONPs simultaneously for 24 h. The levels of TGF-ß1 (A), HIF1α (D), LOX (F), PAI-1 (G) and VEGF (H) were detected by qPCR and Protein expressions of TGF-ß1 (B) and P-Smad3 (C) were analyzed by western blot (B) and normalized to GAPDH. (I) EMSA was performed to analyze the binding of proteins from nuclear extracts of human fetal lung fibroblasts treated with 2 μg/cm2 NiONPs plus 10 nM SB431542 or 1 ng/ml TGF-ß1 for 24 h to the consensus HIF-1α binding site. Lane: 1, negative control; 2, ctl; 3, TGF-ß1; 4, SB431542; 5, NiONPs; 6, NiONPs+TGF-ß1; 7, NiONPs+SB431542; 8, cold-competitive control.
Discussion
In this study, we found that NiONPs exposure could contribute to pulmonary fibrosis in vivo and in vitro. Furthermore, we proposed a model of crosstalk between HIF-1α and TGF-ß1 in human fetal lung fibroblasts which might be a potential mechanism for pulmonary fibrosis induced by NiONPs. Specifically, we suggested that TGF-ß1 facilitated HIF-1α signaling by accumulating HIF-1α protein and enhancing DNA binding activity of HIF-1α. Moreover, activated HIF-1α promoted expression of TGF-ß1 at the level of mRNA and protein.
Nowadays, Nanomaterial is widely used in a range of new industrial applications because of its unique characteristics [58,59]. But the potential damage to human body limits its utilities [8,58,60,61]. Lung is the most susceptible organ to inhalation exposure of dust nanoparticles. Increasing analyses aim at assessing the toxicity of nanoparticles to lungs [53,62,63]. But profibrotic effect of NiONPs received little attention. Inhalation of nickel hydroxide nanoparticles was reported to induce significant oxidative stress and inflammation in lungs [11]. Oxidative stress is implicated as an important molecular mechanism underlying fibrosis in a variety of organs, including the lungs [64]. Thus, we inferred that NiONPs could promote pulmonary fibrosis. Although soluble nickel can not induce such a phenomenon, Ni2+ derived from NiONPs which are phagocytosed by cells because of their nano-size plays a role in the whole process [65]. Ni2+ can mimic hypoxia to up-regulate HIF-1 and HIF-1-dependent transcription [66,67]. The possible explanation of HIF-1α activation involves an iron-containing flavoheme protein which can sense the hypoxia. Ni2+ can activate a signaling cascade leading to HIF-1α stabilization by substituting for iron in this sensor [68,69]. Subsequently, HIF-1α will promote fibrogenesis via several different mechanisms [70].
Accumulation of ECM in the lung is an important characteristic of pulmonary fibrosis [53]. And collagen deposition from fibroblasts results in ECM accumulation. Increased transcriptional activation of collagen-1 was induced by NiONPs (Figure 1A), so as to αSMA, which was a marker of EMT. The point of the most increments of mRNA expressions of collagen-1 and αSMA in time-dependent experiments was at 24 h (Figure 1E, 1G). But protein levels in the same experiments were increased in a time-dependent manner (Figure 1F, 1H). We assume that mRNA expressions were limited by the total amount of NiONPs which was relatively decreased when cells were increased and protein level changes always tended to be lagging.
TGF-ß1 is an established mediator of pulmonary fibrosis. Recent evidence shows that TGF-ß1 is mainly secreted from macrophages in lung. Elevated TGF-ß1 can interact with ALK5 on fibroblasts to cause TGF-ß/Smad signaling activation in fibroblasts [53]. TGF-ß1 was reported to increase HIF-1α expression through binding to ALK5 on human renal tubular epithelial cells and mouse embryonic fibroblasts [33]. Our experiments showed that inhibition of ALK5 can attenuate HIF-1α protein expression in human lung fibroblasts. The potential mechanism by which TGF-ß1 increases HIF-1α activity was supplemented in the present analysis. Exogenous TGF-ß1 and ALK5 inhibitor can positively and negatively regulate the nuclear translocations of HIF-1α without affecting the transcription activity of HIF-1α. Therefore, our data further suggest that autocrine TGF-ß1 from NiONPs treatment fibroblasts causes TGF/Smad3 signaling and then regulates HIF-1α activity.
