

Abstract
Stem cell-based neuronal differentiation has provided a unique opportunity for disease modeling and regenerative medicine. Neurospheres are the most commonly used neuroprogenitors for neuronal differentiation, but they often clump in culture, which has always represented a challenge for neurodifferentiation. In this study, we report a novel method and defined culture conditions for generating sub-type or region-specific neurons from human embryonic and induced pluripotent stem cells derived neurosphere without any genetic manipulation. Round and bright-edged neurospheres were generated in supplemented knockout serum replacement medium (SKSRM) with 10% CO2, which doubled the expression of the NESTIN, PAX6 and FOXG1 genes compared to those cultured with 5% CO2. Furthermore, an additional step (AdSTEP) was introduced to fragment the neurospheres and facilitate the formation of a neuroepithelial-type monolayer that we termed the “neurosphederm”. The large neural tube-type rosette (NTTR) structure formed from the neurosphederm, and the NTTR expressed higher levels of the PAX6, SOX2 and NESTIN genes compared to the neuroectoderm-derived neuroprogenitors. Different layers of cortical, pyramidal, GABAergic, glutamatergic, cholinergic neurons appeared within 27 days using the neurosphederm, which is a shorter period than in traditional neurodifferentiation-protocols (42–60 days). With additional supplements and timeline dopaminergic and Purkinje neurons were also generated in culture too. Furthermore, our in vivo results indicated that the fragmented neurospheres facilitated significantly better neurogenesis in severe combined immunodeficiency (SCID) mouse brains compared to the non-fragmented neurospheres. Therefore, this neurosphere-based neurodifferentiation protocol is a valuable tool for studies of neurodifferentiation, neuronal transplantation and high throughput screening assays.
Keywords: Human embryonic stem cells, Induced pluripotent stem cells, Neuroectoderm, Neurosphere, Neurons, Neurogenesis
Introduction
Human embryonic and induced pluripotent stem cells (h/iPSCs) are of considerable interest in developmental biology and regenerative medicine, representing an enormous opportunity for generating patient-specific cells for screening drugs and cell therapies for various diseases. Stem cell neuronal differentiation has been used as an in vitro model for a number of genetic conditions, such as spinal muscular atrophy1 and familial dysautonomia2, as well as inherited and sporadic forms of various human neurodegenerative conditions, including motor neuron disease, Neiman-Pick disease (NPD), Huntington disease (HD), Parkinson’s disease (PD) and Alzheimer’s disease (AD)3–9. In all cases, h/iPSCs are being used to generate large populations of healthy neurons to explore the therapeutic potential of neurotransplantation. The two basic methods for generating neurons from h/iPSCs are adherent (neuroectoderm)10,11 and non-adherent (embryoid body or neurosphere)12–14 culture conditions. Adherent methods (neuroectoderm) using dual inhibition of SMAD signaling promote efficient neuronal differentiation10,15. Another method is to generate neurons from non-adherent neurospheres or embryoid bodies12–14. In neural transplantation, neurospheres are the most commonly used neuroprogenitors that are injected into the brain, due to their easy delivery and ability to rapidly migrate to the neurogenic areas of the brain16–18. Neurospheres, as dynamic three-dimensional physiological microincubators for human neural precursor cells (NPCs), have many advantages over the neuroectoderm (19). In 1992, Reynold and Weiss showed that free-floating NPCs can divide and form multicellular spheres in vitro19. These neurospheres have self-renewal ability, can be cultured over 10 passages, and can be easily maintained and expanded without losing the expression of neural progenitor markers18,20.
Neurospheres have the potential to generate sub-type or region-specific neurons (22). However, their tendency to clump in culture makes them very difficult to study and to identify the types of neurons that can be derived after neurosphere transplantation18,20. It is also difficult to precisely monitor the morphology of single neurons from neurosphere-derived neuronal aggregates. Moreover, generating sub-type-specific or region-specific functional neurons from h/iPSCs takes more than 6–8 weeks with the traditional neuronal generation protocols11,21,22. Here, we present novel culture conditions and methods to rapidly and efficiently generate functional human sub-type or region-specific neurons from neurospheres. This method involves a combination of supplemented knockout serum replacement medium (SKSRM) with 10% CO2 and a mechanical procedure termed “AdSTEP,” which involves breaking the neurospheres into smaller fragments to increase the efficiency of neuronal production. Furthermore, we injected the fragmented neurospheres into the severe combined immunodeficiency (SCID) mouse brains to investigate the effect of AdSTEP on neurogenesis in vivo, which might have significant impacts on neuronal transplantation and regenerative medicine.
Materials and Methods
Maintenance of the H9 lines and generation of the human induced pluripotent stem cell (iPSC) line HFS-1
A human embryonic cell line (H9 line, National Stem Cell Bank code WA09) ordered from Wicell was cultured and maintained in mTeSR™-1 medium (Stemcell Technologies) (Supplementary Fig. 1A).
The HFS-1 human iPS cell line was generated from human foreskin fibroblasts by transfection with the 7F-2 combination of episomal plasmids (pEP4EO2SEN2K, pEP4EO2SET2K and pCEP4-M2L), as previously reported23. Briefly, approximately 3.0 μg of pEP4EO2SEN2K, 3.2 μg of pEP4EO2SET2K and 2.4 μg of the pCEP4-M2L plasmids were co-transfected into ~1.0×10ˆ6 human neonatal foreskin fibroblasts via Nucleofector™ (VPD-1001 with program U-20, Lonza). The transfected fibroblasts were then plated and maintained in fibroblast culture medium. One day after transfection, the fibroblast medium was replaced with a reprogramming medium consisting of DMEM/F12 supplemented with N-2 supplement (Life Technologies), B-27 supplement (Life Technologies), 0.1 mM non-essential amino acids (NEAA), 1 mM GlutaMAX™, 0.1 mM β-mercaptoethanol, 0.5 μM PD0325901, 3 μM CHIR99021, 0.5 μM A-83-01, 1000 U/ml human LIF and 10 μM HA-100. The putative iPSC colonies were then picked and plated onto Matrigel-coated plates in mTeSR™-1 after approximately 20 days in culture, and the pluripotency of the iPSCs was verified as previously described23.
Neuronal initiation to generate neurospheres and neuroectoderm from the h/iPSCs
The h/iPSCs (70–80% confluent) were treated with collagenase IV (2 mg ml−1, Life Technologies), and harvested for neural differentiation. To generate the neuroectoderm, the collagenase IV-treated h/iPSC fragments were resuspended in knockout serum replacement medium (KSRM, Life Technologies) supplemented with 10 ng/ml bFGF (Life Technologies) and 10 μM ROCK inhibitor (Y-27632, Tocris Bioscience) and then equally distributed onto Matrigel-coated plates. To generate neurospheres, the collagenase IV-treated h/iPSC fragments were plated onto a low adhesion suspension culture plate (Olympus) with KSRM supplemented with 10 ng ml−1 bFGF, 10 μM ROCK inhibitor, 50 ng ml−1 EGF (R&D Systems), 1000 Unit ml−1 LIF (Millipore) and 1 μg ml−1 heparin (Sigma-Aldrich), which was termed “SKSRM”. Both of the cultures were incubated with 10% CO2 in a 37°C incubator for 3 days. A duplicate set of cultures were maintained in a 5% CO2/37°C incubator to compare the effects of the culture conditions on neuronal initiation.
Neuronal induction of the neurospheres and neuroectoderm
After the neuronal initiation, the neurospheres and neuroectoderm were maintained in neuronal induction medium (NIM). NIM is neuronal maintenance medium (NMM) supplemented with 10 μM SB431542 (Tocris Bioscience) and 1 μM dorsomorphin (Tocris Bioscience). NMM is a 1:1 mixture of supplemented DMEM/F12 (Life Technologies) and supplemented Neurobasal (Life technologies) media (detailed description in supplementary table 1). During the 7 days of neuronal induction, the SKSRM media was gradually replaced with NIM media by increasing the ratio of NIM versus SKSRM by 20% every two days. The media shifted from 0% NIM and 100% SKSRM to 100% NIM and 0% SKSRM over 10 days and 5 media changes.
Generation of the neurosphederm from neurospheres using “AdSTEP”
After neuronal induction, the neurospheres were collected in a 15 ml tube and centrifuged for 5 min at 400 × g. The supernatant was aspirated, the pellets were gently resuspended with cell dissociation solution (Stem Cell Technologies), and incubated for 10 min at 37°C. The neurospheres were collected again and resuspended in NMM. The neurospheres were then broken down into smaller fragments by 20–30 pounding motions using a 5 ml polystyrene serological pipette, which we termed the “AdSTEP” mechanical procedure. Finally, the neurosphere fragments were mixed thoroughly and transferred to Matrigel-coated plates. After 3–5 days at 37°C and 5% CO2, a neuroepithelial sheet appeared and was termed the “neurosphederm”.
