

Abstract
The communesin alkaloids are a diverse family of Penicillium-derived alkaloids. Their caged-polycyclic structure and intriguing biological profiles have made these natural products attractive targets for total synthesis. Similarly, the ascidian-derived alkaloid, perophoramidine, is structurally related to the communesins and has also become a popular target for total synthesis. This review serves to summarize the many elegant approaches that have been developed to access the communesin alkaloids and perophoramidine. Likewise, strategies to access the communesin ring system are reviewed.
Keywords: alkaloids, total synthesis, communesin, perophoramidine, vicinal quaternary stereocenters
Graphical Abstract

1. Introduction
In 1993, Numata and coworkers disclosed the isolation and structural elucidation of two complex alkaloid natural products, communesins A (1) and B (2) (Figure 1), from a strain of Penicillium sp. found growing on the mycelium of the algal species Enteromorpha intestinalis. 1 These alkaloids displayed cytotoxicity against cultured lymphocytic leukemia cells with ED50 values of 3.5 and 0.45 μg/mL respectively. The structures of communesins A (1) and B (2) were elucidated using a combination of 1D and multidimensional NMR techniques, IR, and LC/MS. The absolute configuration of the two alkaloids was not determined and neither was the relative stereochemistry at C(11).
Figure 1.

Communesins A (1), B (2), and nomofungin (3)
In 2001 Hemscheidt et al. reported the isolation of an alkaloid from an unidentified fungal organism growing on the bark of the Hawaiian banyan tree Ficus microcarpa. 2 This fungus died soon after its isolation and could not be obtained again from the tree bark. As a result, the authors named the novel natural product “nomofungin” (3) (no more fungus). Bioassays of nomofungin (3) revealed that it exhibited cytotoxicity against LoVo and KB cells with MICs (minimal inhibitory concentration) of 2 and 4.5 μg/mL respectively. The mode of action for this cytotoxicity was posited to occur through microfilament network disruption. Structurally, nomofungin (3) was elucidated to having a structure very similar to the already know communesin B (2). However, in place of the N,N aminal functionality present at C(6) of communesin B (2), Hemscheidt and coworkers proposed that an N,O aminal was present at C(6) in nomofungin (3). Additionally, the absolute configuration of the natural product was assigned using the exciton chirality method. The proposed structure of nomofungin (3) gained substantial attention from the synthetic community because it seemed to Additionally, the structural assignment was found to be incorrect (vide infra).
1.1. Isolation of Other Communesin Alkaloids
Since their initial isolation by Numata et al. the family of communesin alkaloids has grown substantially to include many new members (Figure 2). Jadulco and coworkers isolated communesins C (4) and D (5) from Penicillium sp., which was found on the Mediterranean sponge Axinella verrucosa along with the already known communesin B (2).3 The newly isolated alkaloids differed structurally from communesin B (2) by the identity of the substituent present at N(15). All three alkaloids were tested for antiproliferative activity against several leukemia cell lines by MTT assay including Burkett's Lymphoma and Hodgkin's lymphoma, and displayed ED50 values in the 7-15 μM range. In addition to their activity against cancer lines, communesins B-D (2-5) also displayed toxicity against the brine shrimp, Artemia salina, with activities ranging from 0.3-2.0 μg/mL.
Figure 2.

Structures of isolated communesin alkaloids
Hayashi and coworkers reported the isolation and structural elucidation of communesins D (5), E (6), and F (7), from the fungus Penicillium expansum Link MK-57 along with the already known communesins A (1) and B (2).4 Communesin E (6) contained an acyl chain at N(16) in place of the sorbyl chain present in communesins B-D (2-5). Communesin F (7) was the structurally simplest member isolated and lacked the epoxide functionality present in communesins A-E (1-6). Communesins E (6) and F (7) displayed moderate insecticidal activity against instar silkworms larvae, displaying toxicities of 80-300 μg/g of diet fed to the organism.
Christophersen et al. reported the structures of communesins G (8) and H (9) from the psychrotolerant organism, Penicillium rivulum.5 These two alkaloids contained propionyl and butanoyl substituents respectively at N(16). Unlike the other penicillium organisms, Penicillium rivulum did not produce communesin B (2) as a secondary metabolite. Communesins G (8) and H (9) were tested for antiviral, antibacterial, and antiproliferative activity against murine leukemia cells and were found to be inactive in such assays.
A related natural product, perophoramidine (10), was isolated in 2002 by Ireland and coworkers from Philippine ascidian organism, Perophora namei (Figure 3). Structurally, this alkaloid contained a novel hexacyclic carbon skeleton similar in connectivity to that of the communesin alkaloids. However, unlike the communesins, perophoramidine (10) contained 3 points of halogenation on the aromatic nuclei, possessed two amidine functionalities in place of two aminals, lacked the benzazepine ring and possessed a trans relationship between the two vicinal quaternary stereocenters. The absolute configuration of the natural product was not determined. Initial biological assays revealed that this natural product was active against a HCT116 colon carcinoma cell line with an IC50 of 60 μM, inducing apoptosis via (poly(adenosine-5’-diphosphateribose)polymerase (PARP) cleavage within 24 h.
Figure 3.

Communesin B (2) and perophoramidine (10).
1.2. Biosynthesis of the Communesin Alkaloids and Perophoramidine
Stoltz and coworkers have studied the biosynthesis of the communesin alkaloids, proposing that this family of natural products could arise from the inverse-electron-demand hetero-Diels-Alder reaction of the ergot alkaloid aurantioclavine 11 and a tryptamine-derived aza-o-xylylene 12 (Scheme 1). In order to arrive at the correct relative stereochemistry between the two quaternary stereocenters, the Diels-Alder reaction would need to proceed through an exo transition state. Due to the large amount of strain, intermediate 13 would undergo transamidation with the pendant amine to deliver lactam 14. Subsequent reduction of the lactam, aminal closure, epoxidation and amination would deliver the communesin alkaloids.
Scheme 1.

Proposed biosynthetic pathway for the communesin alkaloids
A similar type of dimerization reaction can be envisioned for the biosynthesis of perophoramidine (10), utilizing tryptamine 15 and methyltryptamine 16 (Scheme 2). However, an endo transition state would have to be operative in the inverse-electron-demand hetero-Diels-Alder reaction in order to obtain the trans-relationship between the two quaternary stereocenters. Halogenation of the aromatic nuclei could occur prior to the cycloaddition or in subsequent enzymatic steps during biosynthesis.
Scheme 2.

Proposed biosynthesis of perophoramidine (10)
Stoltz and coworkers have also proposed that the communesin alkaloids share structural and biosynthetic similarity to the calycanthaceous family of alkaloids. 6 The calycanthaceous alkaloids are a broad class of natural products found in a number of plants and animals. Due to their variegated biological activities and intriguing skeletal structure, these natural products have received extensive attention from the synthetic community 7 and are believed to arise from the oxidative dimerization of two tryptamine units (17), providing dimer 18 (Scheme 3). From this intermediate, ring opening can give rise to a hypothetical intermediate 19, from which 5 possible skeletal isomers can be generated by rearrangement of the aminal moieties (20-24). Of these 5 possibilities, only 4 have been observed in natural products.
Scheme 3.

The Biosynthetic Relationship Between Cyclotryptamine Alkaloids and Communesin Alkaloids
Calycanthine 25 (Figure 4) corresponds to skeletal arrangement 20 and was first isolated in the late 1800's,8 but its structural identity remained a long-standing question until Woodward 9 and Robinson 10 utilized chemical degradation methods to assign its correct structure. Later, Robertson and coworkers were also able to establish the structure of 25 using X-ray crystallography.11
Figure 4.

