

Abstract
The coupling of tertiary carbon radicals with alkene acceptors is an underdeveloped strategy for uniting complex carbon fragments and forming new quaternary carbons. The scope and limitations of a new approach for generating nucleophilic tertiary radicals from tertiary alcohols and utilizing these intermediates in fragment coupling reactions is described. In this method, the tertiary alcohol is first acylated to give the tert-alkyl N-phthalimidoyl oxalate, which in the presence of visible-light, catalytic Ru(bpy)3(PF6)2, and a reductant fragments to form the corresponding tertiary carbon radical. In addition to reductive coupling with alkenes, substitution reactions of tertiary radicals with allylic and vinylic halides is described. A mechanism for the generation of tertiary carbon radicals from tert-alkyl N-phthalimidoyl oxalates is proposed that is based on earlier pioneering investigations of Okada and Barton. Deuterium labeling and competition experiments reveal that the reductive radical coupling of tert-alkyl N-phthalimidoyl oxalates with electron-deficient alkenes is terminated by hydrogen-atom transfer.
INTRODUCTION
The bimolecular coupling of tertiary nucleophiles with carbon-based electrophiles is a straightforward, yet underdeveloped, approach for fragment coupling with concomitant formation of sterically encumbered quaternary carbons. Recent total synthesis endeavors in our laboratories have suggested the potentially broad utility of both trialkyl-tertiary cuprates and nucleophilic carbon radicals in such constructions.1,2,3 Tertiary carbon radicals are often generated from halide precursors. However, the synthesis of structurally elaborate tertiary halides can be complicated by competing elimination and rearrangement reactions. As a result, methods to generate tertiary carbon radicals from other common functional groups have particular significance. Barton’s methods for forming carbon radicals by homolytic fragmentation of carboxylic acid-derived thiohydroxamate esters4 or alcohol-derived thiohydroxamate oxalates,5 introduced more than 25 years ago, are pioneering examples.6,7
In an early application of visible-light photocatalysis in organic synthesis, Okada reported in 1991 the utility of carboxylic acid-derived (N-acyloxy)phthalimides for generating nucleophilic carbon radicals and their use in radical conjugate addition reactions to construct carbon-carbon bonds.8,9 These precursors are particularly attractive as they are readily prepared, highly stable, and often crystalline. This method for generating structurally complex tertiary radicals was a central feature of our total syntheses of the diterpenoid (−)-aplyviolene1a,b and several trans-clerodane diterpenoids.1c To extend this approach to other readily available precursors, we recently described the utility of N-phthalimidoyl oxalate derivatives of tertiary alcohols for generating tertiary radicals using visible-light photocatalysis.3b
Since our recent studies of photoredox-catalyzed generation of tertiary radicals and their use in fragment coupling reactions,1b,c,3b several other research programs contributed important new methods for accessing tertiary carbon radical intermediates. Baran described the reaction of substituted alkenes with Fe(acac)3/PhSiH3 to generate secondary and tertiary radicals and their use in both intramolecular and bimolecular C–C bond-formation.3c In addition, recent reports by the MacMillan group detail the generation of radical intermediates by single-electron oxidation and decarboxylation of carboxylates.3d Although heteroatom-stabilized secondary radicals are most easily formed under these conditions, the adamantyl radical was also produced in this way.
Even though several approaches for producing tertiary carbon radicals from common precursors are now available, the prevalence and accessibility of tertiary alcohols warrants comprehensive investigation of the utility of tert-alkyl N-phthalimidoyl oxalates as precursors of these intermediates. Presented herein is a detailed exploration of the preparation and visible-light photocatalytic coupling of tert-alkyl N-phthalimidoyl oxalates with alkenes.10,11 In addition, studies revealing how small modifications in reaction conditions and resulting changes in termination steps can affect the outcome of coupling reactions of these radical precursors are also disclosed. A comparison of tert-alkyl N-phthalimidoyl oxalates to (N-acyloxy)phthalimide derivatives of tertiary carboxylic acids for initiating coupling reactions of tertiary radicals is provided in the accompanying paper.12
RESULTS AND DISCUSSION
Synthesis of tert-alkyl N-phthalimidoyl oxalates
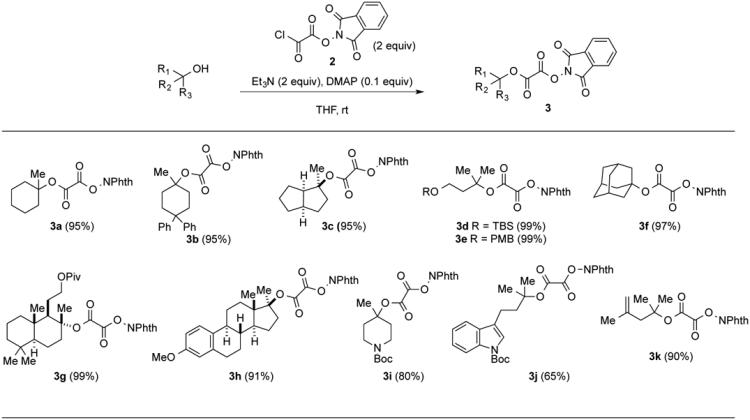
A general strategy for synthesizing tert-alkyl N-phthalimidoyl oxalates from tertiary alcohols consists of the direct acylation of the tertiary alcohol with N-phthalimidoyl chlorooxalate 2, as exemplified in the formation of N-phthalimidoyl oxalate 3a from 1-methylcyclohexanol (1) (eq 1). After careful optimization, the following procedure was developed. Chlorooxalate 2 is initially prepared as a colorless solid by adding an excess of oxalyl chloride (5 equiv) to a THF solution of N-hydroxyphthalimide at − 78 °C, allowing the reaction to warm to room temperature (typically overnight), and then concentrating the reaction mixture under reduced pressure. This reactive acylating agent is then most easily manipulated as a 0.06 M solution in THF. The subsequent acylation of tertiary alcohols takes place efficiently in THF in the presence of 2 equiv each of chlorooxalate 2 and triethylamine and a catalytic amount of DMAP.
 |
The sensitivity of tertiary N-phthalimidoyl oxalates towards protic nucleophiles complicates their isolation and purification. For example, oxalate 3a could not be purified by chromatography on silica gel. Washing a dilute Et2O solution of crude 3a with saturated aqueous NaHCO3 did remove unreacted N-hydroxyphthalimide from the product mixture. However, resubjection of pure phthalimidoyl oxalate 3a to this extraction procedure resulted in substantial cleavage to release N-hydroxyphthalimide, suggesting that even this mild workup was problematic. tert-Alkyl N-phthalimidoyl oxalates are best isolated by diluting a concentrated CH2Cl2 solution of the crude acylation reaction products with hexanes (typically a 25-fold excess by volume) and filtering the resulting suspension. After concentration of the filtrate, the mixed oxalate diesters 3 are obtained in good purity (typically >95%) and high yield. As illustrated in Table 1, this method proved quite general, converting even highly hindered tertiary alcohols to the corresponding mixed oxalates efficiently (e.g., formation of 3g and 3h). Solid tert-alkyl N-phthalimidoyl oxalates prepared in this way could be stored indefinitely at −20 °C, whereas those that are oils could be stored at this temperature for only one week before decomposition was apparent.
Table 1.
Synthesis of tert-Alkyl N-Phthalimidoyl Oxalates (NPhth = N-Phthalimidoyl)
|
The structure of the adamantyl N-phthalimidoyl oxalate (3f) was confirmed by single-crystal X-ray analysis.3b The reduction potential of N-phthalimidoyl oxalate 3a measured by CV in acetonitrile is −1.14 V vs SCE, and, as expected, is slightly less negative than that of the (N-acyloxy)phthalimide derivative of 1-methylcyclohexanecarboxylic acid (−1.26 V vs SCE) under identical conditions (see Supporting Information).13
Coupling of tert-alkyl N-phthalimidoyl oxalates with conjugate acceptors
We anticipated that tert-alkyl N-phthalimidoyl oxalates would react in the presence of a photocatalyst and a stoichiometric reductant via single-electron transfer to the tetracarbonyl substrate, followed by homolytic cleavage of the N–O σ-bond with ejection of phthalimide and two equivalents of CO2 to generate the radical intermediate (Scheme 1).5,8 Addition of the nucleophilic tertiary radical to an electron-deficient olefin and hydrogen-atom transfer to the resulting radical intermediate would yield the product.
Scheme 1. Proposed Mechanism for the Coupling N-Phthalimidoyl Oxalates 3 with Conjugate Acceptors.