Differentiation of neural progenitors, sub-type specific neurons and region specific neurons
After the neuroectoderm and neurosphederm reached confluence, they were further incubated for 3–5 days to allow the neural progenitors to form. Then, the neural progenitors were treated with collagenase IV (2 mg ml−1) and equally distributed onto polyornithine/laminin-coated plates and maintained with NMM until all of the neural subtypes and functional synapses were generated. The neurons were maintained with NMM to generate the basal forebrain cholinergic neurons. The cholinergic neurons appeared in the culture between 19–27 days. Cerebellar Purkinje neurons appeared when the culture was maintained in NMM for approximately 40–45 days. To generate the mid/hindbrain dopaminergic neurons, a neuronal culture was maintained in NMM supplemented with 200 ng ml−1 SHH (sonic hedgehog, R&D Systems) and 20 ng ml−1 Fibroblast Growth Factor-8 (FGF8, Life Technologies) for 7 days. Then, the neurons were maintained for an additional 10 days with NMM supplemented with 10 ng ml−1 brain-derived neurotrophic factor (BDNF)Life Technologies), 10 ng ml−1 glial cell line-derived neurotrophic factor (GDNF) (Life Technologies), 1 ng ml−1 TGF-β3 (transforming growth factor-β3, Life Technologies), 100 μM cAMP (cyclic adenosine monophosphate) and 200 μM AA (L-Ascorbic acid, Sigma).
Immunocytochemistry and microscopy
The cells were grown on coverslips coated with polyornithine/laminin in 24-well plates, and then cells were washed with PBS and fixed with 4% paraformaldehyde (PFA) for 10 minutes at 25°C. After additional washes in PBS, the cells were permeabilized in 0.1% Triton X-100 (Sigma-Aldrich) for 5 min at 25°C, followed by blocking with 5% BSA in PBS containing 10% normal goat serum (NGS, Abcam) for 1 h at 25°C. The cells were incubated overnight with primary antibodies diluted in blocking solution at 4°C. The details regarding the dilutions of the primary antibodies that were used can be found in Supplementary Table 2. The next day, the cells were washed three times with washing buffer (1× PBS containing 0.05% Tween 20 and 1% NGS), and then cells were incubated for 2 h at 25°C with a fluorescent secondary antibody (Life Technologies). After incubation with the secondary antibody and washing, the coverslips with the cells were placed onto slides with Fluoromount-G mounting medium containing DAPI (DAPI-FG, Southern Biotech). All the fluorescent immunostaining was validated by comparing the staining without primary antibody but with IgG control secondary fluorescent antibody to ensure there are no non specifics or background staining (Supplementary Fig. 1 G–I). An EVOS fluorescence microscope (Life Technologies) was used to capture the fluorescent images of the neural precursor cells and contrast images of various other cells with a phase-contrast lens. A Nikon Ti-E inverted confocal microscope (Nikon A1Rsi Laser Scanning Confocal Microscope, Nikon Instruments Inc., Melville, NY) was used to obtain images to identify the subtype- and region-specific neurons.
The images of the glutamatergic neurons and punctate synaptic staining of PSD95 and synaptophysin were captured on a Nikon confocal microscope using a higher magnification lens and the NIS-Elements C software. ImageJ software was used to quantitate the number of MAP2-, GFAP-, SOX2-, NESTIN- and PAX6-positive cells, as well as the number of nuclei (DAPI) (http://imagej.nih.gov/ij/).
Flow cytometry
Flow cytometry analysis was performed to quantify the OCT4-, BMPRII-, SOX2- and NESTIN-positive cells. The cells were prepared as previously described24. Briefly, the neurosphere- and neuroectoderm-derived cells were blocked with 2% normal mouse serum (NMS, Abcam) in PBS for 30 mins at 4°C. The cells were permeabilized with 0.1% Triton X-100 in PBS and then incubated with antibodies conjugated with fluorophores (BD Biosciences, see supplementary table 2 for dilution) for 3 h at 4°C. After the incubation, the cells were thoroughly washed and post-fixed with 4% PFA for analysis using an Accuri C6 flow cytometer (BD Biosciences). The data and flow histograms were analyzed and prepared by the De Novo software (De Novo Software, Glendale, CA). The positive events were determined by comparing the gating population to an IgG control.
Quantitative real-time polymerase chain reaction (qRT-PCR)
The total RNA was extracted from the neurospheres using Trizol and a Direct-zol RNA purification kit (Zymo Research). The cDNAs were synthesized from 1 μg of RNA using a Superscript VILO® cDNA Synthesis Kit (Invitrogen) according to the manufacturer’s instructions. All qRT-PCR reactions were performed using the Fast SYBR Green Master Mix (Applied Biosystems). The reactions were performed on a StepOnePlus RT-PCR system (Applied Biosystems) using 1 μl of the cDNA (1:10 dilution), and 0.5 μM of the gene-specific primers for a total reaction volume of 20 μl. See supplementary Table 3 for the annealing temperatures and primer sequences. The levels of the SOX2, NESTIN, PAX6, and FOXG1 mRNAs were normalized to the mRNA levels of the housekeeping gene GAPDH to allow comparisons among the different experimental groups using the delta delta Ct method25.
NanoString CodeSet design and gene expression quantification
The NanoString CodeSet for the expression of 48 genes was designed by NanoString Technology (http://www.nanostring.com). A total of 100 ng of RNA from fresh-frozen tissue of the neurosphederm- and neuroectoderm-derived neurons were analyzed using the NanoString nCounter analysis system at the University of California, Irvine Genomics High Throughput Facility (http://ghtf.biochem.uci.edu/content/genomics-services, Irvine, CA).
NanoString data processing and gene expression was analyzed using the nSolver analysis software (Settle, WA), as previously described26. Briefly, the raw NanoString counts for each gene within each experiment were subjected to a technical normalization using the counts obtained for the positive control probe sets prior to a biological normalization using the three housekeeping genes included in the CodeSet. The normalized data were log2-transformed using the nSolver analysis software and then used as the input for the class prediction analysis. Finally, the neurosphederm-derived neuronal gene expression data were compared with the neuroectoderm-derived neuronal data and the percentage of genes that only exhibited a fold increase in the neurosphederm-derived neurons was shown in the graph.
Assay of neuronal function with the Fluo-4 Ca2+ fluorescence indicator
The neurons were grown on Matrigel-coated flat bottom 96-well plates to perform the functional assay. The neurons were first washed with Neurobasal medium (low Ca2+ and Mg2+) and washed again with 1× PBS (without Ca2+ and Mg2+). Next, a 5 μM Fluo-4 Ca2+ AM ester (Life Technologies) solution containing 0.001% pluronic F-127 (Life Technologies) was loaded into each well, except for the negative control and blank. The treated cells were incubated for 1 h in the dark at 37°C and 5% CO2. The Fluo-4 dye solution was removed and the cells were washed twice with 1× PBS (without Ca2+ and Mg2+). Then, 0.001, 0.01, 0.1 and 1.0 mM glutamate (glutamate receptor agonist) were added to the cells to examine the increase in the Ca2+-dependent electrical activity with the Fluo-4 dye. Finally, the fluorescence was read on a fluorescent microplate reader (POLARstar Omega, BMG LABTECH) with excitation at 485 nm and emission at 520 nm. The data were analyzed by the Omega software and normalized to the blank values. The intraneuronal calcium concentrations [Ca2+]i were calculated using the following previously described equation:[Ca2+]i=Kd·(F−Fmin)/(Fmax−F) Briefly, Kd is the dissociation constant for Ca2+ and F is the fluorescence (in arbitrary units) of the unknown sample. The values for Fmax and Fmin were determined using previously described calibration procedures27,28. Fmax and Fmin are the ratios at saturating Ca2+ and min zero Ca2+, respectively. The maximum fluorescence intensity (Fmax) was obtained by adding the Ca2+ ionophore ionomycin (Life Technologies, 10 μM). The concentration of the indicators in the calibration solution was selected to provide a similar fluorescence intensity to that of the dye-loaded neurons.
Transplantation and histological analysis
Cell transplantation
hESC-derived neurospheres were labeled with a Qtracker 585 Cell Labeling Kit (Life Technologies). One set of the labeled neurospheres was fragmented using the AdSTEP procedure and another set of labeled neurospheres was kept intact. The fragmented (n=5) and non-fragmented (n=4) Qtracker-labeled neurospheres were then transplanted into the SCID mouse brains. Briefly, adult postnatal day 79–80 SCID mice were anesthetized using isoflurane. The surgical area was cleansed with isopropyl alcohol followed by betadine and a cut was made through the skin to expose the skull. A hole was then made through the skull to allow the cells to be injected into the cortex and subcortical areas. The mice were then given 1.5 μl of cells or a PBS vehicle over the course of two minutes. We injected the cells in a more ventral part of the brain so that the cells would still be dispersed in the cortex, despite the back-up pressure from the needle being removed. The needle was left in situ for three minutes before being slowly removed. All procedures involving animals were conducted according to the NIH guidelines and approved by the Institutional Animal Care and Use Committee (IACUC) at Western University of Health Sciences (Pomona, CA).
