

Abstract
Mutations in voltage-gated sodium channel genes cause several types of human epilepsies. Often, individuals with the same sodium channel mutation exhibit diverse phenotypes. This suggests that factors beyond the primary mutation influence disease severity, including genetic modifiers. Mouse epilepsy models with voltage-gated sodium channel mutations exhibit strain-dependent phenotype variability, supporting a contribution of genetic modifiers in epilepsy. The Scn2aQ54 (Q54) mouse model has a strain-dependent epilepsy phenotype. Q54 mice on the C57BL/6J strain exhibit delayed seizure onset and improved survival compared to [B6xSJL/J]F1.Q54 mice. We previously mapped two dominant modifier loci that influence Q54 seizure susceptibility and identified Hlf (hepatic leukemia factor) as a candidate modifier gene at one locus. Hlf and other PAR bZIP transcription factors had previously been associated with spontaneous seizures in mice thought to be caused by down-regulation of the pyridoxine pathway. An Hlf targeted knockout mouse model was used to evaluate the effect of Hlf deletion on Q54 phenotype severity. Hlf KO/KO;Q54 double mutant mice exhibited elevated frequency and reduced survival compared to Q54 controls. To determine if direct modulation of the pyridoxine pathway could alter the Q54 phenotype, mice were maintained on a pyridoxine-deficient diet for 6 weeks. Dietary pyridoxine deficiency resulted in elevated seizure frequency and decreased survival in Q54 mice compared to control diet. To determine if Hlf could modify other epilepsies, Hlf KO/+ mice were crossed with the Scn1aKO/+ Dravet syndrome mouse model to examine the effect on premature lethality. Hlf KO/+;Scn1aKO/+ offspring exhibited decreased survival compared to Scn1aKO/+ controls. Together these results demonstrate that Hlf is a genetic modifier of epilepsy caused by voltage-gated sodium channel mutations and that modulation of the pyridoxine pathway can also influence phenotype severity.
1. INTRODUCTION
Epilepsy is a common neurological disorder affecting approximately 50 million people worldwide (WHO, 2012). Over 1000 mutations identified in voltage-gated sodium channel genes result in several human epilepsy syndromes (Meisler et al., 2010). Often, individuals with the same mutation can exhibit strikingly different clinical severity. This suggests that the effect of the primary mutation is influenced by other factors, which may include genetic modifiers.
Several mouse models have been generated in order to study genetic epilepsies. Frequently, strain background alters the disease phenotype, supporting a contribution of genetic modifiers in epilepsy. The Scn2aQ54 (Q54) mouse model has a gain-of-function mutation that results in persistent sodium current and epilepsy (Kearney et al., 2001). On the C57BL/6J (B6) strain, Q54 mice have infrequent, adult-onset focal motor seizures and a 75% survival rate beyond six months of age (Bergren et al., 2005). When crossed with the SJL/J (SJL) strain, resulting F1.Q54 mice experience high seizure frequency with juvenile onset and less than 25% survival at six months of age (Bergren et al., 2005). We mapped two dominant loci that modify seizure susceptibility of Q54, designated Moe1 (Modifier of Epilepsy) on chromosome 11 and Moe2 on chromosome 19 (Bergren et al., 2005). In contrast to the overall effect, at the Moe1 locus B6 alleles confer increased seizure risk (Hawkins and Kearney, 2012). Fine-mapping and candidate gene analysis by RNA-seq suggested hepatic leukemia factor (Hlf ) as a candidate modifier at the Moe1. B6 mice express lower levels of Hlf transcript compared to SJL (Hawkins and Kearney, 2012), leading us to hypothesize that deletion of Hlf would increase seizure susceptibility.
Hlf is a member of the PAR bZIP transcription factor family, which includes Hlf, Dbp and Tef. Deletion of this family in a knock-out mouse model resulted in spontaneous generalized tonic-clonic and absence seizures, and premature lethality (Gachon et al., 2004). Two-fold reduction in expression of pyridoxal kinase (PDXK) was implicated as a likely contributor to the epilepsy phenotype. PDXK is essential for conversion of pyridoxine to pyridoxal 5′ phosphate (PLP), a key coenzyme involved in amino acid and neurotransmitter metabolism, including GABA and glutamate (John, 1995). Pyridoxine deficiency in humans results in epilepsy that is successfully treated by PLP administration (Plecko and Stockler, 2009). Based on its prior association with seizures and the pyridoxine pathway, we hypothesized that genetic variation in Hlf could modify the Q54 epilepsy phenotype (Hawkins and Kearney, 2012).