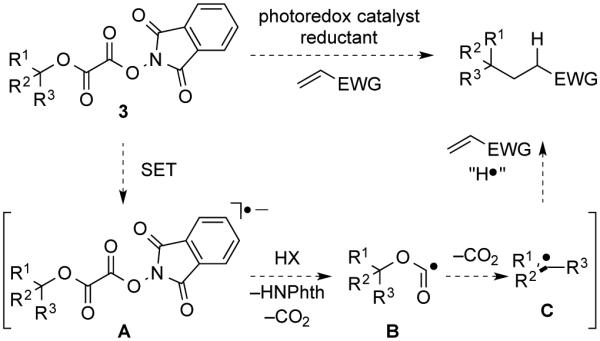
In our earlier investigations of the use of carboxylic acid-derived (N-acyloxy)phthalimides in fragment coupling reactions,1b we found that aprotic conditions similar to those reported by Gagné for the generation and reaction of radicals derived from glucosyl halides—visible light, Ru(bpy)3(BF4)2, diethyl 1,4-dihydro-2,6-dimethyl-3,5-pyridinedicarboxylate (4), i-Pr2NEt, in CH2Cl214—were preferable to the aqueous conditions originally described by Okada.8,12 The reaction of tert-alkyl N-phthalimidoyl oxalate 3a with methyl vinyl ketone (MVK) under related conditions in CH2Cl2 employing 1–2 mol% of the commercially available bis(hexafluorophosphate) salt of Ru(bpy)32+ indeed did provide the coupled product 5, albeit in low yield (eq 2). The low yield was easily traced to competitive decomposition of the N-phthalimidoyl oxalate under these conditions, as control experiments
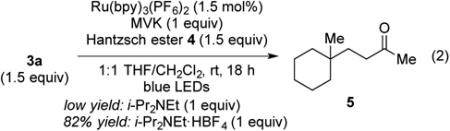 |
established that the red conjugate base of N-hydroxyphthalimide was rapidly formed when oxalate 3a was exposed at room temperature to amines such as i-Pr2NEt, Bu3N, or Cy2NMe in CH2Cl2. As a result of these observations, further experimentation was carried out using i-PrNEt•HBF4 instead of i-Pr2NEt. Additional improvement was realized by replacing CH2Cl2 with a 1:1 mixture of CH2Cl2/THF. This solvent change was necessitated because oxalate 3a slowly decomposed in CH2Cl2 overnight, presumably by ionization to form the 1-methylcyclohexyl cation. In addition to commercially available blue LEDs, a commercially available compact fluorescent light was found to be equally effective as the light source.15
We then performed a series of reactions to evaluate the necessity of each reaction component (Table 2). Although we initially carried out the coupling reactions for 18 h, because the oxalate substrates were not amenable to TLC analysis, the yield of 5 was nearly identical when the reaction was run for only 2 h (entries 1 and 2). Visible light proved to be essential for reactivity, as no conversion of oxalate 3a was observed when the reaction was conducted in the dark (entry 3). Omission of Hantzsch ester 4 also resulted in complete recovery of 3a (entry 4). In contrast, we observed significant product formation in the absence of the photocatalyst (entries 5 and 6). This background reaction was slower than the photocatalyzed reaction, producing product 5 in only 28% yield after 2 h, although the yield was improved to 67% after 18 h. This background reaction, which we believe is mediated by Hantzsch ester 4,16 was also observed by Okada and co-workers when the photocatalyst was not present.8 Omitting the ammonium additive resulted in a somewhat depressed yield of 5 (entry 7). Finally, revisiting the stoichiometry of the coupling substrates (entries 8–10) confirmed that the highest yield of 5 was obtained when oxalate 3a was used in a slight (50%) excess.
Table 2.
Coupling of N-Phthalimidoyl Oxalate 3a with Methyl Vinyl Ketone (MVK) Under Various Reaction Conditionsa
| Entry | Modification | Yield of 5 (%)b |
|---|---|---|
| 1 | none | 82 |
| 2 | reaction time of 2 h | 81 |
| 3 | no light | <5% |
| 4 | no Hantzsch ester | ND |
| 5 | no photocatalyst (2 h) | 28 |
| 6 | no photocatalyst (18 h) | 67 |
| 7 | no i-Pr2NEt•HBF4 | 72 |
| 8 | oxalate 3a (1 equiv) | 67 |
| 9 | oxalate 3a (1.1 equiv) | 68 |
| 10 | oxalate 3a (1 equiv) MVK (1.5 equiv) |
57 |
Conditions of eq 2 using i-Pr2NEt•HBF4; [3a] = 0.15 M.
Isolated yield after silica gel chromatography.
ND = not detected.
Several other photoredox catalysts were examined to promote the formation of adduct 5 from N-phthalimidoyl oxalate 3a and MVK (Table 3). Although no catalyst performed as well as Ru(bpy)32+ (entry 1), several strongly reducing iridium photoredox catalysts were nearly as effective (entries 2–4).17 Even Ru(bpz)32+ (E1/2II/I −0.80 V vs SCE),18 a significantly weaker reductant than Ru(bpy)32+ (E1/2II/I −1.33 V vs SCE), gave a 62% yield (by NMR analysis) of ketone 5 (entry 5).
Table 3.
Coupling of N-Phthalimidoyl Oxalate 3a with MVK in the Presence of Various Photocatalysts and Visible Lighta
| Entry | Photocatalyst | Yield of 5
(%)b |
|---|---|---|
| 1 | Ru(bpy)3(BF4)2 | 82 |
| 2 | Ir(ppy)3 | 75 |
| 3 | Ir(df(CF3)ppy)2(dtbbpy)PF6 | 74 |
| 4 | Ir(dtbbpy)ppy2PF6 | 76 |
| 5 | Ru(bpz)3(PF6)2 | 62 |
Conditions of eq 2 using i-Pr2NEt•HBF4 and a reaction time of 2 h; [3a] = 0.15 M.
Yield measured by 1H NMR relative to an internal standard (1,4-dimethoxybenzene).
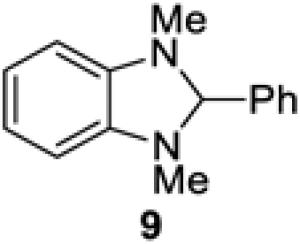
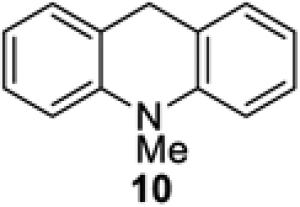
We also explored replacing Hantzsch ester 4 with other potential stoichiometric reductants (Table 4). Introduction of a substituent at the 4-position of the Hantzsch ester rendered the dihydropyridine unreactive under the reaction conditions (entries 2 and 3). The use of 2-phenylbenzothiazoline (8) or N,N’-dimethyl-2-phenylbenzimidazoline (9) successfully resulted in the formation of product 5, although the yield was somewhat less than that observed under identical conditions using Hantzsch ester 4 (entries 4 and 5).19 No conversion to adduct 5 was observed using N-methylacridane (10), a known reductive quencher of Ru(bpy)32+* (entry 6).20
Table 4.
Visible-Light Photocatalytic Coupling of N-Phthalimidoyl Oxalate 3a with MVK in the Presence of Potential Stoichiometric Reductantsaa

Conditions of eq 2 using i-Pr2NEt•HBF4; [3a] = 0.15 M.
Isolated yield after silica gel chromatography.
Yield measured by 1H NMR relative to an internal standard (1,4-dimethoxybenzene);
ND = not detected.
The results of our initial survey of the coupling of tert-alkyl N-phthalimidoyl oxalates with various electron-deficient alkenes are summarized in Table 5. A wide variety of N-phthalimidoyl oxalate substrates and conjugate acceptors are tolerated in the transformation, which generally gives good yields of the coupled products. The highest yields were realized in the coupling reactions with alkenes such as MVK, acrylonitrile and phenyl vinyl sulfone, which are unsubstituted at the β-carbon. The three chiral N-phthalimidoyl oxalates that we examined coupled with MVK from the less-sterically hindered face of the tertiary radical to give products 14, 15 and 16 with >20:1 diastereoselection, with coupled products 14 and 15 being formed in >80% yield. The yield was somewhat lower in the construction of a quaternary center at C17 in the estrone series (16, 68%), likely reflecting the steric demand in forming vicinal quaternary carbon centers. Many of these transformations require only 2–3 h, as shown by the entries in the first row of Table 5. As expected from our exploratory studies, yields were somewhat lower when equal amounts of the N-phthalimidoyl oxalate and alkene were employed: 63% vs 85% in forming 14 and 43% vs 68% in forming 16. In a number of the coupling reactions summarized in Table 5, we observed that the yield was nearly the same when i-Pr2NEt•HBF4 was omitted. Nonetheless, we have found that product yields were more reproducible when this additive was present.
Table 5.
Optimized Visible-Light Photocatalytic Coupling of N-Phthalimidoyl Oxalates with Various Electron-Deficient Alkenesa

|
Isolated yield based upon the radical acceptor after purification by silica gel chromatography (average of two experiments). The yield of 27 was measured by 1H NMR relative to an internal standard (1,4-dimethoxybenzene).
bIsolated yield using 1 equiv each of the phthalimidoyl oxalate and the alkene acceptor.
ND = not detected.
Several radical acceptors having a substituent at the β-carbon also provided coupled products in useful yields. Dimethyl fumarate gave adduct 25 in 85% yield from N-phthalimidoyl oxalate 3a and cyclopenten-2-one coupled with 3a to give product 13 in 55% yield. The yield of adduct 26 resulting from the coupling of 3a with 2-carbomethoxycyclopenten-2-one (62%) was only slightly higher than that observed with cyclopenten-2-one.21 Butenolides were also competent acceptors, with the presence of a γ-methoxy substituent enhancing the yield of the coupled product (72% for 23 and 52% for 22) and completely regulating face-stereoselectivity. Unfortunately, radical additions to acceptors possessing electron-donating alkyl groups at the α-position were much less successful under these conditions. Benzyl methacrylate coupled with 3a to provide product 27 in only 41% yield, whereas no product was detected in the coupling of 3a with methacrylonitrile.22
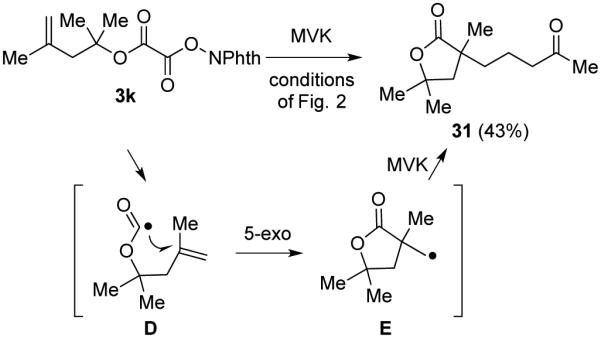
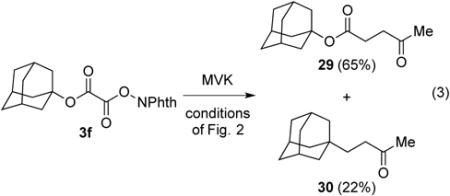
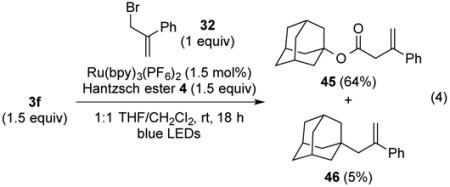
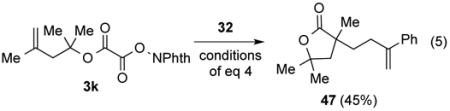
It is well known that alkoxycarbonyl radicals decarboxylate many powers of ten more slowly than carboxy radicals,23,24 with the activation barrier decreasing with the stability of the carbon radical produced.25 In two cases, the alkoxycarbonyl radical (intermediate B of Scheme 1) was trapped by the radical acceptor more rapidly than it underwent decarboxylation to generate the tertiary radical intermediate. Adamantyl N-phthalimidoyl oxalate (3f) when coupled with MVK gave γ-ketoester 29 and product 30 of trapping the adamantyl radical in a 3:1 ratio (eq 3). Trapping of the adamantyl oxycarbonyl radical by MVK to give 29 as the major product is a reflection of the higher energy of the non-planar tertiary adamantyl radical than the tertiary radicals generated in the reactions reported in Table 5. In the second example illustrated in Scheme 2, it is the high rate of 5-exo radical cyclizations (D → E) that results in capture of the intermediate alkoxycarbonyl radical to yield lactone 31 from phthalimidoyl oxalate 3k.26
Scheme 2. Trapping of an Alkoxycarbonyl Radical by a 5-Exo Cyclization.
