

Abstract
Aims
To identify a female mouse model of diabetic peripheral neuropathy (DPN), we characterized DPN in female BTBR ob/ob mice and compared their phenotype to non-diabetic and gender-matched controls. We also identified dysregulated genes and pathways in sciatic nerve (SCN) and dorsal root ganglia (DRG) of female BTBR ob/ob mice to determine potential DPN mechanisms.
Methods
Terminal neuropathy phenotyping consisted of examining latency to heat stimuli, sciatic motor and sural sensory nerve conduction velocities (NCV), and intraepidermal nerve fiber (IENF) density. For gene expression profiling, DRG and SCN were dissected, RNA was isolated and processed using microarray technology and differentially expressed genes were identified.
Results
Similar motor and sensory NCV deficits were observed in male and female BTBR ob/ob mice at study termination; however, IENF density was greater in female ob/ob mice than their male counterparts. Male and female ob/ob mice exhibited similar weight gain, hyperglycemia, and hyperinsulinemia compared to non-diabetic controls, although triglycerides were elevated more so in males than in females. Transcriptional profiling of nerve tissue from female mice identified dysregulation of pathways related to inflammation.
Conclusions
Similar to males, female BTBR ob/ob mice display robust DPN, and pathways related to inflammation are dysregulated in peripheral nerve.
Keywords: Type 2 Diabetes, Animal Models, Diabetic Peripheral Neuropathy, Dyslipidemia, Gene Expression
1. Introduction
The diabetes epidemic is a major medical concern of the 21st century, affecting over 387 million people globally (IDF Diabetes Atlas 2014). The majority of cases (>90%) have type 2 diabetes (T2D) and display insulin resistance that is typically acquired from poor lifestyle choices combined with genetic susceptibility. T2D is associated with increased morbidity, and the debilitating nature of this disease stems from an array of macrovascular and microvascular complications. Diabetic peripheral neuropathy (DPN) is one such complication that presents in approximately 50% of diabetic patients (Edwards, Vincent et al. 2008) and is a leading cause of diabetes-related hospital admissions and non-traumatic foot amputations in the USA (CDC 2011).
Current therapeutic options for DPN rely on controlling and treating T2D; however, significant gender dimorphisms in the responsiveness of patients to anti-diabetic drugs have been reported (Kim, Cha et al. 2005, Donnelly, Doney et al. 2006, Osterbrand, Fahlen et al. 2007). These observations highlight the importance of elucidating gender-specific differences in diabetes disease manifestation; a decree which has been iterated by the National Institutes of Health (Clayton and Collins 2014). To date, clinical studies investigating the role of gender in relation to obesity and diabetes are limited (Gale and Gillespie 2001) but demonstrate that although T2D does not discriminate between gender, typically affecting males and females alike, young males are at a higher risk of developing insulin resistance (Geer and Shen 2009, Macotela, Boucher et al. 2009) and T2D (Wild, Roglic et al. 2004, Ding, Song et al. 2006). Clearly, with differences in prevalence and drug-responses between males and females, treatments tailored to gender-type are needed. Moreover, pre-clinical research utilizing both male and female models of diabetes is required, as the varying responses of gender to anti-diabetic drugs is a likely consequence of pre-clinical studies primarily utilizing male mouse models to avoid the distinct differences in female physiology and metabolism. Although female mouse models of diabetes are readily available, reports on the gender-specific differences observed in diabetic complications are either limited, or in the case of DPN, absent. Thus, characterization of DPN in female mice is important to establish the differences in physiological profiles between the sexes, including time of onset, degree of severity, and response to disease modifying agents.
In the current study, effects of gender were investigated in female and male BTBR ob/ob mice, with an emphasis on identifying differences in DPN severity in the context of comprehensive diabetes phenotyping. Both male and female BTBR ob/ob mice present with a condition similar to T2D (Clee, Nadler et al. 2005), and we recently confirmed that male BTBR ob/ob mice display a robust neuropathic phenotype as early as 9 weeks (O’Brien, Hur et al. 2014). Previous examination of diabetes phenotypes in male and female BTBR ob/ob mice have revealed marked deficits in metabolic homeostasis between gender, with more severe metabolic perturbations in males that include increased hyperglycemia, hypertriglyceridemia, insulin resistance, and dyslipidemia (Clee, Nadler et al. 2005, Hudkins, Pichaiwong et al. 2010). Thus, as these components of the metabolic syndrome are known to be involved in DPN pathogenesis, we hypothesized that females would display a milder neuropathic phenotype, similar to observations seen in the human population (Aaberg, Burch et al. 2008). As this was the first instance of DPN characterization in a female model, we also performed gene expression profiling on dorsal root ganglia (DRG) and sciatic nerve (SCN) of female mice to identify differentially expressed genes (DEGs) that contribute to DPN in female mice and may provide insight into underlying disease mechanisms.
2. Materials and Methods
2.1 Animals
Male and female BTBR ob/+ and ob/ob mice (n=4; BTBR.Cg-Lepob/WiscJ, Jackson Laboratory, Bar Harbor, ME) were fed a standard diet (5LOD; 13.4% kcal fat; Research Diets, NJ). All procedures complied with protocols established by the Diabetic Complications Consortium (DCC) (Sullivan, Lentz et al. 2008) and approved by the University of Michigan (U-M) University Committee on Use and Care of Animals (UCUCA). Daily monitoring and maintenance of mice was provided by the U-M Unit for Laboratory Animal Medicine (ULAM).
2.2 Metabolic and Neuropathic Phenotyping
Male and female BTBR ob/+ and ob/ob mouse phenotyping included both metabolic and neurological measures at ~24 wks. Terminal body weights and fasting blood glucose (FBG; 4 hr fast) were measured. Percent glycosylated hemoglobin (%GHb) was measured by the Chemistry Core at the Michigan Diabetes Research and Training Center (MDRTC), while plasma insulin, cholesterol and triglyceride measurements were performed by the National Mouse Metabolic Phenotyping Center (MMPC; Vanderbilt, TN and University of Washington, WA). Nerve conduction velocities (NCVs) were measured according to published protocols (Sullivan, Hayes et al. 2007, Vincent, Hayes et al. 2009), and at study termination, intraepidermal nerve fiber (IENF) density profiles were determined as previously described (Sullivan, Hayes et al. 2007).
2.3 Affymetrix Microarray
RNA isolated from DRG and SCN of five female BTBR ob/ob and BTBR ob/+ mice was used for microarray hybridization. Total RNA (75 ng) from each sample was amplified and biotin-labeled using the Ovation™ Biotin-RNA Amplification and Labeling System (NuGEN Technologies Inc., San Carlos, CA) according to the manufacturer’s protocol. Amplification and hybridization was performed at the University of Michigan DNA Sequencing Core’s Affymetrix and Microarray Core Group (Ann Arbor, MI) using the Affymetrix GeneChip Mouse Genome 430 2.0 Array. To validate microarray data, DEGs were ranked by fold-change (Tables 1 and 2) and several of the most highly altered DEGs were analyzed by real time RT-PCR (RT-qPCR) using Ywhaz as the endogenous reference gene as previously described (O’Brien, Hur et al. 2014). The genes chosen for validation along with fold-change compared to controls are provided (Supplemental Table 1). Primers were designed in house, optimized, and purchased from Integrated DNA Technologies (Supplemental Table 2).
