

Abstract
Guanine nucleotide exchange factors directly activated by cAMP (Epacs) have emerged as important signaling molecules mediating persistent hypersensitivity in animal models of inflammation, by augmenting the excitability of sensory neurons. Although Epacs activate numerous downstream signaling cascades, the intracellular signaling which mediates Epac-induced sensitization of capsaicin-sensitive sensory neurons remains unknown. Here, we demonstrate that selective activation of Epacs with 8-CPT-2′-O-Me-cAMP-AM (8CPT-AM) increases the number of action potentials (APs) generated by a ramp of depolarizing current and augments the evoked release of calcitonin gene-related peptide (CGRP) from isolated rat sensory neurons. Internal perfusion of capsaicin-sensitive sensory neurons with GDP-βS, substituted for GTP, blocks the ability of 8CPT-AM to increase AP firing, demonstrating that Epac-induced sensitization is G-protein dependent. Treatment with 8CPT-AM activates the small G-proteins Rap1 and Ras in cultures of sensory neurons. Inhibition of Rap1, by internal perfusion of a Rap1-neutralizing antibody or through a reduction in the expression of the protein using shRNA does not alter the Epac-induced enhancement of AP generation or CGRP release, despite the fact that in most other cell types, Epacs act as Rap-GEFs. In contrast, inhibition of Ras through expression of a dominant negative Ras (DN-Ras) or through internal perfusion of a Ras-neutralizing antibody blocks the increase in AP firing and attenuates the increase in the evoked release of CGRP induced by Epac activation. Thus, in this subpopulation of nociceptive sensory neurons, it is the novel interplay between Epacs and Ras, rather than the canonical Epacs and Rap1 pathway, that is critical for mediating Epac-induced sensitization.
Keywords: Peripheral sensitization, calcitonin gene-related peptide (CGRP), excitability, exchange proteins activated by cAMP (Epacs), Ras, Rap1, sensory neurons
INTRODUCTION
Exchange proteins directly activated by cAMP (Epacs) are key downstream signaling effectors of cAMP that function as guanine nucleotide exchange factors (GEFs) for the Ras superfamily of small GTPases (Holz et al., 2006). Recently, an important role of Epacs in mediating pain hypersensitivity in animal models of chronic inflammation has emerged. This hypersensitivity is a consequence of increased excitability of sensory neurons that convey noxious information to the spinal cord (Gold and Gebhart, 2010; Richardson and Vasko, 2002; Woolf and Ma, 2007), a process termed peripheral sensitization. Although much work has described the signal transduction cascades in sensory neurons that mediate sensitization under acute inflammation (Basbaum et al., 2009; Hucho and Levine, 2007; Richardson and Vasko, 2002), less is known about signaling that maintains sensitization under chronic inflammatory conditions. Recent evidence suggests, however, that the transition from acute to persistent hypersensitivity is linked to a switch in signaling pathways that mediate sensitization (Villarreal et al., 2009). For example, acute exposure to prostaglandin E2 (PGE2) produces hyperalgesia and an increase in the excitability of sensory neurons, which are both mediated by increases in cAMP and activation of protein kinase A (PKA) (Aley and Levine, 1999; England et al., 1996; Hingtgen et al., 1995; Lopshire and Nicol, 1998; Sachs et al., 2009). With inflammation, or when sensory neurons are maintained in the presence of the inflammatory mediator, nerve growth factor (NGF), the sensitization induced by subsequent administration of PGE2 shifts from using PKA as the primary effector to activation of Epacs (Eijkelkamp et al., 2013; Hucho et al., 2005; Vasko et al., 2014; Wang et al., 2007).
The question remains, however as to the downstream signaling by Epacs that contributes to persistent sensitization of sensory neurons. Activation of the Ras family of GTPases by Epac1 (RapGEF3) or Epac2 (RapGEF4) (Bos, 2003; de Rooij et al., 1998) is linked to a number of downstream signaling effectors that may contribute to persistent hypersensitivity, including but not limited to phospholipase Cε (PLCε), phospholipase D (PLC), mitogen-activated protein kinases (MAPKs), and phosphatidylinositol 3-kinase (PI3K) (Baviera et al., 2010; Holz et al., 2006; Schmidt et al., 2013; Yano et al., 2007). Although Epacs were initially discovered as GEFs for Rap1, controversy exists, however, as to whether Epacs may also act as GEFs for Ras and whether Ras mediates some of the physiological effects regulated by activation of Epacs. (Li et al., 2006; Lopez De Jesus et al., 2006; Zheng and Quilliam, 2003).
Therefore, determining the downstream signaling pathways that mediate sensitization of nociceptive sensory neurons after activation of Epacs is of high importance in understanding signaling in chronic pain. Consequently, we investigated whether activation of the canonical Epac target Rap1, or the less conventional target, Ras were necessary for the sensitizing actions of Epacs on capsaicin-sensitive sensory neurons. We demonstrate that exposing sensory neuronal cultures to the Epac agonist, 8-CPT-2′-O-Me-cAMP-AM (8CPT-AM) enhances the activity of the small G proteins, Rap1 and Ras. Epac activation with 8CPT-AM also augments the number of action potentials generated by a ramp of depolarizing current and the evoked release of immunoreactive calcitonin gene-related peptide (iCGRP) from sensory neurons. Surprisingly, these actions are not attenuated by inhibition of Rap1, but rather by inhibition of Ras. Our findings demonstrate that Ras is a critical downstream effector of Epac-induced sensitization in capsaicin-sensitive sensory neurons, suggesting a unique signaling pathway for maintaining sensitization of these neurons during chronic inflammation.
METHODS
Materials
Tissue culture supplies were obtained from Invitrogen (Carlsbad, CA) and normocin from Invivogen (San Diego, CA). Capsaicin, potassium chloride, 1-methyl-2-pyrrolidinone (MPL), poly-D-lysine, laminin, and routine chemicals were purchased from Sigma-Aldrich (St. Louis, MO). MPL was the vehicle used to prepare water-insoluble drugs. The maximal concentration of MPL used was 0.01%. Nerve growth factor was purchased from Harlan Bioproducts for Science (Indianapolis, IN). The Gβγ inhibitor gallein (catalog # 3090) was purchased from Tocris Biosciences (Bristol, UK). The Epac agonist, 8-pCPT-2′-O-Me-cAMP-AM (8CPT-AM, catalog # NC9940185), was purchased from ThermoFisher Scientific (Fremont, CA), whereas the phosphate trisacetoxymethyl ester (PO4-AM3, catalog # BLG-P030-003) and N6-Benzoyladenosine-3′, 5′-cAMP-AM (6BNZ-AM catalog # BLG-P079-01) were purchased from Axxora (San Diego, CA). The Rap1 shRNA lentiviral particles (LV) (catalog # sc-270358), control shRNA LV (catalog # sc-108080), rabbit anti-GAPDH antibody for Western blotting (catalog # sc-25778), and HRP-conjugated goat anti-rabbit secondary antibody for Western blotting (catalog # sc-2030) were purchased from Santa Cruz Biotechnology (Dallas, TX). The antibodies used for internal perfusion into sensory neurons were also purchased from Santa Cruz Biotechnology: rabbit anti-Rap1 antibody (catalog # sc-65), normal rabbit IgG (catalog # sc-2027), and normal rat IgG (catalog # sc-2026). HRP-conjugated goat anti-mouse antibody (catalog # 170-6516) was purchased from Bio-Rad (Hercules, CA). The following were purchase from EMD Millipore (Billerican, MA): the Y13-259 rat anti-Ras antibody for internal perfusion into sensory neurons (catalog # OP01A), the rabbit anti-Rap1 antibody for Western blotting (catalog # 07-916), the Ras antibody for Western blotting (catalog # 5-516), the Rap1 activation kit (catalog # 17-321) and the Ras activation kit (catalog # 17-218). The CGRP antibody was a generous gift from Dr. M. Iadarola at the NIH. The Animal Care and Use Committee at Indiana University School of Medicine, Indianapolis, IN approved all procedures used in these studies.
Isolation and cell culture of adult rat sensory neurons
Dorsal root ganglia (DRGs) from male Sprague-Dawley rats (100–200 g, Harlan, Indianapolis, IN) were dissected and sensory neurons were isolated as previously described (Burkey et al., 2004; Zhang et al., 2012). Briefly, the animals were sacrificed via CO2 asphyxiation and the DRGs from the entire spinal column (for release experiments) or from the lumbar region (for electrophysiology experiments) were collected into an ice-cold solution of Puck’s saline and subsequently trimmed to remove nerve fibers. For release experiments, the Puck’s saline was replaced with F-12 medium supplemented with 10% heat-inactivated horse serum, 2 mM glutamine, 100 μg/ml normocin-O™, 50 μg/ml penicillin, 50 μg/ml streptomycin, 50 μM 5-fluoro-2′-deoxyuridine, and 150 μM uridine (F-12 growth medium) containing ~3 mg/ml collagenase and the tissues were incubated for 90 min at 37°C in a 3% CO2 incubator. For patch clamp electrophysiology experiments, the Puck’s solution was replaced with F-12 growth medium containing 10 U/ml of papain and the DRGs were incubated for 10 min at 37°C, followed by a 50 min incubation at 37 °C with F-12 medium containing 1 mg/ml collagenase and 2.5 mg/ml dispase. After a brief centrifugation (1000 × g for 1 min), the supernatants from either preparation were aspirated; the DRGs were resuspended in 2 ml of F-12 growth medium containing 30 ng/ml of nerve growth factor (NGF) and dissociated using mechanical agitation. For release experiments, cells were plated at an approximate density of 30,000 cells per well of a 12-well plate precoated with 0.1 mg/mL of poly-D-lysine and 5 μg/ml of laminin. For patch clamp experiments, cells were plated at an approximate density of 7,500 cells per well of a 48-well plate containing plastic coverslips precoated with 0.1 mg/ml of poly-D-lysine and 10 μg/ml of laminin. The isolated cells were maintained in culture at 37 °C and 3% CO2. The F-12 growth medium supplemented with NGF was changed 24 hours after plating, and every other day thereafter. Cells were used 3–8 days after plating for patch clamp experiments in order to minimize space clamp issues, and 10–12 days after plating for release experiments in order to optimize the expression of CGRP and thus the ability to measure basal release of the peptide. In all instances, controls for release and electrophysiology experiments were from wells of cells harvested at the same time as those treated with various experimental manipulations.
Release of immunoreactive calcitonin gene-related peptide (iCGRP) from sensory neurons
Release experiments were performed on sensory neurons as previously described (Vasko et al., 1994). Briefly, the neuronal cultures were washed once with 0.4 ml of HEPES buffer consisting of (in mM): 25 HEPES, 135 NaCl, 3.5 KCl, 2.5 CaCl2, 1 Mg2Cl2, 3.3 dextrose, and 0.1% (w/v) bovine serum albumin, pH 7.4. Thereafter, the cells were incubated in 10 min sequential periods in 0.4 ml of the HEPES buffer at 37 °C. In order to determine basal neuropeptide release, the cells were exposed to HEPES buffer alone for 10 min during the first incubation. The second 10 min incubation occurred in HEPES buffer in either the absence or presence of drug to assess the effect of treatment on basal release. The third 10 min incubation occurred in HEPES buffer containing either 30 nM capsaicin or 30 mM KCl (substituted for equimolar NaCl) in the absence or presence of drug. The fourth 10 min incubation was with HEPES buffer alone in order to demonstrate a return to resting levels of release. After each of the incubations, the buffer was removed and aliquoted for iCGRP radioimmunoassay (RIA). At the conclusion of the release protocol, each well of cells was incubated in 0.4 ml of 0.1 N HCl for 10 min, scraped and an aliquot assayed for iCGRP to determine the remaining amount of peptide in the cells.
For the RIA, 300 μl of buffer from the aliquoted samples was incubated with 25 μl of a CGRP antibody (1:70,000 dilution) and 25 μl of 125I-[Tyr0] CGRP. After 16 hrs, 0.5 ml of 1% charcoal in 0.1 M phosphate buffer, pH 7.4 was added to each tube. The tubes were centrifuged at 1500 × g for 20 min and the supernatant containing radiolabeled peptide bound to antibody was decanted into separate tubes. Radioactivity was measured using gamma scintillation spectrometry. The counts generated from a CGRP standard curve were used to calculate the amount of iCGRP in each sample. Total CGRP content was determined by adding the amount released to the total remaining after the release. Release data are presented as percent of total content.
Electrophysiology
Current clamp recordings were obtained using the whole-cell patch-clamp technique (Hamill et al., 1981; Zhang et al., 2012). Briefly, plastic coverslips containing sensory neurons were placed in a recording chamber (Model RC-25, Warner Instruments, Hamden, CT, USA) which contained a normal Ringers solution composed of (in mM): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES and 10 glucose, pH at 7.4 with NaOH. Recording pipettes were pulled from glass capillary tubes using a Sutter Instruments pipette puller (Model G85165T-4). The pipette resistance was 2–5 MΩ when filled with an intracellular solution composed of the following (in mM): 140 KCl, 5 MgCl2, 4 ATP, 0.3 GTP, 2.5 CaCl2 2, 5 EGTA (free Ca concentration calculated at ~100 nM), and 10 HEPES, at pH 7.3 with KOH. Upon establishing the whole-cell configuration in normal Ringers solution, voltages were recorded with an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA, USA). Recordings were filtered at 5 kHz and sampled at 2 kHz. The data were acquired and analyzed using either pCLAMP 8.0 or pCLAMP 10.0.