 |
Further investigations of the reaction scope
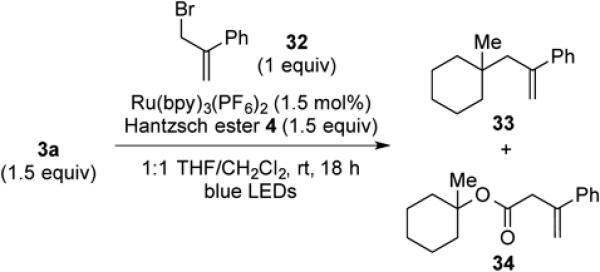
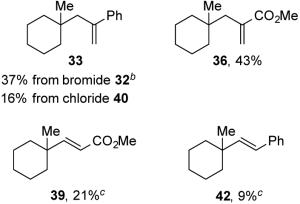
To expand the scope of C–C bond-formation using nucleophilic tertiary carbon radicals generated by visible-light photocatalysis, we examined the coupling of tertiary N-phthalimidoyl oxalates with several allylic and vinylic halide substrates to engage tertiary radical intermediates in addition-fragmentation reactions.27 The coupling of oxalate 3a with α-(bromomethyl)styrene (32) was examined in detail (Table 6). Using the reaction conditions that we had optimized for the reductive coupling reactions, substitution product 33 was obtained in 60% yield (entry 1). To our surprise, minor product 34, arising from allylation of the intermediate alkoxycarbonyl radical, was isolated in 16% yield. Formation of this product must be the result of an unusually fast rate of addition of the alkoxycarbonyl radical to α-(bromomethyl)styrene (32). Attempted reactions in the absence of light (entry 2) or the Hantzsch ester (entry 3) gave no product formation or only traces of the coupled product. The photocatalyst also appears to be essential to achieve useful yields (entry 4). Stopping the reaction after 2 h led to incomplete conversion and lower yields (entries 5 and 6). It is also advantageous to use an excess of the oxalate (1.5 equiv), since reactions in which an excess of acceptor was present gave lower overall yields (entries 7–9). Finally, the additive i-Pr2NEt•HBF4 has no influence on the outcome of the reaction of 3a with α-(bromomethyl)styrene (32) (entry 10).
Table 6.
Optimization and Control Experiments for the Reaction of N-phthalimidoyl Oxalate 3a with α-(Bromomethyl)styrene (32).
| |||
|
| |||
| Entry | Modification | Yield of 33
(%)a |
Yield of 34 (%)a |
|
| |||
| 1 | none | 60 | 16 |
| 2 | no light | NDb | NDb |
| 3 | no Hantzsch ester | traces | ND |
| 4 | no photocatalyst (18 h) |
37 | 7 |
| 5 | no photocatalyst (2 h) |
24 | 3 |
| 6 | reaction time of 2 h | 47 | 9 |
| 7 | oxalate (1 equiv) | 48 | 8 |
| 8 | oxalate (1.1 equiv) | 50 | 9 |
| 9 | oxalate (1 equiv) acceptor (1.5 equiv) |
49 | 16 |
| 10 |
i-Pr2NEt•HBF4
(1 equiv) |
59 | 16 |
Isolated yield after silica gel chromatography.
These products were formed when the light was turned on after 18 h.
ND = not detected.
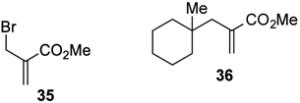
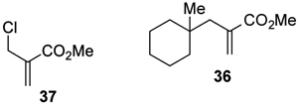
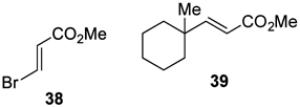
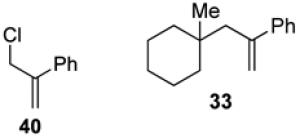
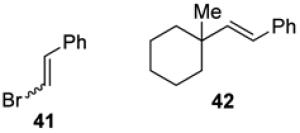
With optimized reaction conditions in hand, the coupling of tert-alkyl N-phthalimidoyl oxalate 3a with three additional allylic halides and two vinylic halides was surveyed (Table 7). Methyl 2-(bromomethyl)acrylate (35) coupled with 3a in 69% yield (entry 1). However, the reaction with the chlorine analog 37 provided no coupled product (entry 2). In lower yield, methyl 3-bromoacrylate (38) gave methyl (E)-3-(1-methylcyclohexyl)acrylate (39) with high E stereoselectivity (entry 3). α-(Chloromethyl)styrene (40) and β-bromostyrene (41) also reacted successfully with N-phthalimidoyl oxalate 3a, albeit in yields of only 47% and 19%, respectively (entries 4 and 5). The efficiency of vinylic substitution reactions was enhanced by the use of 5 equiv of the radical acceptor (entries 3 and 5). Allylic substitution products 36 and 33 are potentially good radical acceptors themselves, nonetheless products resulting from a second addition of the tertiary radical were not observed. We attribute this selectivity to the steric shielding provided by the quaternary carbon fragment in these products. Furthermore, products resulting from the addition of the intermediate alkoxycarbonyl radical to the acceptor were not detected with any substrate other than α-(bromomethyl)styrene (32).
Table 7.
Visible-Light Photocatalytic Allylic and Vinylic Substitution Reactions of N-phthalimidoyl Oxalate 3aa
| Entry | Acceptor | Product | Yield (%)b |
|---|---|---|---|
| 1 |
|
69 | |
| 2 |
|
ND | |
| 3 |
|
35c
(E/Z > 20:1) |
|
| 4 |
|
47 | |
| 5 |
|
19c
(E/Z > 20:1) |
|
| 6 |
|
42 (dr 1:1) |
|
Reaction conditions of Table 6, entry 1.
Isolated yield after silica gel chromatography (average of two experiments).
5 equiv of the acceptor and 1 equiv of the oxalate were used.
ND = not detected.
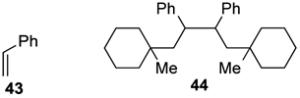
The reaction of N-phthalimidoyl oxalate 3a with styrene (43) was also investigated (entry 6). In this case, only product 44, resulting from recombination of two benzylic radical intermediates, was formed in 42% as a 1:1 mixture of stereoisomers.28 Efforts to capture the benzylic radical in different fashions by modifying the reaction conditions or by adding common hydrogen atom transfer reagents such as Bu3SnH, Et3SiH, Ph3SiH, and PhSH were unsuccessful.29 As expected, couplings of adamantyl N-phthalimidoyl oxalate (3f) and homoallylic N-phthalimidoyl oxalate 3k with α-(bromomethyl)styrene (32) resulted in the first case in predominant formation of ester 45 (eq 4) and in the latter of γ-butyrolactone 47 (eq 5).
 |
 |
Radical coupling in the absence of Ru(bpy)32+
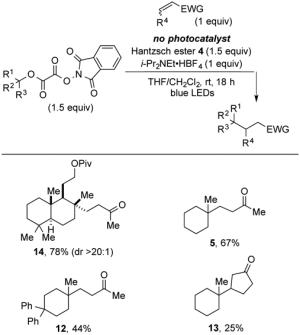
To pursue whether the reductive coupling reaction in the absence of the photocatalyst could be a suitable method, we repeated four of the reductive radical coupling reactions reported earlier with omission of the photocatalyst. In all cases the yield of the coupled product was lower than that obtained using the photocatalyst (compare results in Tables 2 and 8). Useful yields of products after 18 h were obtained in the coupling of two oxalates with MVK: formation of product 14 in 78% yield and 5 in 67% yield. In general, the conversion to coupled products was sluggish and yields were inconsistent in the absence of the photocatalyst.
Table 8.
Coupling of N-Phthalimidoyl Oxalates with Conjugate Acceptors in the Presence of Visible Light and Absence of a Photocatalyst
|
To further investigate the influence of the photocatalyst, addition-fragmentation reactions were also carried out in the absence of Ru(bpy)3(PF6)2 (Table 9). Using five different acceptors, allylic substitution products were formed in yields approximately half of that (47–62%) realized in the presence of the photocatalyst.30
Table 9.
Products and Yields Obtained in Reactions of N-phthalimidoyl Oxalate 3a With Allylic and Vinylic Halides in the Presence of Visible Light and Absence of a Photocatalysta
|
Reaction conditions of Table 8, but no i-Pr2NEt•HBF4; isolated yield after silica gel chromatography.
bEster 34 is formed in 7% yield.
c5 equiv of the acceptor and 1 equiv of the oxalate were used.
Similar radical coupling reactions of (N-acyloxy)phthalimides carried out in the absence of a photocatalyst and a discussion of possible mechanisms of such reactions of tertiary radicals generated from both N-phthalimidoyl oxalate and (N-acyloxy)phthalimide precursors is provided in the accompanying article.12
Mechanistic investigations and discussion
The results of our exploratory studies and substantial precedent are consistent with the generation of tertiary carbon radicals from the visible-light photoredox catalyzed fragmentation of both tertiary N-phthalimidoyl oxalates and (N-acyloxy)phthalimide derivatives of tertiary carboxylic acids. With the oxalate precursors, the slower rate of the second decarboxylation step (B → C, Scheme 1) can lead to the alkoxycarbonyl radical being intercepted by competing fast intramolecular or bimolecular reactions. In the reductive coupling reactions discussed herein, the radical intermediate F produced upon addition of a tertiary radical to an alkene could in principle be terminated by hydrogen-atom transfer (path A, Scheme 3) or by a two-step process involving single-electron reduction to give the resonance-stabilized anion G followed by protonation to give product H (path B).
Scheme 3. Two Potential Termination Pathways.