The mammalian TGF-ß family consists of three members. TGF-ß1 is a profibrotic growth factor and TGF-ß3 is anti-fibrotic [71,72]. There is contention about the effect of TGF-ß2 [71,73]. Previous studies have demonstrated that TGF-ß3 is a potential HIF-1α-regulated growth factors [72,74]. Upregulation of HIF-1α will enhance TGF-ß3 expression through binding to TGF-ß3 promoter [72]. Thus, we wonder TGF-ß1, which has a reverse function compared with TGF-ß3, might be regulated by HIF-1α. In our studies, activation of TGF-ß1/Smad3 was inhibited by HIF-1α downregulation. While further studies are needed to define the specific mechanism by which HIF-1α mediates TGF-ß1 expression.
Finally, our results support that NiONP is an important factor in pulmonary fibrosis induced by dust inhalation. Furthermore, our results suggest TGF-ß1 may provide a positive feedback loop that maintains a high level of HIF-1α activity during the process of pulmonary fibrosis. Inhibition of the cross talk of HIF-1α and TGF-ß1 may be a new strategy for anti-fibrotic manipulation of pulmonary fibrosis induced by NiONPs.
Acknowledgements
This work was supported by the Outstanding Young Scientist Project of National Natural Science Foundation of China (No. 81422039), National Natural Science Foundation of China (No. 81102108) and the Grant for Outstanding Young Scientist from Chongqing Science and Technology Commission (No. CSTC2013jcyjjq10002).
Disclosure of conflict of interest
None.
References
- 1.Gross TJ, Hunninghake GW. Idiopathic pulmonary fibrosis. N Engl J Med. 2001;345:517–525. doi: 10.1056/NEJMra003200. [DOI] [PubMed] [Google Scholar]
- 2.Yaguchi T, Fukuda Y, Ishizaki M, Yamanaka N. Immunohistochemical and gelatin zymography studies for matrix metalloproteinases in bleomycin-induced pulmonary fibrosis. Pathol Int. 1998;48:954–963. doi: 10.1111/j.1440-1827.1998.tb03866.x. [DOI] [PubMed] [Google Scholar]
- 3.Wilson MS, Wynn TA. Pulmonary fibrosis: pathogenesis, etiology and regulation. Mucosal Immunol. 2009;2:103–121. doi: 10.1038/mi.2008.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baumgartner KB, Samet JM, Stidley CA, Colby TV, Waldron JA. Cigarette smoking: a risk factor for idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1997;155:242–248. doi: 10.1164/ajrccm.155.1.9001319. [DOI] [PubMed] [Google Scholar]
- 5.Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ahamed M, Akhtar MJ, Siddiqui MA, Ahmad J, Musarrat J, Al-Khedhairy AA, AlSalhi MS, Alrokayan SA. Oxidative stress mediated apoptosis induced by nickel ferrite nanoparticles in cultured A549 cells. Toxicology. 2011;283:101–108. doi: 10.1016/j.tox.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 7.Ahamed M. Toxic response of nickel nanoparticles in human lung epithelial A549 cells. Toxicol In Vitro. 2011;25:930–936. doi: 10.1016/j.tiv.2011.02.015. [DOI] [PubMed] [Google Scholar]
- 8.Lu S, Duffin R, Poland C, Daly P, Murphy F, Drost E, Macnee W, Stone V, Donaldson K. Efficacy of simple short-term in vitro assays for predicting the potential of metal oxide nanoparticles to cause pulmonary inflammation. Environ Health Perspect. 2009;117:241–247. doi: 10.1289/ehp.11811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saputra D, Yoon JH, Park H, Heo Y, Yang H, Lee EJ, Lee S, Song CW, Lee K. Inhalation of carbon black nanoparticles aggravates pulmonary inflammation in mice. Toxicol Res. 2014;30:83–90. doi: 10.5487/TR.2014.30.2.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cho WS, Duffin R, Poland CA, Duschl A, Oostingh GJ, Macnee W, Bradley M, Megson IL, Donaldson K. Differential pro-inflammatory effects of metal oxide nanoparticles and their soluble ions in vitro and in vivo; zinc and copper nanoparticles, but not their ions, recruit eosinophils to the lungs. Nanotoxicology. 2012;6:22–35. doi: 10.3109/17435390.2011.552810. [DOI] [PubMed] [Google Scholar]