The whole-cell configuration was established in small-diameter sensory neurons (20–25 μm in diameter). The cells were held at their resting membrane potentials (RMP), which ranged between −45 and −65 mV. The cells were then injected with a 1000 msec ramp of depolarizing current sufficient to generate 2–4 action potentials (APs) under control conditions. Depending on the experiment, control recordings were either obtained once at 0 minutes, or twice at 0 minutes and 2, 10, 20, or 30 minutes after obtaining the whole-cell configuration. The same ramp of depolarizing current was used on an individual cell throughout the experiment. Cells were next externally superfused with a normal Ringers solution containing phosphate-trisacetoxymethyl ester (PO4-AM3), the Epac agonist, 8CPT-2′-O-Me-cAMP (8CPT-AM), or the PKA agonist, N6-Benzoyladenosine-3′, 5′-cAMP-AM (6BNZ-AM). We used PO4-AM3 as our control since at the same molar concentration as 8CPT-AM it exposes the neurons to 3 times the amount of acetoxymethyl ester when hydrolyzed by esterases in the cell. Action potentials were then recorded at 2, 4, 6, and 10 minutes after exposure to the drug. Upon completion of the recording session, cells were externally superfused with a normal Ringers solution containing 300 nM capsaicin for a period of 1 minute. Capsaicin sensitivity was established as either a membrane depolarization of at least 10 mV and/or the generation of capsaicin-evoked APs. The results described in this report represent those from capsaicin-sensitive neurons only.
Lentiviral infection
The development and characterization of the dominant negative (17N) Ras (CMV-Ras17N-IRES-EGFP) and eGFP (CMV-IRES-EGFP) lentiviral particles (LV) has been described previously (Park et al., 2010). In experiments measuring neurotransmitter release, sensory neurons were treated with 10–50 μl of a solution containing 109–1010 LV on the second day after plating. In patch clamp electrophysiology experiments, sensory neurons were similarly infected with LV on the first day after plating. F-12 growth medium containing virus was removed after 48 hrs; cells were then grown in normal F-12 growth medium. Neurotransmitter release experiments were conducted 10–12 days after infection and electrophysiology experiments occurred 7–8 days after infection. For studies using reduced expression of Rap1, cultures of sensory neurons were treated with 30,000 infectious units (IFUs) of Rap1 shRNA LV on the second day after plating. Another group of cells was infected in parallel with a universal scramble shRNA LV as a negative control. Virus was removed after 48 hrs and release experiments were performed 10–12 days after infection.
Rap1 and Ras activity assays
After various treatments, cells were scraped in the presence of 0.5 ml of ice-cold phosphate buffered saline (PBS) per well of a 12-well culture dish in a 4 °C cold room. The cell suspensions were centrifuged at 16,000 × g for 1 minute at 4 °C. The supernatants were discarded and 50 μl of ice-cold 1X EMD Millipore RIPA Lysis Buffer (catalog # 20-188) containing 100-fold dilutions of protease inhibitor solution (EMD Millipore, catalog # 539134) and phosphatase inhibitor solution (Sigma-Aldrich, catalog # P0044) was added to the pellets. After sonication, the lysates were centrifuged at 1,000 × g for 30 sec at ~2 °C and the supernatants were transferred to new 1.5 ml tubes. Total protein content was quantified from an aliquot using a modified Bradford Assay. Two-hundred μg of protein from each sample was added to 250 μl of ice-cold 1X RIPA lysis buffer followed by the addition of 30 μg of Ral GDS-RBD agarose beads for the Rap1 activity assay or 30 μg of Raf1 RBD agarose beads for the Ras activity assay. Affinity precipitation of the GTP-bound form of the proteins occurred by allowing the agarose beads to incubate with protein at 4 °C for 45 minutes. For positive and negative controls, either 7.5 μl of 100 mM GTP-γS or 7.5 μl of 100 mM GDP-βS (final dilution of 30 mM), respectively, was added to the 250 μl reaction volume and incubated at 30 °C prior to addition of the agarose beads. The binding of GTP/GDP to small G proteins was stopped by the addition of 15 μl of 1 M MgCl2 (final dilution of 60 mM). Upon completion of the affinity precipitation, the agarose beads were pelleted for 30 s at 14,000 × g and 4 °C. The supernatants were discarded and the beads were washed three times with 100 μl of ice cold 1X RIPA lysis buffer. Thereafter 4 μl of 10X NuPAGE Sample Reducing Agent, 10 μl of 4X NuPAGE LDS Sample Buffer, and 26 μl of double distilled water were added to the beads. The beads were boiled for 5 min, centrifuged for 1 min at 14,000 × g, and the supernatants used in Western blotting.
Western blotting
Collection for cell lysates and quantification of total protein content using the modified Bradford Assay was performed as described above. One-hundred μg of protein was added to 2 μL of 10X NuPAGE Sample Reducing Agent, 5 μL of 4X NuPAGE LDS Sample Buffer, and a sufficient quantity of double distilled water to bring the total volume up to 20 μL. The solution was boiled for 5 min, spun for 30 s in a quick spin microcentrifuge tube, and subsequently used for Western blotting. Lysates from cells or supernatants from Rap1 and Ras activity assays were loaded onto NuPAGE 4–12% Bis-Tris Gels (Life Technologies, Grand Island, NY, catalog # NP0335) and proteins separated using the XCell SureLock™ Mini-Cell Electrophoresis System (Life Technologies catalog # EI0001) at 150 V for 1 hr. Proteins in the gel were transferred onto a 0.2 μm nitrocellulose membrane (Life Technologies catalog # LC2000) at 30 V for 16 hr in a 4 °C cold room using the Bio-Rad Mini Trans-Blot® Cell system (BioRad, Hercules, CA, catalog # 170-3930). Membranes were blocked overnight in a solution of 5% Tris buffered saline (TBS)-MILK and subsequently probed for 16 hr with respective primary antibodies in 5% TBS-MILK containing 1% Tween 20 to minimize nonspecific binding. Immunoblotting was performed using the following antibodies: rabbit anti-Rap1 (1:1000 dilution), mouse anti-Ras (1:1000), and rabbit anti-GAPDH (1:10,000 dilution). Secondary antibodies were also diluted in 5% TBST-MILK and included HRP-conjugated goat anti-rabbit (1:10,000 dilution) and goat anti-mouse (1:5000 dilution). The Western Lightning® Chemiluminescent HRP kit (ThermoFisher Scientific, Fremont, CA, catalog # 50-904-9325) was used to detect protein bands on radiographic film. Quantification of bands was carried out using NIH Image J Freeware Program (Arevalo et al., 2006).
Data analysis
Data are presented as mean ± standard error of the mean (S.E.M). To calculate the parameters of excitability, pClamp was used to differentiate the voltage trace from current clamp recordings with respect to time at a sampling frequency of 0.5 kHz to obtain dV/dt values. A baseline dV/dt value was calculated as the average between the 135 and 185 msec time points of the ramp stimulus. The firing threshold (FT) of the first AP was the membrane potential at which the value of the dV/dt exceeded the baseline average by >20-fold. The rheobase (Rheo) was the amount of ramp current delivered at the FT. The resistance at threshold (RTh) was calculated as the difference between the FT and the RMP divided by the Rheo. The Rheo and RTh values were normalized to their respective before-treatment-controls.
All statistical analyses were performed using GraphPad Prism 6.0. Depending on the experiment, either one- or two-way ANOVA was used to analyze the release data and repeated measures (RM) two-way ANOVA was used to analyze patch clamp electrophysiology data. Significant ANOVA findings were followed a Holm-Sidak all pairwise post hoc analysis. Concentration response curves were fit by nonlinear regression analysis using a variable slope four parameter protocol of GraphPad Prism, wherein a significant difference was found between EC50 values. The significance level was set at p < 0.05.
RESULTS
Activation of Epacs sensitizes sensory neurons
Previous studies have shown that activation of Epacs enhances nociceptive behaviors (Hucho et al., 2005) and mediates the prostaglandin-induced sensitization of sensory neurons after inflammation or exposure to NGF (Vasko et al., 2014). To determine whether direct activation of Epacs sensitizes peptidergic sensory neurons, we assessed whether treatment with the selective Epac agonist, 8CPT-AM (Holz et al., 2008) augmented the evoked release of iCGRP from sensory neurons. Treating sensory neurons with 8CPT-AM for 10 min prior to and during a 30 nM capsaicin or 30 mM high extracellular potassium stimulus significantly increased the release of iCGRP in a concentration-dependent manner. Although concentrations of 8CPT-AM up to 10 μM did not alter basal release of iCGRP, 30 μM 8CPT-AM did increase basal release (data not shown). Consequently, the y-axis in Fig. 1A represents evoked release of peptide, i.e. the release during the 10 min exposure to 30 nM capsaicin or 30 mM KCl minus the release in the 10 min incubation prior to stimulation. The 8CPT-AM compound enhanced capsaicin- and potassium-evoked release of iCGRP with an EC50 of 1.8 μM and 2.0 μM, respectively (Fig. 1C). Therefore in all subsequent studies, we used a concentration of 3 μM of the Epac agonist. Exposing sensory neurons to 30 nM capsaicin significantly increased the release of iCGRP from a basal level of 2.1 ± 0.1 to 12.0 ± 0.5% of total content/well/10 min (Fig. 1B). Similarly, exposing sensory neurons to 30 mM potassium chloride significantly increased the release of iCGRP from basal levels of 0.7 ± 0.1 to 12.2 ± 0.5% of total content/well/10 min (Fig. 1C). Treatment with 3 μM of 8CPT-AM did not alter the basal release of iCGRP; however treatment with the compound for 10 min prior to and throughout either the capsaicin or the high extracellular potassium stimulus significantly increased the capsaicin- and potassium-stimulated release of iCGRP to 28.3 ± 0.8% and 28.7 ± 0.8% of total content/well/10 min (Fig 1B and 1C, respectively). The PO4-AM3 compound served as a negative control for the conjugated acetoxymethyl ester. Exposure to 3 μM PO4-AM3 in 0.01% of MPL vehicle for 10 min prior to and throughout the stimulus period had no effect on basal, or stimulated release of iCGRP (Fig 1B and 1C).
Figure 1. Acute exposure to the Epac agonist 8CPT-AM augments evoked release of iCGRP from sensory neurons.
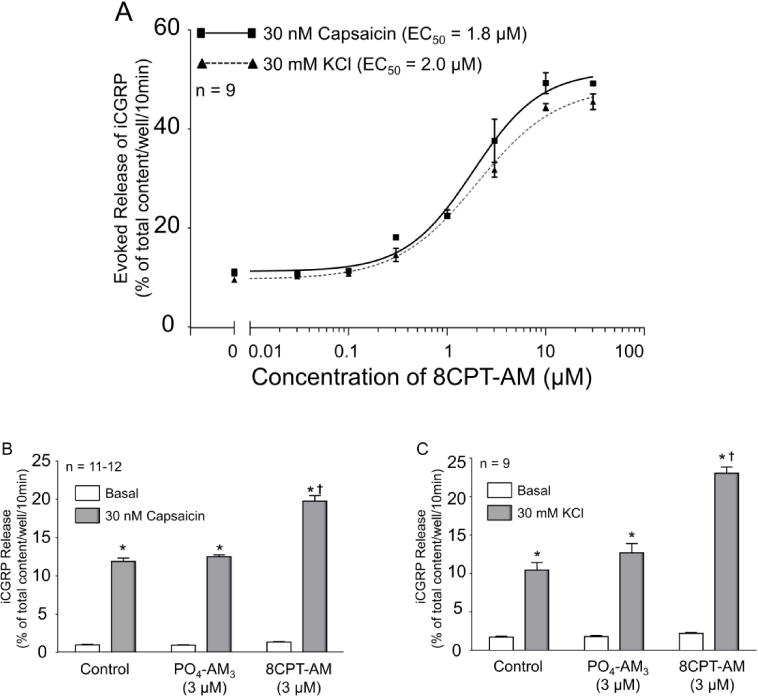
A: Each point represents the mean ± SEM of evoked release of iCGRP as the percent of the total iCGRP content/well/10min. The dotted line with triangles shows the concentration-response curve for release evoked by 30 nM capsaicin, whereas the dashed line with circles shows the concentration-response curve for release evoked by 30 mM KCl. B and C: Each column represents the mean ± SEM of iCGRP released from sensory neuronal cultures as the percent of the total peptide content/well/10min. The light gray columns represent basal release for cells exposed to HEPES buffer alone (vehicle), buffer containing 3 μM PO4AM3, or buffer containing 3 μM 8CPT-AM, as indicated. The dark gray columns represent iCGRP release after exposure to 30 nM capsaicin (A) or 30 mM potassium chloride (B) in the absence or presence of drug, as indicated. Asterisks indicate a significant difference from basal release; whereas a cross indicates a significant difference in stimulated release of iCGRP compared to neurons treated with PO4-AM3 (One-way ANOVA, Holm-Sidak post-hoc: capsaicin: F(2,32) = 76.27, P < 0.0001; potassium: F(2,24) = 43.41, P < 0.0001).