Tissue Processing
Four weeks post-transplantation, the mice were transcardially perfused with PBS (50 ml) followed by chilled 4% PFA (60–70 ml). The brains were immediately placed in fresh 4% PFA for 24 hours, and then in 30% sucrose solution at 4°C. The brains were then sliced (30μm thick) using a cryostat (Leica, Model CM 3050S-3-1-1, Germany) and stored as free-floating sections in cryoprotectant at −20°C.
Immunohistochemical staining and imaging
The free-floating brain sections were washed thoroughly with PBS and then blocked using 10% Triton-X 100, 10% Tween 20, 1% BSA, and 1.5% NGS in PBS. The sections were then incubated with a primary antibody overnight at 4°C (see supplementary table 2 for the primary antibodies). Next, the sections were washed with PBS, incubated with a secondary antibody for two hours, and then washed with PBS and dried before being coverslipped with DAPI-FG. The in vivo images were captured on an Olympus FluoView™ FV1000 confocal microscope with Olympus FluoView software (Olympus America, Inc. Central Valley, PA). ImageJ software was used to quantify the βIII Tubulin staining.
Statistical analysis
At least three (n=3) samples were used for each statistical evaluation. Significances were assessed by one-way ANOVA using the post hoc test. In all cases, p<.05 was considered to be significant. The statistical analyses were performed using StatView (Abacus, Berkeley, CA; discontinued) and GraphPad InStat 3.1 (La Jolla, CA).
Results
Ten percent CO2 facilitated the formation of neurospheres from h/iPSCs
The procedures and various stages of neuronal differentiation from h/iPSCs with different culture conditions and time courses are shown in Fig. 1A. Briefly, the h/iPSCs were exposed to 10% CO2 in SKSRM for the first 3 days of neuronal initiation. At day 3, distinct round and bright-edged neurospheres were formed (Fig. 1B). In comparison, unevenly aggregated neurospheres and non-uniform neuroepithelial sheets were formed from the cells cultured with 5% CO2 (Fig. 1D). The neurospheres were further examined for the expression of neuronal precursor markers (SOX2, NESTIN, PAX6, and FOXG1) via qRT-PCR. The qRT-PCR results showed that with 10% CO2, the neurospheres expressed twice as much NESTIN, PAX6, and FOXG1 compared to the 5% CO2 culture condition, although SOX2 changed less significantly (Fig. 1F). In addition, flow cytometry showed that the expression of both SOX2 and NESTIN was higher in neurospheres on days 10, 20 and 30 compared to the IgG-treated control (ISO; Fig. 1G, H). However, in a similar flow cytometry run, the expression of SOX2 and NESTIN in the neuroectoderm-derived neuronal progenitors started to decline after 20 days, and on day 30, there was no difference in the expression of SOX2 and NESTIN compared to ISO (Fig. 3 I, J). These results indicated that the neurospheres derived with our culture conditions were more stable over a longer period of time compared to the adherent neuroectoderm culture method.
Figure 1.
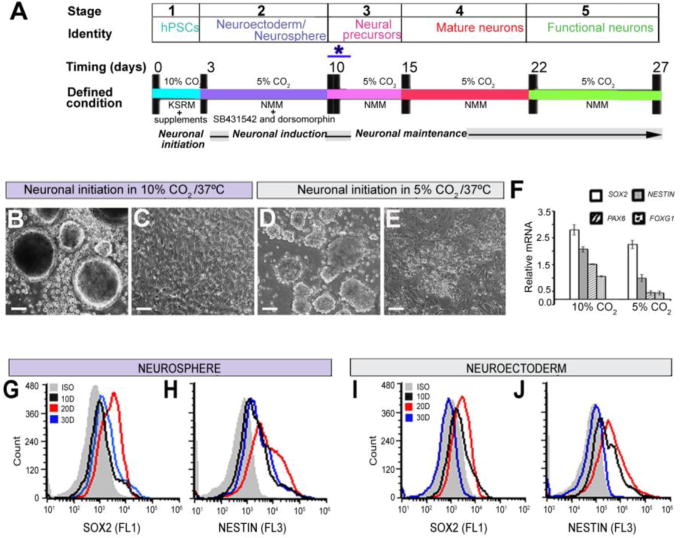
Flow diagram of the neurodifferentiation procedure from h/iPSCs and the characterization and stability of the neurospheres using defined culture conditions. (A) The five major stages and the corresponding cell types generated from these cultures. Neuronal initiation with SKSRM and 10% CO2 at 37°C takes 3 days. Neuronal induction took one week in NMM with SB431542 and dorsomorphin in 5% CO2 at 37°C. The AdSTEP (*) procedure was introduced to generate the neurosphederm from the neurosphere. After generating the neurosphederm and plating the cells, distinct neuronal rosettes appeared at 3–5 days in culture, and mature neurons and other sub type specific neurons appeared at 15–22 days in culture. At day 27, the neurons have fully functional synapses, as shown in the functional assays. (B, D) Generation of neurospheres from h/iPSCs with 10% and 5% CO2, respectively. (C, E) Generation of the neurosphederm from h/iPSCs with 10% and 5% CO2, respectively. (F). Graph of the relative expression levels of the SOX2, NESTIN, PAX6 and FOXG1 genes from neurospheres with 10% and 5% CO2, respectively. The data are presented as the means ± SD. (G, H & I, J) Flow cytometry histogram of the time-dependent expression of SOX2 and NESTIN in the neurosphere and neuroectoderm cultures, respectively. Scale bars, 50 μm.
Figure 3.
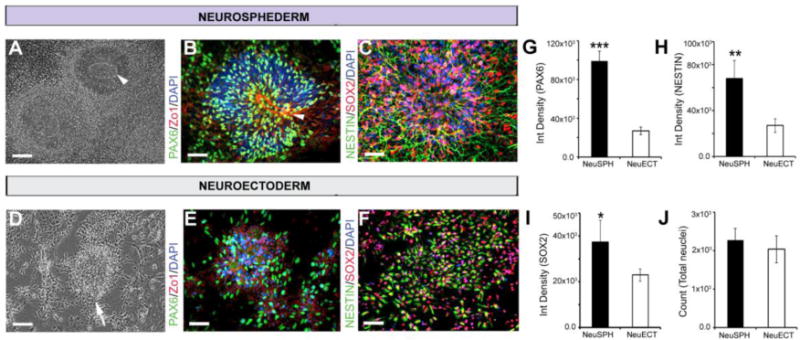
Comparison of the expression of neuronal progenitor genes in the neurosphederm and neuroectoderm. (A) A distinct NTTR structure appeared (white arrowhead) 10 days after neuronal induction. (B) PAX6/Zo1 staining of the neurosphederm- and neuroectoderm-derived neuronal cultures and (C) SOX2/NESTIN staining of the neuroectoderm cultures. Scale bars, 100 and 50 μm, respectively. (D) The neuroectoderm-derived neuronal cultures lacked NTTR structures. (E) PAX6/Zo1 staining of the neuroectoderm-derived cells, and (F) SOX2/NESTIN staining of the neuroectoderm-derived cells. Scale bars, 100 and 50 μm, respectively. The nuclei are stained with DAPI. The bar graph shows the ImageJ quantification of (G–I) the PAX6-, NESTIN- and SOX2-positive cells. (J) The DAPI quantification represents the total number of cells in these experiments, which confirmed that the cultures were similarly confluent. ISO and D represent the isotype control and number of days, respectively.
The AdSTEP mechanical procedure facilitated the generation of neurosphederm and neural stem cells from the neurospheres
Clumping has always been a challenge for examining neuronal differentiation in neurosphere-derived cultures (Fig 2B,C). To facilitate neurosphere-derived neuronal differentiation, an AdSTEP mechanical procedure was introduced to break the neurospheres (Fig. 2A) into smaller fragments (Fig. 2D), which were then plated onto matrigel-coated plates to form a monolayer of neuroepithelial-like cells that was termed the “neurosphederm” (Fig. 2E). After transferring the neurosphederm onto a polyornithine/laminin-coated plate, a large number of neurons was generated that expressed MAP2, a marker for mature neurons (Fig. 2F). On the other hand, without the AdSTEP mechanical procedure, the neurospheres still clumped together and produced much fewer neurons (Fig. 2C). Moreover, the neurosphere-derived neurosphederms were multipotent and were able to differentiate into astrocytes (Fig. 2G). Flow cytometry analysis determined that the culture was approximately 86% mature neurons and 8% astrocytes, as determined by MAP2 and GFAP staining, respectively (Fig. 2H). Image J quantification also confirmed that the neurosphederm generated both neurons and astrocytes at a ratio similar to the flow cytometric analysis (Fig. 2I). In addition, we further examined the neurosphederm-derived rosettes and rosette cores compared to the neuroectoderm-derived neuronal progenitors using immunostaining. The results showed that the neurosphederm-derived rosettes were larger and their core (Fig. 3A, white arrowhead) differed from that of a neuroectoderm-derived culture (Fig. 3D, white arrow). Furthermore, the neurosphederm-derived neural tube-type rosette NTTR structures expressed significantly higher levels of PAX6 and the tight junction protein Zo1 (arrowhead, Fig. 3B) compared to the neuroectoderm-derived cells (Fig. 3D & E), which was confirmed by ImageJ quantification of the PAX6-positive integrated cell density (Fig. 3G). In addition, the neural stem cells (NSCs) derived from the neurosphederm expressed higher levels of SOX2 and NESTIN (Fig. 3C) compared to those derived from the neuroectoderm (Fig. 3F, H, I). The total cell number, determined by counting the nuclei (blue, DAPI, Fig. 3J), confirmed that there was no significant difference in the confluence of either culture.