To determine if Hlf could modify the Q54 phenotype, we evaluated the effect of Hlf deletion on seizures and survival in Q54 mice. Additionally, we tested whether manipulation of the pyridoxine pathway could modify the Q54 phenotype. Finally, to determine whether Hlf would modify epilepsy in another model, we evaluated the effect of Hlf deletion on phenotype of the Scn1a+/− Dravet syndrome model.
2. MATERIAL AND METHODS
2.1 Mice
Hlf tm1Schb (Hlf KO/+) knockout embryos congenic on C57BL/6 were obtained from the European Mouse Mutant Archive (www.emmanet.org) (Gachon et al., 2004). Q54 transgenic mice [Tg(Eno2-Scn2a1*)Q54Mm] congenic on C57BL/6J were previously described (Bergren et al., 2005; Kearney et al., 2001). Scn1atm1Kea (Scn1aKO/+) heterozygous knockout mice congenic on 129S6/SvEvTac (129) were previously described (Miller et al., 2014). Genotyping was performed by PCR of DNA isolated from postnatal day 14 (P14) tail biopsies. Mice were group-housed with access to food and water ad libitum. All studies were approved by the local Animal Care and Use Committees.
Hlf;Q54 double mutant mice were generated by crossing Hlf KO/+ females with Q54 males to generate Hlf KO/+;Q54 offspring, which were then crossed with Hlf KO/+. Resulting offspring included Hlf KO/KO;Q54 and Hlf KO/+;Q54 test mice, and single mutant Q54, Hlf KO/+, and Hlf KO/KO control littermates. Hlf;Scn1aKO/+ double mutant mice were generated by crossing B6. Hlf KO/+ and 129.Scn1aKO/+ mice. Resulting F1 offspring included double mutant Hlf KO/+;Scn1aKO/+ test mice, and Scn1aKO/+, and Hlf KO/+control littermates.
2.2 Phenotyping
Mice were phenotyped as previously described (Hawkins and Kearney, 2012). Briefly, littermates underwent 30 minute video-taping at three and six weeks of age. Behavioral seizures were scored offline by a blinded observer using ObserverXT software (Noldus). Prior extensive video-electroencephalogram (EEG) monitoring of Q54 mice demonstrated strong correlation between behavioral and EEG seizures (κ=0.988) (Anderson et al., 2014; Kearney et al., 2001). The number of focal motor seizures with forelimb clonus and repetitive movements lasting 1–5 seconds, and generalized tonic-clonic seizures (GTCS) with rearing and falling lasting approximately 1 min were counted. Seizure counts from the three and six week observations were combined to obtain the total number of focal motor seizures and GTCS in 60 minutes for each animal.
2.2 Video-EEG Monitoring
Hlf KO/KO;Q54 (n=4) and Hlf KO/+;Q54 (n=2) mice were implanted with headmounts for video-EEG monitoring (Pinnacle Technology, Inc.) as previously described (Hawkins et al., 2011). After ≥ 24 hours of recovery, EEG was recorded from freely moving mice. Hlf KO/KO;Q54 and Hlf KO/+;Q54 mice were monitored for a total of 151 and 52 hours, respectively. Acquisition and analysis was performed with Sirenia software along with contemporaneous video recordings (Pinnacle Technology, Inc.). Epileptiform activity was scored manually.
2.3 Pyridoxine Deficient Diet
Diets pellets were formulated by Research Diets, Inc. to contain a standard dietary amount of pyridoxine HCl (AIN-93G/5.8 ppm) or with no added pyridoxine (AIN-93G/no pyridoxine HCl). Wildtype and Q54 littermates were randomly assigned to pyridoxine deficient or control diet at 3 weeks of age and maintained on the diet for 6 weeks. At nine weeks of age, surviving mice underwent 30 minute video-taping for seizures, as described above.