To determine whether activation of Epacs directly augments excitability of capsaicin-sensitive sensory neurons, we assessed whether treatment with 8CPT-AM could alter the number of APs generated by a ramp of depolarizing current. As shown by representative recordings in Fig. 2A, when an isolated sensory neuron was treated with a bath application of 3 μM 8CPT-AM, there was an increase in the number of APs evoked by the ramp of current, whereas exposure of another neuron to 3 μM PO4-AM3 did not elicit an increase in APs. Summary data for 7 capsaicin-sensitive neurons are shown in Fig 2B. Exposure to 3 μM 8CPT-AM resulted in a time-dependent increase in the number of APs (Fig. 2B). The number of APs increased from a control value of 2.3 ± 0.4 to 8.9 ± 0.7 APs at 10 min after drug. In contrast, exposure to 3 μM PO4-AM3 did not elicit an increase in the number of APs over the 10 minute recording period (Fig. 2B). Consistent with these observations, exposure to the Epac agonist significantly depolarized the resting membrane potential, hyperpolarized the AP firing threshold, decreased the rheobase current, and increased the membrane resistance at threshold (Table 1). Together, the results measuring AP firing and transmitter release show that activation of Epacs with 8CPT-AM does not directly excite sensory neurons, but augments excitability induced by various depolarizing stimuli.
Figure 2. The Epac agonist, 8CPT-AM augments action potentials generated by a ramp of depolarizing current.
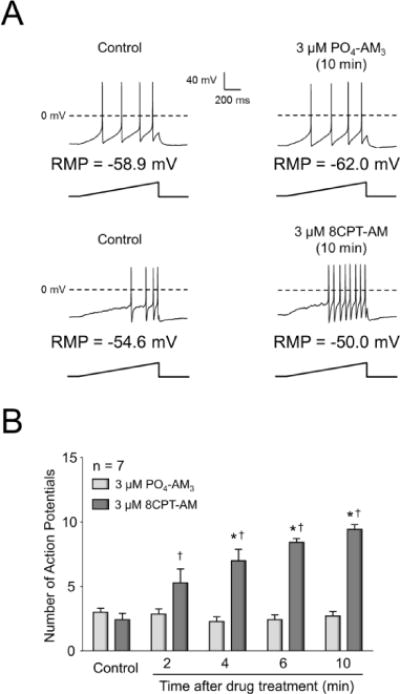
A: Representative recordings of action potentials (APs) fired in response to a ramp of depolarizing current as indicated before and after 10 minutes of treatment with either 3 μM PO4AM3 or 3 μM 8CPT-AM. The resting membrane potentials (RMP) are indicated for each neuron. B: Each column represents the mean ± SEM of the number of APs in 7 capsaicin-sensitive neurons at different times after exposure to 3 μM PO4AM3 or 3 μM 8CPT-AM. Ramp amplitudes were the same for each individual cell at each time point prior to and after treatment. Asterisks indicate a statistically significant difference from each group’s respective control (RM two-way ANOVA, Holm-Sidak post-hoc, F(4,48) = 15.06, P < 0.0001), whereas a cross indicates a statistically significant difference between treatment groups (RM two-way ANOVA, Holm-Sidak post-hoc, F(1,12) = 28.54, P = 0.0002).
Table 1.
Effect of the Epac agonist, 8CPT-AM, on parameters of excitability in capsaicin-sensitive sensory neurons
| Minutes after 3 μM PO4-AM3
|
|||||
|---|---|---|---|---|---|
| 2 | 4 | 6 | 10 | ||
|
Minutes after establishing whole-cell configuration | |||||
| 0 Control |
2 | 4 | 6 | 10 | |
| RMP (mV) | −60.6 ± 2.1 | −61.1 ± 1.7 | −61.6 ± 1.8 | −62.0 ± 2.6 | −61.1 ± 2.4 |
| FT (mV) | −25.4 ± 2.6 | −27.9 ± 2.6 | −25.9 ± 2.5 | −27.6 ± 3.0 | −27.1 ± 2.9 |
| Rheo (pA) | 182 ± 143 | 191 ± 152 | 204 ± 162 | 197 ± 160 | 198 ± 161 |
| Norm Rheo | 1.00 ± 0.00 | 1.01 ± 0.08 | 1.08 ± 0.06 | 1.04 ± 0.08 | 1.04 ± 0.07 |
| RTh (MΩ) | 335 ± 113 | 385 ± 181 | 359 ± 154 | 352 ± 147 | 363 ± 158 |
| Norm RTh | 1.00 ± 0.00 | 1.00 ± 0.12 | 0.96 ± 0.08 | 0.96 ± 0.08 | 0.95 ± 0.09 |
| Minutes after 3 μM 8CPT-AM
|
|||||
|---|---|---|---|---|---|
| 2 | 4 | 6 | 10 | ||
|
Minutes after establishing whole-cell configuration | |||||
| 0 Control |
2 | 4 | 6 | 10 | |
| RMP (mV) | −54.6 ± 2.5 | −54.0 ± 2.6 | −48.9 ± 1.9*† | −48.8 ± 1.1*† | −48.3 ± 1.5*† |
| FT (mV) | −19.9 ± 2.9 | −22.1 ± 3.3 | −23.6 ± 2.4 | −24.4 ± 2.2* | −24.8 ± 1.6* |
| Rheo (pA) | 493 ± 304 | 351 ± 206 | 355 ± 235 | 247 ± 150* | 173 ± 86* |
| Norm Rheo | 1.00 ± 0.00 | 0.75 ± 0.09*† | 0.59 ± 0.08*† | 0.52 ± 0.06*† | 0.44 ± 0.07*† |
| RTh (MΩ) | 199 ± 29 | 248 ± 34 | 287 ± 61 | 308 ± 61 | 363 ± 67* |
| Norm RTh | 1.00 ± 0.00 | 1.28 ± 0.10* | 1.38 ± 0.20*† | 1.52 ± 0.14*† | 1.81 ± 0.20*† |
Values are means ± SEM for the 7 small-diameter, capsaicin-sensitive sensory neurons from Fig. 1B. RMP, resting membrane potential; FT, firing threshold; Rheo, rheobase; RTh, membrane resistance at threshold; Norm Rheo, Normalized Rheo = Rheo at given time point divided by Rheo at Min 0; Norm RTh = RTh at given time point divided by RTh at Min 0. An asterisk indicates a significant difference in the excitability parameter relative to each group’s control recording at Min 0, whereas a cross indicates a significant difference in the excitability parameter between treatment groups using RM two-way ANOVA and the Holm-Sidak post hoc test.
Internal perfusion of GDP-βS into small-diameter sensory neurons blocks the sensitizing effects of 8CPT-AM, but not that of the PKA agonist, 6BNZ-AM
A predominant effect of Epacs is to enhance the exchange of GDP for GTP on small GTPases (de Rooij et al., 1998), but it is uncertain as to whether this mechanism mediates sensitization secondary to activation of Epacs. To examine whether Epac-induced sensitization involves GTPases, we internally perfused sensory neurons for 10 minutes with a pipette solution substituting 3 mM GDP-βS for 0.3 mM GTP. It has been previously shown that internal perfusion with GDP-βS blocks the sensitization produced by PGE2 (Li et al., 2012), an inflammatory mediator known to signal via G-protein coupled receptors (Southall and Vasko, 2001). As illustrated by representative recordings in Fig. 3A, in a sensory neuron internally perfused with 0.3 mM GTP, exposure to 3 μM 8CPT-AM significantly increased the number of evoked APs (upper panel), whereas in a neuron internally perfused with 3 mM GDP-βS, 8CPT-AM did not increase the number of APs generated by the ramp of current. The summary data for 7 capsaicin-sensitive cells from each group are shown in Fig 3C. In sensory neurons internally perfused with normal pipette solution (0.3 mM GTP), exposure to 3 μM 8CPT-AM resulted in a significant increase in ramp-evoked AP firing from a control value of 3.6 ± 0.3 APs at 0 min to 12.6 ± 1.5 APs after a 10 min exposure to the Epac agonist (Fig. 3C). In contrast, in sensory neurons internally perfused for 10 min with 3 mM GDP-βS, exposure to 3 μM 8CPT-AM did not increase the number of APs (Fig. 3C). Internal perfusion with either GTP or with GDP-βS for 10 min prior to treatment with 8CPT-AM did not alter the number of APs fired in response to the ramp of depolarizing current given immediately after establishing the whole cell configuration (Fig. 3C; left panel).
Figure 3. Internal perfusion of GDP-βS blocks the 8CPT-AM-induced increase in APs, but not the increase in APs induced by the PKA agonist, 6BNZ-AM.
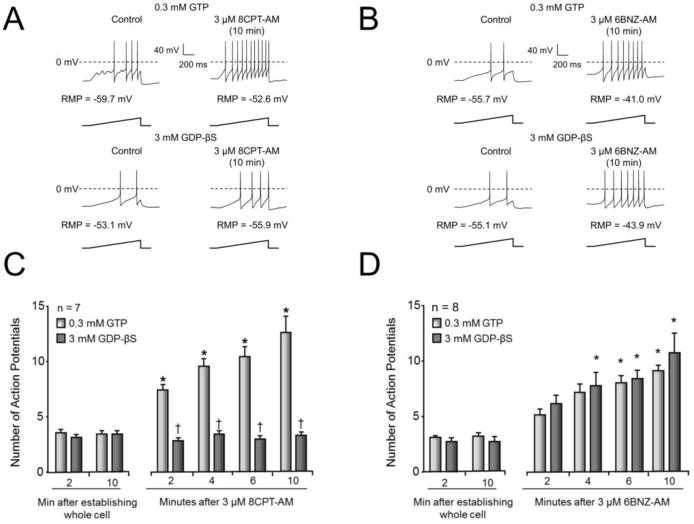
A and B: Representative traces of APs generated by ramps of current as indicated in sensory neurons treated for 10 min with 3 μM 8CPT-AM (A) or with 3 μM 6BNZ-AM (B) and internally perfused with either 0.3 mM GTP (upper traces) or 3 mM GDP-βS (lower traces). The resting membrane potentials (RMP) are indicated for each neuron. C and D: Summary data representing the mean ± SEM of the number of APs in small-diameter capsaicin-sensitive neurons prior to and at the noted times after exposure to 3 μM 8CPT-AM (C) or 3 μM 6BNZ-AM (D). The abscissa represents the time after establishing the whole cell configuration and the onset of internal perfusion. Control recordings were obtained at 0 and 10 min after establishing the whole-cell configuration, then, cells were exposed to bath application of 8CPT-AM (C) or 6BNZ-AM (D). Ramp amplitudes were the same for each individual cell at each time point prior to and after treatment. Asterisks indicate a statistically significant difference from each group’s respective control at 0 and 10 min (Panel C: RM two-way ANOVA, Holm-Sidak post-hoc, F(5,60) = 26.54, P < 0.0001, n = 7 cells; Panel D: RM two-way ANOVA, Holm-Sidak post-hoc, F(5,70) = 46.24, P < 0.0001, n = 8 cells). Crosses indicate a statistically significant difference between treatment groups at the given time point (Panel C: RM two-way ANOVA, Holm-Sidak post-hoc, F(1,12) = 76.71, P < 0.0001; Panel D: RM two-way ANOVA, Holm-Sidak post-hoc, F(1,14) = 0.3229, P = 0.5789).
To determine that GDP-βS does not alter the overall ability of sensory neurons to be sensitized, we examined whether the PKA-selective cAMP analogue, 6BNZ-AM, could enhance AP firing in cells internally perfused with GDP-βS. In this and subsequent experiments, we used activation of PKA as an alternative way to sensitize sensory neurons since previous studies showed that the acute sensitizing actions of PGE2 on excitability are mediated by PKA (Lopshire and Nicol, 1998; Smith et al., 2000). As illustrated by representative recordings (Fig. 3B) and the data from 8 capsaicin-sensitive neurons (Fig. 3D) when cells were internally perfused with 0.3 mM GTP, a 10 min exposure to 3 μM 6BNZ-AM significantly increased the number of evoked APs. In a similar manner when neurons were internally perfused with 3 mM GDP-βS, treatment with 3 μM 6BNZ-AM also significantly increased AP firing (Fig 3D). For example, after a 10 min exposure to 6BNZ-AM, the number of APs generated was 10.6 ± 1.7 in cells internally perfused with GDP-βS and 9.0 ± 0.5 in cells internally perfused with GTP (Fig. 3D).