Representative Cyclotryptamine Alkaloids
Skeletal isomer 21 is observed in the alkaloid chimonanthine 26, which has been isolated in both its enantiomers as well as in its meso form. This natural product can be found in many different plants 12 and on the skin of the poison dart frog Phyllobates terribilis. 13 The structure of (−)-chimonanthine 26 was elucidated using X-ray crystallography.14 Interestingly, (−)-chimonanthine 26 undergoes rearrangement to (+)-calycanthine 25 upon exposure to aqueous acid, raising the question of whether calycanthine 25 is in fact a natural product itself or an isolation artifact. Iso-calycanthine (27) was isolated from the South American shrub Psychotria forsteriana and corresponds to skeletal arrangement 23.15 An oxidized variant of this meso natural product (28), which contains two amidines, was also later isolated from P. forsteriana. 16 Finally, skeletal arrangement represented by 22 is observed in the communesin alkaloids and perophoramidine (10).
2. Studies Towards the Total Syntheses of the Communesin Alkaloids and Perophoramidine
Structurally, the communesin alkaloids contain a unique, densely functionalized, cage-like heptacylic skeleton making them a formidable synthetic endeavor (Figure 5). The southern portion of these alkaloids consists of a fused 6,5,6,6-ring system represented by rings C, D, E, and F which contains an indoline/tetrahydroquinoline aminal. The northern portion, represented by rings A, B, and G, contains an additional 6,5-aminal and a 7-membered benzazepine attached to the 4-position of the indoline ring. Additionally, the communesins bear two vicinal quaternary all carbon stereocenters at C(7) and C(8) which are challenging to construct, especially in an asymmetric fashion.17,18
Figure 5.
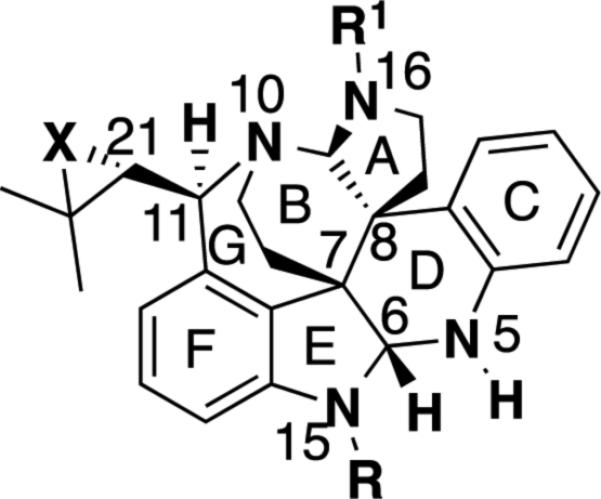
The communesin skeleton
The communesins have attracted extensive research interests from the synthetic community, and many unique strategies have been developed to access these alkaloids in both racemic and enantioenriched form. 19 The following sections of this review article will focus on highlighting the methods that have been developed to access the communesin skeleton and the total syntheses of the communesin alkaloids as well as perophoramidine (10).
2.1 Resolving the Structures of Communesin B and Nomofungin
Although communesins A (1) and B (2) were first isolated in 1993, they received little attention from the synthetic community until Hemscheidt et al. reported the structure of nomofungin (3). The first synthetic studies performed towards the communesin alkaloids were focused on clarifying the structure of communesin B (2) and demonstrating that the structure of nomofungin (3) was erroneously elucidated. In 2003, Stoltz 20 and Funk 21 independently demonstrated that the structure of nomofungin 3 was incorrect, and that the compound that was isolated by Hemscheidt et al. was in fact communesin B (2). Stoltz and coworkers examined the reported NMR data for both communesin B (2) as well as nomofungin (3) and found that the compounds shared nearly identical reported 1H and 13C NMRs. Inspection of the chemical shifts for the C(6) protons and carbons revealed that the N,N-aminal hydrogen in communesin B (2) appeared at 4.70 ppm, while nomofungin (3) appeared at 4.69 ppm. Additionally, both compounds were reported to have a chemical shift in the 13C spectrum for C(6) at 82.5 ppm. A comparison of these shifts for the two natural products to known N,N- and N,O-aminals from the chemical literature revealed that both the 1H and 13C chemical shifts for the N,N-aminal were within normal range for such functionalities. On the other hand, the chemical shifts for the N,O-aminal were outside the typical range reported in the chemical literature.
Additionally, Stoltz and coworkers prepared two N,N-aminals similar in structure to those found in communesin B and compared their chemical shifts at the N,N-aminal to that of the two natural products (Scheme 4). The preparation of these compounds commenced with the cycloaddition of 1,3-dimethyl indole (29) with an aza-o-xylylene generated from aniline 30, affording tetracyclic product 31 in 86% yield. Cleavage of the tosyl group with Mg(0) delivered the requisite aminal 32 in 89% yield. The 1H and 13C chemical shifts for the N,N-aminal were examined and appeared at 4.14 and 83.9 ppm, respectively. While the aminal 1H NMR shifts were outside the expected range for what was expected, the 13C chemical shifts for the aminal carbon were in good agreement with communesin B. To access a system more closely related in structure to communesin B (2), the authors prepared aurantioclavine-derived pentacyclic aminal 35 from the hetero-Diels-Alder reaction of aurantioclavine derivative 33 and aniline 30 followed by cleavage of the tosyl group with Mg(0). The aminal 13C signals for the two diastereomeric compounds appeared at 84.8 and 83.9 ppm, again supporting the notion that communesin B (2) and nomofungin (3) were identical compounds and that the proposed structure of nomofungin (3) was incorrect. The 1H chemical shifts were not discussed for these compounds.
Scheme 4.

Stoltz's Synthesis of Communesin-like N,N-Aminals
To demonstrate that communesin B (2) and nomofungin (3)were the same compound, Funk and coworkers prepared and analyzed N,O- and N,N-aminals 41 and 45 (Scheme 5 and 6). N,O-aminal 41 was prepared via intramolecular hetero-Diels-Alder reaction of an indole and an aza-o-xylylene. Acyl chloride 37 was prepared in three steps from ester diol 36 by formation of the acetonide, saponification of the ethyl ester and treatment of the carboxylic acid with oxalyl chloride. Benzazepine 39 was prepared by hydrogenation of the nitro group of indole 38 and reduction of the resulting lactam with LiAlH4. Coupling of acyl chloride 37 with benzazepine 39 followed by reduction of the amide with LiAlH4 provided benzodioxin 40. To facilitate the [4+2] process, benzodioxin 40 was heated in decalin at 195 °C providing N,O-aminal 41 in 63% yield. Analysis of the 1H and 13C NMR signals of this compound at the N,O-aminal revealed that the signals appeared at 5.4 and 101.0 ppm, respectively. Comparison of these chemical shifts to those of the N,O-aminal present in nomofungin 3 revealed a large inconsistency. The 1H and 13C shifts for nomofungin appeared at 4.7 ppm and 82.4 ppm, respectively, which are both substantially further upfield from those observed in N,O-aminal 41. Conformational effects were very unlikely to lead to such a difference, suggesting that the reported structure for nomofungin (3) was incorrect and that nomofungin was in fact communesin B (2).
Scheme 5.

Funk's Synthesis of N,O-aminal (41)
Scheme 6.

Funk's Synthesis of N,N-Aminal (45)
To further validate their structural hypothesis a similar analysis was performed on N,N-aminal 45, which was prepared in 4 steps (Scheme 6). Benzazepine 39 was alkylated with styrene oxide 42 and the resulting secondary alcohol was converted to carbonate 43. Heating this compound at 160 °C in dichlorobenzene resulted in formation of the aza-o-xylylene intermediate and facilitated the desired [4+2] cycloaddition, providing endo-cycloadduct 44 in 70% yield as a single diastereomer. This compound was converted to N,N-aminal 45 by hydrolysis of the ethyl carbamate with KOH and hydrazine in 67%. Inspection of the NMR spectra of compound 45 revealed that the N,N-aminal 1H and 13C signals appeared at 4.5 and 84.4 ppm respectively. These were in accord with the NMR spectra of communesin B (2) and nomofungin (3), whose signals appeared at 4.7 and 82.4 ppm, suggesting that both communesin B (2) and nomofungin (3) were the same molecule containing an N,N-aminal.
2.2. Studies on the Construction of the Communesin Ring System
Building off of their thermolysis studies, the Funk group reported acid and fluoride-promoted protocols for generating aza-o-xylylenes and utilized these methods to rapidly assemble the hexacyclic substructure of the communesin alkaloids.22 Due to their ready availability, initial studies utilized a tryptamine starting material that lacked a functional handle at the 4-position of the indole nucleus (Scheme 7). Functionality at this position would later be necessary to install the benzazepine ring present in the natural product. To test the feasibility of employing an aza-o-xylylene cycloaddition to synthesize the natural product, aziridine 48 was prepared from N-methyl tryptamine 46 and dibromide 47 in 65% yield. The relative stereochemistry of this compound was confirmed by comparison of the vicinal coupling constants of the aziridine protons, which were in agreement with other trans-aziridines in the chemical literature.23 Treatment of aziridine 48 with triflimide (Tf2NH) delivered cycloadduct 52 in 70% yield in which the carboethoxyl group migrated from the aniline to the lactam. The relative stereochemistry of this compound was assigned based on the coupling constants observed between the α and β-methine protons of the ester and NOEs between the aminal protons and the β-methine proton of the ester. The structure was also confirmed by X-ray crystallography. To rationalize the observed migration, the authors proposed that aziridine 48 undergoes nucleophilic attack into the carboethoxyl group forming aziridinium 49, which retains the carboethoxyl group upon collapse of the tetrahedral intermediate. At this point, the aziridinium fragments to the aza-o-xylylene 51 and undergoes the endo-hetero-Diels-Alder reaction delivering 52. The migration of the carboethoxyl group proved problematic, since substitution at the lactam nitrogen would preclude subsequent studies on the benzazepine formation.
Scheme 7.