The role of the Hantzsch dihydropyridine 4 in the termination mechanism is readily examined by using its 4,4-dideuterio derivative 48.31 The coupling of N-phthalimidoyl oxalate 3a with MVK under our standard conditions using Hantzsch dihydropyridine 48 gave coupled product 5 with nearly quantitative incorporation of deuterium at carbon 2 of the ketone side chain (eq 6).32 This result is consistent with hydrogen atom-transfer being the predominant termination step in reductive coupling of tertiary N-phthalimidoyl oxalates.33
 |
To further explore whether hydrogen-atom transfer or single-electron reduction is involved in the termination steps of radical coupling reactions of N-phthalimidoyl oxalate precursors, we examined coupling reactions of N-phthalimidoyl oxalate 3a with an α,β-unsaturated nitrile
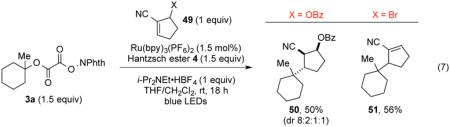 |
containing a leaving group at the allylic α' position (eq 7). The reaction of 3a with allylic benzoate 49a (X = OBz) under our optimized reaction conditions provided addition products 50 in 50% yield, with the depicted isomer being formed predominantly. No trace of the allylic substitution product 51 was seen. In contrast, the coupling of 3a under identical conditions with allylic bromide 49b (X = Br) gave exclusively allylic substitution product 51 in 56% yield. Both results are fully consistent with the absence of single-electron reduction of the radical intermediate I, which undergoes β-scission only when the leaving group is bromide (Scheme 4).
Scheme 4. Potential Homolytic Termination Pathways of the Carbon Radical Intermediate Generated Upon Coupling in the Absence of Single-Electron Transfer.

The overall mechanism that we suggest for the reductive coupling reactions of tertiary N-phthalimidoyl oxalates is summarized in Scheme 5. Visible-light excitation of Ru(bpy)32+ provides access to the excited state Ru(bpy)32+*. This complex is quenched by Hantzsch ester 4, which produces radical cation L and the strongly reducing Ru(bpy)3+. Single-electron transfer from Ru(bpy)3+ to the substrate oxalate then occurs to form radical anion A and regenerate the ground-state photocatalyst. Alternatively, electron transfer to the oxalate substrate could be accomplished by the strongly reducing dihydropyridine radical M, formed by deprotonation of L at C4.34 Homolytic fragmentation of A next occurs to release phthalimide anion, CO2, and alkoxycarbonyl radical intermediate B; alternatively, fragmentation of the N–O bond occurs subsequent to proton donation to the phthalimide fragment by pyridinium acid P.35 A second slower decarboxylation then ensues, forming tertiary carbon radical intermediate C. Addition of this radical to an acceptor provides stabilized radical N, which abstracts a hydrogen atom from Hantzsch ester 4.36 This event forms the product O and regenerates intermediate M, which is capable of propagating the reaction as a radical chain carrier. In the absence of a photoredox catalyst, we propose that a radical chain mechanism mediated by Hantzsch ester 4 is operative. This sequence is discussed in more detail in the accompanying article.12
Scheme 5. Proposed Mechanism for Reductive Coupling of N-Phthalimidoyl Oxalates with MVK in the Presence of the Ru(bpy)32+.

CONCLUSION
A new method for directly transforming a tertiary alcohol to a quaternary carbon was developed. In this method, the tertiary alcohol is first acylated to give a tert-alkyl N-phthalimidoyl oxalate derivative, which in the presence of visible light, 1.5 mol% Ru(bpy)3(PF6)2, and Hantzsch dihydropyridine 4 fragments at room temperature to form the corresponding tertiary carbon radical. Nucleophilic tertiary radicals generated in this way add to electrophilic alkenes to give reduced products in good to moderate yield, and react with allylic and vinylic bromides to provide allylation and vinylation products in moderate yield. With chiral precursors, stereoselection in forming a new quaternary stereocenter can be high (>20:1). This method offers advantages over the original Barton method for forming tertiary radicals from tertiary alcohols,5 as a result of the higher stability of the intermediate mixed oxalate diester in the present method.37
A mechanism for the generating tertiary carbon radical from tert-alkyl N-phthalimidoyl oxalates is proposed that is based on the earlier pioneering investigations of Okada8 and Barton (Scheme 5).5 The high incorporation of deuterium in a coupled product when using 4,4-dideuterio Hantzsch ester 48 (eq 6), and the observation that allylic substitution products are formed from allylic bromide but not allylic ester reactants (eq 7), are consistent with the reductive radical coupling of tert-alkyl N-phthalimidoyl oxalates with electron-deficient alkenes being terminated by hydrogen-atom transfer. The importance of the reaction conditions in determining the fate of the coupled radical intermediate is discussed in detail in the accompanying article.12 In the absence of the photocatalyst, a slower background reaction has been identified that appears to be mediated by Hantzsch ester 4. This sequence is also considered in more detail in the accompanying article.12
EXPERIMENTAL
Materials and Methods
Unless stated otherwise, reactions were conducted in oven-dried glassware under an atmosphere of nitrogen or argon using anhydrous solvents (either freshly distilled or passed through activated alumina columns). For all radical coupling reactions, THF and CH2Cl2 were sparged with argon for 5 min prior to use. All commercially obtained reagents were used as received. Ru(bpy)3(PF6)2 and other photocatalysts were obtained from Sigma Aldrich. Methyl vinyl ketone (MVK), acrylonitrile, benzyl methacrylate and methacrylonitrile were distilled from neat solutions prior to use. All reaction components Hantzsch ester 4,38 4,4-d2-Hantzsch ester 48,27a 4-Me Hantzsch ester 6,39 4-Ph Hantzsch ester 7,39 2-Ph-benzothiazoline 8,40 N,N’-dimethyl-2-phenylbenzimidazoline 9,19b N-methylacridane 10,41, i-Pr2NEt•HBF4,14 methyl 2-(bromomethyl)acrylate (35),42 methyl 2-(chloromethyl)acrylate (37),43 (E)-methyl 3-bromoacrylate (38),44 α-(chloromethyl)styrene (40),45 α-(bromomethyl)styrene (32)45 were prepared according to literature procedures. Usually one representative coupling reaction and yield of the product is described in detail; isolated yields reported in the Results section are the average yields obtained from duplicate experiments. Reaction temperatures were controlled using a temperature modulator, and unless stated otherwise, reactions were performed at room temperature (rt, approximately 23 °C). Thin-layer chromatography (TLC) was conducted with E. Merck silica gel 60 F254 pre-coated plates, (0.25 mm) and visualized by exposure to UV light (254 nm) or by anisaldehyde, ceric ammonium molybdate, iodine, and potassium permanganate staining. EMD silica gel 60 (particle size 0.040–0.063 mm) was used for flash column chromatography. 1H NMR spectra were recorded at 500 or 600 MHz and are reported relative to deuterated solvent signals. Data for 1H NMR spectra are reported as follows: chemical shift (δ ppm), multiplicity, coupling constant (Hz) and integration. 13C NMR spectra were recorded at 125 or 150 MHz. Data for 13C NMR spectra are reported in terms of chemical shift. IR spectra were recorded on a FT-IR spectrometer and are reported in terms of frequency of absorption (cm−1). Optical rotations were measured with a Jasco P-1010 polarimeter. High-resolution mass spectra were obtained from the UC Irvine Mass Spectrometry Facility with a Micromass LCT spectrometer. Blue LEDs (30 cm, 1 watt) were purchased from http://www.creativelightings.com (product code CL-FRS5050-12WP-12V) and powered by 8 AA batteries.
4-(1-Methylcyclohexyl)butan-2-one (5). (Table 2, entry 1 and general procedure for optimization experiments, photocatalyst screen (Table 3) and reductant screen (Table 4))
A 1-dram vial was charged with oxalate 3a (100 mg, 0.30 mmol, 1.5 equiv), Ru(bpy)3(PF6)2 (3 mg, 3.0 μmol, 0.015 equiv), Hantzsch ester 4 (76 mg, 0.30 mmol, 1.5 equiv), i-Pr2NEt•HBF4 (44 mg, 0.20 mmol, 1 equiv) and a magnetic stir bar under argon. After sequential addition of CH2Cl2, (1 mL, sparged with Ar for 5 min), THF (1 mL, sparged with Ar for 5 min), and methyl vinyl ketone (17 μL, 0.20 mmol, 1 equiv), the vial was capped and placed in the center of a 30 cm loop of blue LEDs. The reaction mixture was stirred for 18 h, after which time it was concentrated under reduced pressure. For all experiments reported in Table 2, the crude residue was purified by silica gel chromatography (3% EtOAc/hexanes) to yield ketone 5 as a colorless oil. For all experiments reported in Tables 3 and 4, the yield of product obtained was determined by comparison of diagnostic 1H NMR signals in the crude reaction mixture to those of an internal standard (1,4-dimethoxybenzene). Characterization data for 5 matched those previously reported.3b
(±)-Benzyl 2-methyl-3-(1-methylcyclohexyl)propanoate (27)
Following the general procedure for optimization experiments, oxalate 3a (100 mg, 0.30 mmol, 1.5 equiv), Ru(bpy)3(PF6)2 (3 mg, 3.0 μmol, 0.015 equiv), Hantzsch ester 4 (76 mg, 0.30 mmol, 1.5 equiv), i-Pr2NEt•HBF4 (44 mg, 0.20 mmol, 1 equiv) and benzyl methacrylate (34 μL, 0.20 mmol, 1 equiv) in a mixture of CH2Cl2, (1 mL, sparged with Ar for 5 min) and THF (1 mL, sparged with Ar for 5 min) gave a crude product mixture. The yield of 27 obtained (41%) was determined by comparison of diagnostic 1H NMR signals to those of an internal standard (1,4-dimethoxybenzene). An analytically pure sample was obtained by silica gel chromatography (2.5% EtOAc/hexanes) to provide 27 as a colorless oil. Rf 0.57 (10% EtOAc/hexanes); 1H NMR (600 MHz, CDCl3): δ 7.38–7.30 (m, 5H), 5.12–5.07 (m, 2H), 2.61–2.55 (m, 1H), 1.90 (dd, J = 14.2, 9.0, 1H), 1.44–1.31 (m, 5H), 1.28–1.15 (m, 9H), 0.83 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 178.0, 136.3, 128.6, 128.4, 128.2, 66.3, 38.1, 37.7, 35.4, 33.3, 26.5, 22.11, 22.10, 20.7; IR (thin film): 2925, 2850, 1736, 1455, 1145 cm−1; HRMS-ESI (m/z) [M + Na]+ calculated for C18H26O2Na 297.1830, found 297.1835.