3. RESULTS AND DISCUSSION
3.1 Loss of Hlf Exacerbates Q54 Phenotype
To determine whether Hlf could act as a modifier of the Q54 epilepsy phenotype, we evaluated the effect of Hlf deletion on Q54 seizure severity using an Hlf knockout model. The number and type of seizures at 3 and 6 weeks of age were compared between Hlf;Q54 double mutant mice and littermate controls. There was no difference in the number of mice exhibiting focal motor seizures (Figure 1a) or in the average number of focal motor seizures observed (Figure 1b). GTCS were only observed in Hlf KO/KO;Q54 mice (Figure 1a &1b). This more severe seizure type has never been observed in Q54 mice of this age in our previous studies (Bergren et al., 2009; Hawkins and Kearney, 2012; Jorge et al., 2011; Kearney et al., 2001). Video-EEG monitoring performed on additional Hlf KO/KO;Q54 mice confirmed the generalized electrographic seizure activity of Hlf KO/KO;Q54 mice (Figure 1d).
Figure 1. Loss of Hlf exacerbates the Scn2aQ54 phenotype.

A) Mice were observed for focal motor seizures (FMS) and generalized tonic clonic seizures (GTCS) in 30 minute sessions at 3 and 6 weeks of age. The percentages of mice exhibiting FMS or GTCS during the observation period are shown (n=30–32 per group). Significant differences between groups were determined by Fisher’s exact test. *p < 0.0134 compared to Q54 littermate controls. B) The average number of FMS (±S.E.M) and total number of GTCS observed is shown (n=30–32 per group). C) Kaplan-Meier plot of survival to 6 weeks of age. Hlf KO/KO;Q54 had a significant decrease in survival compared to Q54 mice (p=0.0215, LogRank Mantel-Cox). D) Representative EEG recording from a Hlf KO/KO;Q54 during a seizure. Time span of plot is 3 minutes. Top trace is right posterior to left posterior; bottom trace is right anterior to left posterior.
Hlf KO/KO;Q54 mice exhibited a significant reduction in survival compared to Q54 (Figure 1c). By six weeks of age, 25% of Hlf KO/KO;Q54 died, compared to only 3% of Q54 mice. Consistent with previous reports, Hlf KO/+ and Hlf KO/KO mice had 100% survival throughout the study (data not shown) (Gachon et al., 2004). Together, our seizure and survival data demonstrate that complete loss of Hlf exacerbated the Q54 epilepsy phenotype.
3.2 Effect of Pyridoxine Deficient Diet on Q54
We hypothesized that B6.Q54 mice might be sensitized to pyridoxine deficiency based on previous data that showed reduced Hlf brain transcript in B6 mice (Hawkins and Kearney, 2012), and involvement of Hlf in the transcriptional regulation of PDXK, a key enzyme in the pyridoxine pathway (Gachon et al., 2004). To determine if direct modulation of the pyridoxine pathway would modify their phenotype, Q54 mice were maintained on either a pyridoxine deficient or control diet for six weeks, beginning at 3 weeks of age. Survival was monitored during this time, and seizure frequency was evaluated in a 30 minute session at nine weeks of age. Q54 mice maintained on a pyridoxine deficient diet had significantly more focal motor seizures compared to those on control diet (Figure 2a). No GTCS were observed in the pyridoxine deficient or control diet group. Q54 mice maintained on the pyridoxine deficient diet had a significant reduction in lifespan compared to controls (Figure 2b). Only 69% of Q54 mice on the deficient diet survived, compared to 94% on control diet. Thus, pyridoxine deficiency exacerbated the Q54 epilepsy phenotype, although this was not as dramatic as Hlf deletion since we did not observe GTCS. This suggests that Hlf deletion may have additional effects. Nonetheless, these results suggest that maintaining adequate pyridoxine levels is important in the context of epilepsy caused by a sodium-channel mutation.
Figure 2. Pyridoxine deficiency exacerbates the Scn2aQ54 phenotype.

A) Average FMS frequency of Q54 mice maintained on pyridoxine deficient or control diets from 3 to 9 weeks of age. The number of FMS was counted in a 30 minute session at 9 weeks of age. Significant differences were determined by Student’s T-test (*p < 0.03). Data are represented as mean ± SEM. B) Survival of Q54 mice maintained on pyridoxine deficient or control diets was monitored until nine weeks of age. Kaplan Meier survival plot is shown. Q54 mice on the deficient diet had a significant decrease in survival compared to control diet (p<0.002, LogRank Mantel-Cox).