Table 1.
DRG DEGs
| Description | Gene Symbol | Gene ID# | Fold Change |
|---|---|---|---|
| corticotropin releasing hormone | Crh | 12918 | 22.43 |
| stefin A3 | Stfa3 | 20863 | 20.86 |
| small proline-rich protein 1A | Sprr1a | 20753 | 17.29 |
| solute carrier family 6 (neurotransmitter transporter, serotonin), member 4 | Slc6a4 | 15567 | 10.57 |
| Wilms tumor 1 homolog | Wt1 | 22431 | 10.36 |
| cholecystokinin B receptor | Cckbr | 12426 | 10.18 |
| stefin A2 like 1 | Stfa2l1 | 268885 | 10.05 |
| endothelin converting enzyme-like 1 | Ecel1 | 13599 | 8.68 |
| neuropeptide Y | Npy | 109648 | 7.62 |
| serum amyloid A 3 | Saa3 | 20210 | 7.54 |
| mast cell protease 4 | Mcpt4 | 17227 | −3.33 |
| carboxylesterase 1D | Ces1d | 104158 | −3.54 |
| complement factor D (adipsin) | Cfd | 11537 | −3.54 |
| cytochrome P450, family 2, subfamily e, polypeptide 1 | Cyp2e1 | 13106 | −3.82 |
| cytochrome c oxidase subunit VIIIb | Cox8b | 12869 | −3.89 |
| somatostatin | Sst | 20604 | −4.25 |
| growth differentiation factor 10 | Gdf10 | 14560 | −4.32 |
| peripheral myelin protein 2 | Pmp2 | 18857 | −4.79 |
| mast cell protease 2 | Mcpt2 | 17225 | −4.80 |
| cytochrome P450, family 2, subfamily f, polypeptide 2 | Cyp2f2 | 13107 | −5.04 |
Table 2.
SCN DEGs
| Description | Gene Symbol | Gene ID# | Fold Change |
|---|---|---|---|
| immunoglobulin heavy constant gamma 1 (G1m marker) | Ighg1 | 16017 | 24.97 |
| immunoglobulin heavy constant gamma 2C | Ighg2c | 404711 | 19.75 |
| immunoglobulin heavy constant gamma 2B | Ighg2b | 16016 | 18.85 |
| matrix metallopeptidase 12 | Mmp12 | 17381 | 18.40 |
| chitinase-like 3 | Chil3 | 12655 | 11.96 |
| uncoupling protein 1 (mitochondrial, proton carrier) | Ucp1 | 22227 | 11.55 |
| S100 calcium binding protein A9 (calgranulin B) | S100a9 | 20202 | 9.63 |
| S100 calcium binding protein A8 (calgranulin A) | S100a8 | 20201 | 8.36 |
| brain expressed gene 1 | Bex1 | 19716 | 7.78 |
| pentraxin related gene | Ptx3 | 19288 | 7.39 |
| apelin receptor | Aplnr | 23796 | −3.17 |
| melan-A | Mlana | 77836 | −3.17 |
| enoyl-Coenzyme A delta isomerase 3 | Eci3 | 69123 | −3.21 |
| Ras association (RalGDS/AF-6) domain family member 6 | Rassf6 | 73246 | −3.28 |
| macrophage galactose N-acetyl-galactosamine specific lectin 2 | Mgl2 | 216864 | −3.30 |
| dickkopf homolog 2 (Xenopus laevis) | Dkk2 | 56811 | −3.35 |
| spondin 1, (f-spondin) extracellular matrix protein | Spon1 | 233744 | −3.91 |
| paraoxonase 1 | Pon1 | 18979 | −11.67 |
| aldo-keto reductase family 1, member C-like | Akr1cl | 70861 | −17.46 |
| cytochrome P450, family 2, subfamily f, polypeptide 2 | Cyp2f2 | 13107 | −21.13 |
2.4 Data and Microarray Analyses
Body weight, blood glucose and %GHb levels, plasma cholesterol and triglyceride levels, thermal latency measures, IENF density, and NCVs of BTBR ob/+ and ob/ob mice for each gender were compared using two-tailed T-test in GraphPad Prism version 6 for Windows (San Diego, California).
Microarray data were analyzed using our established in-house microarray data analysis pipeline (Hur, Sullivan et al. 2011, Pande, Hur et al. 2011, O’Brien, Hur et al. 2014). Briefly, Affymetrix raw data files (CEL files) were processed using a local copy of GenePattern, a bioinformatics platform from the Broad Institute (Reich, Liefeld et al. 2006). The samples were Robust Multi-array Average (RMA) normalized using the BrainArray Custom Chip Definition File (CDF) ENTREZG version 16 (Dai, Wang et al. 2005). The raw and processed microarray data have been deposited into the NCBI Gene Expression Omnibus data repository (http://www.ncbi.nlm.nih.gov/geo, accession # GSE70852). Intensity-Based Moderated T-test (IBMT) (Sartor, Tomlinson et al. 2006) identified DEGs using a false discovery rate (FDR) < 5% cutoff. DEGs were obtained between control (ob/+) and diabetic mice (ob/ob) in DRG and SCN. Database for Annotation, Visualization and Integrated Discovery (DAVID; http://david.abcc.ncifcrf.gov/) (Huang da, Sherman et al. 2009, Huang da, Sherman et al. 2009) identified significantly enriched biological functions among the DEGs in terms of Gene Ontology (GO; http://www.geneontology.org/) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG; http://www.genome.jp/kegg/) pathways. Heat-maps were generated using the top 10 most enriched biological functions in each DEG set based on significance values (log-transformed Benjamini-Hochberg (BH)-corrected P-values) to visually represent the overall similarity and differences between the DEG sets. DEGs from our previously published 5- and 13-week BTBR ob/ob mice were included in the comparison identify commonly dysregulated pathways between male and female DPN models.