(3-(1-Methylcyclohexyl)prop-1-en-2-yl)benzene (33) and 1-methylcyclohexyl 3-phenylbut-3-enoate (34). (Table 6, entry 1, and general procedure for allylic and vinylic substitution)
A 1-dram vial was charged with oxalate 3a (50 mg, 0.15 mmol, 1.5 equiv), Ru(bpy)3(PF6)2 (1 mg, 1.5 μmol, 0.015 equiv), Hantzsch ester 4 (38 mg, 0.15 mmol, 1.5 equiv), and a magnetic stir bar under argon. After sequential addition of CH2Cl2, (0.5 mL, sparged with Ar for 5 min), THF (0.5 mL, sparged with Ar for 5 min) and α-(bromomethyl)styrene (32, 15 μL, 0.10 mmol, 1 equiv), the vial was capped and placed in the center of a 30 cm loop of blue LEDs. The reaction mixture was stirred for 18 h, after which it was concentrated under reduced pressure. The crude residue was purified by silica gel chromatography (0-5% diethyl ether/pentane) to provide 33 (13 mg, 0.062 mmol, 62%) and 34 (3.4 mg, 0.013 mmol, 13%) as colorless oils.
Data for 33: Rf 0.74 (100% pentane); 1H NMR (500 MHz, CDCl3): δ 7.38 (d, J = 7.5, 2H), 7.30 (t, J = 7.4, 2H), 7.25-7.22 (m, 1H), 5.23 (d, J = 1.7, 1H), 5.03 (bs, 1H), 2.49 (s, 2H), 1.45-1.32 (m, 5H), 1.27-1.12 (m, 5H), 0.74 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 147.3, 144.3, 128.2, 127.0, 126.7, 116.7, 38.4, 34.3, 26.5, 22.3; IR (thin film): 3079, 2924, 2855, 1622, 1491, 1445 cm−1; HRMS-CI (m/z) [M + H]+ calculated for C16H22H 215.1800, found, 215.1798.
Data for 34: Rf 0.29 (5% EtOAc/hexanes); 1H NMR (500 MHz, CDCl3): δ 7.46 (d, J = 7.8, 2H), 7.32 (t, J = 7.7, 2H), 7.29–7.26 (m, 1H), 5.52 (s, 1H), 5.23 (s, 1H), 3.47 (s, 2H), 2.07–2.02 (m, 2H), 1.46–1.40 (m, 1H), 1.38–1.33 (m, 4H), 1.31–1.22 (m, 5H), 1.20-1.12 (m, 1H); 13C NMR (125 MHz, CDCl3): δ 170.6, 141.7, 140.1, 128.4, 127.8, 126.1, 116.0, 82.5, 43.0, 36.6, 25.4, 21.9; IR (thin film): 2932, 2860, 1726, 1447, 1243, 1146 cm−1; HRMS-CI (m/z) [M + NH4]+ calculated for C17H22O2NH4 276.1964, found 276.1956.
Methyl 2-((1-methylcyclohexyl)methyl)acrylate (36)
In an identical fashion, oxalate 3a (50 mg, 0.15 mmol, 1.5 equiv) was coupled with methyl 2-(bromomethyl)acrylate (35, 12 μL, 0.10 mmol, 1 equiv) to give a crude residue, which was purified by silica gel chromatography (3% diethyl ether/pentane) to provide 36 (14 mg, 0.069 mmol, 70%) as a colorless oil. Rf 0.78 (10% diethyl ether/pentane); 1H NMR (500 MHz, CDCl3): δ 6.16 (d, J = 1.3, 1H), 5.44 (bs, 1H), 3.73 (s, 3H), 2.30 (s, 2H), 1.50-1.38 (m, 5H), 1.26-1.21 (m, 5H), 0.81 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 169.1, 138.4, 127.4, 52.0, 37.5, 34.0, 26.5, 22.2; IR (thin film): 2926, 2850, 2359, 2342, 1726, 1457, 1445, 1201, 1154 cm−1; HRMS-CI (m/z) [M + NH4]+ calculated for C12H20NO2NH4 214.1807, found, 214.1813.
Methyl (E)-3-(1-methylcyclohexyl)acrylate (39)
In an identical fashion, oxalate 3a (50 mg, 0.15 mmol, 1.5 equiv) was coupled with (E)-methyl 3-bromoacrylate (38, 72 μL, 0.75 mmol, 5 equiv) to give a crude residue, which was purified by silica gel chromatography (3% diethyl ether/pentane) to provide 39 (11 mg, 0.058 mmol, 39%) as a colorless oil. Rf 0.51 (10% diethyl ether/pentane); 1H NMR (500 MHz, CDCl3): δ 6.96 (d, J = 16.2, 1H), 5.77 (d, J = 16.1, 1H), 3.73 (s, 3H), 1.55-1.31 (m, 10H), 1.02 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 167.9, 159.1, 117.7, 51.6, 37.2, 26.2, 22.4; IR (thin film): 2929, 2853, 1727, 1650, 1435, 1310, 1274, 1171 cm−1; HRMS-CI (m/z) [M + NH4]+ calculated for C11H18O2NH4 200.1651, found 200.1648.
Preparation of 33 from α-(chloromethyl)styrene) (40)
In an identical fashion, oxalate 3a (50 mg, 0.15 mmol, 1.5 equiv) was coupled with α-(chloromethyl)styrene (40, 14 μL, 0.10 mmol, 1 equiv) to give 33 (10 mg, 0.048 mmol, 48%).
(E)-(2-(1-Methylcyclohexyl)vinyl)benzene (42)
In an identical fashion, oxalate 3a (50 mg, 0.15 mmol, 1.5 equiv) was coupled with β-bromostyrene (41, 97 μL, 0.75 mmol, 5 equiv) to give a crude residue, which was purified by silica gel chromatography (100% pentane) to provide 42 (6 mg, 0.029 mmol, 19%) as a colorless oil. Rf 0.78 (100% pentane); 1H NMR (500 MHz, CDCl3): δ 7.37 (d, J = 7.1, 2H), 7.32-7.28 (m, 2H), 7.21-7.17 (m, 1H), 6.32 (d, J = 16.5, 1H), 6.22 (d, J = 16.3, 1H), 1.62-1.58 (m, 1H), 1.55-1.49 (m, 5H), 1.44-1.35 (m, 4H), 1.07 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 128.6, 126.8, 126.1, 38.1, 26.5, 22.6; IR (thin film): 2926, 2855, 1491, 1447, 1260 cm−1; HRMS-CI (m/z) [M]+ calculated for C15H20 200.1565, found, 200.1558.
(±)-(1,4-Bis(1-methylcyclohexyl)butane-2,3-diyl)dibenzene (44)
A 1-dram vial was charged with oxalate 3a (50 mg, 0.15 mmol, 1.5 equiv), Ru(bpy)3(PF6)2 (1 mg, 1.5 μmol, 0.015 equiv), Hantzsch ester 4 (38 mg, 0.15 mmol, 1.5 equiv), and a magnetic stir bar under argon. After sequential addition of CH2Cl2, (0.5 mL, sparged with Ar for 5 min), THF (0.5 mL, sparged with Ar for 5 min) and styrene (43, 12 μL, 0.10 mmol, 1 equiv), the vial was capped and placed in the center of a 30 cm loop of blue LEDs. The reaction mixture was stirred for 18 h, after which it was concentrated under reduced pressure. The crude residue was purified by silica gel chromatography (100% pentane) to provide 44 (9 mg, 0.022 mmol, 44%), a 1:1 mixture of stereoisomers, as a colorless solid.
Data for diastereomer 1: Rf 0.66 (100% hexanes); 1H NMR (500 MHz, CDCl3): δ 7.25-7.21 (m, 4H), 7.16 (d, J = 7.3, 2H), 7.12 (d, J = 7.4, 4H), 2.72 (d, J = 9.0, 2H), 1.58-1.53 (m, 2H), 1.39 (d, J = 14.1, 2H), 1.26-1.19 (m, 6H), 1.14-1.08 (m, 5H), 1.05-0.98 (m, 5H), 0.83 (bs, 4H), 0.46 (s, 6H); 13C NMR (125 MHz, CDCl3): δ 146.7, 129.5, 127.9, 125.8, 49.2, 38.8, 38.4, 33.4, 29.9, 26.4, 22.1, 21.9; IR (thin film): 2924, 2854, 1494, 1452 cm−1; HRMS-CI (m/z) [M + NH4]+ calculated for C30H46N 420.3630, found, 420.3647.
Data for diastereomer 2: Rf 0.50 (100% hexanes); 1H NMR (500 MHz, CDCl3): δ 7.10-7.07 (m, 4H), 7.04-7.00 (m, 2H), 6.95 (d, J = 7.1, 4H), 2.82 (d, J = 7.3, 2H), 1.79-1.70 (m, 4H), 1.37-1.32 (m, 6H), 1.22-1.17 (m, 10H), 1.00-0.95 (m, 4H), 0.63 (s, 6H); 13C NMR (125 MHz, CDCl3): δ 145.4, 129.7, 127.3, 125.4, 49.1, 38.6, 33.8, 29.9, 26.6, 22.2, 22.1; IR (thin film): 2924, 2858, 1494, 1452 cm−1; HRMS-CI (m/z) [M + NH4]+ calculated for C30H46N 420.3630, found, 420.3627.
Adamantan-1-yl 3-phenylbut-3-enoate (45) and 1-(2-phenylallyl)adamantane (46)
In an identical fashion, oxalate 3f (55 mg, 0.15 mmol, 1.5 equiv) was coupled with α-(bromomethyl)styrene (32, 15 μL, 0.10 mmol, 1 equiv), to give a crude residue, which was purified by silica gel chromatography (0-3% diethyl ether/pentane) to provide 45 (20 mg, 0.066 mmol, 66%) and 46 (1 mg, 5.0 μmol, 5%) as colorless solids.