We have previously demonstrated that activation of receptors linked to heterotrimeric G proteins increases the sensitivity of sensory neurons (Lopshire and Nicol, 1998; Southall et al., 2002; Southall and Vasko, 2001). Thus, to address the possibility that the Epac agonist could augment excitability through heterotrimeric G-proteins, we examined whether inhibition of adenylyl cyclases (linked to the Gαs subunit of heterotrimeric G proteins) or inhibition of the Gβγ heterodimer subunit would alter Epac agonist-induced sensitization of sensory neurons. When sensory neurons were treated overnight with 30 μM gallein, a treatment known to block Gβγ-mediated effects in sensory neurons (Belkouch et al., 2011), 8CPT-AM increased the number of APs in a manner analogous to effects seen in untreated neurons (Table 2). In a similar manner, when cells were treated with 100 μM THFA, a treatment shown to inhibit adenylyl cyclase-mediated effects in sensory neurons (Nicolson et al., 2007), there was no significant inhibition of the Epac-induced augmentation of APs (Table 2). These results indicate that the sensitizing actions of the Epac agonist on excitability are not mediated through the activation of Gαs or Gβγ.
Table 2.
The effects of the adenylyl cyclase inhibitor, THFA or the Gβγ inhibitor, gallein on the ability of 8CPT-AM to augment the firing of action potentials in capsaicin-sensitive sensory neurons
| Minutes after 3 μM 8CPT-AM
|
||||||
|---|---|---|---|---|---|---|
| 2 | 4 | 6 | 10 | |||
|
Minutes after establishing whole-cell configuration | ||||||
| 0 Control |
2 Control |
4 | 6 | 8 | 12 | |
| Untreated | 2. 5 ± 0.2 | 3.0 ± 0.4 | 6.2 ± 0.7* | 8.0 ± 1.2* | 9.5 ± 1.1* | 11.5 ± 1.0* |
| 100 μM THFA | 2.8 ± 0.3 | 3.3 ± 0.3 | 6.7 ± 0.8* | 7.5 ± 0.6* | 9.8 ± 0.9* | 11.0 ± 1.2* |
| 30 μM Gallein | 3.0 ± 0.4 | 3.0 ± 0.4 | 5.5 ± 0.5* | 7.7 ± 0.6* | 8.2 ± 0.7* | 11.3 ± 1.0* |
Values are means ± SEM for the 7 small-diameter, capsaicin-sensitive sensory neurons. An asterisk indicates a significant difference in the number of APs after exposure to 3 μM 8-CPT-AM compared to controls at 2 min after establishing the whole cell configuration using RM two-way ANOVA and the Holm-Sidak post hoc test.
The Epac agonist, 8CPT-AM, augments the activity of Ras and Rap1 in cultures of sensory neurons
Because Epacs regulate activation of the Ras family of small GTPases, (de Rooij et al., 1998; Lopez De Jesus et al., 2006), we next asked whether exposing sensory neuronal cultures to 8CPT-AM augments Ras and/or Rap1 activity as measured by an increase in Ras-GTP and/or Rap1-GTP. In lysates from neurons treated for 10 min with 3 μM 8CPT-AM, there was a significant increase in the ratio of Ras-GTP to total Ras from control values of 0.07 ± 0.01 to 0.28 ± 0.03 (Fig. 4A); there was also an increase in the ratio of Rap1-GTP to total Rap from control values of 0.13 ± 0.08 to 0.42 ± 0.03 (Fig. 4B). The activation of Ras and Rap1 by 3 μM 8CPT-AM was not maximal, because in control lysates treated with 3 mM GTP-γS, the Ras-GTP to total Ras ratio was 0.69 ± 0.03 (Fig. 4A) and Rap1-GTP to total Rap1 ratio was 0.63 ± 0.05 (Fig. 4B). A 10 min treatment of sensory neuron cultures with 3 μM PO4-AM3 did not alter the Ras-GTP to total Ras ratio (Fig. 4A) or the Rap1-GTP to total Rap1 ratio (Fig. 4B) compared to the lysates from untreated controls or those lysates treated for 10 min with 3 mM GDP-βS.
Figure 4. The Epac agonist, 8CPT-AM, increases Ras and Rap1 activity in cultures of rat sensory neurons.
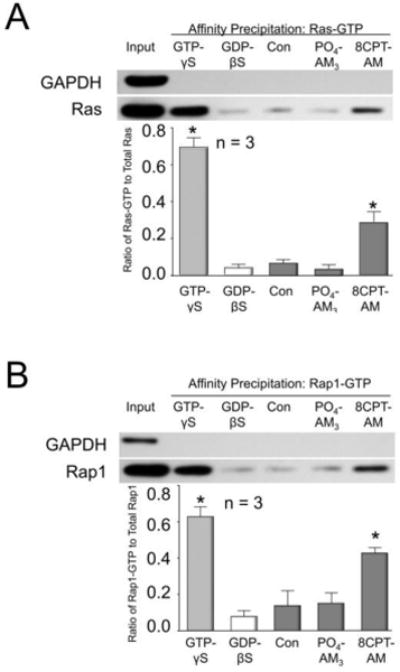
The upper panels show representative Western blots from an affinity precipitation experiment assessing the level of Ras-GTP (A) or Rap1-GTP (B) in lysates from sensory neuronal cultures. The input lanes represent loading of 200 μg of lysate from untreated sensory neuronal cultures wherein the Ras bands (A) and Rap1 bands (B) represent total Ras and Rap1 content, respectively. The remaining lanes represent 200 μg of lysate that underwent affinity precipitation. The sole presence of the GAPDH bands in the input lanes serve as evidence that the affinity precipitation does not pull down other proteins. The GTP-γS, and GDP-βS lanes represent the addition of 3 mM GTP-γS or 3mM GDP-βS, respectively, to 200 μg of lysate from untreated controls for 30 min prior to pull down. The next three lanes represent Ras-GTP (A) or Rap1-GTP (B) from 200 μg of lysate obtained from either untreated cultures (Con), cultures treated for 10 min with 3 μM PO4-AM3, or cultures treated for 10 min with 3 μM 8CPT-AM, as indicated. In the lower panels, each column represents the mean ± SEM of the densitometry of the ratio of Ras-GTP to total Ras (A) or of the ratio of Rap1-GTP to total Rap1 (B) from three independent experiments for the treatments indicated. Asterisks indicate a statistically significant increase in Ras-GTP or Rap1-GTP compared to untreated controls. (A: One-way ANOVA, Holm-Sidak post-hoc, F(4,10) = 164.3, P < 0.0001; B: One-way ANOVA, Holm-Sidak post-hoc, F(4,10) = 18.73, P = 0.0001).
Overexpression of dominant negative Ras (DN-Ras) attenuates the sensitization produced by Epac activation
Since activation of Epacs increases Ras activity in sensory neuronal cultures, we next determined whether inhibiting Ras activation by overexpression of DN-Ras in sensory neurons would attenuate the sensitization caused by the Epac agonist, 8CPT-AM. We chose to examine the effects of DN-Ras since this mutant version of the Ras protein prevents activation of wild type Ras by forming complexes with Ras exchange factors (Feig and Cooper, 1988; Quilliam et al., 1994). To overexpress DN-Ras, cells were infected with lentiviral particles containing the sequences to the CMV promoter, DN-Ras, an internal ribosome entry site, and enhanced green fluorescent protein (DN-Ras LV). As a vector control, cells were infected with lentiviral particles containing the sequences to the CMV promoter and enhanced green fluorescent protein (GFP LV). In green fluorescent neurons infected with DN-Ras-LV, exposure to 3 μM 8CPT-AM failed to augment the number of evoked APs (2.6 ± 0.3 APs for the control vs. 2.3 ± 0.4 APs after a 10 min treatment with 8CPT-AM, n=7, Fig. 5A, left panel). In contrast, there was a significant time-dependent increase in AP firing in response to 3 μM 8CPT-AM in neurons infected with the vector control (GFP LV), (Fig. 5A, right panel). Although neurons expressing DN-Ras were unresponsive to 8CPT-AM, a subsequent 10 min exposure to the PKA agonist, 6BNZ-AM, significantly increased the number of APs (Fig. 5A, left panel). Taken together, these results demonstrate that cells expressing DN-Ras were not sensitized by the Epac agonist but were sensitized by the PKA agonist.
Figure 5. Over expression of DN-Ras attenuates 8CPT-AM-induced sensitization of sensory neurons.
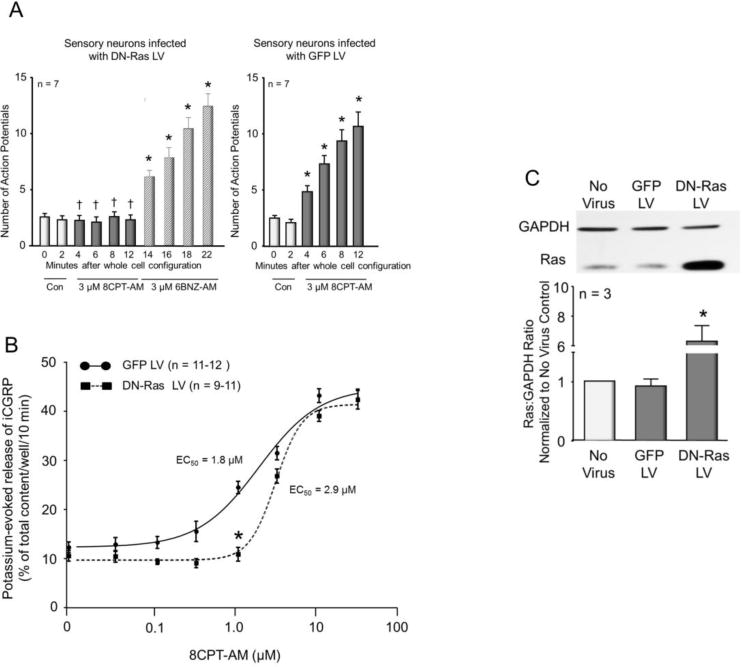
A: Each column represents the mean ± SEM of the number of APs generated by a ramp of depolarizing current in 7 capsaicin-sensitive neurons infected with DN-Ras LV (left panel) or GFP LV (right panel). Light columns show number of APs at 0 and 2 min after establishing the whole-cell configuration. Dark columns show number of APs after bath application of 3 μM 8CPT-AM, whereas hatched columns show the number of APs recorded after bath application of 3 μM 6BNZ-AM. Ramp amplitudes were the same for each individual cell at each time point prior to and after treatment. Asterisks indicate a statistically significant difference from each group’s respective control (For 8CPT-AM: RM two-way ANOVA, Holm-Sidak post-hoc, F(5,60) = 36.56, P < 0.0001; for 6BNZ-AM: RM one-way ANOVA, Holm-Sidak post-hoc, F(6,54) = 70.99, P < 0.0001), whereas a cross indicates a statistically significant difference between the DN-Ras LV and GFP LV treatment groups at the given time point (RM two-way ANOVA, Sidak’s post-hoc, F(1,12) = 28.14, P = 0.0002). B: Each point represents the mean ± SEM of potassium-evoked release of iCGRP as the percent of the total iCGRP content/well/10min. The solid line shows the concentration-response curve for neurons infected with GFP LV, whereas the dashed line shows the concentration-response curve for cells infected with DN-Ras LV. An asterisk indicates a statistically significant difference in iCGRP release between treatment groups (Two-way ANOVA, Holm-Sidak post-hoc, F(1,158) = 43.46, P < 0.0001). C. The top panel shows a representative Western blot of Ras and GAPDH expression in control sensory neuronal cultures (no virus), cultures infected with GFP LV, or cultures infected with DN-Ras LV. In the lower panel, each column represents the mean ± SEM of the densitometry of the ratio of Ras to GAPDH normalized to no virus controls from 3 independent experiments for the treatments indicated. An asterisk indicates a statistically significant difference from GFP-LV controls using one-way ANOVA and Holm-Sidak post-hoc test (F(2,6) = 25.79, P = 0.0011).
We next asked whether expression of DN-Ras in cultures of sensory neurons could alter the Epac agonist-induced increase in potassium-stimulated release of iCGRP. We used high extracellular potassium to stimulate release because Ras can regulate the expression of TRPV1 (Bron et al., 2003) and we previously showed that expression of DN-Ras in sensory neuronal cultures attenuates capsaicin-stimulated release of iCGRP (Park et al., 2010). In neuronal cultures treated with GFP-LV, exposure to 8CPT-AM enhanced the potassium-evoked release of iCGRP in a concentration-dependent manner with an EC50 value of 1.8 μM and a Hill slope of 1.1 (see Fig. 5B). In cultures infected with DN-Ras LV, there was a significant rightward shift in the 8CPT-AM concentration-response curve relative to cultures infected with GFP LV. In cultures infected with DN-Ras, the EC50 of 8CPT-AM on potassium-stimulated release of iCGRP was 2.9 μM with a Hill slope of 2.7 (Fig. 5B). In neurons treated with DN-Ras LV, 1 μM 8CPT-AM did not increase potassium-evoked release (10.9 ± 1.4% of total content/well/10 min) relative to controls not exposed to the Epac agonist (10.6 ± 1.1 % of total content/well/10 min). Conversely, in neuronal cultures treated with GFP-LV, 1 μM 8CPT-AM significantly increased potassium-evoked release to 24.5 ± 1.3% of total iCGRP content/well/10 min, relative to controls in the absence of the agonist (12.3 ± 1.1 % of total iCGRP content/well/10 min). At higher concentrations of 8CPT-AM, there was no difference in the potassium-evoked release between cultures infected with DN-Ras LV versus those infected with GFP LV. Using green fluorescence as an indicator of infection, we found that approximately 70% of our cells expressed EGFP. Thus, it seems possible that higher concentrations of the Epac agonist enhanced release from uninfected cells which masked the inhibitory effect of DN-Ras.