3. Results
3.1 Female and Male BTBR ob/ob Mice Develop Robust DPN
We recently reported that male BTBR ob/ob mice rapidly develop considerable peripheral nerve deficits after diabetes onset (O’Brien, Hur et al. 2014). Thus, our initial goal for this study was to assess phenotypic differences associated with gender using this robust mouse model of diabetes with established DPN. Male mice display signs of extreme morbidity sooner than females; therefore, male and female mice were sacrificed at 22 wk and 26 wk, respectively. At terminal stages, electrophysiological testing and quantification of IENF densities confirmed the presence of neuropathy in both obese male and female mice compared to gender-matched non-diabetic controls (Figure 1). NCVs of the sciatic nerve (motor; MNCV) and sural nerve (sensory; SNCV) were both significantly decreased (Figure 1A,B), with obese male mice demonstrating a ~1.7-fold decrease in MNCV and obese females displaying a ~1.5-fold decrease compared to their respective controls. Similar SNCV deficits were also observed; males exhibited a ~1.3-fold decrease while females exhibited a ~1.4-fold decrease relative to lean control mice. Morphological analysis was performed to assess IENF innervation (Supplementary Figure 1) and quantification of IENF densities revealed significantly lower IENF levels in diabetic mice compared to the respective non-diabetic controls (Figure 1C). Further assessment indicated that IENF loss was greater in obese males (~2.7-fold decrease) than females (~1.4-fold decrease). Additional behavioral testing of nerve function confirmed a loss of sensation in female ob/ob mice, as evidenced by decreased sensitivity to thermal stimuli in tail flick and hind-paw testing (Supplementary Figure 2A,B). Combined, these physiological and anatomical DPN measures confirm a peripheral neuropathic phenotype in both male and female ob/ob mice that is similar to that seen in T2D patients, although female BTBR ob/ob mice display a less severe loss of IENFs despite being 4 wk older than males.
Figure 1. Female BTBR ob/ob mice display robust neuropathy characterized by electrophysiological and morphological deficits.
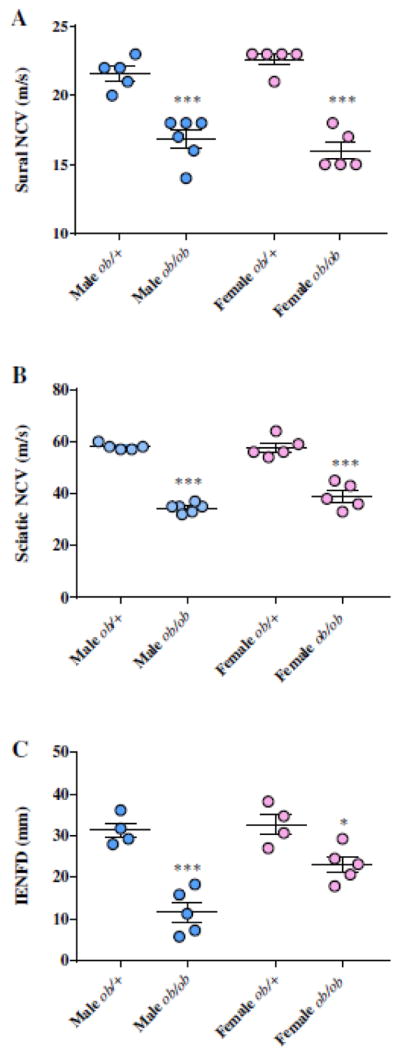
Analysis of sciatic (A; Motor NCV) and sural (B; Sensory NCV) nerve conduction velocity in male and female BTBR ob/+ and ob/ob mice ~24 weeks of age. C. Quantification of IENF density in male and female BTBR ob/+ and ob/ob mice. Male BTBR mice are represented by blue filled circles while female mice are shown in pink filled circles. Means ± SEM, n = 4–5 per group. * P < 0.05; *** P < 0.0001 vs. gender-matched, non-diabetic ob/+ mice.
3.2 Female and Male BTBR ob/ob Mice Share a Similar, Robust Diabetic Phenotype
Both male and female BTBR ob/ob mice exhibit the key physical and metabolic features of obesity and T2D (Figure 2). At study termination, both male and female BTBR ob/ob mice were severely obese (68.1±4.04 and 61.8±3.56 g, respectively), exhibiting a ~1.8- and 1.4-fold increase in body weight compared to non-diabetic controls, respectively (Figure 2A). Likewise, %GHb was 1.9-fold higher in males and 1.65-fold higher in female obese mice relative to controls, reflecting severe hyperglycemia (Figure 2B). Early during the course of diabetes, male BTBR ob/ob mice have increased FBG compared to females (unpublished observations), but by 24 wk both males and females display similar levels of hyperglycemia, in agreement with previous studies (Hudkins, Pichaiwong et al. 2010). Fasting plasma lipids were also measured, as dyslipidemia has previously been strongly associated with DPN progression (Vincent, Hayes et al. 2009, Hur, Sullivan et al. 2011). As expected, total plasma triglycerides were elevated in obese BTBR ob/ob mice compared to controls (Figure. 2C). Male BTBR ob/ob exhibited a higher level of hypertriglyceridemia than their female counterparts (256±17.6 vs. 119±45.5 mg/dL, respectively), however both males and females had a similar relative fold-change increase in respect to non-diabetic controls (3.9- and 4.4-fold increase, respectively). Measures of fasting plasma insulin in both male and female BTBR ob/ob mice further confirmed severe hyperinsulinemia compared to control mice (female ob/+ = 1.32 ± 0.13 S.E.M.; male ob/+ mice = 1.28± 0.16 S.E.M.), with levels exceeding the threshold of detection (maximum threshold = 10 ng/ml).
Figure 2. Diabetic phenotype of BTBR ob/ob mice.
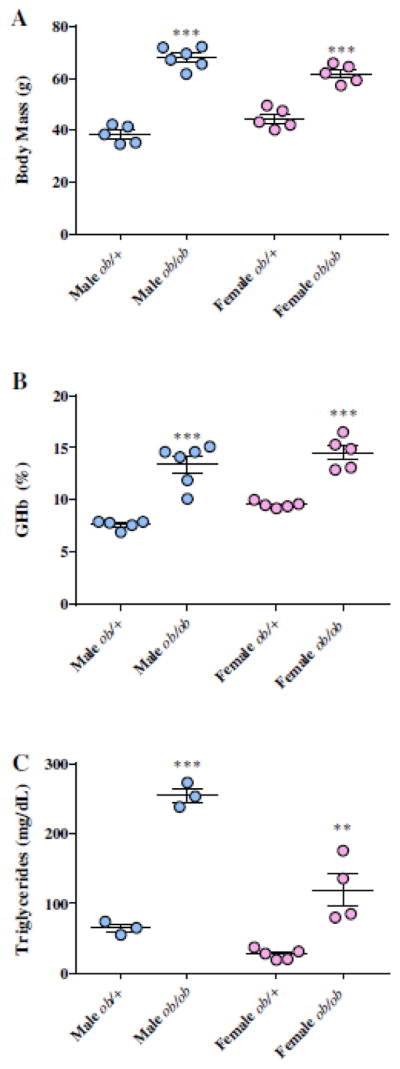
Terminal body weights (A) of male and female BTBR ob/+ and ob/ob mice. At study completion, either blood or plasma of fasting mice was analyzed for glycosylated hemoglobin (%GHb) and (B) triglycerides (C), respectively. Means ± SEM, n = 4–5 per group. Male BTBR mice are represented by blue filled circles while female mice are shown in pink filled circles. *** P < 0.0001 vs. non-diabetic ob/+ mice.