Data for 45: Rf 0.36 (4% EtOAc/hexanes); 1H NMR (500 MHz, CDCl3): δ 7.43 (d, J = 7.1, 2H), 7.32 (t, J = 7.6, 2H), 7.29–7.25 (m, 1H), 5.50 (s, 1H), 5.21 (s, 1H), 3.43 (s, 2H), 2.11 (s, 3H), 2.00 (d, J = 3.1, 6H), 1.61 (s, 6H); 13C NMR (125 MHz, CDCl3): δ 170.5, 141.8, 140.3, 128.4, 127.7, 126.0, 115.8, 80.9, 42.9, 41.2, 36.3, 30.9; IR (thin film): 2911, 2853, 1727, 1455, 1342, 1252, 1165, 1057 cm−1; HRMS-CI (m/z) [M + NH4]+ calculated for C20H24O2NH4 314.2120, found 314.2130.
Data for 46: Rf 0.72 (100% hexanes); 1H NMR (500 MHz, CDCl3): δ 7.40 (d, J = 7.1, 2H), 7.30 (t, J = 7.4, 2H), 7.25–7.21 (m, 1H), 5.26 (d, J = 2.0, 1H), 4.97 (s, 1H), 2.34 (s, 2H), 1.86 (s, 3H), 1.62 (d, J = 12.1, 3H), 1.53 (d, J = 13.3, 3H), 1.38 (s, 6H); 13C NMR (125 MHz, CDCl3): δ 146.1, 144.0, 128.2, 127.0, 126.6, 116.3, 49.9, 43.1, 37.1, 33.8, 28.9; IR (thin film): 2901, 2846, 1450 cm−1; HRMS-CI (m/z) [M]+ calculated for C19H24 252.1878, found 252.1870.
3,5,5-Trimethyl-3-(3-phenylbut-3-en-1-yl)dihydrofuran-2(3H)-one (47)
In an identical fashion, oxalate 3k (50 mg, 0.15 mmol, 1.5 equiv) was coupled with α-(bromomethyl)styrene (32, 15 μL, 0.10 mmol, 1 equiv). After 18 h, the reaction mixture was diluted with CH2Cl2. The resulting organic phase was washed with 4 M HCl (3 × 10 mL) and 2 N NaOH (3 × 10 mL), dried over Na2SO4, and evaporated under reduced pressure. The crude residue was purified by silica gel chromatography (10% acetone/hexanes) to provide 47 (12 mg, 0.047 mmol, 47%) as a colorless oil. Rf 0.53 (10% acetone/hexanes); 1H NMR (500 MHz, CDCl3): δ 7.38 (d, J = 7.1, 2H), 7.33 (t, J = 7.5, 2H), 7.29–7.27 (m, 1H), 5.29 (s, 1H), 5.09 (s, 1H), 2.67–2.60 (m, 1H), 2.48-2.41 (m, 1H), 2.15 (d, J = 13.3, 1H),1.95 (d, J = 13.5, 1H), 1.77–1.72 (m, 2H), 1.46 (s, 3H), 1.42 (s, 3H), 1.35 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 181.3, 147.8, 140.9, 128.6, 127.7, 126.2, 112.9, 81.1, 46.7, 45.1, 38.4, 30.6, 30.3, 30.2, 25.9; IR (thin film): 2974, 2934, 2874, 1758, 1455, 1376, 1268, 1187, 1090 cm−1; HRMS-CI (m/z) [M + NH4]+ calculated for C17H22O2NH4 276.1964, found 276.1962.
General procedure for coupling reactions in the absence of a photocatalyst (Tables 8 and 9). Preparation of 5
A 1-dram vial was charged with oxalate 3a (100 mg, 0.30 mmol, 1.5 equiv), Hantzsch ester 4 (76 mg, 0.30 mmol, 1.5 equiv), i-Pr2NEt•HBF4 (44 mg, 0.20 mmol, 1 equiv) and a magnetic stir bar under argon. After sequential addition of CH2Cl2, (1 mL, sparged with Ar for 5 min), THF (1 mL, sparged with Ar for 5 min), and methyl vinyl ketone (17 μL, 0.20 mmol, 1 equiv), the vial was capped and placed in the center of a 30 cm loop of blue LEDs. The reaction mixture was stirred for 18 h, after which time it was concentrated under reduced pressure. The crude residue was purified by silica gel chromatography (3% EtOAc/hexanes) to yield ketone 5 (23 mg, 0.14 mmol, 67%) as a colorless oil. Characterization data for 5 matched those previously reported.3b
Deuterium incorporation in product 5 using 4,4-d2-Hantzsch ester 48
A 1-dram vial was charged with oxalate 3a (100 mg, 0.30 mmol, 1.5 equiv), Ru(bpy)3(PF6)2 (3 mg, 3.0 μmol, 0.015 equiv), 4,4-d2-Hantzsch ester 48 (77 mg, 0.30 mmol, 1.5 equiv), i-Pr2NEt•HBF4 (44 mg, 0.20 mmol, 1 equiv) and a magnetic stir bar under argon. After sequential addition of CH2Cl2, (1 mL, sparged with Ar for 5 min), THF (1 mL, sparged with Ar for 5 min), and methyl vinyl ketone (17 μL, 0.20 mmol, 1 equiv), the vial was capped and placed in the center of a 30 cm loop of blue LEDs. The reaction mixture was stirred for 18 h, after which time it was concentrated under reduced pressure. Purification of the crude residue by silica gel chromatography (2.5% EtOAc/hexanes) provided ketone 5 (13 mg, 0.070 mmol, 37%)46 as a colorless oil. Integration of all 1H NMR signals determined the deuterium incorporation to be >95%. Rf 0.43 (10% EtOAc/hexanes); 1H NMR (600 MHz, CDCl3): δ 2.39–2.30 (m, 1H), 2.15 (s, 3H), 1.49 (d, J = 8.4, 2 H), 1.46–1.38 (m, 5H), 1.33–1.20 (m, 5H), 0.83 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 210.2, 38.1 (t, J = 20.2), 37.7, 32.3, 30.0, 29.8, 26.5, 24.7, 22.1; IR (thin film): 2925, 2853, 1716, 1356 cm−1; HRMS-ESI (m/z) [M + Na]+ calculated for C11H19DONa 192.1475, found 192.1484.
2-Cyanocyclopent-2-en-1-yl benzoate (49a)
A round bottom flask was charged with 5-hydroxycyclopent1-ene 1-carbonitrile47 (500 mg, 4.58 mmol, 1 equiv) and Et3N (1.3 mL, 9.17 mmol, 2 equiv), DMAP (56 mg, 0.45 mmol, 0.1 equiv) and THF (15 mL) were added sequentially under argon. The solution was cooled to 0 °C and benzoyl chloride (1.1 mL, 9.17 mmol, 2 equiv) was added dropwise. The cloudy suspension was stirred while warming to rt over 18 h. After this time, the reaction mixture was cooled to 0 °C and quenched with saturated aqueous NH4Cl solution (10 mL). The contents of the flask were transferred to a separatory funnel, diluted with Et2O (50 mL) and the layers separated. The organic layer was washed with saturated aqueous NH4Cl solution (3 × 40 mL), aqueous 1 N NaOH (3 × 40 mL), and brine (2 × 40 mL). The organic layer was dried over MgSO4, filtered and concentrated under reduced pressure. The crude residue was purified by silica gel chromatography (10-20% EtOAc/hexanes) to provide 49a (862 mg, 4.04 mmol, 88%) as a colorless oil that solidified after storage at −20 °C. Rf 0.14 (10% EtOAc/hexanes); 1H NMR (500 MHz, CDCl3): δ 8.04 (d, J = 7.9, 2H), 7.57 (t, J = 7.9, 1 H), 7.44 (t, J= 7.9, 2H), 7.03–7.00 (m, 1H), 6.08–6.04 (m, 1H), 2.81–2.73 (m, 1H), 2.64–2.56 (m, 2H), 2.09–2.02 (m, 1H); 13C NMR (125 MHz, CDCl3): δ 165.8, 154.3, 133.3, 129.7, 129.5, 128.4, 115.1, 114.9, 79.3, 32.1, 30.6; IR (thin film): 3069, 2950, 2226, 1972, 1915, 1720 cm−1; HRMS-ESI (m/z) [M + Na]+ calculated for C13H11NO2Na 236.0687, found 236.0678.
Preparation of reductive-coupling product 50
A 1-dram vial was charged with oxalate 3a (100 mg, 0.30 mmol, 1.5 equiv), Ru(bpy)3(PF6)2 (3 mg, 3.0 μmol, 0.015 equiv), Hantzsch ester 4 (76 mg, 0.30 mmol, 1.5 equiv), i-Pr2NEt•HBF4 (44 mg, 0.20 mmol, 1 equiv) and a magnetic stir bar under argon. After sequential addition of CH2Cl2 (1 mL, sparged with Ar for 5 min), THF (1 mL, sparged with Ar for 5 min), and acceptor 49a (43 mg, 0.20 mmol, 1 equiv), the vial was capped and placed in the center of a 30 cm loop of blue LEDs. The reaction mixture was stirred for 18 h, after which time it was diluted with Et2O (30 mL) and transferred to a separatory funnel. The ether layer was washed with aqueous 4 N HCl (4 × 20 mL) and aqueous 2 N NaOH (3 × 20 mL) and was dried over MgSO4. The organic layer was filtered and concentrated under reduced pressure. The crude residue was subjected to silica gel chromatography (4% acetone/hexanes) to provide 50 (34 mg, 0.11 mmol, 55%, dr 8:2:1:1) as a colorless oil.
Data for 50 (major diastereomer, 8:2:1:1): Rf 0.40 (10% EtOAc/hexanes); 1H NMR (600 MHz, CDCl3): δ 8.10 (d, J = 8.0, 2 H), 7.58 (t, J = 7.4, 1 H), 7.45 (t, J = 8.0, 2H), 5.30 (app. q, J = 7.6, 1H), 3.42–3.39 (m, 1H), 2.31–2.24 (m, 1H), 2.10–1.96 (m, 3H), 1.82–1.76 (m, 1H), 1.60–1.55 (m, 2H), 1.53–1.45 (m, 6H), 1.31–1.22 (m, 2H), 1.14 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 166.2, 133.5, 130.0, 129.6, 128.6, 118.6, 74.4, 37.01, 37.0, 35.3, 34.5, 28.2, 26.2, 21.9, 21.62, 21.61; IR (thin film): 2925, 2853, 1722, 1271 cm−1; HRMS-ESI (m/z) [M + Na]+ calculated for C20H25NO2Na 334.1783, found 334.1789.