Figure 2.
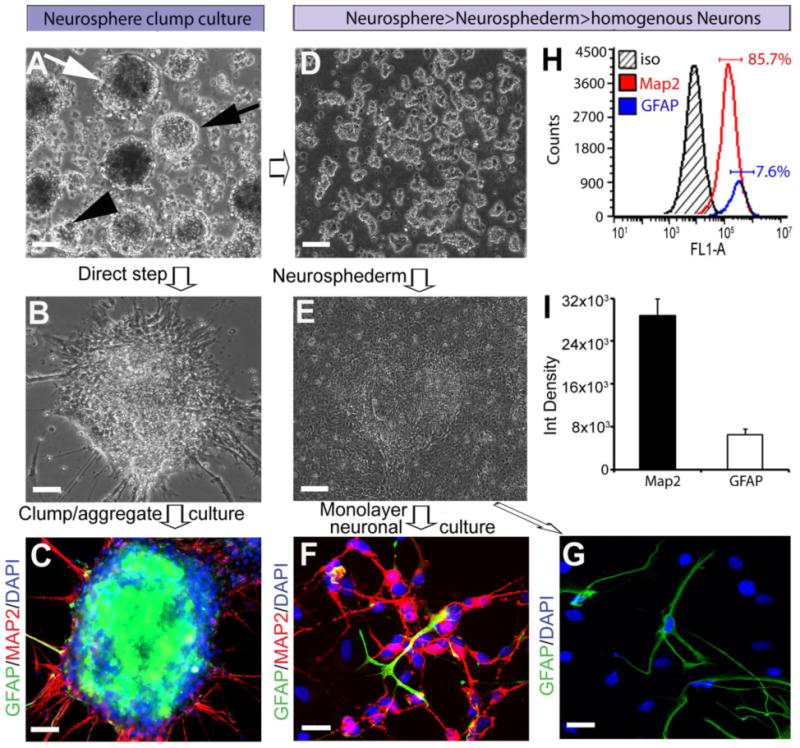
Comparison of neurogenesis with or without the AdSTEP mechanical procedure. (A). One week after neuronal induction, round, different sized spheres were observed. The arrows indicate the different shapes of the neurospheres, including a contrast sphere (white arrow), bright neurosphere (black arrow), and small sized spheres (black arrowhead). Scale bar, 100 μm. (B) Neuronal processes emanating from the neurospheres were observed after transferring the neurospheres to polyornithine/laminin-coated plates. Scale bar, 50 μm. (C) The neuronal cultures of the neurospheres (21 days) remained as clumps. These neurons express the mature neuronal marker MAP2 (red) and also the astrocyte marker GFAP (green), but we were unable to identify the neuronal or astrocytic morphology due to the tight clumping of the cells in the neurosphere. Scale bar, 50 μm. (D) The AdSTEP procedure (see materials and methods for details) dissociated the neurosphere into neurosphere fragments. Scale bar, 100 μm. (E) After the AdSTEP procedure, a neuroepithelial-type sheet appeared in the culture, which is termed the “neurosphederm”. Scale bars, 100 μm. (F) The neurosphederm was then transferred onto polyornithine/laminin-coated plates and neural stem cells were generated as single monolayers of cells. The cells were double stained with MAP2 and GFAP (red and green, respectively) antibodies. (G) The astrocyte marker GFAP (green) was observed in these cultures. Scale bars, 50 μm. The nuclei are stained with DAPI (blue) and the images were captured by confocal microscopy. (H) The flow histogram of MAP2 and GFAP indicated that 85.7% of the cells were MAP2-positive and approximately 8% were GFAP-positive. (I) The bar graph showed the quantification of the MAP2/GFAP-positive cells using the ImageJ software.
Generation of sub-type and region-specific neurons from neurosphere-derived neurosphederm
Different layers of cortical, pyramidal, GABAergic interneurons and excitatory glutamatergic neurons were generated using our culture method in this neurogenesis model. In our neuronal culture, the TBR1 cortical neurons appeared (Fig. 4A) within 17 days, and then the deep layer cortical neurons, the FOXP2- and EMX1-positive pyramidal neurons, emerged from the culture (Figure 4B, D) at approximately 22–27 days. Finally, the SATB2-positive layer 5 cortical neurons were identified (Fig. 4C). GABAergic interneurons and excitatory glutamatergic neurons (Fig. 4E, F) also appeared one week after neuronal induction (17 days) and their populations increased at approximately day 27, which is earlier than that observed in previously reported protocols11,22. We also analyzed the expression of specific neuronal genes from the progenitors, mature neurons, cortical neurons and specific sub-types using the NanoString Technologies nCounter system, and the bar graph represents the fold change percentage of neuronal gene expression in the neurosphederm-derived neurons compared to the neuroectoderm-derived neurons (Fig. 4G). Our results indicated that the neurosphederm protocol can not only generate very similar neural sub-types as the neuroectoderm-derived method (Supplementary Fig. 2), but it is also a more efficient neurogenesis method compared to the neuroectoderm method.
Figure 4.
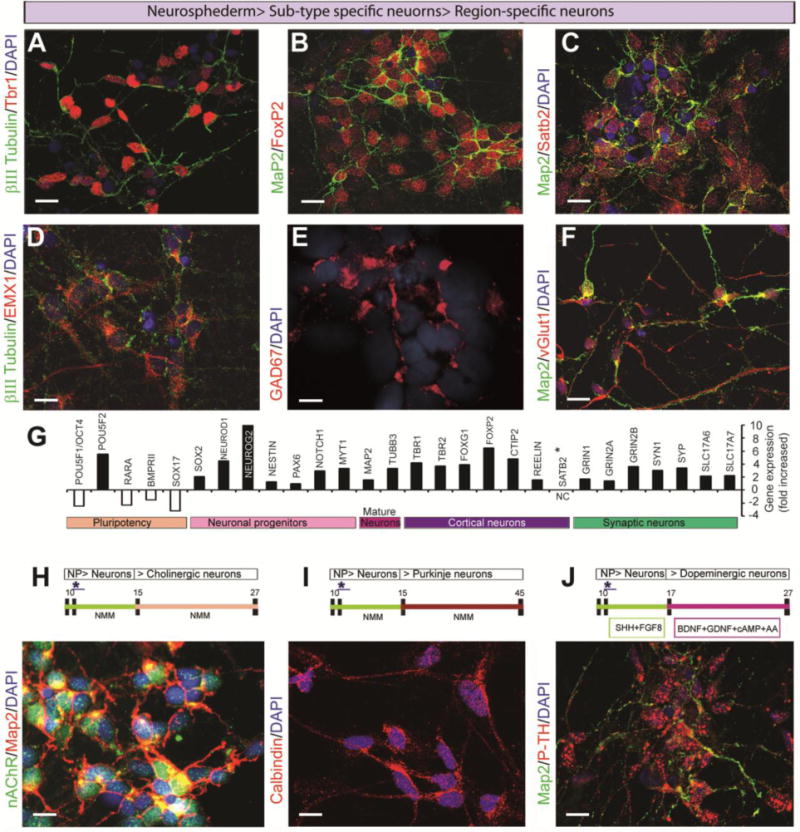
Generation of sub-type- and region-specific neurons from the neurospheres. The confocal images of the cortical layer-specific neurons were produced by double staining with (A) βIII tubulin/TBR1 (green/red), (B) MAP2/FOXP2 (green/red), and (C) cortical layer 2 (SATB2, red). These neurons appeared over 2 weeks after neuronal induction, and (D) cortical pyramidal neurons (EMX1, red) also appeared from the βIII tubulin-stained (green) neurosphederm-derived neuronal cultures. (E) Interneuron expression was analyzed by staining with the GAD67 marker (red). (F) The excitatory glutamatergic neuronal cells were observed with double staining for vGLUT1 (red) and MAP2 (green). (G) The bar graph represents the percentage of the fold increase in gene expression in the neurosphederm-derived neurons compared to the neuroectoderm-derived neurons. Gene expression analysis was performed using NanoString Technologies software as described in detail in the materials and methods. (H) The forebrain cholinergic neurons were detectable between 19–27 days in culture. These cells were confirmed by the presence of the nicotine acetyl choline receptor (nAChR, green) and a mature neuron marker (MAP2, red). (I) Purkinje neurons appeared between 40–45 days and expressed high levels of calbindin, a human Purkinje cell marker (red). (J) Dopaminergic neurons were detected between 25–27 days in the presence of additional supplements (see materials and methods). The dopaminergic neurons were identified by staining with antibodies to p-tyrosine hydroxylase (p-TH, red) and MAP2 (green). All of the images in this panel were captured by confocal microscopy. The nuclei are stained with DAPI. All scale bars, 25 μm.