3.3 Loss of Hlf exacerbates phenotype of Scn1aKO/+ Dravet mice
To determine if the Hlf modifier effect would translate to other epilepsy models, we assessed survival of Hlf KO/+;Scn1aKO/+ double mutant mice. We observed a significant reduction in lifespan between Hlf KO/+;Scn1aKO/+ and Scn1aKO/+ mice (Figure 3). By four weeks of age, only 45% of Hlf KO/+;Scn1aKO/+ mice survived, compared to 71% of Scn1aKO/+ mice. Scn1aKO/+ mice recapitulate many features of Dravet syndrome, including development of early onset epilepsy, spontaneous seizures and premature death (Kalume et al., 2013; Mistry et al., 2014; Yu et al., 2006). It is hypothesized that reduced GABAergic inhibitory function responsible for the Scn1aKO/+ seizure phenotype (Yu et al., 2006). This mechanism differs from Q54 mice, where seizures result from hyperexcitability of principal excitatory neurons (Kearney et al., 2001). Exacerbation of the epilepsy phenotype in a second model with a different underlying mechanism suggests that loss of Hlf alters excitability in a common pathway, perhaps by altering neurotransmitter synthesis.
Figure 3. Loss of Hlf exacerbates premature lethality phenotype of Scn1aKO/− Dravet mice.
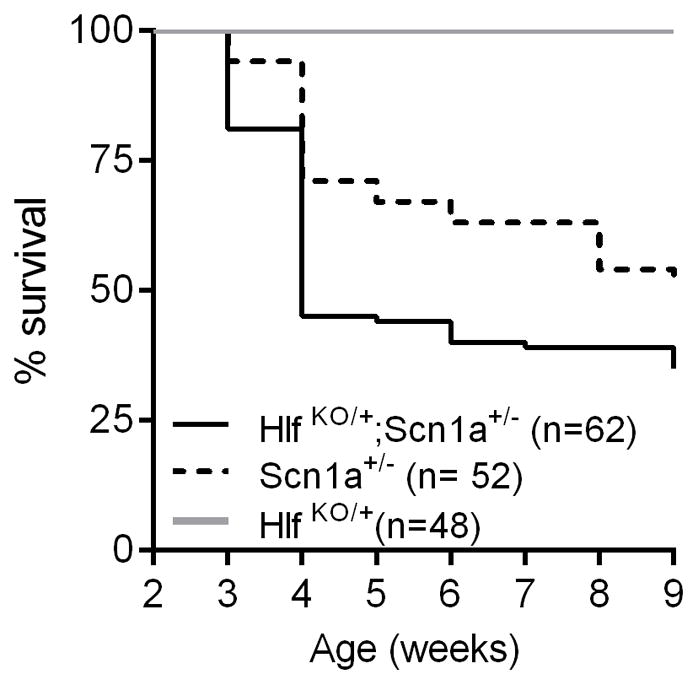
Survival of Hlf KO/+, Scn1aKO/+ and Hlf KO/+;Scn1aKO/+ mice was monitored until nine weeks of age. Kaplan Meier survival plot is shown. Hlf KO/+;Scn1aKO/+ mice had reduced survival compared to Scn1aKO/+ mice (p<0.006, LogRank Mantel-Cox).
3.4 Conclusions
We demonstrated that loss of Hlf modifies phenotypes in two genetic epilepsy models, Q54 and Scn1aKO/+ mice. The mechanism whereby Hlf exerts its effect remains to be determined in future studies. Elucidation of the underlying mechanism may suggest novel therapeutic strategies for improved treatment of epilepsy.
HIGHLIGHTS.
Genetic deletion of Hepatic leukemia factor (Hlf) exacerbated epilepsy in Scn2aQ54 mice.
Pyridoxine deficiency exacerbated the epilepsy phenotype of Scn2aQ54 mice.
Genetic deletion of Hlf worsened survival in the Scn1a+/− model of Dravet syndrome.
Hlf acts as a genetic modifier of epilepsy, possibly by pyridoxine pathway modulation.