Data for 50 (minor diastereomer, 8:2:1:1): Rf 0.44 (10% EtOAc/hexanes); 1H NMR (600 MHz, CDCl3): δ 8.00 (d, J = 8.1, 2H), 7.6 (t, J = 7.6, 1H), 7.46 (t, J = 7.6, 2H), 5.52–5.50 (m, 1H), 3.08–3.05 (m, 1H), 2.53–2.46 (m, 1H), 2.25–2.19 (m, 1H), 1.96–1.78 (m, 3H), 1.60–1.45 (m, 8H), 1.34–1.23 (m, 2H), 1.13 (s, 3H); 13C NMR (125 MHz, CDCl3): δ 165.6, 133.5, 129.8, 129.7, 128.6, 119.9, 78.5, 37.5, 37.1, 36.9, 34.1, 30.6, 26.3, 23.1, 22.0, 21.7.
5-Bromocyclopent-1-enecarbonitrile (49b)
A round-bottom flask was charged with 5-hydroxycyclopent1-ene 1-carbonitrile47 (500 mg, 4.58 mmol, 1 equiv) and Et2O (20 mL) and CBr4 (3.04 g, 9.17 mmol, 2 equiv) were added sequentially under argon. The solution was cooled to 0 °C and PPh3 (2.40 g, 9.17 mmol, 2 equiv) was added in one portion. The resulting heterogeneous mixture was warmed to rt and stirred for 2 h. After this time, the reaction mixture was filtered through a plug of Celite and the filtrate was concentrated under reduced pressure. The crude residue was purified by silica gel chromatography (10-15% EtOAc/hexanes) to yield 49b (481 mg, 2.81 mmol, 61%) as a colorless oil. Rf 0.26 (10% EtOAc/hexanes); 1H NMR (500 MHz, CDCl3): δ 6.87–6.82 (m, 1H), 5.09–5.04 (m, 1H), 2.86–2.75 (m, 1H), 2.63–2.52 (m, 2H), 2.50–2.42 (m, 1H); 13C NMR (125 MHz, CDCl3): δ 151.9, 119.1, 114.7, 53.1, 35.5, 32.3; IR (thin film): 2927, 2227, 1607, 1189, 1010 cm−1; HRMS-ESI (m/z) [M + H]+ calculated for C6H7BrN 193.9581, found 193.9582.
Preparation of allylated product 51
A 1-dram vial was charged with oxalate 3a (100 mg, 0.30 mmol, 1.5 equiv), Ru(bpy)3(PF6)2 (3 mg, 3.0 μmol, 0.015 equiv), Hantzsch ester 4 (76 mg, 0.30 mmol, 1.5 equiv), i-Pr2NEt•HBF4 (44 mg, 0.20 mmol, 1 equiv) and a magnetic stir bar under argon. After sequential addition of CH2Cl2 (1 mL, sparged with Ar for 5 min), THF (1 mL, sparged with Ar for 5 min), and acceptor 49b (43 mg, 0.20 mmol, 1 equiv), the vial was capped and placed in the center of a 30 cm loop of blue LEDs. The reaction mixture was stirred for 18 h, after which time it was concentrated under reduced pressure. The crude residue was subjected to silica gel chromatography (4% acetone/hexanes) to provide 51 (22 mg, 0.11 mmol, 57%) as a colorless oil: Rf 0.47 (10% EtOAc/hexanes); 1H NMR (500 MHz, CDCl3): δ 6.79–6.76 (m, 1H), 2.89–2.83 (m, 1H), 2.50–2.34 (m, 2H), 2.01–1.92 (m, 1H), 1.87–1.79 (m, 1H), 1.64–1.38 (m, 7H), 1.36–1.18 (m, 3H), 0.88 (s, 3H);13C NMR (125 MHz, CDCl3): δ 152.4, 118.7, 116.7, 36.7, 36.1, 36.0, 33.0, 26.3, 25.2, 22.0, 21.8 ; IR (thin film): 2926, 2853, 2215, 1459 cm−1; HRMS-ESI (m/z) [M + Na]+ calculated for C13H19NNa 212.1415, found 212.1405.
Supplementary Material
Acknowledgement
Financial support was provided by the National Science Foundation (CHE1265964) and the National Institute of General Medical Sciences (R01-GM098601). We thank the Alexander von Humboldt Foundation for the support of G.P. by a Feodor Lynen Postdoctoral Research Fellowship and the ACS Organic Chemistry Division for partial support of G.L.L. by a Graduate Fellowship. NMR and mass spectra were determined at UC Irvine using instruments purchased with the assistance of NSF and NIH shared instrumentation grants. We are grateful to Juliet F. K. Kotyk and Professor Jenny Y. Yang for assistance in conducting cyclic voltammetry measurements.
Footnotes
Supporting Information. Image of a typical photocatalysis reaction setup, copies of 1H and 13C NMR spectra of new compounds, nOe data for compound 50, and and cyclic voltammetry data. This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interest.
References
- 1.(a) Schnermann MJ, Untiedt NL, Jimenez-Oses G, Houk KN, Overman LE. Angew. Chem., Int. Ed. 2012;51:9581–9586. doi: 10.1002/anie.201205001. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Schnermann MJ, Overman LE. Angew. Chem., Int. Ed. 2012;51:9576–9580. doi: 10.1002/anie.201204977. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Müller DS, Untiedt NL, Dieskau AP, Lackner GL, Overman LE. J. Am. Chem. Soc. 2015;137:660–663. doi: 10.1021/ja512527s. [DOI] [PubMed] [Google Scholar]
- 2.(a) Gaoni Y. Tetrahedron. 1989;45:2819–2840. For selected examples of recently disclosed methods for generating tertiary organometallic reagents and using them for C–C bond formation, see. [Google Scholar]; (b) Mandelt K, Meyer-Wilmes I, Fitjer L. Tetrahedron. 2004;60:11587–11595. [Google Scholar]; (c) Hodgson DM, Chung YK, Nuzzo I, Freixas G, Kulikiewicz KK, Cleator E, Paris JM. J. Am. Chem. Soc. 2007;129:4456–4462. doi: 10.1021/ja0672932. [DOI] [PubMed] [Google Scholar]; (d) Joshi-Pangu A, Wang C-Y, Biscoe MR. J. Am. Chem. Soc. 2011;133:8478–8481. doi: 10.1021/ja202769t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ren P, Stern L-A, Hu X. Angew. Chem., Int. Ed. 2012;51:9110–9113. doi: 10.1002/anie.201204275. [DOI] [PubMed] [Google Scholar]; (f) Iwasaki T, Takagawa H, Singh SP, Kuniyasu H, Kambe N. J. Am. Chem. Soc. 2013;135:9604–9607. doi: 10.1021/ja404285b. [DOI] [PubMed] [Google Scholar]; (g) Leonori D, Aggarwal VK. Angew. Chem., Int. Ed. 2015;54:1082–1096. doi: 10.1002/anie.201407701. [DOI] [PubMed] [Google Scholar]
- 3.(a) Furst L, Narayanam JMR, Stephenson CRJ. Angew. Chem., Int. Ed. 2011;50:9655–9659. doi: 10.1002/anie.201103145. For recently disclosed methods for generating tertiary radicals and using them for C–C bond formation, see. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lackner GL, Quasdorf KW, Overman LE. J. Am. Chem. Soc. 2013;135:15342–15345. doi: 10.1021/ja408971t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lo JC, Yabe Y, Baran PS. J. Am. Chem. Soc. 2014;136:1304–1307. doi: 10.1021/ja4117632. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chu L, Ohta C, Zuo Z, MacMillan DWC. J. Am. Chem. Soc. 2014;136:10886–10889. doi: 10.1021/ja505964r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Chinzei T, Miyazawa K, Yasu Y, Koike T, Akita M. RSC Adv. 2015;5:21297–21300. [Google Scholar]
- 4.(a) Barton DHR, Serebryakov EPA. Proc. Chem. Soc. 1962:309. [Google Scholar]; (b) Barton DHR, Dowlatshahi HA, Motherwell WB, Villemin D. J. Chem. Soc., Chem. Commun. 1980:732–733. [Google Scholar]; (c) Barton DHR, Crich D, Motherwell WB. Tetrahedron Lett. 1983;24:4979–4982. [Google Scholar]; (d) Barton D, Chern C-Y, Jaszberenyi J. Tetrahedron. 1995;51:1867–1886. [Google Scholar]; (e) Saraiva MF, Couri MRC, Hyaric ML, de Almeida MV. Tetrahedron. 2009;65:3563–3572. For a brief review, see. [Google Scholar]
- 5.(a) Barton DHR, Crich D. Tetrahedron Lett. 1985;26:757–760. [Google Scholar]; (b) Barton DHR, Crich D, Kretzschmar G. J. Chem. Soc., Perkin Trans. 1. 1986:39–53. [Google Scholar]
- 6.Hasebe M, Tsuchiya T. Tetrahedron Lett. 1986;27:3239–3242. Other methods include, generation of tertiary radicals by photolysis of benzophenone oxime esters, see. [Google Scholar]
- 7.(a) Nugent WA, RajanBabu TV. J. Am. Chem. Soc. 1988;110:8561–8562. The ring-opening of substituted epoxides is a notably useful method for generating tertiary radicals, with intramolecular reactions of β-alkoxy tertiary radicals generated in this way being widely developed. See. [Google Scholar]; (b) Gansäuer A, Lauterbach T, Narayan S. Angew. Chem., Int. Ed. 2003;42:5556–5573. doi: 10.1002/anie.200300583. [DOI] [PubMed] [Google Scholar]; (c) Cuerva JM, Justica J, Oller–Löpez JLV, Enrique Oltra J. Top. Curr. Chem. 2006;264:63–91. [Google Scholar]
- 8.Okada K, Okamoto K, Morita N, Okubo K, Oda M. J. Am. Chem. Soc. 1991;113:9401–9402. [Google Scholar]
- 9.(a) Mariano PS, Stavinoha JL. In: Synthetic Organic Photochemistry. Horspool WM, editor. Plenum Press; NY, USA: 1984. pp. 145–257. The formation of carbon radicals by photochemically induced single-electron transfer using UV light has a long history. For selected reviews of early work in this area, see. [Google Scholar]; (b) Eberson L. Electron Transfer Reactions in Organic Chemistry. Springer-Verlag; NY, USA: 1987. [Google Scholar]; (c) Fagnoni M, Dondi D, Ravelli D, Albini A. Chem. Rev. 2007;107:2725–2756. doi: 10.1021/cr068352x. [DOI] [PubMed] [Google Scholar]
- 10. A portion of this study was described in a preliminary communication.3b.