Region-specific neurons were generated using the same protocol with different supplements or time courses. Forebrain cholinergic neurons (Fig. 4H) were generated by simply following our neuronal generation timeline up to day 27. The cholinergic neurons appeared between 19–27 days, as shown by double staining (green, nAChR) with MAP2 (red). Purkinje neurons, which expressed a high level of the Purkinje marker calbindin, appeared in the culture between 40–45 days (Fig. 4I)29,30. Midbrain or hindbrain dopaminergic neurons can be generated with additional supplements, as mentioned in the materials and methods. At approximately 27 days, the neurons expressed high levels of phospho-tyrosine hydroxylase (p-TH) (Fig. 4J), which is an active form of tyrosine hydroxylase. Therefore, the neurosphere-derived neurosphederm is an effective method for differentiating neural stem cells.
Generation of functional synapses and neural networks from neurosphederm in vitro
It is important to test whether the neurosphederm-derived neurons can produce functional synapses6,31,32. At day 27, PSD95 and synaptophysin were expressed in the neurons derived from both the neurosphere-derived neurosphederm (Fig. 5A) and the neuroectoderm (Fig. 5C). On the same day, the neurosphederm-derived neurons also showed higher expression of PSD95 and vGLUT1 (Fig. 5B). Therefore, the neuronal functional assay was performed on this day. For the functional assay, Fluo-4, a Ca2+ indicator dye, was added to the neuronal culture for 1 h. Prominent green fluorescence was detected in the neurons derived from both the neurosphederm (Fig. 5D) and neuroectoderm (Fig. 5E), indicating ongoing, spontaneous Ca2+ activity in those neurons. When glutamate, a major excitatory neurotransmitter, was added to the neurons, it elicited a dose-dependent increase in Fluo-4 fluorescence, with the exception of 1 mM glutamate (Fig. 5F). Time-lapse imaging showed that the neurosphederm-derived neurons had higher spontaneous Ca2+ activity (supplementary video 1) compared to the neuroectoderm-derived neurons (supplementary video 2). In addition, the cells were treated with the iontropic glutamate receptors (iGluRs) antagonists (+)-MK-801 (Abcam) and NBQX (Abcam) to block the NMDA and AMPA/and receptors, respectively, in the presence of 100 μM glutamate. The result showed that the inhibitors block the spontaneous Ca2+ activity of the neurosphederm-derived neurons (Fig. 5G). The ability of these neurons to actively respond to glutamate receptor antagonists and agonists indicates that the neurosphederm-derived neurons can form neural networks in vitro.
Figure 5.
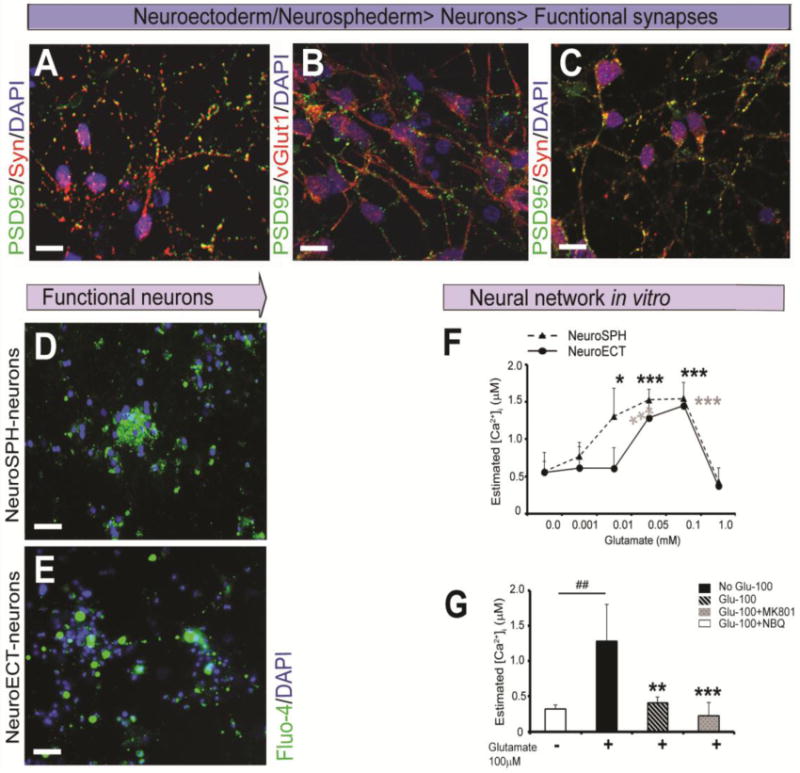
Generation of functional synapses and a neural network in vitro from the neuroectoderm- and neurosphederm-derived neurons. The confocal images of the excitatory synapses were obtained from day 27 neurosphederm-derived neurons (A,B). The expression of the post-synaptic density protein PSD95 (green) and presynaptic marker synaptophysin (SYN, red) appeared punctate staining that were expressed on opposing neurons. PSD95 (green) and vGLUT1 (red) were co-localized in the neurosphederm-derived neurons. Scale bars, 20 μm. (C) For comparison, the neuroectoderm-derived neurons were stained with PSD95 (green) and the presynaptic marker synaptophysin (SYN, red). Scale bars, 20 μm. (D, E) The functional activity of the neurosphederm- and neuroectoderm-derived neurons was determined using the Fluo-4 Ca2+ AM ester dye, which indicated the spontaneous Ca2+ activity through green fluorescence. Scale bars, 80 μm. The nuclei are stained with DAPI. (F) The graph shows a dose-dependent increase in the intraneuronal Ca2+ activity (μM) in response to glutamate, with the exception of 1 mM glutamate, which shows no activity in either the neurosphederm (dotted line)- and neuroectoderm (solid line)-derived neuronal cultures. (G) The bar graph shows the glutamate-dependent neuronal activity and specific inhibition via the glutamate receptor antagonists iGluRs, MK801 and NBQX. The black and white bars show the neurons from the neurosphederm and neuroectoderm cultures, respectively. The data are presented as the means ± SD, N=3. *p< 0.05 or *p < 0.05 represent significant differences. neuroSPH and neuroECT represented the neurosphederm and neuroectoderm, respectively.
Comparing neurogenesis using the fragmented and non-fragmented neurospheres in the mouse brain
The fragmented and non-fragmented neurospheres were labeled with Qtracker and injected into SCID mouse brains. Six weeks after the neurosphere transplantation, the mice were sacrificed and a histological analysis was performed to determine whether the neurospheres had integrated into the mouse brains. The arrow and insert box represent the site of injection and image acquisition, respectively (Fig. 6A & B). We used an antibody against human-specific nuclear antigen (HumN) to identify the transplanted human cells, which were clearly overlaid with the Qtracker-labeled cells (the arrowhead shows the Qtracker-labeled cells that colocalized with HumN). The results showed that the transplanted neurospheres differentiated into PAX6-positive cells (arrowhead, Fig. 6C). The PAX6-positive cells are the predominant neural progenitors for the developing cortex during neurogenesis33,34, as shown in the illustration (Fig. 6D)33,34. Most importantly, a large number of βIII Tubulin-positive neuronal cells were observed in the fragmented neurosphere sections (Fig. 6E) compared to the sections with the non-fragmented neurospheres (Fig. 6G). Most of the βIII Tubulin-positive neurons from the fragmented neurospheres overlaid with the Qtracker-labeled cells (indicated by an arrowhead, Fig. 6F), but those in the non-fragmented neurosphere sections exhibited little overlap and lacked distinct neuronal soma (indicated by an arrowhead, Fig. 6G & H). ImageJ quantification of the βIII Tubulin staining showed significant differences in the fragmented and non-fragmented engrafted sections (Fig. 6L). GFAP staining also showed that the transplanted cells expressed a large number of astrocytes (Fig. 6I) and that some of them were overlaid with the Qtracker-labeled cells (arrowhead, Fig. 6 J,K), suggesting that that the engrafted AdSTEP-fragmented neurospheres facilitated the differentiation of neurospheres into multipotent neural stem cells and mature neurons in vivo compared to the non-fragmented neurospheres.
Figure 6.