- 11.Kang GS, Gillespie PA, Gunnison A, Moreira AL, Tchou-Wong KM, Chen LC. Long-term inhalation exposure to nickel nanoparticles exacerbated atherosclerosis in a susceptible mouse model. Environ Health Perspect. 2011;119:176–181. doi: 10.1289/ehp.1002508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song Y, Li X, Du X. Exposure to nanoparticles is related to pleural effusion, pulmonary fibrosis and granuloma. Eur Respir J. 2009;34:559–567. doi: 10.1183/09031936.00178308. [DOI] [PubMed] [Google Scholar]
- 13.Morimoto Y, Ogami A, Todoroki M, Yamamoto M, Murakami M, Hirohashi M, Oyabu T, Myojo T, Nishi K, Kadoya C, Yamasaki S, Nagatomo H, Fujita K, Endoh S, Uchida K, Yamamoto K, Kobayashi N, Nakanishi J, Tanaka I. Expression of inflammation-related cytokines following intratracheal instillation of nickel oxide nanoparticles. Nanotoxicology. 2010;4:161–176. doi: 10.3109/17435390903518479. [DOI] [PubMed] [Google Scholar]
- 14.Glista-Baker EE, Taylor AJ, Sayers BC, Thompson EA, Bonner JC. Nickel nanoparticles cause exaggerated lung and airway remodeling in mice lacking the T-box transcription factor, TBX21 (T-bet) Part Fibre Toxicol. 2014;11:7. doi: 10.1186/1743-8977-11-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelly BD, Hackett SF, Hirota K, Oshima Y, Cai Z, Berg-Dixon S, Rowan A, Yan Z, Campochiaro PA, Semenza GL. Cell type-specific regulation of angiogenic growth factor gene expression and induction of angiogenesis in nonischemic tissue by a constitutively active form of hypoxia-inducible factor 1. Circ Res. 2003;93:1074–1081. doi: 10.1161/01.RES.0000102937.50486.1B. [DOI] [PubMed] [Google Scholar]
- 16.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 17.Cockman ME, Masson N, Mole DR, Jaakkola P, Chang GW, Clifford SC, Maher ER, Pugh CW, Ratcliffe PJ, Maxwell PH. Hypoxia inducible factor-alpha binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J Biol Chem. 2000;275:25733–25741. doi: 10.1074/jbc.M002740200. [DOI] [PubMed] [Google Scholar]
- 18.Tanimoto K, Makino Y, Pereira T, Poellinger L. Mechanism of regulation of the hypoxia-inducible factor-1 alpha by the von Hippel-Lindau tumor suppressor protein. Embo J. 2000;19:4298–4309. doi: 10.1093/emboj/19.16.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamura T, Sato S, Iwai K, Czyzyk-Krzeska M, Conaway RC, Conaway JW. Activation of HIF1alpha ubiquitination by a reconstituted von Hippel-Lindau (VHL) tumor suppressor complex. Proc Natl Acad Sci U S A. 2000;97:10430–10435. doi: 10.1073/pnas.190332597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, Pavletich N, Chau V, Kaelin WG. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol. 2000;2:423–427. doi: 10.1038/35017054. [DOI] [PubMed] [Google Scholar]
- 21.Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, Hohenstein B, Saito Y, Johnson RS, Kretzler M, Cohen CD, Eckardt KU, Iwano M, Haase VH. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest. 2007;117:3810–3820. doi: 10.1172/JCI30487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Copple BL, Kaska S, Wentling C. Hypoxiainducible factor activation in myeloid cells contributes to the development of liver fibrosis in cholestatic mice. J Pharmacol Exp Ther. 2012;341:307–316. doi: 10.1124/jpet.111.189340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moon JO, Welch TP, Gonzalez FJ, Copple BL. Reduced liver fibrosis in hypoxia-inducible factor-1alpha-deficient mice. Am J Physiol Gastrointest Liver Physiol. 