3.3 Gene Set Enrichment Analysis Indicates Inflammation as a Prominent Dysregulated Pathway in the Peripheral Nerve of Female BTBR ob/ob Mice
To complement our neuropathy phenotyping, microarray analysis was performed on RNA isolated from SCN and DRG to identify dysregulated genes in female BTBR ob/ob mice. Using a 5% FDR cutoff, 584 and 1,105 DEGs were identified in SCN and DRG, respectively. The 10 most up-regulated and down-regulated DEGs identified in DRG and SCN tissues are listed in Tables 1 & 2, respectively. Subsequently, gene set enrichment analysis identified numerous biological functions dysregulated in female mice with established DPN. Overrepresented pathways in female BTBR ob/ob mice included functions related to inflammation and the immune response (Figure 3), similar to our findings in 13 wk male BTBR ob/ob mice (O’Brien, Hur et al. 2014). There were 131 common DEGs between the DRG and SCN sets, 124 of which show concordant changes in gene expression. The common DEGs are listed in Supplementary Table 3, while Supplementary Figure 3 illustrates the most over-represented (enriched) biological functions among these DEGs. We found numerous significantly enriched terms among the concordant DEGs, including complement activation, humoral immune response, and inflammatory response (Supplementary Figure 3), suggesting that inflammatory processes occur within both tissues.
Figure 3. Gene expression analysis of nerve tissue isolated from 26 wk female BTBR ob/ob mice.

Functional enrichment analysis was performed using DAVID on each of the DEG data sets from either DRG or SCN. Benjamini-Hochberg (BH)-corrected P-values of the top 10 most significant functional terms are represented by heat-map with a log10-based color and number index.
4. Discussion
The rising interest in gender-based differences in diabetes (Clayton and Collins 2014) has highlighted the need for further study into the effects of sex on T2D pathogenesis and the development of diabetic complications. We report here the first instance of a female T2D mouse model presenting with a neuropathic phenotype. Similar to male BTBR ob/ob mice, female BTBR ob/ob mice exhibit robust peripheral neuropathy, including decreased IENF density, impaired motor and sensory NCVs, and thermal hypoalgesia. Moreover, application of our established microarray and gene set enrichment analysis approach (Hur, Sullivan et al. 2011, Pande, Hur et al. 2011, O’Brien, Hur et al. 2014, Hur, Dauch et al. 2015) to DRG and SCN of female BTBR ob/ob mice identified genes and biological functions related to inflammation and the immune response. These functions are similar to our previously reported findings in male BTBR ob/ob mice (O’Brien, Hur et al. 2014), providing further evidence that these pathways are implicated in DPN pathogenesis.
We are the first to report that female BTBR ob/ob mice display significant deficits in nerve function as well as decreased IENF, thus indicating a similar neuropathic phenotype to that observed in T2D patients. With the general consensus that male BTBR ob/ob mice are at greater risk for development of hyperglycemia, insulin resistance, and related metabolic abnormalities (Clee, Nadler et al. 2005, Hudkins, Pichaiwong et al. 2010) whereas females display mild protection (Gale and Gillespie 2001), we anticipated a more robust DPN phenotype in males compared to females. However, upon examination of terminal electrophysiological measures, we observed no noticeable differences in MNCV and SNCV between genders at study termination (Figure 1, A,B), suggesting that electrophysiological functions are similarly impaired across gender. It is unknown, however, whether metabolic imbalances between genders at disease onset may impact nerve function earlier than 26 wk. Thus, further assessment of sex-dimorphisms in peripheral nerve function is warranted, as differences may be identified during early stages of DPN development. Future studies will include side-by-side NCV assessments earlier in the disease course to determine if gender-related differences are present. Although no discernable nerve electrophysiology differences were observed, assessment of IENF density, considered the most accurate measure of small fiber neuropathy (Pittenger, Ray et al. 2004), revealed that male BTBR ob/ob mice exhibited greater IENF loss than females (Figure 1C). These findings suggest that the greater small fiber loss in male BTBR ob/ob mice may possibly be a result of more robust increased metabolic perturbations (described below). In support of this idea, small fiber neuropathy in humans is associated with obesity and dyslipidemia, as seen in the male BTBR ob/ob mice, more than hyperglycemia (Smith and Singleton 2013).
Our terminal metabolic phenotyping involved measuring weight, %GHb, plasma triglycerides, and fasting plasma insulin (Figure 2), hallmarks typically increased in diabetes and obesity. BTBR ob/ob mice were profoundly heavier than their controls (Figure 2A) and had hyperglycemia and poor glycemic control (Figure 2B), hypertriglyceridemia (Figure 2C), and hyperinsulinemia. %GHb, a marker of glucotoxicity and a hallmark of diabetes, was only mildly more elevated in male than in female BTBR ob/ob mice at study termination when comparing fold-change in relation to non-diabetic controls (Figure 2B). This mild increase in plasma glucotoxicity was expected due to the increased FBG typically found in males and is consistent with previous reports in this model (Clee, Nadler et al. 2005, Askari, Wietecha et al. 2014).
As dyslipidemia has recently been identified as a contributory factor in DPN progression in both humans and mice (Vincent, Hinder et al. 2009), plasma triglycerides were measured in male and female BTBR ob/ob mice. Although male and female BTBR ob/ob mice similarly display an abnormal lipid profile compared to controls (Figure 2), male mice exhibited a greater degree of hypertriglyceridemia which agrees with previous reports (Hudkins, Pichaiwong et al. 2010). As the increase in triglycerides correlates with decreased IENF, which was more profound than the difference in %GHb between genders, our findings are in line with the hypothesis that lipids, and not just glycemia, may contribute to DPN progression. A recent Cochrane review investigating blood glucose control in the prevention and treatment of DPN in man found that targeting hyperglycemia has little effect on neuropathy outcomes in T2D (Callaghan, Little et al. 2012). The observed sex-based dimorphism in our study was anticipated due to greater metabolic imbalances seen in male mice that, as a consequence, would promote DPN and greater IENF loss. Indeed, studies have reported that elevated plasma triglycerides significantly correlate with both decreased sural nerve fiber density in T2D patients (Wiggin, Sullivan et al. 2009) and a loss of small unmyelinated nerve fibers (Smith and Singleton 2013). Although the morphology of sural nerves was not measured, our findings support that decreased IENFs in ob/ob mice may similarly be the result of lipotoxicity and elevated triglycerides (Figure 2C), and that lipid lowering therapies may therefore halt DPN progression. Indeed evidence suggests that such therapies may alter the progression of diabetic complications, including diabetic neuropathy (Vincent, Hinder et al. 2009). Results from the FIELD trial demonstrates that fibrates are effective treating diabetic nephropathy and retinopathy (Keech, Mitchell et al. 2007, Forsblom, Hiukka et al. 2010), while a report from the Fremantle Diabetes Study suggests that fibrates and statins may protect against the development of sensory neuropathy in patients with type 2 diabetes (Davis, Yeap et al. 2008). Furthermore, we have recently published data from C57BKS db/db mice treated with pioglitazone, a PPAR-γ agonist, in which decreased plasma triglycerides are associated reduced small fiber neuropathy, as determined by IENF density and latency to thermal response (Hur, Dauch et al. 2015). We contend that insulin resistance in peripheral nerves is another potential contributor to DPN (Grote, Groover et al. 2013). Our results demonstrate that both male and female mice have profound hyperinsulinemia, as fasting plasma insulin exceeded the threshold for detection (10 ng/ml). A comprehensive assessment of insulin resistance in peripheral nerves was beyond the scope of this study; however, as men are at greater risk of developing systemic insulin resistance (Gale and Gillespie 2001, Geer and Shen 2009), confirmed in the BTBR ob/ob mouse model (Clee, Nadler et al. 2005), this sex dimorphism would suggest a similar situation in peripheral nerves where insulin resistance is more pronounced in males, thus promoting greater nerve dysfunction. Furthermore, the increased risk of developing insulin resistance in males may explain the higher fold-change of fasting plasma triglycerides when compared to females. As a consequence of systemic insulin resistance, impaired insulin action on adipocytes results in decreased uptake of circulating triglycerides (Guilherme, Virbasius et al. 2008), an important feature in maintaining lipid homeostasis. Interestingly, female BTBR ob/ob mice retain adipose tissue insulin sensitivity (Clee, Nadler et al. 2005). Thus, our findings demonstrating higher levels of circulating triglycerides in male mice may be due to increased adipose tissue insulin resistance, thus contributing even further to nerve dysfunction that is brought upon by peripheral nerve insulin resistance.