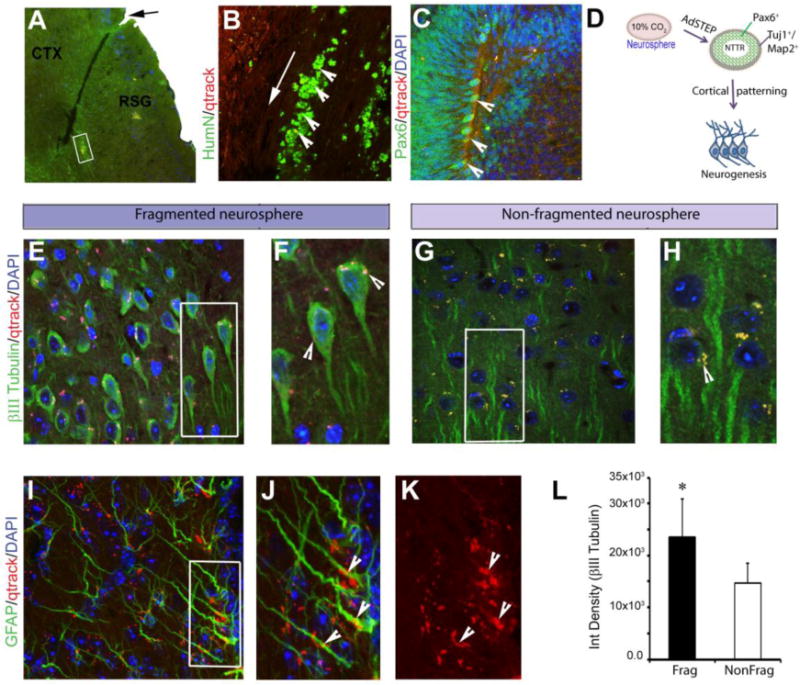
Comparison of neurogenesis of the AdSTEP fragmented and non-fragmented neurospheres in the mouse brain. (A) Representative image of the fragmented Qtracker-labeled neurospheres in a mouse; the implanted site is indicated by an arrow. Dispersion of the fragmented neurospheres into the cortex (CTX, cortex; and RSG, Retrosplenial granular). Scale bars, 200 μm. (B) Staining of the area with antibodies against human-specific nuclear antigen (HumN) (an arrow shows the injection site) that overlaid with the Qtracker-labeled (red) cells (arrowhead). Scale bars, 50 μm. (C) After transplantation, a significant number of PAX6-positive cells were observed in the AdSTEP-fragmented neurosphere-implanted graft, and they co-localized with the Qtracker-labeled cells (Qtracker-labeled cell colocalized with the PAX6 cell indicated by the arrowhead). Scale bars, 50 μm. (D) Illustration of the mechanism of neurogenesis from the neurospheres. The cells cultured with 10% CO2 and SKSRM produce large and bright neurospheres. After introducing the AdSTEP process to the neurospheres, NTTR structures with increased numbers of PAX6-positive cells appeared in the culture within 3–5 days. (E) βIII Tubulin-positive neurons were abundant in the graft from the AdSTEP-fragmented neurospheres. (F) The arrowhead in the inset showed that the implanted cells overlaid with the Qtracker-labeled cells had differentiated into neurons. Scale bars, 25 μm. (G) The non-fragmented neurospheres displayed very few βIII tubulin-positive cells. (H) The arrowhead in the inset showed the Qtracker-labeled cells with no overlaid neurons. Scale bars, 25 μm. (I) GFAP-positive astrocytes in the graft, and the arrowhead showed the overlaid Qtracker-labeled implanted cells (J) with or (K) without GFAP staining. Scale bars, 25 μm. The nuclei of all images are stained with DAPI and all of the images in this panel were captured by confocal microscopy. (L) The bar diagram represents the ImageJ quantification of the integrated density of the βIII tubulin-positive cells from the implanted AdSTEP-fragmented and non-fragmented neurospheres. The data are presented as the means ± SD, N=4. *p< 0.05 represents a significant difference. Frag and NonFrag represented the implanted AdSTEP-fragmented and non-fragmented neurospheres, respectively.
Discussion
The neurosphere-derived cultures for neuronal differentiation are a valuable model system for studying neurogenesis and understanding the molecular mechanisms associated with neurodegenerative diseases18,20. Recent studies on iPSC-derived neurospheres and 3D cultures showed significant promise for the development of disease-specific cells with the desired genetic backgrounds, which would facilitate the study of many important diseases, such as Timothy syndrome, Fragile X syndrome or NPD35–38. Here, we present a new defined culture medium and conditions: SKSRM medium and 10% CO2. This new culture condition doubled the expression of the neuroprogenitor genes NESTIN, PAX6, and FOXG1 compared to the traditional 5% CO2 culture conditions. The molecular mechanism by which the higher CO2 levels facilitate neurogenesis is still not clear. It could be due to reduced oxygenation or hypoxia, as previously reported39–42. There are several groups that have reported that hypoxia or reduced oxygenation enhances neural stem cell colony survival and increased NESTIN, SOX1, SOX2, and FOXG1 expression40,42,43, similar to our study (Fig. 4G).
Neurospheres are also a good source of neural progenitors for neural transplantation due to their easy delivery and ability to migrate12,16,17,44. However, clumping has been a challenge for neurodifferentiation both in vivo and in vitro. AdSTEP was another important mechanical procedure that enabled us to overcome the challenge of neurosphere-based neurodifferentiation. AdSTEP was able to facilitate the generation of the monolayer neuroepithelium, which we call the ‘neurosphederm’. The neurosphederm not only allowed us to increase the number neuroprogenitor cells but also generated a robust multipotent NSC population and enabled us to clearly identify the neuronal phenotypes. In addition, AdSTEP increased the expression of PAX6 fivefold, and also increased the expression of FOXG1 (Fig. 1F). PAX6 is a downstream effector (a transcription factor) of Wnt/β-catenin signaling in the proliferation and neuronal differentiation of cortical radial glia, a major NSC population in the developing cortex31,45. FOXG1 is a telencephalic marker that induces the expression of pallial determinants. PAX6 and NGN2 are involved in cortical patterning (FOXG1>PAX6, NGN2>TBR1>Cortical layers, e.g., FOXP2, SATB2, EMX1)34,46,47. An interesting aspect of these gene expression results is the fact that the 10% CO2 culture condition, which mimics reduced oxygenation, was sufficient to induce and give rise to the different layers of cortical neurons, suggesting that this culture method is a promising potential neural model system for studying cortical development.
Furthermore, our neurosphederm can not only generate more neuroprogenitors, but it can also give rise to all of the subtype-specific neurons and synapses, e.g., glutamatergic, GABAergic and cortical neurons. In addition, the neurosphederm-derived neurons also showed stronger spontaneous neuronal activity, as shown by the Ca2+ fluorescence activity (supplementary viedo 1), which was accelerated by glutamate in a dose-dependent manner. Therefore, it is clear that the neurospheres-derived neurons respond via glutamatergic neurotransmission, and the enhancement or suppressed electrical activity with glutamate receptor agonists or antagonist, respectively, confirmed the development of an excitatory neural network in vitro (Fig. 5G). Here, we used calcium-dependent fluorescent indicator dyes that allowed us to measure the synchronized activity across a network of cells33,48. In contrast, it is also possible to determine single-cell resolution neuronal activity using patch-clamp electrophysiology, but the ability to measure a network is limited to typically one or two neurons. Therefore, the Fluo-4 Ca2+ indicator dye was used in this study to identify the neural networks created by the neurosphederm-derived neurons.
Another important feature of this system is the generation of region-specific neurons from the neurosphederm, which provides a model system for future studies of various neurological disorders. The early and substantial loss of basal forebrain cholinergic neurons (BFCNs) is a constant feature of AD6,49. BFCNs were rapidly generated within 3 weeks using the neurosphederm, making it a useful model system for studying AD. Mid/hindbrain dopaminergic neurons (DNs) play a critical role in PD4,8. DNs were generated using the same timeline over 27 days (3–4 weeks). However, SHH and FGF8 were added after neuronal induction to generate these neurons. Cerebellar Purkinje neurons, which are known to have a crucial role in NPD36 and HD50,51, appeared in our culture between 45–48 days, which is relatively longer and is a similar pattern as that observed in the developing cerebellum in vivo29,30. Thus, our method can rapidly generate subtype-specific or region-specific neurons compared to the other currently available neurodifferentiation methods10,11,21,22.
In addition, we also injected the neurospheres into SCID mouse brains. The confocal images showed that the AdSTEP-fragmented neurospheres had abundant PAX6-positive cells in the cerebral cortex (Fig. 6C) that contained larger NTTR structures, similar to those observed in vitro. These results suggested that the engrafted neurospheres were further differentiated to neural precursor cells, which further contributed to neurogenesis (Fig. 6D). Therefore, our AdSTEP neurospheres, generated under defined culture conditions, easily integrated into the mouse brains, demonstrating great promise for neurogenesis studies and stem cell therapy. Overall, this novel and rapid virus-free method for generating neuronal populations from neurospheres has many advantages, all of which will have a great impact on our understanding of neuronal identity after neurosphere transplantation as well as the mechanisms of disease.