2009;296:G582–592. doi: 10.1152/ajpgi.90368.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Distler JH, Jungel A, Pileckyte M, Zwerina J, Michel BA, Gay RE, Kowal-Bielecka O, Matucci-Cerinic M, Schett G, Marti HH, Gay S, Distler O. Hypoxia-induced increase in the production of extracellular matrix proteins in systemic sclerosis. Arthritis Rheum. 2007;56:4203–4215. doi: 10.1002/art.23074. [DOI] [PubMed] [Google Scholar]
- 25.Tzouvelekis A, Harokopos V, Paparountas T, Oikonomou N, Chatziioannou A, Vilaras G, Tsiambas E, Karameris A, Bouros D, Aidinis V. Comparative expression profiling in pulmonary fibrosis suggests a role of hypoxia-inducible factor-1alpha in disease pathogenesis. Am J Respir Crit Care Med. 2007;176:1108–1119. doi: 10.1164/rccm.200705-683OC. [DOI] [PubMed] [Google Scholar]
- 26.Pietruska JR, Liu X, Smith A, McNeil K, Weston P, Zhitkovich A, Hurt R, Kane AB. Bioavailability, intracellular mobilization of nickel, and HIF-1alpha activation in human lung epithelial cells exposed to metallic nickel and nickel oxide nanoparticles. Toxicol Sci. 2011;124:138–148. doi: 10.1093/toxsci/kfr206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sampson N, Berger P, Zenzmaier C. Therapeutic targeting of redox signaling in myofibroblast differentiation and age-related fibrotic disease. Oxid Med Cell Longev. 2012;2012:458276. doi: 10.1155/2012/458276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Warburton D, Shi W, Xu B. TGF-beta-Smad3 signaling in emphysema and pulmonary fibrosis: an epigenetic aberration of normal development? Am J Physiol Lung Cell Mol Physiol. 2013;304:L83–85. doi: 10.1152/ajplung.00258.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harris WT, Kelly DR, Zhou Y, Wang D, MacEwen M, Hagood JS, Clancy JP, Ambalavanan N, Sorscher EJ. Myofibroblast differentiation and enhanced TGF-B signaling in cystic fibrosis lung disease. PLoS One. 2013;8:e70196. doi: 10.1371/journal.pone.0070196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jester JV, Huang J, Petroll WM, Cavanagh HD. TGFbeta induced myofibroblast differentiation of rabbit keratocytes requires synergistic TGFbeta, PDGF and integrin signaling. Exp Eye Res. 2002;75:645–657. doi: 10.1006/exer.2002.2066. [DOI] [PubMed] [Google Scholar]
- 31.Liu J, Wang Y, Pan Q, Su Y, Zhang Z, Han J, Zhu X, Tang C, Hu D. Wnt/beta-catenin pathway forms a negative feedback loop during TGFbeta1 induced human normal skin fibroblast-to-myofibroblast transition. J Dermatol Sci. 2012;65:38–49. doi: 10.1016/j.jdermsci.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 32.Glista-Baker EE, Taylor AJ, Sayers BC, Thompson EA, Bonner JC. Nickel nanoparticles enhance platelet-derived growth factor-induced chemokine expression by mesothelial cells via prolonged mitogen-activated protein kinase activation. Am J Respir Cell Mol Biol. 2012;47:552–561. doi: 10.1165/rcmb.2012-0023OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Basu RK, Hubchak S, Hayashida T, Runyan CE, Schumacker PT, Schnaper HW. Interdependence of HIF-1alpha and TGF-beta/Smad3 signaling in normoxic and hypoxic renal epithelial cell collagen expression. Am J Physiol Renal Physiol. 2011;300:F898–905. doi: 10.1152/ajprenal.00335.