We confirmed that overexpression of DN-Ras occurred in DN-Ras-LV infected cultures in two ways. First, cultures infected with DN-Ras LV had a significantly lower total content of iCGRP (496.8 ± 7.8 fmol/well) compared to cultures infected with GFP LV (1873.7 ± 51.2 fmol/well). This confirms our previous findings that DN-Ras suppresses production of iCGRP in sensory neurons (Park et al., 2010). Second, we observed an approximately 6-fold increase in the expression of Ras in cultures infected with DN Ras LV (Fig. 5C).
Internal perfusion of a Ras-neutralizing antibody blocks the 8CPT-AM-induced increase in the number of action potentials generated by a ramp of current
To further substantiate that Epac-induced sensitization of sensory neurons is mediated by Ras, we examined the effects of acute suppression of Ras activity by internally perfusing a Ras-neutralizing antibody into sensory neurons. For these studies we utilized the rat monoclonal Ras antibody, Y13-259, because it blocks activation of Ras (Furth et al., 1982; Sigal et al., 1986) and because we previously showed that a 30 μg/ml internal perfusion of the antibody attenuates the NGF-induced increase in excitability of mouse sensory neurons (Duan et al., 2011). Using a treatment paradigm similar to our previous study, we first observed that internal perfusion of 30 μg/ml rat IgG or Ras antibody for 20 min after establishing the whole-cell configuration does not alter the number of APs generated by a ramp of depolarizing current (see Figs 6A and 6B, controls), demonstrating that these antibodies do not alter baseline excitability. In sensory neurons internally perfused with the Ras-neutralizing antibody, bath application of 3 μM 8CPT-AM did not augment the number of ramp-evoked APs, whereas in sensory neurons internally perfused with IgG, bath application of 8CPT-AM significantly increased the number of APs in a time-dependent manner (Fig. 6A). In cells internally perfused with IgG and subsequently treated with 8CPT-AM, the Epac agonist significantly depolarized the resting membrane potential, hyperpolarized the firing threshold, reduced the rheobase current, and increased the membrane resistance at threshold, whereas internal perfusion with the Ras antibody blocked the effects of 8CPT-AM on these parameters of excitability (Table 3).
Figure 6. Internal perfusion with a Ras-neutralizing antibody blocks the Epac agonist-induced, but not the PKA agonist-induced, increase in APs.
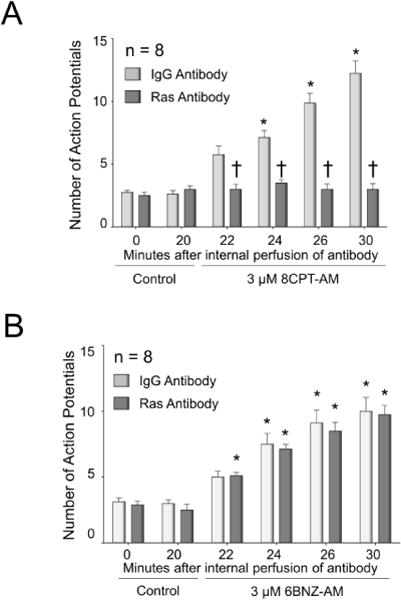
Each column represents the mean ± SEM of the number of APs in small-diameter capsaicin-sensitive neurons internally perfused with either 30 μg/ml IgG (light gray columns) or 30 μg/ml Ras antibody (dark gray columns) for 20 min prior to treatment with 3 μM 8CPT-AM (A) or 3 μM 6BNZ-AM (B). The antibody perfusion continued for the entirety of the experiment. Asterisks indicate a statistically significant difference from each groups respective controls at 0 and 20 min (Panel A: RM two-way ANOVA, Holm-Sidak post-hoc, F(5,70) = 48.39, P < 0.0001, n = 8 cells; Panel B: RM two-way ANOVA, Holm-Sidak post-hoc, F(5,70) = 78.02, P < 0.0001, n = 8 cells), whereas a cross indicates a statistically significant difference between treatment groups at the given time point (Panel A: RM two-way ANOVA, Holm-Sidak post-hoc, F(1,14) = 52.71, P < 0.0001; Panel B: RM two-way ANOVA, Holm-Sidak post-hoc, F(1,14) = 0.2674, P = 0.6132).
Table 3.
Effect of internal perfusion of Ras antibody on 8CPT-AM-induced alterations in the parameters of excitability in capsaicin-sensitive sensory neurons.
| Minutes after 3 μM 8CPT-AM
|
||||||
|---|---|---|---|---|---|---|
| 2 | 4 | 6 | 10 | |||
|
Minutes after internal perfusion of antibody | ||||||
| 0 Control |
20 Control |
22 | 24 | 26 | 30 | |
| IgG antibody | ||||||
| RMP (mV) | −61.9 ± 1.3 | −62.1 ± 1.3 | −59.6 ± 1.6 | −56.5 ± 1.7*† | −53.8 ± 1.3*† | −51.0 ± 1.2*† |
| FT (mV) | −19.7 ± 1.6 | −20.5 ± 1.5 | −25.1 ± 2.0* | −27.2 ± 1.9* | −28.5 ± 2.1* | −28.6 ± 2.1* |
| Rheo (pA) | 195 ± 29 | 180 ± 27 | 138 ± 22* | 102 ± 25* | 82 ± 20* | 70 ± 20* |
| Norm Rheo | 1.00 ± 0.00 | 0.92 ± 0.06 | 0.71 ± 0.07*† | 0.50 ± 0.09*† | 0.41 ± 0.07*† | 0.35 ± 0.07*† |
| RTh (MΩ) | 242 ± 26 | 274 ± 45 | 292 ± 44 | 394 ± 73* | 390 ± 55* | 394 ± 73* |
| Norm RTh | 1.00 ± 0.00 | 1.12 ± 0.09 | 1.23 ± 0.14 | 1.63 ± 0.24*† | 1.62 ± 0.17*† | 1.73 ± 0.20*† |
|
| ||||||
| Ras antibody | ||||||
| RMP (mV) | −62.6 ± 1.5 | −63.3 ± 1.3 | −64.0 ± 1.4 | −64.7 ± 1.7 | −65.4 ± 1.6 | −65.0 ± 1.3 |
| FT (mV) | −22.0 ± 2.3 | −21.1 ± 2.5 | −22.1 ± 2.6 | −23.7 ± 2.7 | −23.7 ± 2.7 | −22.4 ± 2.6 |
| Rheo (pA) | 215 ± 71 | 244 ± 75 | 227 ± 74 | 213 ± 73 | 212 ± 72 | 224 ± 74 |
| Norm Rheo | 1.00 ± 0.00 | 1.19 ± 0.14 | 1.01 ± 0.07 | 0.95 ± 0.06 | 0.94 ± 0.10 | 1.02 ± 0.09 |
| RTh (MΩ) | 264 ± 57 | 231 ± 46 | 271 ± 57 | 289 ± 68 | 316 ± 78 | 293 ± 77 |
| Norm RTh | 1.00 ± 0.00 | 0.93 ± 0.07 | 1.04 ± 0.06 | 1.07 ± 0.05 | 1.16 ± 0.13 | 1.06 ± 0.06 |
Values are means ± SEM for the 8 small-diameter, capsaicin-sensitive sensory neurons from Fig 5A. An asterisk indicates a significant difference in the excitability parameter relative to each group’s control recording at Min 0, whereas a cross indicates a significant difference in the excitability parameter between IgG and Ras antibody treatment groups using RM two-way ANOVA with the Holm-Sidak post hoc test.
Internal perfusion with either 30 μg/ml normal rat IgG or rat anti-Ras antibody had no effect on the sensitization produced by 3 μM 6BNZ-AM (Fig. 6B). In cells perfused with IgG, a 10 min exposure to 3 μM 6BNZ-AM significantly increased the number of APs from 3.1 ± 0.3 to 10.0 ± 1.1 (n=8), and in cells perfused with Ras antibody, a 10 min exposure to 3 μM 6BNZ-AM increased the number of APs from 2.9 ± 0.3 to 9.8 ± 0.7 (n=8). These results substantiate that inhibition of Ras attenuates Epac-induced sensitization without altering the ability of the sensory neurons to be sensitized by activation of PKA.
Internal perfusion of a Rap1 antibody does not alter the 8CPT-AM-induced increase in the number of action potentials generated by a ramp of current
Because Epacs are exchange factors for Rap GTPases (Gloerich and Bos, 2010) and because activation of Epacs increases Rap1 activity in sensory neurons (see Fig. 4B), we examined whether inhibiting Rap1 activity in sensory neurons by internal perfusion of a Rap1-neutralizing antibody could alter the ability of the Epac agonist, 8CPT-AM, to augment AP firing. To establish that the Rap1 antibody could block Rap1 activation, lysates acquired from cultures of sensory neurons were treated with 3 mM GTP-γS in the absence or presence of 100 μg/ml normal rabbit IgG or increasing amounts of rabbit anti-Rap1 antibody for 20 min at room temperature. Affinity precipitation and Western blot densitometry were then used to quantify the ratio of Rap1-GTP to total Rap1. As can be seen in a representative Western blot in Fig. 7A (top panel) and in summarized results from 4 independent experiments (lower panel), exposing lysates to 3 mM GTP-γS in the absence or presence of IgG significantly increased the Rap1-GTP to total Rap1 ratio compared to lysates exposed to 3 mM GDP-βS. A 20 min pretreatment with 10 μg/ml of Rap1 antibody did not reduce the ratio of Rap1-GTP to total Rap1, whereas 30 μg/ml of the antibody significantly decreased the ratio by approximately 50%, and 100 μg/ml of the Rap1 antibody completely blocked the GTP-γS-induced increase in Rap1-GTP (Fig. 7A). Since 100 μg/ml of the Rap1 antibody blocked Rap1 activation, this concentration was used for internal perfusion into sensory neurons.
Figure 7. Internal perfusion of a Rap1 antibody blocks Rap1 activation, but does not block the Epac agonist-induced increase in APs.
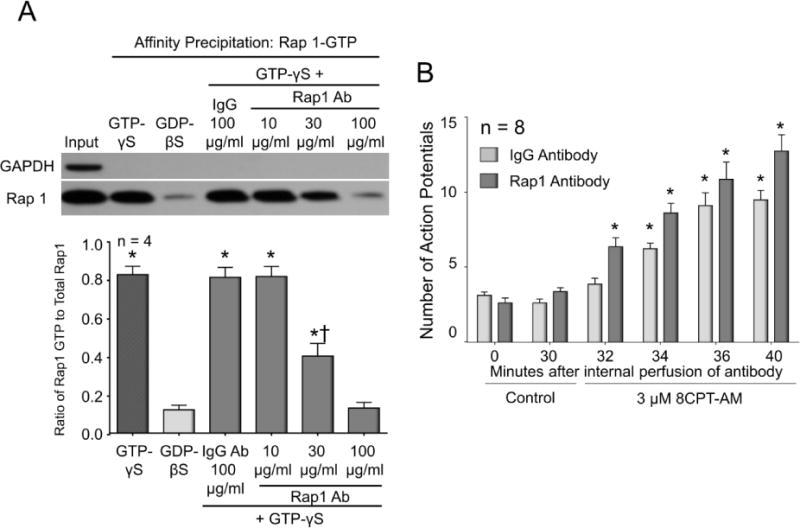
A: The upper panel shows a representative Western blot from affinity precipitation experiments assessing the level of Rap1-GTP in lysates taken from sensory neuronal cultures and subsequently exposed to IgG or Rap1 antibody and GTP-γS. The input lane signifies loading of 200 μg of lysate wherein the Rap1 signal represents total Rap1 content. The remaining lanes represent 200 μg of lysate that underwent affinity precipitation. The sole presence of the GAPDH band in the input lane serves as evidence that the affinity precipitation does not pull down other proteins. The GTP-γS and GDP-βS lanes represent the addition of 3 mM GTP-γS or GDP-βS, respectively, to the 200 μg of lysate for 30 min prior to the Rap1-GTP pulldown. The remaining lanes represent treatment of lysates with either 100 μg/ml IgG or various amounts of Rap1 antibody for 20 minutes prior to and throughout exposure to 3 mM GTP-γS. In the lower panel, each column represents the mean ± SEM of the densitometry ratio of Rap1-GTP to total Rap1 from 4 independent experiments for the treatments indicated. An asterisk indicates a statistically significant difference in Rap1-GTP compared to the GDP-βS group, whereas a cross indicates a significant difference in Rap1-GTP between the 10 and 30 μg/ml Rap1 antibody groups (One-way ANOVA, Holm-Sidak post-hoc, F(5,18) = 54.51, P < 0.0001). B: Each column represents the mean ± SEM of number of APs generated by a ramp of current in 8 capsaicin-sensitive neurons internally perfused with either 100 μg/ml normal rabbit IgG (light columns) or 100 μg/ml rabbit anti-Rap1 antibody (dark columns) prior to (controls; 0 and 30 min after establishing the whole cell configuration) and at the various times after exposure to 3 μM 8CPT-AM. An asterisk indicates a statistically significant difference from each group’s respective control recordings at 30 min (RM two-way ANOVA, Holm-Sidak post-hoc, F(5,70) = 86.14, P < 0.0001).