Supplementary Material
Highlights.
This research involves a rapid neurogeneration method and can form neural network within day 27.
A new culture condition with 10% CO2 during the first 3 days of neural initiation promotes generation of neurospheres from human pluripotent stem cells.
An additional procedure (AdSTEP) was introduced which increased neurogenesis in vitro and in vivo.
This neurodifferentiation method is completely viral free and there has no genetic modification.
Acknowledgments
This work was supported by the National Institutes of Environmental Health Sciences (1R15 ES019298-01A1).
Abbreviation
- h/iPSCs
human embryonic and induced pluripotent stem cells
- SKSRM
supplemented knockout serum replacement medium
- NPCs
neural precursor cells
- AdSTEP
additional step
- NTTR
neural tube-type rosette
- SCID
Severe combined immunodeficiency
- NIM
Neuronal induction medium
- NMM
Neuronal maintenance medium
- p-TH
phospho-tyrosine hydroxylase
- nAChR
Nicotinic acetylcholine receptor
- iGluRs
iontropic glutamate receptors
- HumN
human-specific nuclear antigen
- SHH
sonic hedgehog
Appendix A. Supplementary data
Supplementary data to this article can be found online.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHORS’ CONTRIBUTIONS
Aynun N. Begum: Performed most of the experiments, prepared the figures and wrote the manuscript.
Caleigh Guoynes: Performed the brain tissue processing and immunohistochemistry.
Jane Cho: Involved in the qPCR experiment and edited the manuscript.
Kabirullah Lutfy: Performed the stem cell transplantation and edited the manuscript.
Jijun Hao: Generated the iPSC lines from foreskin fibroblasts using an episomal vector and edited the manuscript
Yiling Hong: Directed the research, wrote and edited the manuscript.
References
- 1.Ebert AD, Yu J, Rose FF, Jr, Mattis VB, Lorson CL, Thomson JA, Svendsen CN. Induced pluripotent stem cells from a spinal muscular atrophy patient. Nature. 2009;457(7227):277–280. doi: 10.1038/nature07677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee G, Papapetrou EP, Kim H, Chambers SM, Tomishima MJ, Fasano CA, Ganat YM, Menon J, Shimizu F, Viale A, Tabar V, Sadelain M, Studer L. Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature. 2009;461(7262):402–406. doi: 10.1038/nature08320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dimos JT, Rodolfa KT, Niakan KK, Weisenthal LM, Mitsumoto H, Chung W, Croft GF, Saphier G, Leibel R, Goland R, Wichterle H, Henderson CE, Eggan K. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008;321(5893):1218–1221. doi: 10.1126/science.1158799. [DOI] [PubMed] [Google Scholar]
- 4.Park IH, Arora N, Huo H, Maherali N, Ahfeldt T, Shimamura A, Lensch MW, Cowan C, Hochedlinger K, Daley GQ. Disease-specific induced pluripotent stem cells. Cell. 2008;134(5):877–886. doi: 10.1016/j.cell.2008.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yagi T, Ito D, Okada Y, Akamatsu W, Nihei Y, Yoshizaki T, Yamanaka S, Okano H, Suzuki N. Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum Mol Genet. 2011;20(23):4530–4539. doi: 10.1093/hmg/ddr394. [DOI] [PubMed] [Google Scholar]
- 6.Shi Y, Kirwan P, Smith J, MacLean G, Orkin SH, Livesey FJ. A human stem cell model of early Alzheimer’s disease pathology in Down syndrome. Sci Transl Med. 2012;4(124):124ra29. doi: 10.1126/scitranslmed.3003771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Israel MA, Yuan SH, Bardy C, Reyna SM, Mu Y, Herrera C, Hefferan MP, Van GS, Nazor KL, Boscolo FS, Carson CT, Laurent LC, Marsala M, Gage FH, Remes AM, Koo EH, Goldstein LS. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature. 2012;482(7384):216–220. doi: 10.1038/nature10821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Devine MJ, Ryten M, Vodicka P, Thomson AJ, Burdon T, Houlden H, Cavaleri F, Nagano M, Drummond NJ, Taanman JW, Schapira AH, Gwinn K, Hardy J, Lewis PA, Kunath T. Parkinson’s disease induced pluripotent stem cells with triplication of the alpha-synuclein locus. Nat Commun. 2011;2:440. doi: 10.1038/ncomms1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jeon I, Lee N, Li JY, Park IH, Park KS, Moon J, Shim SH, Choi C, Chang DJ, Kwon J, Oh SH, Shin DA, Kim HS, Do JT, Lee DR, Kim M, Kang KS, Daley GQ, Brundin P, Song J. Neuronal properties, in vivo effects, and pathology of a Huntington’s disease patient-derived induced pluripotent stem cells. Stem Cells. 2012;30(9):2054–2062. doi: 10.1002/stem.1135. [DOI] [PubMed] [Google Scholar]
- 10.Chambers SM, Fasano CA, Papapetrou EP, Tomishima M, Sadelain M, Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat Biotechnol. 2009;27(3):275–280. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi Y, Kirwan P, Livesey FJ. Directed differentiation of human pluripotent stem cells to cerebral cortex neurons and neural networks. Nat Protoc. 2012;7(10):1836–1846. doi: 10.1038/nprot.2012.116. [DOI] [PubMed] [Google Scholar]
- 12.Matigian N, Abrahamsen G, Sutharsan R, Cook AL, Vitale AM, Nouwens A, Bellette B, An J, Anderson M, Beckhouse AG, Bennebroek M, Cecil R, Chalk AM, Cochrane J, Fan Y, Feron F, McCurdy R, McGrath JJ, Murrell W, Perry C, Raju J, Ravishankar S, Silburn PA, Sutherland GT, Mahler S, Mellick GD, Wood SA, Sue CM, Wells CA, Mackay-Sim A. Disease-specific, neurosphere-derived cells as models for brain disorders. Dis Model Mech. 2010;3(11–12):785–798. doi: 10.1242/dmm.005447. [DOI] [PubMed] [Google Scholar]
- 13.Koehler KR, Tropel P, Theile JW, Kondo T, Cummins TR, Viville S, Hashino E. Extended passaging increases the efficiency of neural differentiation from induced pluripotent stem cells. BMC Neurosci. 2011;12:82. doi: 10.1186/1471-2202-12-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bez A, Corsini E, Curti D, Biggiogera M, Colombo A, Nicosia RF, Pagano SF, Parati EA. Neurosphere and neurosphere-forming cells: morphological and ultrastructural characterization. Brain Res. 2003;993(1–2):18–29. doi: 10.1016/j.brainres.2003.08.061. [DOI] [PubMed] [Google Scholar]
- 15.Lindvall O, Kokaia Z. Stem cells in human neurodegenerative disorders–time for clinical translation? J Clin Invest. 2010;120(1):29–40. doi: 10.1172/JCI40543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Englund U, Fricker-Gates RA, Lundberg C, Bjorklund A, Wictorin K. Transplantation of human neural progenitor cells into the neonatal rat brain: extensive migration and differentiation with long-distance axonal projections. Exp Neurol. 2002;173(1):1–21. doi: 10.1006/exnr.2001.7750. [DOI] [PubMed] [Google Scholar]
- 17.Flax JD, Aurora S, Yang C, Simonin C, Wills AM, Billinghurst LL, Jendoubi M, Sidman RL, Wolfe JH, Kim SU, Snyder EY. Engraftable human neural stem cells respond to developmental cues, replace neurons, and express foreign genes. Nat Biotechnol. 1998;16(11):1033–1039. doi: 10.1038/3473. [DOI] [PubMed] [Google Scholar]
- 18.Jensen JB, Parmar M. Strengths and limitations of the neurosphere culture system. Mol Neurobiol. 2006;34(3):153–161. doi: 10.1385/MN:34:3:153. [DOI] [PubMed] [Google Scholar]
- 19.Reynolds BA, Weiss S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science. 1992;255(5052):1707–1710. doi: 10.1126/science.1553558. [DOI] [PubMed] [Google Scholar]
- 20.Reynolds BA, Rietze RL. Neural stem cells and neurospheres–re-evaluating the relationship. Nat Methods. 2005;2(5):333–336. doi: 10.1038/nmeth758. [DOI] [PubMed] [Google Scholar]
- 21.Goulburn AL, Stanley EG, Elefanty AG, Anderson SA. Generating GABAergic cerebral cortical interneurons from mouse and human embryonic stem cells. Stem Cell Res. 2012;8(3):416–426. doi: 10.1016/j.scr.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 22.Liu Y, Liu H, Sauvey C, Yao L, Zarnowska ED, Zhang SC. Directed differentiation of forebrain GABA interneurons from human pluripotent stem cells. Nat Protoc. 2013;8(9):1670–1679. doi: 10.1038/nprot.2013.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu J, Chau KF, Vodyanik MA, Jiang J, Jiang Y. Efficient feeder-free episomal reprogramming with small molecules. PLoS One. 2011;6(3):e17557. doi: 10.1371/journal.pone.0017557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lippmann ES, Estevez-Silva MC, Ashton RS. Defined human pluripotent stem cell culture enables highly efficient neuroepithelium derivation without small molecule inhibitors. Stem Cells. 2014;32(4):1032–1042. doi: 10.1002/stem.1622. [DOI] [PubMed] [Google Scholar]