Acknowledgments
We thank Clint McCollom, Alison Miller and Nicole Zachwieja for technical assistance. This work was supported by NIH grants R01-NS053792 (J.A.K.), F31-NS077700 (N.A.H.) and an Epilepsy Foundation Predoctoral Fellowship (N.A.H.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anderson LL, Thompson CH, Hawkins NA, Nath RD, Petersohn AA, Rajamani S, Bush WS, Frankel WN, Vanoye CG, Kearney JA, George AL., Jr Antiepileptic activity of preferential inhibitors of persistent sodium current. Epilepsia. 2014;55:1274–1283. doi: 10.1111/epi.12657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergren SK, Chen S, Galecki A, Kearney JA. Genetic modifiers affecting severity of epilepsy caused by mutation of sodium channel Scn2a. Mamm Genome. 2005;16:683–690. doi: 10.1007/s00335-005-0049-4. [DOI] [PubMed] [Google Scholar]
- Bergren SK, Rutter ED, Kearney JA. Fine mapping of an epilepsy modifier gene on mouse Chromosome 19. Mamm Genome. 2009;20:359–366. doi: 10.1007/s00335-009-9193-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gachon F, Fonjallaz P, Damiola F, Gos P, Kodama T, Zakany J, Duboule D, Petit B, Tafti M, Schibler U. The loss of circadian PAR bZip transcription factors results in epilepsy. Genes Dev. 2004;18:1397–1412. doi: 10.1101/gad.301404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins NA, Kearney JA. Confirmation of an epilepsy modifier locus on mouse chromosome 11 and candidate gene analysis by RNA-Seq. Genes, brain, and behavior. 2012;11:452–460. doi: 10.1111/j.1601-183X.2012.00790.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins NA, Martin MS, Frankel WN, Kearney JA, Escayg A. Neuronal voltage-gated ion channels are genetic modifiers of generalized epilepsy with febrile seizures plus. Neurobiology of disease. 2011;41:655–660. doi: 10.1016/j.nbd.2010.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John RA. Pyridoxal phosphate-dependent enzymes. Biochim Biophys Acta. 1995;1248:81–96. doi: 10.1016/0167-4838(95)00025-p. [DOI] [PubMed] [Google Scholar]
- Jorge BS, Campbell CM, Miller AR, Rutter ED, Gurnett CA, Vanoye CG, George AL, Kearney JA. Voltage-gated potassium channel KCNV2 (Kv8.2) contributes to epilepsy susceptibility. Proc Natl Acad Sci U S A. 2011;108:5443–5448. doi: 10.1073/pnas.1017539108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalume F, Westenbroek RE, Cheah CS, Yu FH, Oakley JC, Scheuer T, Catterall WA. Sudden unexpected death in a mouse model of Dravet syndrome. J Clin Invest. 2013;123:1798–1808. doi: 10.1172/JCI66220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney JA, Plummer NW, Smith MR, Kapur J, Cummins TR, Waxman SG, Goldin AL, Meisler MH. A gain-of-function mutation in the sodium channel gene Scn2a results in seizures and behavioral abnormalities. Neuroscience. 2001;102:307–317. doi: 10.1016/s0306-4522(00)00479-6. [DOI] [PubMed] [Google Scholar]
- Meisler MH, O’Brien JE, Sharkey LM. Sodium channel gene family: epilepsy mutations, gene interactions and modifier effects. J Physiol. 2010;588:1841–1848. doi: 10.1113/jphysiol.2010.188482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AR, Hawkins NA, McCollom CE, Kearney JA. Mapping genetic modifiers of survival in a mouse model of Dravet syndrome. Genes, brain, and behavior. 2014;13:163–172. doi: 10.1111/gbb.12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry AM, Thompson CH, Miller AR, Vanoye CG, George AL, Jr, Kearney JA. Strain- and age-dependent hippocampal neuron sodium currents correlate with epilepsy severity in Dravet syndrome mice. Neurobiology of disease. 2014;65:1–11. doi: 10.1016/j.nbd.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plecko B, Stockler S. Vitamin B6 dependent seizures. Can J Neurol Sci. 2009;36(Suppl 2):73–77. [PubMed] [Google Scholar]
- World Health Organization. Epilepsy Fact sheet N°999. 2015. [Google Scholar]
- Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, Spain WJ, McKnight GS, Scheuer T, Catterall WA. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. 2006;9:1142–1149. doi: 10.1038/nn1754. [DOI] [PubMed] [Google Scholar]