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schnaper HW, Jandeska S, Runyan CE, Hubchak SC, Basu RK, Curley JF, Smith RD, Hayashida T. TGF-beta signal transduction in chronic kidney disease. Front Biosci (Landmark Ed) 2009;14:2448–2465. doi: 10.2741/3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Reilly S, Ciechomska M, Cant R, van Laar JM. Interleukin-6 (IL-6) trans signaling drives a STAT3-dependent pathway that leads to hyperactive transforming growth factor-beta (TGFbeta) signaling promoting SMAD3 activation and fibrosis via Gremlin protein. J Biol Chem. 2014;289:9952–9960. doi: 10.1074/jbc.M113.545822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tobar N, Toyos M, Urra C, Mendez N, Arancibia R, Smith PC, Martinez J. c-Jun N terminal kinase modulates NOX-4 derived ROS production and myofibroblasts differentiation in human breast stromal cells. BMC Cancer. 2014;14:640. doi: 10.1186/1471-2407-14-640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Murata M, Yoshida K, Yamaguchi T, Matsuzaki K. Linker phosphorylation of Smad3 promotes fibro-carcinogenesis in chronic viral hepatitis of hepatocellular carcinoma. World J Gastroenterol. 2014;20:15018–15027. doi: 10.3748/wjg.v20.i41.15018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khalil N, O’Connor RN, Unruh HW, Warren PW, Flanders KC, Kemp A, Bereznay OH, Greenberg AH. Increased production and immunohistochemical localization of transforming growth factor-beta in idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 1991;5:155–162. doi: 10.1165/ajrcmb/5.2.155. [DOI] [PubMed] [Google Scholar]
- 39.Ueno M, Maeno T, Nomura M, Aoyagi-Ikeda K, Matsui H, Hara K, Tanaka T, Iso T, Suga T, Kurabayashi M. Hypoxia-inducible factor-1alpha mediates TGF-beta-induced PAI-1 production in alveolar macrophages in pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2011;300:L740–752. doi: 10.1152/ajplung.00146.2010. [DOI] [PubMed] [Google Scholar]
- 40.Copple BL. Hypoxia stimulates hepatocyte epithelial to mesenchymal transition by hypoxiainducible factor and transforming growth factor-beta-dependent mechanisms. Liver Int. 2010;30:669–682. doi: 10.1111/j.1478-3231.2010.02205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abdul-Hafez A, Shu R, Uhal BD. JunD and HIF-1alpha mediate transcriptional activation of angiotensinogen by TGF-beta1 in human lung fibroblasts. FASEB J. 2009;23:1655–1662. doi: 10.1096/fj.08-114611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chae KS, Kang MJ, Lee JH, Ryu BK, Lee MG, Her NG, Ha TK, Han J, Kim YK, Chi SG. Opposite functions of HIF-alpha isoforms in VEGF induction by TGF-beta1 under non-hypoxic conditions. Oncogene. 2011;30:1213–1228. doi: 10.1038/onc.2010.498. [DOI] [PubMed] [Google Scholar]
- 43.Hanna C, Hubchak SC, Liang X, Rozen-Zvi B, Schumacker PT, Hayashida T, Schnaper HW. Hypoxia-inducible factor-2alpha and TGFbeta signaling interact to promote normoxic glomerular fibrogenesis. Am J Physiol Renal Physiol. 2013;305:F1323–1331. doi: 10.1152/ajprenal.00155.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McMahon S, Charbonneau M, Grandmont S, Richard DE, Dubois CM. Transforming growth factor beta1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of PHD2 expression. J Biol Chem. 2006;281:24171–24181. doi: 10.1074/jbc.M604507200. [DOI] [PubMed] [Google Scholar]
- 45.Haase VH. The VHL/HIF oxygen-sensing pathway and its relevance to kidney disease. Kidney Int. 2006;69:1302–1307. doi: 10.1038/sj.ki.5000221. [DOI] [PubMed] [Google Scholar]