- 25.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3(6):1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 26.Northcott PA, Shih DJ, Remke M, Cho YJ, Kool M, Hawkins C, Eberhart CG, Dubuc A, Guettouche T, Cardentey Y, Bouffet E, Pomeroy SL, Marra M, Malkin D, Rutka JT, Korshunov A, Pfister S, Taylor MD. Rapid, reliable, and reproducible molecular sub-grouping of clinical medulloblastoma samples. Acta Neuropathol. 2012;123(4):615–626. doi: 10.1007/s00401-011-0899-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Briz V, Galofre M, Sunol C. Reduction of glutamatergic neurotransmission by prolonged exposure to dieldrin involves NMDA receptor internalization and metabotropic glutamate receptor 5 downregulation. Toxicol Sci. 2010;113(1):138–149. doi: 10.1093/toxsci/kfp244. [DOI] [PubMed] [Google Scholar]
- 28.Hyrc K, Handran SD, Rothman SM, Goldberg MP. Ionized intracellular calcium concentration predicts excitotoxic neuronal death: observations with low-affinity fluorescent calcium indicators. J Neurosci. 1997;17(17):6669–6677. doi: 10.1523/JNEUROSCI.17-17-06669.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laure-Kamionowska M, Maslinska D. Calbindin positive Purkinje cells in the pathology of human cerebellum occurring at the time of its development. Folia Neuropathol. 2009;47(4):300–305. [PubMed] [Google Scholar]
- 30.Iritani S, Kuroki N, Ikeda K, Kazamatsuri H. Calbindin immunoreactivity in the hippocampal formation and neocortex of schizophrenics. Prog Neuropsychopharmacol Biol Psychiatry. 1999;23(3):409–421. doi: 10.1016/s0278-5846(99)00005-6. [DOI] [PubMed] [Google Scholar]
- 31.Hansen DV, Rubenstein JL, Kriegstein AR. Deriving excitatory neurons of the neocortex from pluripotent stem cells. Neuron. 2011;70(4):645–660. doi: 10.1016/j.neuron.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shi Y, Kirwan P, Smith J, Robinson HP, Livesey FJ. Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat Neurosci. 2012;15(3):477–86. doi: 10.1038/nn.3041. S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eiraku M, Watanabe K, Matsuo-Takasaki M, Kawada M, Yonemura S, Matsumura M, Wataya T, Nishiyama A, Muguruma K, Sasai Y. Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem Cell. 2008;3(5):519–532. doi: 10.1016/j.stem.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 34.Muzio L, Di BB, Stoykova A, Boncinelli E, Gruss P, Mallamaci A. Emx2 and Pax6 control regionalization of the pre-neuronogenic cortical primordium. Cereb Cortex. 2002;12(2):129–139. doi: 10.1093/cercor/12.2.129. [DOI] [PubMed] [Google Scholar]
- 35.Kumari D, Swaroop M, Southall N, Huang W, Zheng W, Usdin K. High-Throughput Screening to Identify Compounds That Increase Fragile X Mental Retardation Protein Expression in Neural Stem Cells Differentiated From Fragile X Syndrome Patient-Derived Induced Pluripotent Stem Cells. Stem Cells Transl Med. 2015;4(7):800–808. doi: 10.5966/sctm.2014-0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Macauley SL, Sidman RL, Schuchman EH, Taksir T, Stewart GR. Neuropathology of the acid sphingomyelinase knockout mouse model of Niemann-Pick A disease including structure-function studies associated with cerebellar Purkinje cell degeneration. Exp Neurol. 2008;214(2):181–192. doi: 10.1016/j.expneurol.2008.07.026. [DOI] [PubMed] [Google Scholar]
- 37.Pasca SP, Portmann T, Voineagu I, Yazawa M, Shcheglovitov A, Pasca AM, Cord B, Palmer TD, Chikahisa S, Nishino S, Bernstein JA, Hallmayer J, Geschwind DH, Dolmetsch RE. Using iPSC-derived neurons to uncover cellular phenotypes associated with Timothy syndrome. Nat Med. 2011;17(12):1657–1662. doi: 10.1038/nm.2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pasca AM, Sloan SA, Clarke LE, Tian Y, Makinson CD, Huber N, Kim CH, Park JY, O’Rourke NA, Nguyen KD, Smith SJ, Huguenard JR, Geschwind DH, Barres BA, Pasca SP. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat Methods. 2015;12(7):671–678. doi: 10.1038/nmeth.3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heinrich C, Gascon S, Masserdotti G, Lepier A, Sanchez R, Simon-Ebert T, Schroeder T, Gotz M, Berninger B. Generation of subtype-specific neurons from postnatal astroglia of the mouse cerebral cortex. Nat Protoc. 2011;6(2):214–228. doi: 10.1038/nprot.2010.188. [DOI] [PubMed] [Google Scholar]
- 40.Morrison SJ, Csete M, Groves AK, Melega W, Wold B, Anderson DJ. Culture in reduced levels of oxygen promotes clonogenic sympathoadrenal differentiation by isolated neural crest stem cells. J Neurosci. 2000;20(19):7370–7376. doi: 10.1523/JNEUROSCI.20-19-07370.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Putnam RW, Filosa JA, Ritucci NA. Cellular mechanisms involved in CO(2) and acid signaling in chemosensitive neurons. Am J Physiol Cell Physiol. 2004;287(6):C1493–C1526. doi: 10.1152/ajpcell.00282.2004. [DOI] [PubMed] [Google Scholar]
- 42.Clarke L, van der Kooy D. Low oxygen enhances primitive and definitive neural stem cell colony formation by inhibiting distinct cell death pathways. Stem Cells. 2009;27(8):1879–1886. doi: 10.1002/stem.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xie Y, Zhang J, Lin Y, Gaeta X, Meng X, Wisidagama DR, Cinkornpumin J, Koehler CM, Malone CS, Teitell MA, Lowry WE. Defining the role of oxygen tension in human neural progenitor fate. Stem Cell Reports. 2014;3(5):743–757. doi: 10.1016/j.stemcr.2014.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Betarbet R, Zigova T, Bakay RA, Luskin MB. Migration patterns of neonatal subventricular zone progenitor cells transplanted into the neonatal striatum. Cell Transplant. 1996;5(2):165–178. doi: 10.1177/096368979600500207. [DOI] [PubMed] [Google Scholar]
- 45.Gan Q, Lee A, Suzuki R, Yamagami T, Stokes A, Nguyen BC, Pleasure D, Wang J, Chen HW, Zhou CJ. Pax6 mediates ss-catenin signaling for self-renewal and neurogenesis by neocortical radial glial stem cells. Stem Cells. 2014;32(1):45–58. doi: 10.1002/stem.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Muzio L, Mallamaci A. Foxg1 confines Cajal-Retzius neuronogenesis and hippocampal morphogenesis to the dorsomedial pallium. J Neurosci. 2005;25(17):4435–4441. doi: 10.1523/JNEUROSCI.4804-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muzio L, Soria JM, Pannese M, Piccolo S, Mallamaci A. A mutually stimulating loop involving emx2 and canonical wnt signalling specifically promotes expansion of occipital cortex and hippocampus. Cereb Cortex. 2005;15(12):2021–2028. doi: 10.1093/cercor/bhi077. [DOI] [PubMed] [Google Scholar]
- 48.Dawitz J, Kroon T, Hjorth JJ, Meredith RM. Functional calcium imaging in developing cortical networks. J Vis Exp. 2011;(56) doi: 10.3791/3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Duan L, Bhattacharyya BJ, Belmadani A, Pan L, Miller RJ, Kessler JA. Stem cell derived basal forebrain cholinergic neurons from Alzheimer’s disease patients are more susceptible to cell death. Mol Neurodegener. 2014;9:3. doi: 10.1186/1750-1326-9-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dougherty SE, Reeves JL, Lucas EK, Gamble KL, Lesort M, Cowell RM. Disruption of Purkinje cell function prior to huntingtin accumulation and cell loss in an animal model of Huntington disease. Exp Neurol. 2012;236(1):171–178. doi: 10.1016/j.expneurol.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dougherty SE, Reeves JL, Lesort M, Detloff PJ, Cowell RM. Purkinje cell dysfunction and loss in a knock-in mouse model of Huntington disease. Exp Neurol. 2013;240:96–102. doi: 10.1016/j.expneurol.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.