Funk's Synthesis of Pentacycle 52
To circumvent the migration pathway, the fluoride-labile 2-(trimethylsilyl)ethyl carbamate (TEOC) was introduced in place of the ethyl carbamate in aziridine 48 (Scheme 8). To test the compatibility of the TEOC group in the aza-o-xylylene cycloaddition, aziridine 53 was treated with TBAF facilitating the aza-o-xylylene formation. Subsequent endo-cycloaddition with this intermediate provided pentacycle 55 in 80% yield.
Scheme 8.

Funk's Aza-o-Xylylene Cycloaddition with a TEOC-Protected Aziridine 53
With a set of conditions for the aza-o-xylylene cycloaddition, 4-ethynyl indole 56 was prepared, which contains a functional handle at the 4-position of the indole nucleus needed to complete the communesin alkaloids (Scheme 9). Treatment of 56 with TBAF facilitated the [4+2] cycloaddition, providing endo-cycloadduct 57. This intermediate was unstable and slowly underwent 7-exo-dig benzazepine formation to 58, which conveniently was the next intermediate in the synthetic sequence. To accelerate this desired but otherwise slow process, endo-cycloadduct 57 was treated with an Au(I) source ro activate the alkyne providing benzazepine 58 in 89% yield over 2 steps. To install the second aminal present in the communesin system, benzazepine 58 was treated with LiOH to hydrolyze the ester moiety to the carboxylic acid, which was converted to a mixed anhydride and treated with sodium azide facilitating the Curtius rearrangement and delivering aminal 59. Aminal 59 constitutes a formidable partial synthesis, and contains six of the seven rings present in the communesin alkaloids.
Scheme 9.

Funk's Synthesis of Hexacycle 59
Weinreb and coworkers have also tackled the total syntheses of the communesin alkaloids and perophoramidine. Approaches to these natural products have hinged on the use of a Heck reaction to prepare oxindole products bearing one of the two quaternary stereocenters present in the targets. Weinreb's initial studies were aimed at utilizing a halogen-selective carbonylative Heck reaction to forge a quaternary stereocenter in the total synthesis of perophoramidine (Scheme 10). 24 The synthesis of the necessary Heck precursor was initiated with the olefination of lactone ylide 60 and anisaldehyde 61, providing benzylidene lactone 62 exclusively as the E-isomer. Weinreb aminolysis of this compound with the aluminum amide of aniline 63 delivered primary alcohol 64 which was silylated with TBSCl. Attempts to facilitate the carbonylative Heck reaction at this stage did not lead to any of the desired oxindole product. The authors postulated that this lack of reactivity was attributed to the low population of the amide rotamer needed to facilitate the reaction and that substitution of the amide N–H would promote the desired Heck reaction. For this purpose, amide 65 was alkylated with MeI and MOMCl to afford the two substituted Heck reaction precursors 66 and 67 respectively. When the key carbonylative Heck reaction was attempted on these alkylated substrates, the Me (68) and MOM (69) oxindoles were obtained as single diastereomers in 74 and 88% yield, respectively. The observed relative stereochemistry of the products was rationalized by the syn addition of the aryl-Pd intermediate and CO across the E-olefin.
Scheme 10.

Synthesis of Oxindoles 68 and 69
To cleave the silyl ether, the N-methyl oxindole product 68 was treated with TBAF and NH4F under conditions developed by Fürstner25, and the resulting primary alcohol was heated with TsOH to arrive at lactone 70 in 79% yield as a 3:1 mixture of epimers (Scheme 11). Analogously, N-MOM oxindole 69 was treated with HCl in MeOH to cleave the silyl ether and then was heated with TsOH to afford 71 as a single diastereomer. Installation of the second vicinal quaternary stereocenter was pursued via allylation of MOM lactone 71 with NaH and allyl iodide in DMF, which resulted in the formation of allyl lactone 72 in 65% yield as a single diastereomer. While the authors originally assigned the stereochemistry of this allylation as shown in Scheme 11, which agrees with the stereochemistry of perophoramidine (10), this stereochemical assignment was later found to be incorrect and the opposite diastereomer was actually obtained (vide infra).
Scheme 11.

Elaboration of Weinreb's Synthesis
Since perophoramidine (10) contains an aryl bromide on one of the two aromatic nuclei, the authors also explored a similar sequence starting with 4-bromo-benzaldehyde 73 targeting lactone 78 as an intermediate (Scheme 12). Unlike aryl chlorides, aryl bromides can readily undergo Heck reactions and the proposed reaction would be challenging from a chemoselectivity perspective. However, the oxidative insertion of palladium into an aryl iodide should be substantially faster than oxidative insertion into an aryl bromide, and hence this chemoselective reaction should be a viable process. Olefination of 4-bromo benzaldehyde 73 with ylide 60 followed by aminolysis with aniline 63 delivered a primary alcohol (75), which was silylated with TBSCl, treated with LiHMDS and MeI to arrive at Heck precursor 76. Treatment of the Heck precursor 76 under the previously optimized conditions resulted in smooth formation of N-methyl oxindole 77 in 77% yield as a single diastereomer. This compound was treated with HCl in MeOH followed by heating with TsOH in PhH to arrive at lactone 78 as a 5:1 mixture of diasteromers in 80% yield over 2 steps.
Scheme 12.

Synthesis of Bromolactone 78
With the success of a brominated substrate, additional studies were undertaken by Weinreb to synthesize perophoramidine (10) by employing brominated nitrobenzaldehyde 80.26 The authors planned on reducing the nitro group in subsequent steps and utilizing the amine to construct the southern amidine present in the molecule. Applying the previously employed sequence (see Scheme 12), the authors prepared brominated nitrobenzaldehyde 80 by condensation of nitrotoluene 79 with dimethylformamide-dimethyl acetal and periodate cleavage of the resulting enamine (Scheme 13). The aldehyde was olefinated with the lactone ylide 60, which was then opened with the aluminum amide of aniline 63. The resulting primary alcohol 82 was silylated with TBSCl and the amide was protected with a MOM group to deliver Heck precursor 83. Under optimized conditions, the Heck reaction produced the desired oxindole 84 along with undesired reaction byproducts 85 and 86. This mixture was treated directly with TBAF to cleave the TBS group and to cyclize the lactone delivering nitrolactone 87 as a 4:1 mixture of diastereomers in 51% from Heck precursor 83. Nitrolactone was allylated with allyl iodide to deliver allyl lactone 88 in 87% yield as a single diastereomer. Allyl lactone 88 was a crystalline solid whose structure was determined via X-ray crystallography. It was found that the stereochemical course of the allylation was opposite from that present in perophoramidine (10), and the allyl group was installed syn to the oxindole ethylene chain. At this point, the authors re-examined the relative stereochemistry of allyl lactone 72 (Scheme 11) by X-ray crystallography of one of its derivatives and found that the relative stereochemistry in this compound was also opposite of what was originally reported. However, the relative stereochemistry obtained from the allylations was the same of that which is observed in the communesin alkaloids and the Weinreb group chose to utilize this undesired stereochemical outcome to pursue the total synthesis of this family of alkaloids.
Scheme 13.