We previously performed gene expression profiling on male BTBR ob/ob mice which resulted in the identification of numerous biological functions dysregulated in the peripheral nerve of diabetic mice compared to controls (O’Brien, Hur et al. 2014); therefore, we performed an identical analysis on SCN and DRG from 26 wk old female BTBR mice. We identified numerous DEGs with large fold changes in female BTBR ob/ob mice compared to controls (Tables 1 and 2). Among the DEGs identified in DRG tissue, several encode proteins implicated in inflammatory signaling pathways, including Crh (corticotropin releasing hormone), which promotes macrophage foam cell formation (Cho, Kang et al. 2015), and Stfa3 (stefin A3), a cysteine protease inhibitor upregulated in lipopolysaccharide-stimulated glial cells (Hosoi, Suzuki et al. 2005) that has a role in protecting cells from inappropriate proteolysis. Saa3 (serum amyloid A3) is an acute phase protein that was also highly overexpressed and is association with diabetic complications (Hamano, Saito et al. 2004). In addition, SAA3 is also a mediator in diabetic kidney disease (Anderberg, Meek et al. 2015), and was previously identified as overexpressed in male BTBR ob/ob mouse SCN (O’Brien, Hur et al. 2014).
Using the publically available DNMKB database (a repository of our completed gene expression profiling in nerves of diabetic animals; http://jdrf.neurology.med.umich.edu/DNMKB/) to compare the 26 wk old female DEG set with the 13 wk male DEG set, a post-hoc comparison identified 161 commonly dysregulated DEGs between the two genders (Supplementary Table 3). Similar to our findings in female BTBR ob/ob SCN, those with the greatest fold change for both genders included several immunoglobulin family members (Ighg1, Ighg2c and Ighg2b), Mmp12 (matrix metallopeptidase 12), and Ucp1. In addition, S100a8, S1009, Pon1 (paraoxonase 1), and Pmp2 were also similarly dysregulated in both genders. While our current analyses examined gene expression changes in females later in the disease course than our male mouse analyses, we similarly identified dysregulation of biological functions related to inflammation in both the DRG and SCN of female BTBR ob/ob mice, suggesting a common mechanism of nerve injury in both genders (Figure 3). Although several branches of inflammatory response are likely to be involved in the peripheral nerve environment, collectively, our microarray data suggest an increase in peripheral nerve antigens in BTBR ob/ob mice. As antibodies to neural myelin antigens have been identified in demyelinating diseases (Allen, Giannopoulos et al. 2005), an increase of IgG immunoglobulins in SCN may similarly due to an increase in neural autoantigens. For example, neuropeptide y (Npy), a widely expressed protein in the peripheral nervous system that is increased in response to nerve injury (Ji, Zhang et al. 1994) is a DEG in our female BTBR ob/ob mice and is a known autoantigen in T1D and T2D patients (Skarstrand, Dahlin et al. 2013). Interestingly, Mmp12 and S100a8 are involved in inflammation, tissue remodeling, and injury. MMP12 is an extracellular matrix protease involved in collagen degradation and tissue destruction that is produced by Schwann cells (Hughes, Wells et al. 2002). S100 proteins are calcium binding proteins expressed in neural tissues, increased in patients with T2D (Krisp, Jacobsen et al. 2013), that stimulate a local inflammatory response through biding to RAGE receptors causing a release in proinflammatory cytokines.
One notable downregulated shared DEG identified was Pon1 (similarly decreased in BKS db/db mice (Pande, Hur et al. 2011)). PON1 is an anti-atherosclerotic component of high-density lipoprotein (HDL) and plays a role in the prevention of lipid peroxidation. The PON1 gene is activated by PPARγ, whose expression itself has been found to be deceased in the sural nerve of patients with progressive DPN (Hur, Sullivan et al. 2011). Pmp2 was another common DEG decreased in male and female BTBR ob/ob SCN. Pmp2 is one of the most abundant myelin proteins in the peripheral nervous system, with 15% of myelin comprising of Pmp2, and it is predominantly expressed in myelinated Schwann cells where it has a role as a lipid binding protein and is thought to mediate lipid transport. PMP2-deficient mice exhibit decreased NCVs (Zenker, Stettner et al. 2014), so decreased expression of Pmp2 in our mouse models of DPN is not surprising, as they too display decreased NCV.
We acknowledge that this current study has some caveats. First, while there are multiple concerns with leptin-deficient models in diabetes research, these models remain favorable to high fat diet (HFD)-fed models for generating a robust diabetes with a predictable and extensively characterized neuropathic phenotype (O’Brien, Sakowski et al. 2014). However, due to the rapid onset of a severe neuropathic phenotype in BTBR ob/ob mice (O’Brien, Hur et al. 2014), this model may not be suitable for understanding the early subtle changes that occur in the peripheral nerve as a result of metabolic imbalances. In addition, our non-diabetic control animals consisted only of heterozygote BTBR ob/+ mice. As these mice are known to exhibit subtle metabolic differences to wild-type mice (Hudkins, Pichaiwong et al. 2010) that may also manifest in mild neurological differences, the inclusion of wild-type BTBR mice in future in vivo studies is warranted. Second, for reasons related to study design and execution, side-by-side comparisons between gender at exact ages could not be performed. The primary reason for this is that male BTBR ob/ob mice exhibited signs of ill health at 22 weeks requiring study termination, while female mice remained healthy until the prescribed 26 week termination. Lastly, as this was a preliminary investigation, only a small cohort of animals was phenotyped. Despite these caveats, these analyses support the use of female BTBR ob/ob mice as a novel model for evaluating the effects of gender on DPN mechanisms and treatments.