- 46.Kimura K, Iwano M, Higgins DF, Yamaguchi Y, Nakatani K, Harada K, Kubo A, Akai Y, Rankin EB, Neilson EG, Haase VH, Saito Y. Stable expression of HIF-1alpha in tubular epithelial cells promotes interstitial fibrosis. Am J Physiol Renal Physiol. 2008;295:F1023–1029. doi: 10.1152/ajprenal.90209.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sanchez-Elsner T, Ramirez JR, Sanz-Rodriguez F, Varela E, Bernabeu C, Botella LM. A cross-talk between hypoxia and TGF-beta orchestrates erythropoietin gene regulation through SP1 and Smads. J Mol Biol. 2004;336:9–24. doi: 10.1016/j.jmb.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 48.Duan WX, He MD, Mao L, Qian FH, Li YM, Pi HF, Liu C, Chen CH, Lu YH, Cao ZW, Zhang L, Yu ZP, Zhou Z. NiO nanoparticles induce apoptosis through repressing SIRT1 in human bronchial epithelial cells. Toxicol Appl Pharmacol. 2015;286:80–91. doi: 10.1016/j.taap.2015.03.024. [DOI] [PubMed] [Google Scholar]
- 49.Gill SE, Parks WC. Metalloproteinases and their inhibitors: regulators of wound healing. Int J Biochem Cell Biol. 2008;40:1334–1347. doi: 10.1016/j.biocel.2007.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pistollato F, Abbadi S, Rampazzo E, Viola G, Della Puppa A, Cavallini L, Frasson C, Persano L, Panchision DM, Basso G. Hypoxia and succinate antagonize 2-deoxyglucose effects on glioblastoma. Biochem Pharmacol. 2010;80:1517–1527. doi: 10.1016/j.bcp.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 52.Wangpaichitr M, Savaraj N, Maher J, Kurtoglu M, Lampidis TJ. Intrinsically lower AKT, mammalian target of rapamycin, and hypoxiainducible factor activity correlates with increased sensitivity to 2-deoxy-D-glucose under hypoxia in lung cancer cell lines. Mol Cancer Ther. 2008;7:1506–1513. doi: 10.1158/1535-7163.MCT-07-2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang P, Nie X, Wang Y, Li Y, Ge C, Zhang L, Wang L, Bai R, Chen Z, Zhao Y, Chen C. Multiwall carbon nanotubes mediate macrophage activation and promote pulmonary fibrosis through TGF-beta/Smad signaling pathway. Small. 2013;9:3799–3811. doi: 10.1002/smll.201300607. [DOI] [PubMed] [Google Scholar]
- 54.Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, Rifkin DB, Sheppard D. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- 55.Bottinger EP, Bitzer M. TGF-beta signaling in renal disease. J Am Soc Nephrol. 2002;13:2600–2610. doi: 10.1097/01.asn.0000033611.79556.ae. [DOI] [PubMed] [Google Scholar]
- 56.Poncelet AC, de Caestecker MP, Schnaper HW. The transforming growth factor-beta/SMAD signaling pathway is present and functional in human mesangial cells. Kidney Int. 1999;56:1354–1365. doi: 10.1046/j.1523-1755.1999.00680.x. [DOI] [PubMed] [Google Scholar]
- 57.Eul B, Rose F, Krick S, Savai R, Goyal P, Klepetko W, Grimminger F, Weissmann N, Seeger W, Hanze J. Impact of HIF-1alpha and HIF-2alpha on proliferation and migration of human pulmonary artery fibroblasts in hypoxia. FASEB J. 2006;20:163–165. doi: 10.1096/fj.05-4104fje. [DOI] [PubMed] [Google Scholar]
- 58.Borm PJ, Robbins D, Haubold S, Kuhlbusch T, Fissan H, Donaldson K, Schins R, Stone V, Kreyling W, Lademann J, Krutmann J, Warheit D, Oberdorster E. The potential risks of nanomaterials: a review carried out for ECETOC. Part Fibre Toxicol. 2006;3:11. doi: 10.1186/1743-8977-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nohynek GJ, Lademann J, Ribaud C, Roberts MS. Grey goo on the skin? Nanotechnology, cosmetic and sunscreen safety. Crit Rev Toxicol. 2007;37:251–277. doi: 10.1080/10408440601177780. [DOI] [PubMed] [Google Scholar]
- 60.Nel A, Xia T, Madler L, Li N. Toxic potential of materials at the nanolevel. Science. 2006;311:622–627. doi: 10.1126/science.1114397. [DOI] [PubMed] [Google Scholar]