Preparation of the Nitrolactone 88
In their first generation synthetic studies, Weinreb and coworkers planned to utilize iodoaniline 90 and PMP-lactone 91 as aromatic building blocks to assemble the Heck reaction precursor (Scheme 14). The benzylic ether of aniline 90 would serve as a functional handle to introduce the benzazepine ring present in the natural products. For the opposite side of the molecule, the benzylic ether of lactone 91 would serve as an amine surrogate that could be unmasked by oxidation to the carboxylic acid and Curtius rearrangement to the aniline. The synthesis began with the benzylation of alcohol 89 followed by reduction of the nitro group to deliver aniline 90. Conversion of this intermediate to the aluminum amide and treatment with PMP lactone 91 resulted in formation of primary alcohol 92. Silylation of this alcohol with TBSCl and alkylation of the amide with MeI provided Heck precursor 93. Performing the Heck reaction on this substrate under optimized reaction conditions led to formation of the desired oxindole product 94 as a single diastereomer in 71% yield. This Heck product was treated with TBAF to cleave the TBS group, which also resulted in concomitant lactonization delivering lactone 95 as a 2:1 mixture of diastereomers. Allylation of the lactone with allyl iodide led to formation of isolable ketene acetal 96, which could be thermally rearranged to the desired allyl lactone 97 as a 3.3:1 mixture of diastereomers. Due to the disappointing diastereoselectivity of the allylation, the authors chose to modify the aniline starting material for the sequence in hopes of improving the selectivity of the allylation.
Scheme 14.

Synthesis of Allyl Lactone 97
The second generation synthesis began with conversion of phenol 98 to a dimethyl carbamate and reduction of nitro group providing aniline 99 (Scheme 15). Formation of the aluminum amide of this intermediate followed by treatment with PMP lactone 91 provided primary alcohol 100. This alcohol was etherified with a MOM group and the amide was methylated with MeI to arrive at Heck precursor 101. Conducting the Heck reaction under optimized conditions provided the desired oxindole product 102 in 79% yield as a single diastereomer. This material was treated with HCl to cleave the MOM group and to facilitate the lactonization, delivering lactone 103 in 70%. Allylation with allyl iodide again delivered an isolable ketene acetal 104, which was rearranged to allyl lactone 105 by heating in DMF with a disappointing 2.3:1 dr. However, conducting the rearrangement in PhMe in place of DMF led to formation of allyl lactone 105 in an improved 9.8:1 dr. The group later published a sequence similar to the one outlined in Scheme 15 to complete the synthesis of communesin F (7) (vide infra).
Scheme 15.

Synthesis of Allyl Lactone 105
The Garg group has recently developed an interrupted Fischer indolization reaction, which has been applied to assemble indoline substrates (Scheme 16). 27 In a single operation, this transformation could be used to form 3 new bonds, 2 heterocyclic rings, and 2 new stereocenters, one of which is quaternary. Additionally, the Garg group has employed this methodology in the total synthesis of several indoline-containing natural products such as physovenine and aspidophylline A. 28 During their studies, it was found that N-methyl-hydrazine 106 undergoes smooth interrupted Fischer indolization with latent aldehyde 107 to afford tetracycle 108 in 70% yield. This product contains the 6,5,6,6-ring system present in the communesin alkaloids and perophoramidine (10). With this result in hand, the authors envisioned that this methodology could be used as a key step to synthesize the communesin alkaloids and perophoramidine (10) by employing more functionalized coupling partners in the interrupted Fischer indolization.
Scheme 16.

The Interrupted Fischer Indolization Reaction to Form the Southern Communesin Ring System
To this end, latent aldehyde 112 was prepared from acetal 109 by way of an elimination and hetero-Diels-Alder reaction (Scheme 17). 29 This latent aldehyde was treated under the interrupted Fischer indolization conditions, facilitating the desired process and delivering tertracycle 113 in 51% yield.
Scheme 17.

The Interrupted Fischer Indolization Reaction to Form the Southern Perophoramidine Ring System
With this preliminary result in hand, a hydrazine starting material was prepared that would facilitate the formation of the benzazepine ring in the communesin skeleton employing a Pictet-Spengler reaction (Scheme 18). Reaction of para-methoxy-substituted hydrazine 114 with latent aldehyde 115 resulted in formation of tetracycle 116 in 54% yield. Removal of the phthalimide group with hydrazine provided amine 117 in near quantitative yield, which was condensed with 3-methylbut-2-enal (118) to afford imine 119 in 95% yield. Unfortunately, attempts to facilitate the Pictet-Spengler reaction with imine 119 were unsuccessful, and led to either hydrolysis of the starting material or no reaction.
Scheme 18.

The Interrupted Fischer Indolization Reaction to Form the Southern Communesin Ring System
With the obstacles encountered in elaborating the interrupted Fischer indolization products towards the communesin alkaloids, attempts were made to access perophoramidine using the described methodology (Scheme 19). Substituted latent aldehyde 125 was chosen as a substrate for the interrupted Fischer indolization process and was accessed in 6 steps. Commercially available aniline 121 was sulfonated with TsCl and subsequently reduced with LiAlH4 to deliver benzylic alcohol 122. Oxidation to the aldehyde with PCC followed by allyl Grignard addition afforded secondary alcohol 123 which was converted to secondary chloride 124 with SOCl2. Hetero-Diels-Alder reaction of the secondary chloride 124 with enol ether 110 resulted in a low 11% yield of the [4+2] product 125 and resulted primarily in the undesired elimination product. Furthermore, attempts to employ the Diels-Alder product 125 in the interrupted Fischer indolization failed to deliver the desired product 126 with a variety of Lewis- and Brønsted- acids and with different aryl hydrazines. The failure of the interrupted Fischer indolization was posited to have occurred due to the steric interactions of the allyl moiety in latent aldehyde 125.
Scheme 19.

Attempts to Access Tetracycle 126
To determine if steric interactions were precluding the desired reaction between aryl hydrazines and latent aldehyde 125, a latent aldehyde lacking the allyl moiety 128 was prepared from benzylic alcohol 127 in 2 steps via chlorination and hetero-Diels-Alder reaction (Scheme 20). Reaction of latent aldehyde 128 with phenylhydrazine (129) facilitated the desired interrupted Fischer indolization in 68% yield. Furthermore, tetracycle 130 could be treated with NCS in AcOH to deliver dichloride 131 which could potentially serve as an intermediate for the total synthesis of perophoramidine.
Scheme 20.

Synthesis of Dichloride 131
Wu and coworkers reported an acid-catalyzed approach to forming aza-o-xylylenes from 2-aminobenzyl alcohols (133) and utilized these intermediates in annulation reactions with indoles (132) (Scheme 21).30 TFA was found to serve as an efficient catalyst for the transformation, and a variety of 3-substituted indoles (132) could participate in the reaction leading to annulated indoline aminals of general structure 134 in moderate to excellent yields. This structural motif represents a portion of the skeleton present in the communesin alkaloids and perophoramidine.
Scheme 21.

Wu's Amino Alcohol-Based Approach to the Southern Ring System of the Communesin Alkaloids
Adlington and coworkers were able to construct a significant portion of the communesin skeleton utilizing a thermal intramolecular cycloaddition between an aza-o-xylylene and an indole (Scheme 22).31 The authors aimed to utilize a thermal [4+2] cycloreversion of a substituted 1,3-benzoxazin-2-one to generate an aza-o-xylylene upon extrusion of CO2. This dehydrative [4+2]-approach bears similarity to the strategies developed both by Funk and Wu for accessing similar ring systems (vide supra). To test the key aza-o-xylylene cycloaddition, amino alcohol 138 was synthesized. Reaction of tryptamine 135 with isatin 136 led to amide 137, which was treated with vinyl-MgBr to provide amino alcohol 138. Treatment of amino alcohol 138 with CDI led to formation of pentacycle 140 in a disappointing 36% yield. However, the transformation served to construct the two vicinal quaternary stereocenters in a single synthetic step. The reaction likely proceeds through the expected aza-o-xylylene intermediate 139, which is trapped by the pendant indole.
Scheme 22.