Internal perfusion of 100 μg/ml normal rabbit IgG or rabbit anti-Rap1 antibody for 30 min prior to treatment with 8CPT-AM did not alter the number of APs when compared to those recorded immediately after attaining the whole-cell configuration: 3.1 ± 0.2 and 2.6 ± 0.3 APs at 0 and 30 min respectively for IgG and 2.6 ± 0.3 and 3.4 ± 0.3 APs at 0 and 30 min respectively for Rap1 antibody (Fig. 7B). We chose an internal perfusion time of 30 min based on the 20 minutes required for the Rap1 antibody to neutralize Rap1 activity in vitro and based on the 5–9 minutes necessary to achieve a “steady-state” intracellular concentration of a 150 kDa IgG antibody as described by Pusch and Neher (Pusch and Neher, 1988). Internal perfusion with IgG or with Rap1 antibody did not attenuate the increased AP firing produced after exposure to 3 μM 8CPT-AM (Fig. 7B). In cells internally perfused with Rap1 antibody, there was a time-dependent increase in the number of APs after exposure to the Epac agonist, and this increase was not significantly different from that observed in cells internally perfused with IgG. These data show that the Epac-induced sensitization of small-diameter capsaicin-sensitive sensory neurons is not mediated by activation of Rap1, despite the fact that the Epac agonist can increase Rap1 activity (see Fig. 4B).
Reduced expression of Rap1 does not alter the 8CPT-AM-induced increase in capsaicin-stimulated release of iCGRP
We next examined whether reducing the expression of Rap1 in sensory neuronal cultures could prevent the 8CPT-AM-induced augmentation of iCGRP release. For these experiments, cultures of sensory neurons were infected with either lentiviral particles containing a universal scramble shRNA construct (scramble shRNA LV), or lentiviral particles containing a Rap1 shRNA construct (Rap1 shRNA LV). Ten days after infection, basal and capsaicin-stimulated release of iCGRP was measured in the absence or presence of 3 μM 8CPT-AM (see Fig. 8A). As observed in our earlier experiments (Fig. 1A), treating naïve cultures (no virus) for 10 min with 3 μM 8CPT-AM increased the capsaicin-stimulated release of iCGRP from 15.6 ± 1.1 to 28.0 ± 1.0% of total iCGRP content/well/10 min without affecting basal release (Fig. 8A). Similarly, in neurons infected with scramble shRNA LV, a 10 min pretreatment with the Epac agonist significantly augmented capsaicin-stimulated release from 13.5 ± 0.4 to 25.0 ± 1.0% of total iCGRP content/well/10 min. Consistent with the electrophysiological recordings using the Rap1-neutralizing antibody, in neurons infected with Rap1 shRNA LV, a 10 min pretreatment with the Epac agonist also increased the capsaicin-stimulated release of iCGRP from 14.6 ± 0.9 to 25.5 ± 1.2% of total iCGRP content/well/10 min (Fig. 8A).
Figure 8. Reduced expression of Rap1 in sensory neurons prevents 8CPT-AM-induced activation of Rap1, but does not alter the ability of the agonist to augment capsaicin-evoked release of iCGRP.
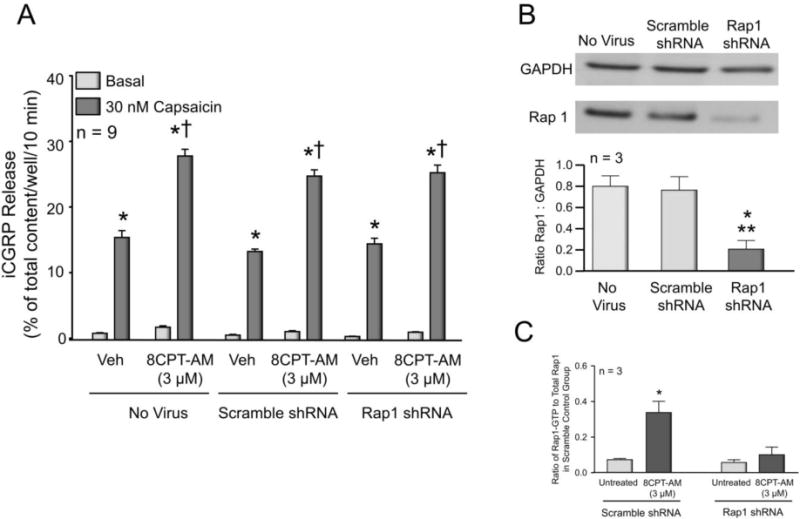
A: Each column represents the mean ± SEM of iCGRP released from sensory neuronal cultures as % of total content/well/10min. The light columns represent basal release when cells were exposed to vehicle or to buffer containing 3 μM 8CPT-AM, as indicated. The dark columns represent iCGRP release after exposure to 30 nM capsaicin in either the absence or presence of 8CPT-AM, as indicated. Cultures were untreated (No virus), treated with 30,000 infectious units (IFU) of lentivirus containing a universal scramble shRNA construct, or 30,000 IFU of virus containing shRNA for Rap1. An asterisk indicates a significant difference from basal release; whereas a cross indicates a significant difference in capsaicin-stimulated release of iCGRP relative to each group’s respective vehicle treated controls (Two-way ANOVA, Holm-Sidak post-hoc, F(1,48) = 229.7, P < 0.0001). B: The upper panel shows a representative Western blot for Rap1 and GAPDH (as a loading control) from lysates of control cultures (no virus), cultures treated with lentivirus containing a scramble shRNA construct, or cultures treated with virus containing shRNA for Rap1. In the lower panel, each column represents the mean ± SEM of the densitometry of the ratio of Rap1 to GAPDH. An asterisk and a double asterisk indicate a statistically significant difference in Rap1 expression compared to untreated cultures and those treated with lentivirus containing scramble shRNA, respectively. (One-way ANOVA, Holm-Sidak post-hoc, F(2,6) = 30.64, P = 0.0007). C: Each column represents the mean ± SEM of the densitometry of the ratio of Rap1-GTP to the total Rap1 in cultures infected with scramble shRNA lentivirus. For both scramble shRNA and Rap1 shRNA lentivirus infected groups, Rap1-GTP expression was assessed in the absence (untreated) or presence of treatment with 3 μM 8CPT-AM as indicated. Results are from 3 independent experiments. An asterisk indicates a statistically significant difference in the ratio of Rap1-GTP to total Rap1 relative to each groups untreated control (Two-way ANOVA, Holm-Sidak post-hoc, F(1,8) = 15.55, P = 0.0043).
In wells of cells harvested at the same time as those used in release experiments, we confirmed that the viral infection reduced both Rap1 expression and activity. In cultures infected with scramble shRNA LV, there was no significant difference in the expression levels of Rap1 compared to untreated controls (Fig. 8B). In contrast, infection with Rap1 shRNA LV significantly reduced the expression of Rap1 by approximately 80% compared to controls (Fig. 8B). Additionally, in order to verify that the reduction in Rap1 expression was sufficient to block the 8CPT-AM-induced increase in Rap1 activity, we determined that in neuronal cultures infected with Rap1 shRNA LV, exposure to 3 μM 8CPT-AM for 10 min did not increase the ratio of Rap1-GTP to total Rap1, whereas in neuronal cultures infected with scramble shRNA LV, exposure to 3 μM 8CPT-AM significantly increased the ratio of Rap1-GTP to total Rap1 (Fig. 8C).
DISCUSSION
Our findings clearly demonstrate that Epac activation sensitizes small-diameter capsaicin-sensitive sensory neurons and that this action is dependent on the small G protein, Ras. Exposing sensory neuronal cultures to the Epac agonist 8CPT-AM increases Ras activity and augments the number of APs generated in response to a ramp of depolarizing current, an effect that is completely blocked by internally perfusing the neurons with GDP-βS or with the Ras neutralizing antibody. Overexpressing DN-Ras also prevents Epac-induced increases in AP firing and attenuates the Epac-induced augmentation of stimulated iCGRP release. There are currently no assays available to measure Ras activity at the single cell level. Consequently, Ras activity cannot be measured directly in the patch clamp experiments. However, the activity assays do offer support for the electrophysiology data as activation of Epacs increases Ras activity and inhibition of Ras activity blocks the Epac agonist from enhancing excitability. These two pieces of data support the conclusion that Epac activation elicits hypersensitivity in a Ras-dependent manner. Our manipulations of Ras did not affect the ability of neurons to generate APs under control conditions or alter the ability of the PKA selective agonist, 6BNZ-AM, to augment ramp-evoked AP firing, i.e., to sensitize sensory neurons. Thus, inhibition of Ras specifically alters the ability of sensory neurons to respond to Epac activation, but does not alter the ability of neurons to be sensitized by activation of PKA, which is a canonical pathway mediating acute sensitization of sensory neurons (Aley and Levine, 1999; England et al., 1996; Hingtgen et al., 1995; Lopshire and Nicol, 1998; Sachs et al., 2009). This is important since activation of Epacs is implicated in long term sensitization of sensory neurons (Eijkelkamp et al., 2013; Hucho et al., 2005; Vasko et al., 2014; Wang et al., 2007). Consequently, inhibition of either Epac or Ras-activation is a useful strategy for blunting chronic hypersensitivity without altering the ability of sensory neurons to be acutely sensitized, i.e., treating chronic inflammatory pain without compromising acute pain perception.
The fact that Epac-induced sensitization of small diameter sensory neurons is Ras-dependent, but does not require Rap1, is a novel finding since a majority of studies show that Epacs are predominately GEFs for Rap GTPases (de Rooij et al., 1998; Kawasaki et al., 1998). For example, in isolated cerebellar granule cells, activation of Rap1 mediates the Epac-induced increase in conductance of calcium-activated potassium channels (BK) (Ster et al., 2007) and in rat cortical neurons, Epac-induced remodeling of synaptic spines is Rap dependent (Woolfrey et al., 2009). In our sensory neuronal cultures, we demonstrate that Epac activation by 8CPT-AM increases Rap1 activity. This increase in activity, however, is not causally linked to Epac-induced sensitization since inhibiting Rap1 activity by either perfusing a Rap1-neutralizing antibody into sensory neurons or by using shRNA to reduce expression of Rap1 did not prevent the increase in evoked APs and transmitter release caused by the Epac agonist. These manipulations of Rap1 blocked 8CPT-AM and GTP-γS induced increases in Rap1-GTP, confirming that they were effective in inhibiting Rap1. Thus, our findings suggest a specificity of Epac signaling that is likely unique and that could provide a specific target for altering peripheral sensitization without affecting Epac signaling in other neuronal populations.