With the global incidence of T2D on the rise and an increase in the aging population predicted, the number of men and women with diabetes and diabetic complications is set to increase. This inevitable crisis highlights the need for further investigation into how gender influences the development of diabetic complications. Though marginal, there is a higher prevalence of T2D in young men than women (Wild, Roglic et al. 2004, Ding, Song et al. 2006), likely attributed to the fact that males are more susceptible to insulin resistance than females (van Genugten, Utzschneider et al. 2006). Studies have also demonstrated that (i) the insulin analogue glargine causes a significantly greater decrease in HbA1c in males than females (Osterbrand, Fahlen et al. 2007) and that (ii) males respond better to sulfonylureas than females (Donnelly, Doney et al. 2006), while (iii) females respond more favorably to rosiglitazone than males (Kim, Cha et al. 2005). Our understanding of sex dimorphisms in diabetes is compounded by underlying physiological differences which are numerous and include differences in glucose control and energy homeostasis (Basu, Dalla Man et al. 2006), insulin disposal and clearance (Jensen, Nielsen et al. 2012), regional fat disposition (Geer and Shen 2009, Macotela, Boucher et al. 2009), and sex steroid hormones (Shi and Clegg 2009). For instance, high levels of estrogen confer protection against diabetes development in women (Margolis, Bonds et al. 2004, Le May, Chu et al. 2006, Shi and Clegg 2009, Tiano and Mauvais-Jarvis 2012), and the decreased estrogen production along with increased longevity in post-menopausal women promotes a greater incidence of T2D in this population compared to males, which is also due to increased longevity in this sex (Gale and Gillespie 2001). Evidence has shown that the incidence of diabetes in females is similar to males prior to puberty or after onset of menopause, suggesting that protection is conferred by female hormone, estrogen. Indeed, estrogen prevents β cell failure in most rodent models of diabetes, demonstrating protection through various pathways (Tiano and Mauvais-Jarvis 2012). Similar to T2D incidence, DPN appears to be more prevalent in men than women. The impact of gender on peripheral nerve function is evidenced by nerve conduction studies and quantitative sensory testing that demonstrate earlier development of DPN in male patients with either type 1 diabetes (T1D) and T2D relative to their female counterparts (Aaberg, Burch et al. 2008). Further studies are needed to verify whether this DPN-predominance persists in men with T2D, as males with T1D are more susceptible to DPN (Gale and Gillespie 2001).
Our findings demonstrate that female BTBR ob/ob mice exhibit a robust DPN phenotype. Although terminal measures of body weight, hyperglycemia, and hyperinsulinemia were relatively similar at the study conclusion irrespective of gender, male mice exhibited a greater degree of dyslipidemia. The hypertriglyceridemia in male BTBR ob/ob mice highlights the presence of gender-specific differences in this T2D mouse model, and based on recent studies, identifies a feature which may explain why male BTBR ob/ob mice exhibit a greater decrease in IENF densities suggesting a more robust small fiber neuropathy in males compared to females. Although we have provided preliminary data into how sex dimorphisms in diabetes may influence DPN progression, further investigation is required to identify the biological components that confer male susceptibility/female resistance so that tailored gender-specific therapeutic strategies can be devised and implemented.
Supplementary Material
Acknowledgments
The authors would like to acknowledge the technical expertise of Ms. Jacqueline Dauch, Mrs. Carey Backus, Ms. Chelsea Lindblad, and Dr. Sang Su Oh in conducting animal experiments and Mrs. Yu Hong for RNA processing. The authors thank the Hormone Core at Vanderbilt University and the Lipid Laboratory at the Mouse Metabolic Phenotyping Center at the University of Washington for mouse plasma insulin and lipid measurements, respectively, and the Chemistry Core of the MDRTC (930DK020572) at the U-M for mouse %GHb measurements. The authors would also like to thank Dr. Charlie Alpers at the University of Washington for expert advice on the BTBR ob/ob mouse strain.
N.J.R. P.D.O.B., and J.M.H conducted animal experiments, researched data, and wrote the manuscript. S.A.S reviewed and edited the manuscript. J.H. researched data and reviewed and edited the manuscript. E.L.F. designed and directed the study, contributed to discussion, and reviewed the manuscript.
Funding was provided by the National Institutes of Health (1DP3DK094292, 1R24082841 to E.L.F.); Novo Nordisk Foundation (NNF14SA0006 to E.L.F.), Juvenile Diabetes Research Foundation (Postdoctoral Fellowship to J.H.), Milstein, Nathan and Rose Research Fund; Sinai Medical Staff Foundation Neuroscience Scholar Fund 2; Robert C Graham Fund; Walbridge Aldinger Graduate Fellowship Fund (Post-doctoral Fellowship to P.D.O.B); American Diabetes Association; Program for Neurology Research & Discovery; and the A. Alfred Taubman Medical Research Institute.
Footnotes
No potential conflicts of interest relevant to this article were reported.