Aldlington's Strategy Towards the Communesin Alkaloids
The relative stereochemistry of the two quaternary stereocenters in pentacycle 140 was opposite from that needed to access the communesin alkaloids. In attempts to address this issue, the authors removed the benzyl group and sulfonated the resulting N–H lactam providing lactam 141 (Scheme 23). This material was reduced with DIBAL-H leading to N,O-aminal 142 which was olefinated to deliver vinyl ether 143.
Scheme 23.

Adlington's Elaboration of Pentacycle 143
In their synthetic studies towards racemic communesin F, Somafai and coworkers were able to access the hexacyclic core of the natural product utilizing a Diels-Alder strategy between butadiene and a functionalized α,β-unsaturated oxindole dimer. 32 This strategy would allow for the installation of both vicinal quaternary stereocenters present in the molecules early in the synthesis in a single operation.
The group's synthesis commenced with the coupling of N-sulfonyl isatin 144 with oxindole to provide isoindigo 145 in 45% yield (Scheme 24). This material could be treated with butadiene to undergo the key Diels-Alder reaction to provide cycloadduct 146 in 63% yield. The authors found that inclusion of molecular sieves in the reaction mixture was crucial to the transformation's success. Otherwise, cleavage of the N-tosyl protecting group was observed. With both vicinal quaternary stereocenters in place, elaboration of the tetracyclic ring system was pursued. Cycloadduct 146 was treated with Meerwein's salt in the presence of 1,6-di-tert-butyl-4-methylpyridine (DTBMP) to provide an ethyl-iminoether in 92% yield. The choice of base proved important in this transformation, as use of Hunig's base provided the undesired N-ethyl product as the major product. Ozonolytic cleavage of the cyclohexene olefin followed by reduction of the dialdehyde provided a diol intermediate, which was converted to diazide 147 under Mitsunobu conditions with hydrazoic acid. This intermediate (147) was reduced with hydrogen and Pd/C to deliver a diamine intermediate that underwent selective cyclization to γ-lactam 148 in 85% yield. Attempts to selectively cyclize the N-tosyl aniline in the presence of the primary amine proved difficult. To this end, the primary amine was converted to an ethyl carbamate by treatment with ethyl chloroformate and this material was treated with LiAlH4. Unfortunately, the desired tetracycle 150 was only obtained as a minor product in a mere 12% yield, while the remainder of the reaction mixture consisted of bridged orthoamide 149. Fortunately, this undesired orthoamide could be converted to pentacycle 150 by treatment with DIBAL-H. With pentacyclic lactam 150 in hand, attempts were made to construct the northern aminal in the natural product. The secondary amine was protected with a Boc group by treatment Boc2O and the γ-lactam was converted to imino ether 151 by treatment with Meerwein's salt. Unfortunately, attempts to cyclize the secondary amine onto the imino ether proved unsuccessful under a variety of conditions examined.
Scheme 24.

Somafai's Synthesis of Pentacycle 151
Takemoto and coworkers have developed a SmI2-mediated reductive cyclization reaction that transforms carbodiimides bearing unsaturated carbonyls to 2-iminoindolines bearing an all-carbon quaternary stereocenter (Scheme 25). 33 This methodology was used to construct the pentacyclic core of dehaloperophoramidine, the dehalogenated analog of perophoramidine in a racemic fashion.34
Scheme 25.

SmI2 Reductive Cyclization of Carbodiimides
Unsaturated lactams 153 and 154 were prepared from commercially-available ethyl 3-(benzylamino)propanoate (152) (Scheme 26). The reactivity of this material in reductive cyclization was explored and was found to be largely dependent on the identity of the carbodiimide aryl group. Utilizing the para-methoxyphenyl imide 153 the authors could facilitate the cyclization sequence in 86% yield utilizing their standard conditions. The relative stereochemistry of the product (155) was confirmed via X-ray crystallography and is rationalized to occur by protonation of the Sm-enolate from the bottom, sterically more accessible face of the molecule. Trying to utilize the same conditions on the para-methoxybenzyl substrate 154, on the other hand led to complete recovery of the starting material, even when the reaction was heated to 60 °C. It was found that adding 10 vol% of HMPA to the reaction mixture led the desired transformation to occur in 86% yield, providing compound 156. The authors posit that addition of HMPA to the reaction mixture leads to an increase in reduction potential in SmI2. To elaborate 155 into the pentacyclic core of perophoramidine, the authors explored Pd-catalyzed amidation reactions. They found that amidine 157 could be accessed in the absence of the metal catalyst simply by heating spirocycle 155 to 120 °C in the presence of NaOtBu, but in a mere 27% yield. Employing Pd(OAc)2 as catalyst in the presence of PCy3 improve this transformation and led to amidine 157 in 86% isolated yield.
Scheme 26.

Synthesis and Reductive Cyclization
Most recently, the Stoltz group reported racemic formal syntheses of both communesin F (7) and perophoamidine (10) utilizing a Pd-catalyzed decarboxylative allylic alkylation as a diastereodivergent reaction.35 By varying the substrate, both the cis and trans vicinal quaternary stereocenter-bearing products could be synthesized to provide communesin F (7) and perophoramidine (10), respectively. The formal synthesis of communesin F (7) began with the coupling of bromooxindole 158 and malonate 159 (Scheme 27). Proceeding through an azaxylylene intermediate, this transformation provided oxindole malonate 160 in 95% yield. This material was methylated with MeI, and the product was subjected to a protiodesilylation/lactonization sequence to deliver lactone 161. At this stage, the key Pd-catalyzed transformation was examined. Treatment of lactone 162 with 5 mol% Pd(PPh3)4 in THF smoothly facilitated the decaboxylative allylation, delivering allyl lactone in 78% yield as a single diastereomer. The relative configuration of this compound was confirmed by single crystal X-ray analysis and corresponded to the diastereomer needed to synthesize communesin F (7). To elaborate the synthesis, the nitro group was reduced leading to a lactamization event delivering bisoxindole 163 in 80% yield. Protection of the primary alcohol and oxindole nitrogens followed by ozonolysis of the olefin provided aldehyde 164 in 94% yield. Reductive amination with benzylamine 165 led to a translactamization event providing anilide 166 in near quantitative yield. To complete the formal synthesis, the lactam was activated with Tf2O the resulting spirocyclic intermediate was reduced, and the nitrobenzyl protecting group was cleaved to deliver pentacycle 167. This intermediate was used by Qin and coworkers (vide infra) to complete the total synthesis of communesin F (7) in 11 steps.
Scheme 27.

Stoltz's Formal Synthesis of (±)-communesin F (7)
For their formal synthesis of perophoramidine (10), the authors would have to invert the diastereoselectivity of the decarboxylative allylation reaction. To this end, bromooxindole 168 was coupled with malonate 169 in 96% yield and the oxindole nitrogen was methylated to deliver oxindole malonate 170 (Scheme 28). This acyclic diallyl diester was treated with Pd(PPh3)4 at −78 °C to facilitate the Pd-catalyzed reaction. The allylated product (171) was obtained from this transformation in 78% yield as a single diastereomer. Single crystal X-ray analysis of the product revealed that the relative stereochemistry in of the product was opposite of that obtained from the allylation of lactone 162 (Scheme 27), and was the required relative stereochemistry present in perophoramidine (10). At this stage, the nitroarene was reduced with TiCl3 leading to a transamidation. The oxindole nitrogen was protected with a Boc group, and the olefin was converted to an aldehyde via ozonolyis, delivering aldehyde 172. Analogous to the communesin system, the aldehyde was subjected to reductive amination, leading to a translactamization providing anilide 173. Unlike the communesin system, reductive cyclization of the oxindole nucleus in 173 could be accomplished in a single step employing alane to deliver aminal 174. To complete the formal synthesis, the indoline methyl group was oxidized with PDC, and the resulting formyl and nitrobenzyl groups were cleaved with aqueous NaOH providing pentacycle 175. This intermediate was used previously by Funk and coworkers to complete the formal synthesis of perophoramidine (10) in 9 steps (vide infra).
Scheme 28.