- 11.(a) Renaud P, Sibi MP. Radicals in Organic Synthesis. Vol. 2. Wiley–VCH; Weinheim: 2001. For selected reviews discussing the addition of carbon radicals to alkenes, see. [Google Scholar]; Zard SZ. Radical Reactions in Organic Synthesis. Oxford; New York: 2003. [Google Scholar]; Srikanth GSC, Castle SL. Tetrahedron. 2005;61:10377–10441. [Google Scholar]; Rowlands GJ. Tetrahedron. 2009;65:8603–8655. [Google Scholar]; (b) Yang J, Zhang J, Oi L, Hu C, Chen Y. Chem. Commun. 2015;51:5275–5278. doi: 10.1039/c4cc06344a. For a recent disclosure of photocatalytic reductive alkynylation of carbon radicals formed from (N-acyloxy)phthalimides, see. [DOI] [PubMed] [Google Scholar]
- 12.Pratsch G, Lackner GL, Overman LE. J. Org. Chem. 2015;80 doi: 10.1021/acs.joc.5b00795. Accompanying article, see. xxxx. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okada K, Okamoto K, Oda M. J. Am. Chem. Soc. 1988;110:8736–8738. Okada reports reduction potentials in the range –1.28 V to –1.37 V (vs SCE) for three (N-acyloxy)phthalimides in acetonitrile, see. [Google Scholar]
- 14.Andrews RS, Becker JJ, Gagné MR. Angew. Chem., Int. Ed. 2010;49:7274–7276. doi: 10.1002/anie.201004311. [DOI] [PubMed] [Google Scholar]
- 15. Conducting the reaction depicted in eq 2 using a compact fluorescent light in place of blue LEDs gave 5 in 80% yield after a reaction time of 2 h.
- 16. Further discussion of the related reaction of (N-acyloxy)phthalimides in the absence of the photocatalyst is provided in the accompanying article.12.
- 17.Prier CK, Rankic DA, MacMillan DWC. Chem. Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. For a comprehensive review of visible-light photocatalysis, which discusses the reactivity and electrochemical potentials of common photoredox catalysts, see. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Reduction potentials are reported vs the saturated calomel electrode (SCE) and were determined in acetonitrile unless noted otherwise.
- 19.(a) Zhu X-Q, Li H-R, Li Q, Ai T, Lu J-Y, Yang Y, Cheng J-P. Chem. Eur. J. 2003;9:871–880. doi: 10.1002/chem.200390108. For a detailed discussion of the reductive properties of several heterocyclic hydrogen donors, see. [DOI] [PubMed] [Google Scholar]; (b) Zhu X-Q, Zhang M-T, Yu A, Wang C-H, Cheng J-P. J. Am. Chem. Soc. 2008;130:2501–2516. doi: 10.1021/ja075523m. [DOI] [PubMed] [Google Scholar]; (c) Zhu X-Q, Tan Y, Cao C-T. J. Phys. Chem. B. 2010;114:2058–2075. doi: 10.1021/jp911137p. [DOI] [PubMed] [Google Scholar]
- 20.Fukuzimi S, Mochizuki S, Tanaka T. J. Phys. Chem. 1990;94:722–726. [Google Scholar]
- 21. The isolation of 26 as a single stereoisomer likely results from the acidic (4 M HCl) workup promoting isomerization to yield the more stable trans stereoisomer.
- 22. This result is discussed in more detail in the accompanying manuscript.
- 23.Togo H. Advanced Free Radical Reactions for Organic Synthesis. Elsevier Science; 2004. The rate constant for the loss of CO2 from a carboxyl radical is estimated to be on the order of 109 s−1, see. [Google Scholar]
- 24.(a) Rüegge D, Fischer H. Int. J. Chem. Kinet. 1986;18:145–158. Decarboxylation rate of 105–106 s−1 for the tert-butoxycarbonyl radical, see. [Google Scholar]; (b) Beckwith ALJ, Bowry V, Moad G. J. Org. Chem. 1988;53:1632–1641. [Google Scholar]; (c) Simakov PA, Martinez FN, Horner JH, Newcomb M. J. Org. Chem. 1998;63:1226–1232. [Google Scholar]
- 25.Morihovitis T, Schiesser CH, Skidmore MA. J. Chem. Soc., Perkin Trans. 1999;2:2041–2047. [Google Scholar]
- 26.(a) Togo H, Yokoyama M. Heterocycles. 1990;31:437–441. [Google Scholar]; (b) Togo H, Fujii M, Yokoyama M. Bull. Chem. Soc. Jpn. 1991;64:57–67. [Google Scholar]
- 27.(a) Larraufie M-H, Pellet R, Fensterbank L, Goddard J-P, Lacôte E, Malacria M, Ollivier C. Angew. Chem., Int. Ed. 2011;50:4463–4466. doi: 10.1002/anie.201007571. For other radical allylations realized using visible-light photocatalysis, see. [DOI] [PubMed] [Google Scholar]; (b) Dai X, Cheng D, Guan B, Mao W, Xu X, Li X. J. Org. Chem. 2014;79:7212–7219. doi: 10.1021/jo501097b. [DOI] [PubMed] [Google Scholar]
- 28.(a) Schäfer H. Angew. Chem., Int. Ed. 1970;9:158–159. Only a few radical addition-dimerization cascades of styrene derivatives are known. [Google Scholar]; (b) Kambe N, Miyamoto M, Terao J, Watabe H. Bull. Chem. Soc. Jpn. 2003;76:2209–2214. [Google Scholar]; (c) Shen Z-L, Cheong H-L, Loh T-P. Tetrahedron Lett. 2009;50:1051–1054. [Google Scholar]
- 29.(a) Bockrath B, Bittner E, McGrewt J. J. Am. Chem. Soc. 1984;106:135–138. [Google Scholar]; (b) Mayer JM. Acc. Chem. Res. 2011;44:36–46. doi: 10.1021/ar100093z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wille U. Chem. Rev. 2013;113:813–853. doi: 10.1021/cr100359d. [DOI] [PubMed] [Google Scholar]; (d) Crich D, Grant D, Krishnamurthy V, Patel M. Acc. Chem. Res. 2007;40:453–463. doi: 10.1021/ar600020v. [DOI] [PubMed] [Google Scholar]
- 30. In the absence of the photocatalyst, the reaction of N-phthalimidoyl oxalate 3a with styrene gave only trace amounts of the dimeric product 44 produced in the photocatalytic reaction. The efficiency of an addition-dimerization cascade leading to 44 would be highly dependent on the concentration of free benzylic radicals in solution. Without the photocatalyst, the generation of this intermediate is apparently sluggish and radical dimerization is disfavored.
- 31.(a) Norcross BE, Klinedinst PE, Jr., Westheimer FH. J. Am. Chem. Soc. 1962;84:797–802. [Google Scholar]; (b) Neumann M, Zeitler K. Chem. Eur. J. 2013;19:6950–6955. doi: 10.1002/chem.201204573. For recent use of 4,4-dideuterio-Hantzsch esters in studies of reactions promoted by visible-light photoredox catalysis, see. [DOI] [PubMed] [Google Scholar]
- 32. The position and percentage of deuterium incorporated in product 5 was determined by 1H NMR analysis.
- 33. As the protic acid generated by oxidation of 48 would be a mixture of protio and deuterio species, this high level of deuterium incorporation would not be observed in the two-step termination sequence, see the accompanying article.12.
- 34.Fukuzimi S, Koumitsu S, Hironaka K, Tanaka T. J. Am. Chem. Soc. 1987;109:305–316. [Google Scholar]
- 35. The potential role of a proton donor in the fragmentation of the N-acyloxyphthalimido fragment is discussed in more detail in the accompanying article.12.
- 36. The corresponding radical cation L is also a potential hydrogen atom source.
- 37. Although not specifically demonstrated in our study, tert-alkyl N-phthalimidoyl oxalates could undoubtedly be employed advantageously to deoxygenate tertiary alcohols by omitting the alkene radical acceptor utilized in our investigations.
- 38.Eey STC, Lear MJ. Org. Lett. 2010;12:5510–5513. doi: 10.1021/ol102390t. [DOI] [PubMed] [Google Scholar]
- 39.Abdel-Mohsen HT, Conrad J, Beifuss U. Green Chem. 2012;14:2686–2690. [Google Scholar]
- 40.Chikashita H, Miyazaki M, Itoh K. Synthesis. 1984;4:308–310. [Google Scholar]
- 41.Pintér Á, Sud A, Sureshkumar D, Klussmann M. Angew. Chem., Int. Ed. 2010;49:5004–5007. doi: 10.1002/anie.201000711. [DOI] [PubMed] [Google Scholar]
- 42.(a) Huang H, Liu X, Deng J, Qiu M, Zheng Z. Org. Lett. 2006;8:3359–3362. doi: 10.1021/ol0612399. [DOI] [PubMed] [Google Scholar]; (b) Kippo T, Fukuyama T, Ryu I. Org. Lett. 2011;13:3864–3867. doi: 10.1021/ol201395p. [DOI] [PubMed] [Google Scholar]
- 43.(a) reference 42a. [Google Scholar]; (b) Young WG, Caserio FF, Jr., Brandon DD., Jr. J. Am. Chem. Soc. 1960;82:6163–6168. [Google Scholar]
- 44.Ma S, Lu X, Li Z. J. Org. Chem. 1992;57:709–713. [Google Scholar]
- 45.Tripathi CB, Mukherjee S. Angew. Chem., Int. Ed. 2013;52:8450–8453. doi: 10.1002/anie.201304173. [DOI] [PubMed] [Google Scholar]
- 46. One representative coupling reaction and yield of the product is described in detail. Isolated yields reported in the Results and Discussion section are the average yields obtained from duplicate experiments.
- 47.Graff M, Al Dilaimi A, Seguineau P, Rambaud M, Villieras J. Tetrahedron Lett. 1986;27:1577–1578. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.