- 61.Nel AE, Madler L, Velegol D, Xia T, Hoek EM, Somasundaran P, Klaessig F, Castranova V, Thompson M. Understanding biophysicochemical interactions at the nano-bio interface. Nat Mater. 2009;8:543–557. doi: 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- 62.Cho WS, Duffin R, Poland CA, Howie SE, MacNee W, Bradley M, Megson IL, Donaldson K. Metal oxide nanoparticles induce unique inflammatory footprints in the lung: important implications for nanoparticle testing. Environ Health Perspect. 2010;118:1699–1706. doi: 10.1289/ehp.1002201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Park EJ, Roh J, Kim SN, Kang MS, Han YA, Kim Y, Hong JT, Choi K. A single intratracheal instillation of single-walled carbon nanotubes induced early lung fibrosis and subchronic tissue damage in mice. Arch Toxicol. 2011;85:1121–1131. doi: 10.1007/s00204-011-0655-8. [DOI] [PubMed] [Google Scholar]
- 64.Cheresh P, Kim SJ, Tulasiram S, Kamp DW. Oxidative stress and pulmonary fibrosis. Biochim Biophys Acta. 2013;1832:1028–1040. doi: 10.1016/j.bbadis.2012.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Horie M, Nishio K, Fujita K, Kato H, Nakamura A, Kinugasa S, Endoh S, Miyauchi A, Yamamoto K, Murayama H, Niki E, Iwahashi H, Yoshida Y, Nakanishi J. Ultrafine NiO particles induce cytotoxicity in vitro by cellular uptake and subsequent Ni(II) release. Chem Res Toxicol. 2009;22:1415–1426. doi: 10.1021/tx900171n. [DOI] [PubMed] [Google Scholar]
- 66.Namiki A, Brogi E, Kearney M, Kim EA, Wu T, Couffinhal T, Varticovski L, Isner JM. Hypoxia induces vascular endothelial growth factor in cultured human endothelial cells. J Biol Chem. 1995;270:31189–31195. doi: 10.1074/jbc.270.52.31189. [DOI] [PubMed] [Google Scholar]
- 67.Salnikow K, An WG, Melillo G, Blagosklonny MV, Costa M. Nickel-induced transformation shifts the balance between HIF-1 and p53 transcription factors. Carcinogenesis. 1999;20:1819–1823. doi: 10.1093/carcin/20.9.1819. [DOI] [PubMed] [Google Scholar]
- 68.Goldberg MA, Dunning SP, Bunn HF. Regulation of the erythropoietin gene: evidence that the oxygen sensor is a heme protein. Science. 1988;242:1412–1415. doi: 10.1126/science.2849206. [DOI] [PubMed] [Google Scholar]
- 69.Zhu H, Bunn HF. Oxygen sensing and signaling: impact on the regulation of physiologically important genes. Respir Physiol. 1999;115:239–247. doi: 10.1016/s0034-5687(99)00024-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Higgins DF, Kimura K, Iwano M, Haase VH. Hypoxia-inducible factor signaling in the development of tissue fibrosis. Cell Cycle. 2008;7:1128–1132. doi: 10.4161/cc.7.9.5804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sgonc R, Wick G. Pro- and anti-fibrotic effects of TGF-beta in scleroderma. Rheumatology (Oxford) 2008;47(Suppl 5):v5–7. doi: 10.1093/rheumatology/ken275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Scheid A, Wenger RH, Schaffer L, Camenisch I, Distler O, Ferenc A, Cristina H, Ryan HE, Johnson RS, Wagner KF, Stauffer UG, Bauer C, Gassmann M, Meuli M. Physiologically low oxygen concentrations in fetal skin regulate hypoxia-inducible factor 1 and transforming growth factor-beta3. FASEB J. 2002;16:411–413. doi: 10.1096/fj.01-0496fje. [DOI] [PubMed] [Google Scholar]
- 73.Talior-Volodarsky I, Connelly KA, Arora PD, Gullberg D, McCulloch CA. a11 integrin stimulates myofibroblast differentiation in diabetic cardiomyopathy. Cardiovasc Res. 2012;96:265–275. doi: 10.1093/cvr/cvs259. [DOI] [PubMed] [Google Scholar]
- 74.Caniggia I, Mostachfi H, Winter J, Gassmann M, Lye SJ, Kuliszewski M, Post M. Hypoxiainducible factor-1 mediates the biological effects of oxygen on human trophoblast differentiation through TGFbeta(3) J Clin Invest. 2000;105:577–587. doi: 10.1172/JCI8316. [DOI] [PMC free article] [PubMed] [Google Scholar]