Stoltz's Formal Synthesis of (±)-perophoramidine
3. Total Syntheses of the Communesin Alkaloids
3.1 Racemic Total Syntheses of Communesin Alkaloids
The Qin group has utilized the cyclopropanation of indoles as a key transformation to construct quaternary stereocenters in the total synthesis of various indoline alkaloid natural products. 36 The group disclosed their efforts to use such a strategy towards to total synthesis of the communesin alkaloids and perophoramidine (10) (Scheme 29).37 Starting with tryptamine derivative 176 and isatin 177, diazoamide 178 could be constructed over several steps. Treatment of this compound with an appropriate Cu(I) source led to formation of a carbene and facilitated a cyclopropanation sequence delivering azido cyclopropane 179. Reduction of the azide functionality led to concomitant cyclopropane opening delivering pentacyclic lactam 180. Pentacyclic lactam 180 contains a large portion of the skeleton present in the communesins and perophoramidine (10).
Scheme 29.

Qin's Early Early Cyclopropanation Studies Towards the Synthesis of Communesin Alkaloids
Following this initial report, Qin and coworkers reported the first total synthesis of the (±)-communesin F (7) in 2007 using a similar cyclopropanation strategy.38 The group was able to utilize a pendant diazoester to install one of the two quaternary stereocenters present in communesin F (7) in a diastereoselective fashion (Scheme 30). Qin's synthesis commenced with the assembly of azido ester 184 in four steps from tryptophol derivate 182 and aryl azide 181. Aryl azide 181 was coupled with tryptophol derivate 182 in 95% yield. The ketoester product was condensed with tosyl hydrazine under acidic conditions to form the tosyl hydrazone, which was converted to diazoester 183 by treatment with DBU. With diazoester 183 in hand, the stage was set for the key cyclopropanation reaction. Treatment of this intermediate with stoichiometric CuOTf in DCM led to formation of cyclopropane intermediate 184 as an inconsequential 1.6:1 mixture of diastereomers. Staudinger reduction of the azide functionality with PBu3 in aqueous THF produced an amine that underwent an intramolecular SN2 reaction/fragmentation cascade delivering N–H aminal 185 in 83% as a single diastereomer. N–H aminal was protected as its methyl carbamate 186 in 93% yield by treatment with methyl chloroformate. At this stage, diastereoselective construction of the second vicinal quaternary stereocenter was performed utilizing an allylation reaction. This transformation was accomplished by treatment of 185 with NaH and allyl bromide in DMF, providing allyl lactone 188 as a single diastereomer in 85% yield. The authors were able to elucidate that the allylation reaction proceeded through an O-allylation [3,3]-sigmatropic rearrangement by observation of the intermediate ketene acetal 189, which underwent rearrangement in situ. To rationalize the stereochemical course of the reaction, it is proposed that allylation proceeds from the sterically more accessible top face of the molecule while the bottom face is shielded by the bromophenyl portion of the molecule.
Scheme 30.

Qin's Synthesis of Pentacycle 188
To elaborate the synthesis, oxidative cleavage of the allyl moiety was accomplished using a two-step protocol in 95% yield (Scheme 31). The resulting aldehyde (189) was converted to primary alcohol 190 in three steps via oxime formation, Ra-Ni reduction, and treatment of the amine product with NaOMe in MeOH to facilitate the transamidation. Conversion of the primary alcohol 190 to the tert-butyl carbamate 191 was performed in four steps via oxidation using the Dess-Martin periodinane (DMP), oxime formation, reduction with Ra-Ni and reaction of the amine with Boc2O. A Heck reaction was performed with 2-methyl-3-buten-2-ol in the presence of 50 mol% Pd(OAc)2 and 200 mol% (o-tol)3P under microwave irradiation providing a handle for subsequent benzazepine formation. The tertiary alcohol product 192 was treated with PPTS facilitating the acid-catalyzed cyclization event providing benzazepine 194 in 66% along with 26% of the undesired elimination product 193. To construct the final ring in communesin F (7), the lactam in benzazepine 194 was activated for nucleophilic attack by the nitrogen in the benzazepine ring. This was accomplished by conversion of the lactam to the ethyl imino ether, which in turn was treated with TFA to liberate the secondary amine. The secondary amine did not undergo spontaneous cyclization to the desired amidine product 195. Instead, the cyclization event was facilitated by treatment with silica gel at 50 °C (81% over 3 steps). Amidine 195 was used to complete the total synthesis by hydrolysis of the carbamate functionality with KOH/MeOH, and reduction of the amidine with sodium borohydride under acidic conditions, which delivered communesin F (7) as a mixture of two rotamers. Treatment of the natural product with TFA in CHCl3 provided the natural product's TFA salt (7•TFA) in 95% yield.
Scheme 31.

Completion of (±)-Communesin F•TFA (7•TFA)
Weinreb and coworkers have been able to successfully apply their Heck reaction conditions to a tetrasubstituted olefin substrate in a route that was used to complete the total synthesis of communesin F (7) (Scheme 32).39 To assemble the Heck precursor, enol triflate 196 was cross-coupled with aryl boronic acid 197. The cross-coupling product 198 was saponified to the carboxylic acid, which was converted to the acyl chloride and subsequently treated with aniline 199 delivering amide 200 in 87% yield. The benzyl group was converted to the ethyl carbamate by treatment of amide 200 with ethyl chloroformate (96%), and the resulting product was methylated with iodomethane in 92% yield delivering the Heck reaction precursor 201.
Scheme 32.

Weinreb's Synthesis of Heck Precursor 201
While both inter- and intramolecular Heck reactions are uncommon for tetrasubstituted olefins, the intramolecular Heck reaction of amide 201 proceeded smoothly employing a Pd(OAc)2/PPh3 system and delivered the oxindole product 202 in 90% yield. (Scheme 33) The synthesis was elaborated by hydrogenation of the nitro group, converting the resulting aniline product to tert-butyl carbamate 203 in 87% yield over 2 steps. To construct the lower aminal, semi-reduction of the oxindole was performed using AlH3•NMe2Et providing a tetracyclic intermediate in 74% yield, which was treated with KOH in EtOH to hydrolyze the ethyl carbamate and afford enamine 204 in 94% yield. Functionalization of the enamine to install the second vicinal quaternary stereocenter proved challenging, and conversion of enamine to the lactam was necessary to achieve this transformation. This was accomplished with the use of cyanogen azide, which afforded cyanoamidine 206 in 93% yield, proceeding through [3 + 2] cycloadduct 205 as an intermediate. Cyanoamidine 206 was hydrolyzed under basic conditions to deliver an N–H lactam, which was converted to N–Boc lactam 207 in 95% yield. At this stage, the second quaternary stereocenter was introduced via diastereoselective allylation. Treatment of lactam 207 with KOtBu and allyl iodide at −78 °C resulted in formation of the desired product 208 in 87% yield as a single diastereomer. The allylation event was rationalized to proceed from the sterically more accessible convex top face of the lactam enolate.
Scheme 33.

Elaboration to Pentacycle 208
To elaborate the northern portion of the molecule, the Boc group was cleaved using KOH in EtOH in 94% yield (Scheme 34). The resulting N–H lactam was converted to mesylate 209 using a three step sequence which included oxidative cleavage, reduction of the aldehyde, and mesylation of the primary alcohol. The BOM group was removed via hydrogenolysis and the resulting alcohol was oxidized using DMP affording aldehyde 210 in 75% over 2 steps. This aldehyde was elaborated into azide 211 in 2 steps using a nucleophilic displacement of the mesylate with sodium azide and a cross aldol reaction with acetone. A Boc group was installed on the δ–lactam, and the azide was reduced under Staudinger conditions leading to a transamidation to the γ-lactam 212. Reaction with methylithium afforded an allylic alcohol, which was treated with PPTS to cyclize azapane 192. From this point, the synthesis was completed by activation of the lactam with Meerwein's reagent to provide an imino ether which was cyclized to amidine 214. Reduction of the amidine and acylation was achieved using NaBH4 in the presence of Ac2O and the product was treated with TFA to provide communesin F•TFA (7•TFA).
Scheme 34.