The authors are deeply saddened by the passing of our dear friend and colleague Lisa L. McLean. Lisa died on July 5, 2015 from a sudden illness. Her selfless dedication facilitated many studies in the field of diabetic complications. She will be deeply missed by everyone who knew her.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aaberg ML, Burch DM, Hud ZR, Zacharias MP. Gender differences in the onset of diabetic neuropathy. J Diabetes Complications. 2008;22(2):83–87. doi: 10.1016/j.jdiacomp.2007.06.009. [DOI] [PubMed] [Google Scholar]
- Allen D, Giannopoulos K, Gray I, Gregson N, Makowska A, Pritchard J, Hughes RA. Antibodies to peripheral nerve myelin proteins in chronic inflammatory demyelinating polyradiculoneuropathy. J Peripher Nerv Syst. 2005;10(2):174–180. doi: 10.1111/j.1085-9489.2005.0010207.x. [DOI] [PubMed] [Google Scholar]
- Anderberg RJ, Meek RL, Hudkins KL, Cooney SK, Alpers CE, Leboeuf RC, Tuttle KR. Serum amyloid A and inflammation in diabetic kidney disease and podocytes. Lab Invest. 2015;95(3):250–262. doi: 10.1038/labinvest.2014.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askari B, Wietecha T, Hudkins KL, Fox EJ, O’Brien KD, Kim J, Nguyen TQ, Alpers CE. Effects of CP-900691, a novel peroxisome proliferator-activated receptor alpha, agonist on diabetic nephropathy in the BTBR ob/ob mouse. Lab Invest. 2014;94(8):851–862. doi: 10.1038/labinvest.2014.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu R, Dalla Man C, Campioni M, Basu A, Klee G, Toffolo G, Cobelli C, Rizza RA. Effects of age and sex on postprandial glucose metabolism: differences in glucose turnover, insulin secretion, insulin action, and hepatic insulin extraction. Diabetes. 2006;55(7):2001–2014. doi: 10.2337/db05-1692. [DOI] [PubMed] [Google Scholar]
- Callaghan BC, Little AA, Feldman EL, Hughes RA. Enhanced glucose control for preventing and treating diabetic neuropathy. Cochrane Database Syst Rev. 2012;6:CD007543. doi: 10.1002/14651858.CD007543.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention. National Diabetes Statistics Report: Estimates of Diabetes and Its Burden in the United States, 2014. Atlanta, GA: US Department of Health and Human Services; 2014. [Google Scholar]
- Cho W, Kang JL, Park YM. Corticotropin-Releasing Hormone (CRH) Promotes Macrophage Foam Cell Formation via Reduced Expression of ATP Binding Cassette Transporter-1 (ABCA1) PLoS One. 2015;10(6):e0130587. doi: 10.1371/journal.pone.0130587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton JA, Collins FS. Policy: NIH to balance sex in cell and animal studies. Nature. 2014;509(7500):282–283. doi: 10.1038/509282a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clee SM, Nadler ST, Attie AD. Genetic and genomic studies of the BTBR ob/ob mouse model of type 2 diabetes. Am J Ther. 2005;12(6):491–498. doi: 10.1097/01.mjt.0000178781.89789.25. [DOI] [PubMed] [Google Scholar]
- Dai M, Wang P, Boyd AD, Kostov G, Athey B, Jones EG, Bunney WE, Myers RM, Speed TP, Akil H, Watson SJ, Meng F. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data. Nucleic Acids Res. 2005;33(20):e175. doi: 10.1093/nar/gni179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis TM, Yeap BB, Davis WA, Bruce DG. Lipid-lowering therapy and peripheral sensory neuropathy in type 2 diabetes: the Fremantle Diabetes Study. Diabetologia. 2008;51(4):562–566. doi: 10.1007/s00125-007-0919-2. [DOI] [PubMed] [Google Scholar]
- Ding EL, Song Y, Malik VS, Liu S. Sex differences of endogenous sex hormones and risk of type 2 diabetes: a systematic review and meta-analysis. JAMA. 2006;295(11):1288–1299. doi: 10.1001/jama.295.11.1288. [DOI] [PubMed] [Google Scholar]
- Donnelly LA, Doney AS, Hattersley AT, Morris AD, Pearson ER. The effect of obesity on glycaemic response to metformin or sulphonylureas in Type 2 diabetes. Diabet Med. 2006;23(2):128–133. doi: 10.1111/j.1464-5491.2005.01755.x. [DOI] [PubMed] [Google Scholar]
- Edwards JL, Vincent AM, Cheng HT, Feldman EL. Diabetic neuropathy: mechanisms to management. Pharmacol Ther. 2008;120(1):1–34. doi: 10.1016/j.pharmthera.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsblom C, Hiukka A, Leinonen ES, Sundvall J, Groop PH, Taskinen MR. Effects of long-term fenofibrate treatment on markers of renal function in type 2 diabetes: the FIELD Helsinki substudy. Diabetes Care. 2010;33(2):215–220. doi: 10.2337/dc09-0621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale EA, Gillespie KM. Diabetes and gender. Diabetologia. 2001;44(1):3–15. doi: 10.1007/s001250051573. [DOI] [PubMed] [Google Scholar]
- Geer EB, Shen W. Gender differences in insulin resistance, body composition, and energy balance. Gend Med. 2009;6(Suppl 1):60–75. doi: 10.1016/j.genm.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grote CW, Groover AL, Ryals JM, Geiger PC, Feldman EL, Wright DE. Peripheral nervous system insulin resistance in ob/ob mice. Acta Neuropathol Commun. 2013;1(1):15. doi: 10.1186/2051-5960-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9(5):367–377. doi: 10.1038/nrm2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamano M, Saito M, Eto M, Nishimatsu S, Suda H, Matsuda M, Matsuki M, Yamamoto S, Kaku K. Serum amyloid A, C-reactive protein and remnant-like lipoprotein particle cholesterol in type 2 diabetic patients with coronary heart disease. Ann Clin Biochem. 2004;41(Pt 2):125–129. doi: 10.1258/000456304322880005. [DOI] [PubMed] [Google Scholar]
- Hosoi T, Suzuki S, Okuma Y, Kawakami A, Ogawa N, Ozawa K, Nomura Y. LPS induces stefin A3 expression in mouse primary cultured glial cells. Brain Res Mol Brain Res. 2005;140(1–2):138–141. doi: 10.1016/j.molbrainres.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic acids research. 2009;37(1):1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Hudkins KL, Pichaiwong W, Wietecha T, Kowalewska J, Banas MC, Spencer MW, Muhlfeld A, Koelling M, Pippin JW, Shankland SJ, Askari B, Rabaglia ME, Keller MP, Attie AD, Alpers CE. BTBR Ob/Ob mutant mice model progressive diabetic nephropathy. J Am Soc Nephrol. 2010;21(9):1533–1542. doi: 10.1681/ASN.2009121290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes PM, Wells GM, Perry VH, Brown MC, Miller KM. Comparison of matrix metalloproteinase expression during Wallerian degeneration in the central and peripheral nervous systems. Neuroscience. 2002;113(2):273–287. doi: 10.1016/s0306-4522(02)00183-5. [DOI] [PubMed] [Google Scholar]
- Hur J, Dauch JR, Hinder LM, Hayes JM, Backus C, Pennathur S, Kretzler M, Brosius FC, 3rd, Feldman EL. The Metabolic Syndrome and Microvascular Complications in a Murine Model of Type 2 Diabetes. Diabetes. 2015;64(9):3294–3304. doi: 10.2337/db15-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur J, Sullivan KA, Pande M, Hong Y, Sima AA, Jagadish HV, Kretzler M, Feldman EL. The identification of gene expression profiles associated with progression of human diabetic neuropathy. Brain. 2011;134(Pt 11):3222–3235. doi: 10.1093/brain/awr228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Diabetes Federation. IDF Diabetes Atlas. 6. Brussels, Belgium: International Diabetes Federation; 2013. [Google Scholar]