For the current studies, we used the selective Epac agonist 8CPT-AM to directly activate the proteins. We chose this pharmacological approach since it allows a direct activation of Epacs rather than using receptor agonists such as PGE2 which could introduce confounding variables. One potential concern, however, is whether 8CPT-AM has off-target actions that could account for its ability to sensitize sensory neurons. For example, at approximately 10-fold higher concentrations than we used in the current work, 8CPT-AM can increase PKA activity. The EC50 of 8CPT-AM for activating PKA is 30 μM, whereas the EC50 for Epacs is just 0.9 μM (Rehmann et al., 2003). Because the EC50 for 8CPT-AM to augment evoked transmitter release was 2 μM (see Fig. 1C), it is unlikely that the effect is mediated by PKA. 8CPT-AM also has been reported to have antagonist actions on the P2Y12 receptor (Herfindal et al., 2013). Since inhibition of Gβγ or adenylyl cyclase did not block the sensitizing actions of 8CPT-AM, it seems unlikely that the effect is mediated by heterotrimeric G proteins. For release studies, we used overexpression of DN-Ras as our means of blocking Ras activity. One potential limitation of DN-Ras (17N) is that the molecule forms complexes with Ras GEFs (Quilliam et al., 1994) and thus could nonspecifically inactivate GEFs for other small G proteins. This seems unlikely, since internal perfusion of the Y13-259 Ras-neutralizing antibody into sensory neurons blocked AP firing in a manner analogous to DN-Ras. Indeed, the Y13-259 Ras-neutralizing antibody blocks Ras activity (Furth et al., 1982; Sigal et al., 1986) without inhibiting the activity of Rap1 (von Lintig et al., 2000). The ability of the DN-Ras to attenuate the Epac-induced increase in potassium-evoked release of iCGRP at lower concentrations of 8CPT-AM, but not at higher concentrations could be explained by the fact that not all the neurons in the cultures expressed DN-Ras. We observed a ~70% infection efficiency as measured by the expression of green fluorescence in sensory neuronal cultures. In uninfected cells, higher concentrations of 8CPT-AM could presumably cause sufficient activation of Epacs to overcome the inhibitory effects of DN-Ras. Consistent with this notion, when we recorded APs from non-fluorescent sensory neurons in cultures infected with DN-Ras LV, we observed that the Epac agonist, 8CPT-AM, enhanced AP firing in response to a ramp of depolarizing current (data not shown), thus demonstrating that suppression of the Epac agonist-induced enhancement of AP firing only occurs in cells that express DN-Ras. It is also possible that activation of Epacs could engage divergent signaling pathways that separately regulate transmitter release and AP firing. For example, it is well established that the small GTPase, Rab3a is localized on synaptic vesicles and is involved in exocytosis (Geppert et al., 1994; Sudhof, 2013). Furthermore, AP firing is largely dependent on voltage-gated sodium and potassium channels, whereas transmitter release is dependent on free calcium levels which could increase through voltage-gated calcium channels, TRP channels, or intracellular release. Thus, modulation of transmitter release involves an integrated response which includes, but is not limited to, alterations in the number of APs.
Although our studies show that Ras activation is necessary for sensitization of a subpopulation of sensory neurons, the question remains whether the Epacs produce their effects by acting as Ras GEFs. Some evidence suggests that Epacs can augment exchange of GTP for GDP in Ras (Lopez De Jesus et al., 2006), but other work suggests that the activation of Ras by Epac agonists is dependent on PLC-ε and a subsequent increase in intracellular calcium (Keiper et al., 2004; Metrich et al., 2010). Interestingly, studies in the H1299 lung carcinoma cell line and in insulin-secreting pancreatic β cells indicate that Ras is involved in trafficking Epac2 to the plasma membrane, thereby promoting an interaction with Rap1 (Idevall-Hagren et al., 2013; Li et al., 2006), although this is unlikely in sensory neurons since inhibiting Rap1 does not alter Epac-induced sensitization. Further studies are warranted, however, to ascertain whether Epacs serve as Ras GEFs in sensory neurons, or whether the ability of the Epac agonists to increase Ras activity is secondary to other, as yet, undetermined actions of Epacs.
It is well established that a number of intracellular signaling pathways mediate sensitization of sensory neurons (Gold and Gebhart, 2010; Richardson and Vasko, 2002). This redundancy could be advantageous since it provides diversity in initiating and maintaining hypersensitivity in response to injury, but can negatively impact the development of potential therapies for chronic pain conditions. For example, in large-diameter capsaicin-insensitive sensory neurons (Drew et al., 2002), activation of Epac1 augments mechanically-evoked currents (Eijkelkamp et al., 2013; Lolignier et al., 2015) whereas our previous work demonstrated that Epac2 is necessary for the increase in evoked transmitter release caused by PGE2 (Vasko et al., 2014) in cultures of capsaicin-sensitive sensory neurons maintained in NGF. It therefore seems plausible that these two Epac isoforms could selectively alter different physiological or pathological responses in different sensory neurons using different downstream signaling effectors. Indeed, Epac-mediated signaling is quite diverse and varies in different cell types (Gloerich and Bos, 2010; Grandoch et al., 2010; Holz et al., 2006). For example, Epac2 mediates the cAMP-dependent potentiation of neurotransmitter release in the hippocampus (Fernandes et al., 2015), whereas Epac1 mediates the cAMP-activated release of chloride from human T84 intestinal cells (Hoque et al., 2010), a mechanism that is important in maintaining hydration of the gastrointestinal tract. Thus, delineating the pathways linked to hypersensitivity in different subpopulations of neurons is critical for understanding mechanisms of different pain modalities.
Our previous work demonstrated that in sensory neurons maintained in the presence of NGF, PGE2 enhances evoked AP firing and neurotransmitter release in an Epac-dependent mechanism. In the current study, we demonstrate that direct activation of Epacs in sensory neurons activates both Ras and Rap1 small GTPase proteins; however, in capsaicin-sensitive sensory neurons, the ability of the Epac agonist to enhance AP firing and neurotransmitter release is blocked by inhibition Ras and not Rap1 (Fig 9). Our demonstration that sensitization of capsaicin-sensitive sensory neurons grown in NGF occurs through activation of the Epac-Ras signaling cascade provides a novel signaling pathway that could serve as an ideal therapeutic target in the treatment of chronic pathological pain without altering the ability to respond to normal physiological sensations.
Figure 9. Diagram for the signaling pathway mediating Epac-induced sensitization of capsaicin-sensitive sensory neurons.
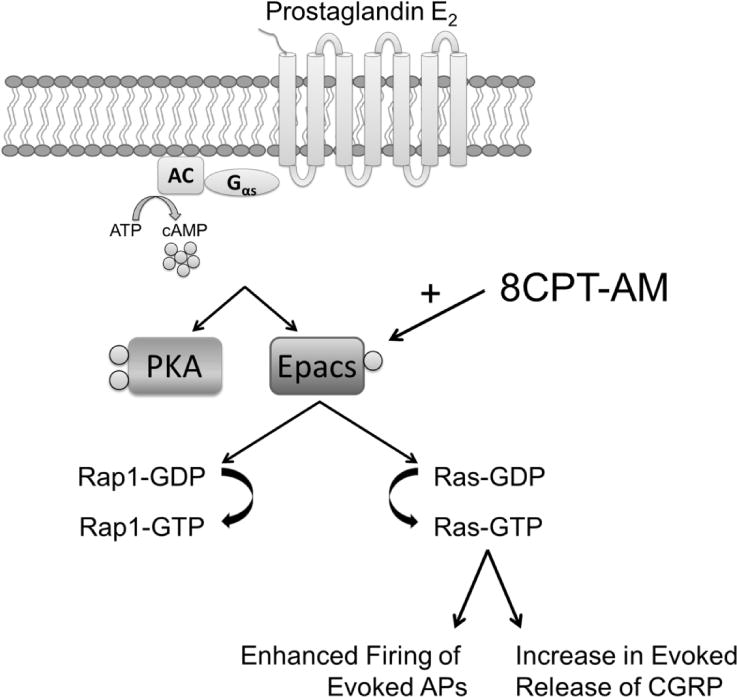
Our current work demonstrates that direct activation of Epacs in sensory neurons with the Epac-selective cAMP analogue, 8CPT-AM, subsequently activates Rap1 and Ras small GTPase proteins. However, in capsaicin-sensitive sensory neurons, the ability of the Epac agonist to enhance evoked AP firing and neuropeptide release is mediated by Ras and not Rap1. (Gαs – The Gα stimulatory subunit of heterotrimeric G proteins coupled to seven transmembrane receptors; AC – Adenylyl Cyclase; ATP – Adenosine Triphosphate).
Highlights.
-
-
Epac activation enhances evoked AP firing and CGRP release in sensory neurons.
-
-
Epac activation enhances activity of Ras and Rap1 in cultures of sensory neurons.
-
-
Inhibition of Ras, but not Rap1 blocks the sensitizing actions of Epac activation.
Acknowledgments
M.R.V. conceived the overall design of the project. B.S., G.D.N., and M.R.V. conceived and designed the experiments. B.S. performed the experiments and analyzed the data. B.S., G.D.N., and M.R.V. interpreted results of experiments. E.L.T. generously provided the dominant-negative Ras lentiviral particles and offered critical feedback. B.S. and M.R.V prepared figures. B.S. drafted the manuscript. G.D.N. and M.R.V. edited and revised the manuscript. We would like to thank Dr. Jill C. Fehrenbacher for valuable suggestions regarding experimental design with dominant negative Ras and Drs. Ruizong Wang and Yihong Zhang for technical assistance. This work was supported by NIH Grant NS069915 to MRV. These studies were conducted, in part, in a facility constructed with the support from the Research Facilities Improvement Program Grant Number C06 RR015481-01 from the National Center for Research Resources, National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
No conflicts of interest, financial or otherwise, are declared by the author(s).
References
- Aley KO, Levine JD. Role of protein kinase A in the maintenance of inflammatory pain. J Neurosci. 1999;19:2181–2186. doi: 10.1523/JNEUROSCI.19-06-02181.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arevalo JC, Waite J, Rajagopal R, Beyna M, Chen ZY, Lee FS, Chao MV. Cell survival through Trk neurotrophin receptors is differentially regulated by ubiquitination. Neuron. 2006;50:549–559. doi: 10.1016/j.neuron.2006.03.044. [DOI] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, Julius D. Cellular and molecular mechanisms of pain. Cell. 2009;139:267–284. doi: 10.1016/j.cell.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baviera AM, Zanon NM, Navegantes LC, Kettelhut IC. Involvement of cAMP/Epac/PI3K-dependent pathway in the antiproteolytic effect of epinephrine on rat skeletal muscle. Mol Cell Endocrinol. 2010;315:104–112. doi: 10.1016/j.mce.2009.09.028. [DOI] [PubMed] [Google Scholar]
- Belkouch M, Dansereau MA, Reaux-Le Goazigo A, Van Steenwinckel J, Beaudet N, Chraibi A, Melik-Parsadaniantz S, Sarret P. The chemokine CCL2 increases Nav1.8 sodium channel activity in primary sensory neurons through a Gbetagamma-dependent mechanism. J Neurosci. 2011;31:18381–18390. doi: 10.1523/JNEUROSCI.3386-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bos JL. Epac: a new cAMP target and new avenues in cAMP research. Nat Rev Mol Cell Biol. 2003;4:733–738. doi: 10.1038/nrm1197. [DOI] [PubMed] [Google Scholar]
- Bron R, Klesse LJ, Shah K, Parada LF, Winter J. Activation of Ras is necessary and sufficient for upregulation of vanilloid receptor type 1 in sensory neurons by neurotrophic factors. Molecular and Cellular Neuroscience. 2003;22:118–132. doi: 10.1016/s1044-7431(02)00022-2. [DOI] [PubMed] [Google Scholar]
- Burkey TH, Hingtgen CM, Vasko MR. Isolation and culture of sensory neurons from the dorsal-root ganglia of embryonic or adult rats. Methods Mol Med. 2004;99:189–202. doi: 10.1385/1-59259-770-X:189. [DOI] [PubMed] [Google Scholar]
- de Rooij J, Zwartikus FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- Drew LJ, Wood JN, Cesare P. Distinct mechanosensitive properties of capsaicin-sensitive and -insensitive sensory neurons. J Neurosci. 2002;22:1–5. doi: 10.1523/JNEUROSCI.22-12-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan JH, Wang Y, Duarte D, Vasko MR, Nicol GD, Hingtgen CM. Ras signaling pathways mediate NGF-induced enhancement of excitability of small-diameter capsaicin-sensitive sensory neurons from wildtype but not Nf1+/− mice. Neurosci Lett. 2011;496:70–74. doi: 10.1016/j.neulet.2011.03.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eijkelkamp N, Linley JE, Torres JM, Bee L, Dickenson AH, Gringhuis M, Minett MS, Hong GS, Lee E, Oh U, Ishikawa Y, Zwartkuis FJ, Cox JJ, Wood JN. A role for Piezo2 in EPAC1-dependent mechanical allodynia. Nat Commun. 2013;4(1682):1–13. doi: 10.1038/ncomms2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England S, Bevan S, Docherty RJ. PGE2 modulates the tetrodotoxin-resistant sodium current in neonatal rat dorsal root ganglion neurones via the cyclic AMP-protein kinase A cascade. J Physiol. 1996;495:429–440. doi: 10.1113/jphysiol.1996.sp021604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feig LA, Cooper GM. Inhibition of NIH 3T3 cell proliferation by a mutant ras protein with preferential affinity for GDP. Mol Cell Biol. 1988;8:3235–3243. doi: 10.1128/mcb.8.8.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes HB, Riordan S, Nomura T, Remmers CL, Kraniotis S, Marshall JJ, Kukreja L, Vassar R, Contractor A. Epac2 Mediates cAMP-Dependent Potentiation of Neurotransmission in the Hippocampus. J Neurosci. 2015;35:6544–6553. doi: 10.1523/JNEUROSCI.0314-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furth ME, Davis LJ, Fleurdelys B, Scolnick EM. Monoclonal antibodies to the p21 products of the transforming gene of Harvey murine sarcoma virus and of the cellular ras gene family. J Virol. 1982;43:294–304. doi: 10.1128/jvi.43.1.294-304.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geppert M, Bolshakov VY, Siegelbaum SA, Takei K, De Camilli P, Hammer RE, Sudhof TC. The role of Rab3A in neurotransmitter release. Nature. 1994;369:493–497. doi: 10.1038/369493a0. [DOI] [PubMed] [Google Scholar]