Completion of (±)-Communesin F•TFA (7•TFA)
Most recently, Funk and coworkers reported an expedient 16-step synthesis of (±)-communesin F (7) by employing a [4+2] cycloaddition between an indol-2-one and functionalized indole-derivative (Scheme 35).40 Previously, the group showed that this methodology could be used as a key step in the total synthesis of (±)-perophoramidine (10) (vide infra) as well as other natural products.41 By using this cycloaddition, the southern portion of communesin F (7) could be assembled rapidly and efficiently. Funk's synthesis commenced with the key reaction between indole 215 and bromooxidole 216. Treatment of these reaction partners with AgCO3 in MeCN at rt smoothly facilitated the desired hetero-Diels Alder reaction, which proceeded through endo transition state 217 delivering dimer 218 in 70% yield as a single diastereomer. Tosylation of dimer 218 and subsequent methanolysis facilitated a rearrangement to tetracycle 219 in 61% yield. This intermediate was suitable for single crystal X-ray diffraction, and was used to confirm the assigned relative stereochemistry of the cycloaddition. Methylation of tetracycle 219 with Meerwein's reagent, followed by hydrogenation of the azide, and cleavage of the tosyl group provided carbamate 220. At this stage, the side chain for the benzazepine was introduced using a Heck reaction between carbamate 220 and 2-methyl-3-buten-2-ol. The allylic alcohol product was cyclized to the benzazepine ring employing Hg(OTf)2 in 86% yield. The Boc group was removed from benzazepine 221 utilizing TBSOTf,42 and the amine product from this transformation was cyclized to the bridged lactam 222 with AlMe3. At this stage, the second quaternary stereocenter was introduced via alkylation with iodoacetonitrile in 77% yield and with complete diastereocontrol. The nitrile product 223 was reduced with LiAlH4 to hemiacetal 224 which, in turn, was converted to aminal 225 via reductive amination. To complete the synthesis, aminal 225 was acylated with acetic anhydride to deliver communesin F (7) in 13 steps from bromotryptophol 215.
Scheme 35.

Funk's Synthesis of (±)-Communesin F (7)
3.2. Asymmetric Total Syntheses of Communesin Alkaloids
The first asymmetric total synthesis of communesin F (7) was reported by Ma and coworkers (Scheme 36). This route also served to establish the absolute configuration of the natural product.43 Earlier work in the Ma group demonstrated that an intramolecular oxidative coupling of indole derivative 226 could be accomplished in high yield when an electron-poor arene (i.e. R = NO2) was present in the substrate. The authors postulated that increased acidity of the α-position of the amide led to the observed trend in reactivity. The product from this transformation constructs the C(7) all carbon quaternary stereocenter present in the communesin alkaloids.
Scheme 36.

Ma's Early Studies on the Oxidative Cyclization for the Synthesis of Communesin Alkaloids
Based on the success of the oxidative coupling reaction, the Ma group chose to utilize this transformation as a key step in the asymmetric total synthesis of communesin F (7) (Scheme 37). To render the synthesis asymmetric, silylated (S)-phenylglycinol was employed as a chiral auxiliary at the amide. The oxidative coupling substrate 231 was assembled in 65% yield over three steps from bromotryptophol 230 via oxidation, reductive amination with (S)-OTBS-phenylglycinol, and a BOP coupling with 2-(2-nitrophenyl)acetic acid. Double deprotonation of coupling substrate 231 with LiHMDS in THF at −78 °C followed by treatment with I2 led to the desired coupling product 232 as a mixture of diastereomers. The nitro group was reduced with iron and the resulting aniline cyclized spontaneous to a tetracycle which was methylated to afford an N–H aminal in 66% over 3 steps. At this stage, the d.r. of the oxidative coupling was determined to be 3:1 as well. The N–H aminal was converted to the tert-butyl carbamate 233 in 89% yield, and the second quaternary stereocenter was introduced via allylation with allyl iodide in >20:1 dr. A three-step sequence was used to remove the (S)-OTBS-phenylglycinol auxiliary from carbamate 233 which included protiodesilylation of the OTBS group, elimination, and hydrolysis, affording lactam 234 in 93% yield over 2 steps.44 The olefin moiety was cleaved oxidatively under modified Johnson-Lemieux conditions to provide an aldehyde, which was reduced to primary alcohol 235 with NaBH4 in 95% yield over 2 steps. At this stage, a Heck reaction similar to the one employed by Qin was used to install the side chain for assembly of the benzazepine ring. This reaction was conducted with Pd(OAc)2 as the precatalyst under microwave irradiation resulting in a 67% yield (80% brsm) of the desired allylic alcohol 236. Surprisingly, mesylation of the diol 236 not only led to formation of the primary mesylate but also resulted in spontaneous cyclization of the benzazepine ring under the reaction conditions in 63% yield. The mesylate product was treated with sodium azide to afford primary azide 237 in 78% yield. An aza-Wittig reaction was used to cyclize the final 5-membered ring in the natural product. Typically, aza-Wittig reactions with amides are uncommon and require high reaction temperatures. However, due to the strained nature of the amide, which cannot have its lone pair donate into the carbonyl group, this transformation could be conducted at 80 °C and delivered the amidine in 83% yield. To complete the synthesis, the amidine was reduced with sodium borohydride in the presence of acetic acid and acetic anhydride and the product was converted to the TFA salt of (−)-communesin F (7). The natural product was accessed in 19 steps (longest linear sequence) from 4-bromotryptophol (230) and in around 6% overall yield. The absolute configuration of communesin F (7) was established by comparing the rotation of the synthetic sample to the natural product.
Scheme 37.

Ma's Synthesis of (−)-Communesin F (7)
While numerous syntheses of communesin F (7) have been disclosed over the last decade, the syntheses of communesins A and B (1 and 2), the epoxide-bearing and biologically most active members of this family of natural products remained unsolved. Recently, Ma and coworkers reported the total syntheses of these two natural products in a catalytic asymmetric manner.45 While direct conversion of communesin F (7) to communesin A (1) was attempted via epoxidation reactions, the natural product was found to be unstable to the reaction conditions examined likely due to the sensitivity of the aminal functionalities towards oxidation. To circumvent this limitation, the olefin side chain was oxidized at an early-stage and was protected as a ketal to avoid undesired side reactions. For this purpose, a different synthesis was devised. Aurantioclavine ketal 243 was prepared in 13 steps from 4-bromotryptophol (230) (Scheme 38). The synthesis commenced with the preparation of tertiary alcohol 238 in 3 steps from 4-bromotryptophol (230), which included TBS protection of the primary alcohol, tosylation of the indole and a Heck reaction to introduce the alcohol side chain. Tertiary alcohol 238 was treated with AD-mix-β under Sharpless dihydroxylation conditions to deliver enantioenriched diol 239 in 94% yield and 96% ee. Diol 239 was converted to the cyclic sulfite, which was subsequently treated with sodium azide to arrive at azide 240 in 64% yield over 2 steps. Ketalization with dimethoxypropane followed by reduction of the azide with LiAlH4 provided an amine that was converted to 2-nitrobenzene sulfonamide 241 in 90% over 2 steps. Protiodesilylation of the TBS group unmasked a primary alcohol, which was cyclized using a Mitsunobu reaction and hydrolyzed to afford benzazepine 242 in 81% over 3 steps. Removal of the tosyl group with Mg(0) delivered aurantioclavine ketal 243 in 92% yield.
Scheme 38.