- Jensen MD, Nielsen S, Gupta N, Basu R, Rizza RA. Insulin clearance is different in men and women. Metabolism. 2012;61(4):525–530. doi: 10.1016/j.metabol.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Zhang X, Wiesenfeld-Hallin Z, Hokfelt T. Expression of neuropeptide Y and neuropeptide Y (Y1) receptor mRNA in rat spinal cord and dorsal root ganglia following peripheral tissue inflammation. J Neurosci. 1994;14(11 Pt 1):6423–6434. doi: 10.1523/JNEUROSCI.14-11-06423.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keech AC, Mitchell P, Summanen PA, O’Day J, Davis TM, Moffitt MS, Taskinen MR, Simes RJ, Tse D, Williamson E, Merrifield A, Laatikainen LT, d’Emden MC, Crimet DC, O’Connell RL, Colman PG F. s. investigators. Effect of fenofibrate on the need for laser treatment for diabetic retinopathy (FIELD study): a randomised controlled trial. Lancet. 2007;370(9600):1687–1697. doi: 10.1016/S0140-6736(07)61607-9. [DOI] [PubMed] [Google Scholar]
- Kim YM, Cha BS, Kim DJ, Choi SH, Kim SK, Ahn CW, Lim SK, Kim KR, Huh KB, Lee HC. Predictive clinical parameters for therapeutic efficacy of rosiglitazone in Korean type 2 diabetes mellitus. Diabetes Res Clin Pract. 2005;67(1):43–52. doi: 10.1016/j.diabres.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Krisp C, Jacobsen F, McKay MJ, Molloy MP, Steinstraesser L, Wolters DA. Proteome analysis reveals antiangiogenic environments in chronic wounds of diabetes mellitus type 2 patients. Proteomics. 2013;13(17):2670–2681. doi: 10.1002/pmic.201200502. [DOI] [PubMed] [Google Scholar]
- Le May C, Chu K, Hu M, Ortega CS, Simpson ER, Korach KS, Tsai MJ, Mauvais-Jarvis F. Estrogens protect pancreatic beta-cells from apoptosis and prevent insulin-deficient diabetes mellitus in mice. Proc Natl Acad Sci U S A. 2006;103(24):9232–9237. doi: 10.1073/pnas.0602956103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macotela Y, Boucher J, Tran TT, Kahn CR. Sex and depot differences in adipocyte insulin sensitivity and glucose metabolism. Diabetes. 2009;58(4):803–812. doi: 10.2337/db08-1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis KL, Bonds DE, Rodabough RJ, Tinker L, Phillips LS, Allen C, Bassford T, Burke G, Torrens J, Howard BV I. Women’s Health Initiative. Effect of oestrogen plus progestin on the incidence of diabetes in postmenopausal women: results from the Women’s Health Initiative Hormone Trial. Diabetologia. 2004;47(7):1175–1187. doi: 10.1007/s00125-004-1448-x. [DOI] [PubMed] [Google Scholar]
- O’Brien PD, Hur J, Hayes JM, Backus C, Sakowski SA, Feldman EL. BTBR ob/ob mice as a novel diabetic neuropathy model: Neurological characterization and gene expression analyses. Neurobiol Dis. 2014;73C:348–355. doi: 10.1016/j.nbd.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien PD, Sakowski SA, Feldman EL. Mouse models of diabetic neuropathy. ILAR J. 2014;54(3):259–272. doi: 10.1093/ilar/ilt052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterbrand M, Fahlen M, Oden A, Eliasson B. A method to predict the metabolic effects of changes in insulin treatment in subgroups of a large population based patient cohort. Eur J Epidemiol. 2007;22(3):151–157. doi: 10.1007/s10654-007-9107-4. [DOI] [PubMed] [Google Scholar]
- Pande M, Hur J, Hong Y, Backus C, Hayes JM, Oh SS, Kretzler M, Feldman EL. Transcriptional profiling of diabetic neuropathy in the BKS db/db mouse: a model of type 2 diabetes. Diabetes. 2011;60(7):1981–1989. doi: 10.2337/db10-1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittenger GL, Ray M, Burcus NI, McNulty P, Basta B, Vinik AI. Intraepidermal nerve fibers are indicators of small-fiber neuropathy in both diabetic and nondiabetic patients. Diabetes Care. 2004;27(8):1974–1979. doi: 10.2337/diacare.27.8.1974. [DOI] [PubMed] [Google Scholar]
- Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nat Genet. 2006;38(5):500–501. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- Sartor MA, Tomlinson CR, Wesselkamper SC, Sivaganesan S, Leikauf GD, Medvedovic M. Intensity-based hierarchical Bayes method improves testing for differentially expressed genes in microarray experiments. BMC Bioinformatics. 2006;7:538. doi: 10.1186/1471-2105-7-538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Clegg DJ. Sex differences in the regulation of body weight. Physiol Behav. 2009;97(2):199–204. doi: 10.1016/j.physbeh.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skarstrand H, Dahlin LB, Lernmark A, Vaziri-Sani F. Neuropeptide Y autoantibodies in patients with long-term type 1 and type 2 diabetes and neuropathy. J Diabetes Complications. 2013;27(6):609–617. doi: 10.1016/j.jdiacomp.2013.06.007. [DOI] [PubMed] [Google Scholar]
- Smith AG, Singleton JR. Obesity and hyperlipidemia are risk factors for early diabetic neuropathy. J Diabetes Complications. 2013;27(5):436–442. doi: 10.1016/j.jdiacomp.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan KA, Hayes JM, Wiggin TD, Backus C, Su Oh S, Lentz SI, Brosius F, 3rd, Feldman EL. Mouse models of diabetic neuropathy. Neurobiol Dis. 2007;28(3):276–285. doi: 10.1016/j.nbd.2007.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan KA, Lentz SI, Roberts JL, Jr, Feldman EL. Criteria for creating and assessing mouse models of diabetic neuropathy. Curr Drug Targets. 2008;9(1):3–13. doi: 10.2174/138945008783431763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiano JP, Mauvais-Jarvis F. Importance of oestrogen receptors to preserve functional beta-cell mass in diabetes. Nat Rev Endocrinol. 2012;8(6):342–351. doi: 10.1038/nrendo.2011.242. [DOI] [PubMed] [Google Scholar]
- van Genugten RE, Utzschneider KM, Tong J, Gerchman F, Zraika S, Udayasankar J, Boyko EJ, Fujimoto WY, Kahn SE G. S. G. American Diabetes Association. Effects of sex and hormone replacement therapy use on the prevalence of isolated impaired fasting glucose and isolated impaired glucose tolerance in subjects with a family history of type 2 diabetes. Diabetes. 2006;55(12):3529–3535. doi: 10.2337/db06-0577. [DOI] [PubMed] [Google Scholar]
- Vincent AM, Hayes JM, McLean LL, Vivekanandan-Giri A, Pennathur S, Feldman EL. Dyslipidemia-induced neuropathy in mice: the role of oxLDL/LOX-1. Diabetes. 2009;58(10):2376–2385. doi: 10.2337/db09-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent AM, Hinder LM, Pop-Busui R, Feldman EL. Hyperlipidemia: a new therapeutic target for diabetic neuropathy. J Peripher Nerv Syst. 2009;14(4):257–267. doi: 10.1111/j.1529-8027.2009.00237.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiggin TD, Sullivan KA, Pop-Busui R, Amato A, Sima AA, Feldman EL. Elevated triglycerides correlate with progression of diabetic neuropathy. Diabetes. 2009;58(7):1634–1640. doi: 10.2337/db08-1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27(5):1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- Zenker J, Stettner M, Ruskamo S, Domenech-Estevez E, Baloui H, Medard JJ, Verheijen MH, Brouwers JF, Kursula P, Kieseier BC, Chrast R. A role of peripheral myelin protein 2 in lipid homeostasis of myelinating Schwann cells. Glia. 2014;62(9):1502–1512. doi: 10.1002/glia.22696. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.