- Gloerich M, Bos JL. Epac: defining a new mechanism for cAMP action. Annu Rev Pharmacol Toxicol. 2010;50:355–375. doi: 10.1146/annurev.pharmtox.010909.105714. [DOI] [PubMed] [Google Scholar]
- Gold MS, Gebhart GF. Nociceptor sensitization in pain pathogenesis. Nat Med. 2010;16:1248–1257. doi: 10.1038/nm.2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandoch M, Roscioni SS, Schmidt M. The role of Epac proteins, novel cAMP mediators, in the regulation of immune, lung and neuronal function. Br J Pharmacol. 2010;159:265–284. doi: 10.1111/j.1476-5381.2009.00458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Herfindal L, Nygaard G, Kopperud R, Krakstad C, Doskeland SO, Selheim F. Off-target effect of the Epac agonist 8-pCPT-2′-O-Me-cAMP on P2Y12 receptors in blood platelets. Biochem Biophys Res Commun. 2013;437:603–608. doi: 10.1016/j.bbrc.2013.07.007. [DOI] [PubMed] [Google Scholar]
- Hingtgen CM, Waite KJ, Vasko MR. Prostaglandins facilitate peptide release from rat sensory neurons by activating the adenosine 3′,5′-cyclic monophosphate transduction cascade. J Neurosci. 1995;15:5411–5419. doi: 10.1523/JNEUROSCI.15-07-05411.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz GH, Chepurny OG, Schwede F. Epac-selective cAMP analogs: New tools with which to evaluate the signal transduction properties of cAMP-regulated guanine nucleotide exchange factors. Cellular Signalling. 2008;20:10–20. doi: 10.1016/j.cellsig.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holz GH, Kang G, Harbeck M, Roe MW, Chepurny OG. Cell physiology of cAMP sensor Epac. J Physiol. 2006;577:5–15. doi: 10.1113/jphysiol.2006.119644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoque KM, Woodward OM, van Rossum DB, Zachos NC, Chen L, Leung GP, Guggino WB, Guggino SE, Tse CM. Epac1 mediates protein kinase A-independent mechanism of forskolin-activated intestinal chloride secretion. The Journal of general physiology. 2010;135:43–58. doi: 10.1085/jgp.200910339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hucho T, Levine JD. Signaling pathways in sensitization: toward a nociceptor cell biology. Neuron. 2007;55:365–376. doi: 10.1016/j.neuron.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Hucho TB, Dina OA, Levine JD. Epac mediates a cAMP-to-PKC signaling in inflammatory pain: an isolectin B4(+) neuron-specific mechanism. J Neurosci. 2005;25:6119–6126. doi: 10.1523/JNEUROSCI.0285-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idevall-Hagren O, Jakobsson I, Xu Y, Tengholm A. Spatial control of Epac2 activity by cAMP and Ca2+-mediated activation of Ras in pancreatic β cells. Sci Signal. 2013;60:S1–6. doi: 10.1126/scisignal.2003932. [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Springett GM, Mochizuki N, Toki S, Nakaya M, Matsuda M, Housman DE, Graybiel AM. A family of cAMP-binding proteins that directly activate Rap1. Science. 1998;282:2275–2279. doi: 10.1126/science.282.5397.2275. [DOI] [PubMed] [Google Scholar]
- Keiper M, Stope MB, Szatkowski D, Bohm A, Tysack K, Vom Dorp F, Saur O, Oude Weernink PA, Evellin S, Jakobs KH, Schmidt M. Epac- and Ca2+ -controlled activation of Ras and extracellular signal-regulated kinases by Gs-coupled receptors. J Biol Chem. 2004;279:46497–46508. doi: 10.1074/jbc.M403604200. [DOI] [PubMed] [Google Scholar]
- Li C, Chi XX, Xie W, Strong JA, Zhang JM, Nicol GD. Sphingosine 1-phosphate receptor 2 antagonist JTE-013 increases the excitability of sensory neurons independently of the receptor. J Neurophysiol. 2012;108:1473–1483. doi: 10.1152/jn.00825.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Asuri S, Rebhun JF, Castro AF, Paranavitana NC, Quilliam LA. The RAP1 guanine nucleotide exchange factor Epac2 couples cyclic AMP and Ras signals at the plasma membrane. J Biol Chem. 2006;281:2506–2514. doi: 10.1074/jbc.M508165200. [DOI] [PubMed] [Google Scholar]
- Lolignier S, Eijkelkamp N, Wood JN. Mechanical allodynia. Pflugers Arch. 2015;467:133–139. doi: 10.1007/s00424-014-1532-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez De Jesus M, Stope MB, Oude Weernink PA, Mahlke Y, Borgermann C, Ananaba VN, Rimmbach C, Rosskopf D, Michel MC, Jakobs KH, Schmidt M. Cyclic AMP-dependent and Epac-mediated activation of R-Ras by G protein-coupled receptors leads to phospholipase D stimulation. J Biol Chem. 2006;281:21837–21847. doi: 10.1074/jbc.M604156200. [DOI] [PubMed] [Google Scholar]
- Lopshire JC, Nicol GD. The cAMP transduction cascade mediates the prostaglandin E2 enhancement of the capsaicin-elicited current in rat sensory neurons: whole-cell and single-channel studies. J Neurosci. 1998;15:6081–6092. doi: 10.1523/JNEUROSCI.18-16-06081.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metrich M, Laurent AC, Breckler M, Duquesnes N, Hmitou I, Courillau D, Blondeau JP, Crozatier B, Lezoualc’h F, Morel E. Epac activation induces histone deacetylase nuclear export via a Ras-dependent signalling pathway. Cell Signal. 2010;22:1459–1468. doi: 10.1016/j.cellsig.2010.05.014. [DOI] [PubMed] [Google Scholar]
- Nicolson TA, Foster AF, Bevan S, Richards CD. Prostaglandin E2 sensitizes primary sensory neurons to histamine. Neuroscience. 2007;150:22–30. doi: 10.1016/j.neuroscience.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Park KA, Fehrenbacher JC, Thompson EL, Duarte DB, Hingtgen CM, Vasko MR. Signaling pathways that mediate nerve growth factor-induced increase in expression and release of calcitonin gene-related peptide from sensory neurons. Neuroscience. 2010;171:910–923. doi: 10.1016/j.neuroscience.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch M, Neher E. Rates of diffusional exchange between small cells and a measuring patch pipette. Pflügers Arch. 1988;411:204–211. doi: 10.1007/BF00582316. [DOI] [PubMed] [Google Scholar]
- Quilliam LA, Kato K, Rabun KM, Hisaka MM, Huff SY, Campbell-Burk S, Der CJ. Identification of residues critical for Ras(17N) growth-inhibitory phenotype and for Ras interaction with guanine nucleotide exchange factors. Mol Cell Biol. 1994;14:1113–1121. doi: 10.1128/mcb.14.2.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehmann H, Schwede F, Doskeland SO, Wittinghofer A, Bos JL. Ligand-mediated activation of the cAMP-responsive guanine nucleotide exchange factor Epac. J Biol Chem. 2003;278:38548–38556. doi: 10.1074/jbc.M306292200. [DOI] [PubMed] [Google Scholar]
- Richardson JD, Vasko MR. Cellular mechanisms of neurogenic inflammation. J Pharmacol Exp Ther. 2002;302:839–845. doi: 10.1124/jpet.102.032797. [DOI] [PubMed] [Google Scholar]
- Sachs D, Villarreal C, Cunha F, Parada C, Ferreira S. The role of PKA and PKCepsilon pathways in prostaglandin E2-mediated hypernociception. Br J Pharmacol. 2009;156:826–834. doi: 10.1111/j.1476-5381.2008.00093.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M, Dekker FJ, Maarsingh H. Exchange protein directly activated by cAMP (epac): a multidomain cAMP mediator in the regulation of diverse biological functions. Pharmacol Rev. 2013;65:670–709. doi: 10.1124/pr.110.003707. [DOI] [PubMed] [Google Scholar]
- Sigal IS, Gibbs JB, A’Alonzo JS, Scolnick EM. Identification of effector residues and a neutralizing epitope of Ha-ras-encoded p21. Proc Natl Acad Sci U S A. 1986;83:4725–4729. doi: 10.1073/pnas.83.13.4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JA, Davis CL, Burgess GM. Prostaglandin E2-induced sensitization of bradykinin-evoked responses in rat dorsal root ganglion neurons is mediated by cAMP-dependent protein kinase A. Eur J Neurosci. 2000;12:3250–3258. doi: 10.1046/j.1460-9568.2000.00218.x. [DOI] [PubMed] [Google Scholar]
- Southall MD, Bolyard LA, Vasko MR. Twenty-four hour exposure to prostaglandin downregulates prostanoid receptor binding but does not alter PGE(2)-mediated sensitization of rat sensory neurons. Pain. 2002;96:285–296. doi: 10.1016/S0304-3959(01)00458-4. [DOI] [PubMed] [Google Scholar]
- Southall MD, Vasko MR. Prostaglandin receptor subtypes, EP3C and EP4, mediate the prostaglandin E2-induced cAMP production and sensitization of sensory neurons. J Biol Chem. 2001;276:16083–16091. doi: 10.1074/jbc.M011408200. [DOI] [PubMed] [Google Scholar]
- Ster J, De Bock F, Guerineau NC, Janossy A, Barrere-Lemaire S, Bos JL, Bockaert J, Fagni L. Exchange protein activated by cAMP (Epac) mediates cAMP activation of p38 MAPK and modulation of Ca2+-dependent K+ channels in cerebellar neurons. Proc Natl Acad Sci U S A. 2007;104:2519–2524. doi: 10.1073/pnas.0611031104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC. Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron. 2013;80:675–690. doi: 10.1016/j.neuron.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasko MR, Campbell WB, Waite KJ. Prostaglandin E2 enhances bradykinn-stimulated release of neuropeptides from rat sensory neurons in culture. J Neurosci. 1994;14:4987–4997. doi: 10.1523/JNEUROSCI.14-08-04987.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasko MR, Habashy Malty R, Guo C, Duarte DB, Zhang Y, Nicol GD. Nerve growth factor mediates a switch in intracellular signaling for PGE2-induced sensitization of sensory neurons from protein kinase A to Epac. PLoS One. 2014;9:e104529. doi: 10.1371/journal.pone.0104529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarreal CF, Funez MI, Figueiredo F, Cunha FQ, Parada CA, Ferreira SH. Acute and persistent nociceptive paw sensitisation in mice: the involvement of distinct signalling pathways. Life Sci. 2009;85:822–829. doi: 10.1016/j.lfs.2009.10.018. [DOI] [PubMed] [Google Scholar]
- von Lintig FC, Pilz RB, Boss GR. Quantitative determination of Rap 1 activation in cyclic nucleotide-treated HL-60 leukemic cells: lack of Rap 1 activation in variant cells. Oncogene. 2000;19:4029–4034. doi: 10.1038/sj.onc.1203741. [DOI] [PubMed] [Google Scholar]
- Wang C, Gu Y, Li GW, Huang LY. A critical role of the cAMP sensor Epac in switching protein kinase signalling in prostaglandin E2-induced potentiation of P2X3 receptor currents in inflamed rats. J Physiol. 2007;584:191–203. doi: 10.1113/jphysiol.2007.135616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Ma Q. Nociceptors–noxious stimulus detectors. Neuron. 2007;55:353–364. doi: 10.1016/j.neuron.2007.07.016. [DOI] [PubMed] [Google Scholar]
- Woolfrey KM, Srivastava DP, Photowala H, Yamashita M, Barbolina MV, Cahill ME, Xie Z, Jones KA, Quilliam LA, Prakriya M, Penzes P. Epac2 induces synapse remodeling and depression and its disease-associated forms alter spines. Nat Neurosci. 2009;12:1275–1284. doi: 10.1038/nn.2386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano N, Suzuki D, Endoh M, Zhao TC, Padbury JF, Tseng YT. A novel phosphoinositide 3-kinase-dependent pathway for angiotensin II/AT-1 receptor-mediated induction of collagen synthesis in MES-13 mesangial cells. J Biol Chem. 2007;282:18819–18830. doi: 10.1074/jbc.M610537200. [DOI] [PubMed] [Google Scholar]
- Zhang YH, Kays J, Hodgdon KE, Sacktor TC, Nicol GD. Nerve growth factor enhances the excitability of rat sensory neurons through activation of the atypical protein kinase C isoform, PKMδ. J Neurophysiol. 2012;107:315–335. doi: 10.1152/jn.00030.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Quilliam LA. Activation of the Ras superfamily of small GTPases. Workshop on exchange factors. EMBO Rep. 2003;4:463–468. doi: 10.1038/sj.embor.embor831. [DOI] [PMC free article] [PubMed] [Google Scholar]