Ma's Asymmetric Synthesis of Aurantioclavine Ketal 243
Coupling of aurantioclavine ketal 243 with 2-(2-nitrophenyl)acetic acid using BOPCl proceeded uneventfully to provide the oxidative coupling substrate 244 in 91% yield (Scheme 39). Double deprotonation of 244 with 2 equivalents of LiHMDS and the addition of iodine at −78 °C resulted in formation of undesired side products and failed to provide the coupling product 246. On the other hand, addition of iodine at room temperature led to smooth formation of the spiro-fused coupling product 246 as a single diastereomer in 66% isolated yield (73% brsm). To rationalize the observed diastereochemical outcome, the authors proposed that the oxidative coupling proceeds through transition state 245, which contains favorable π-stacking interactions between the electron-poor arene and electon-rich indole and contains the bulky ketal on the opposite face from radical combination. To elaborate the synthesis, the nitro group was reduced with H2 and Ra-Ni and the resulting aniline cyclized onto the imine leading to the formation of an N– H aminal in 70% yield, which was methylated to afford methyl aminal 247 in 90%. Installation of the second quaternary stereocenter was accomplished via alkylation with iodoacetonitrile, which occurred in 67% yield and complete diastereoselectivity. The nitrile 248 was reduced with LiAlH4 leading to formation of lactol 249, which was converted to aminal 250 via reductive amination with sodium cyanoborohydride in 92% yield over 2 steps.
Scheme 39.

Formation of Heptacycle 250
Communesin A (1) was completed by acylation of aminal 250 in 65% followed by hydrolysis of the ketal functionality, mesylation of the secondary alcohol and cyclization to the epoxide in 55% yield over 2 steps (Scheme 40). Analogously, communesin B (2) was accessed by coupling aminal with sorbic acid with BOPCl, hydrolysis of the ketal, mesylation and epoxide formation in 69% over 3 steps. In summary, Ma's syntheses accessed both communesins A (1) and B (2) in 24 steps and ~2-3% overall yield from 4-bromotryptophol (230). The routes utilized a diastereoselective oxidative coupling as a key step to forge one of the two quaternary stereocenters in the target.
Scheme 40.

Completion of Communesins A (1) and B (2)
4. Total Syntheses of the Perophoramidine
Like the communesin alkaloids, perophoramidine (10) has attracted the interest of the synthetic community due to its intriguing structure and biological activity. Many groups that have synthesized the communesins have also pursued perophoramidine (10) as a target. While similar in structure to the communesin alkaloids, perophoramidine (10) has unique structural features that make it a synthetic challenge. The natural product's vicinal quaternary stereocenters have the opposite relationship from that encountered in the communesins and any strategy that was applied to the construction of this motif in the communesins would need to be rehashed in order to be used to prepare perophoramidine (10). Additionally, perophoramidine contains three points of halogenation on the aromatic nuclei, one bromide and two chlorides, which need to be incorporated when devising a retrosynthesis. The remainder of this review will outline the elegant syntheses that have been developed to access perophoramidine (10).
4.1 Racemic Total Syntheses of Perophoramidine
The first total synthesis of perophoramidine (10) was reported by Funk and coworkers (Scheme 41). By employing a hetero-Diels-Alder reaction as a key transformation, the authors could construct both vicinal quaternary stereocenters in the target in a single synthetic operation.46 The synthesis commenced with the [4 + 2] cycloaddition between silyl tryptophol 253 and bromooxindole 254 to form cycloadduct 255 in 89% yield and >20:1 dr. This material was converted to the Boc carbamate, which was subsequently treated with triphenylphosphine to reduce the azide functionality and to facilitate a transamidation cascade affording pentacycle 256 in 89% yield. Due to the electronic difference between the two aromatic nuclei in pentacycle 256, NCS could be used to chlorinate the more electron rich aromatic ring chemoselectively in an excellent 86% yield followed by sulfonation to arrive at dichloride 257. The dichloride was protiodesilylated to the primary alcohol, and converted to azide 258 employing a Mitsunobu reaction. Reduction of azide 258 under Staudinger conditions led to a transamidation from the γ- to the δ-lactam in 86% yield. The nosyl sulfonamide was methylated with iodomethane, and the resulting δ-lactam (259) was converted to imino ether 260 in 68% yield. Removal of the nosyl group with thiophenol led to formation of the secondary amine which spontaneously cyclized onto the imino ether functionality leading to an amidine which was oxidized with MnO2 in 65% yield to afford (±)-perophoramidine (10).
Scheme 41.

Funk's Synthesis of (±)-Perophoramidine (10)
A total synthesis of dehaloperophoramidine (274), the dehalogenated analog of perophoramidine, was reported Rainier et al. utilizing their 2-thioindole spirocyclization methodology (Scheme 42).47, 48 The thioindole cyclization precursor 264 was prepared from the condensation of N-Boc isatin (136) and N-benzyl tryptamine (261) followed by thioether installation and sodium borohydride reduction. Secondary alcohol 263 was treated with MsCl, which led to spontaneous spirocyclization providing 265 as a 2:1 mixture of diastereomers. This mixture was treated with DBU, which served to cyclize the amidine and equilibrate the diastereomeric mixture to a single lactam diastereomer 266. Installation of the second quaternary stereocenter was performed via alkylation with allyl iodide and delivered allyl lactam 267 in 89% yield as a single diastereomer.
Scheme 42.

Rainier's Synthesis of Pentacycle 267
With both quaternary stereocenters installed, the synthesis was elaborated by manipulation of the allyl moiety (Scheme 43). Oxidative cleavage under Johnson-Lemieux conditions afforded an aldehyde, which was converted to a secondary amine via reductive amination and then treated with Boc2O to afford carbamate 268. At this stage, the amidine was converted to aminal 269 with sodium borohydride. While this transformation may seem counterintuitive, the authors found that cyclization of the northern amidine functionality could not be performed when the southern amidine was present in the molecule. Treatment of aminal 269 with TFA served to remove both Boc groups from the molecule and afforded 270 which was treated with Boc2O to chemoselectively introduce a Boc group at the secondary amine and not the aminal. Dissolving metal conditions were used to cleave the benzyl group, and the indoline nitrogen was treated with AcCl to provide N-acyl aminal 271 in 88% yield over 2 steps. Exposure of 271 to Meerwein's reagent with NaHCO3 as an acid scavenger led to formation of imino ether 272 in 81% yield, and this compound was treated with TFA to remove the Boc group and cyclize the northern amidine providing hexacycle 273. Finally, the N-acetyl group was hydrolyzed with KOH, and the aminal functionality was oxidized to the amidine delivering dehaloperophoramidine (274).
Scheme 43.

Completion of (±)-Dehaloperophoramidine (274)
Takemoto and coworkers reported a total synthesis of (±)-dehaloperophoramidine (274) employing a unique intramolecular dearomatizing cyclization-allylation sequence to forge both quaternary stereocenters in a single operation (Scheme 44).49 Their synthesis commenced with the preparation of dearomatizing cyclization precursor 283. Suzuki coupling of lactam 275 and boronic acid 276 followed by cleavage of the Boc group provided aniline 277. This material was treated with aryl isothiocyanate 278 delivering thiourea 279 in 85% yield. Thiourea 279 was converted to a carbodiimide by treatment with iodine, cyclized to the quinolone amidine via a 6π-electrocyclic reaction and converted to the tert-butyl carbamate 280 in 69% over 3 steps. At this stage, the amidine was protected with a PMB group using PMBI in 91%. The PMB amidine 281 was treated with TFA to cleave the Boc group, and the resulting N–H lactam was converted to thiolactam 282 with Lawesson's reagent in 97% yield. To complete the synthesis of the dearomatizing cyclization precursor 283, thiolactam 282 was converted to the imino thioether with MeI in 89%, condensed with MeNH2 to provide an amidine, which was protected with a Boc group to deliver 283 in 87% over 2 steps.
Scheme 44.

Synthesis of Dearomatizing Cyclization-Allylation Precursor 283
The key dearomatizing cyclization-allylation was performed by first treating 283 with n-BuLi in THF followed by the addition of allyl iodide (Scheme 45). Under these reaction conditions, the desired allyl bisamidine was obtained in 67% yield as a single diastereomer. The allyl moiety was dihydroxylated and the resulting diol was cleaved with NaIO4 leading to loss of the Boc group and generation of hydroxyamidine 285 in 75% over 2 steps. To complete the synthesis, hydroxyamidine 285 was reduced with NaCNBH3 to amidine 286 and the PMB group was removed with H3PO4 in the presence of anisole delivering (±)-dehaloperophoramidine (274) in 71%.
Scheme 45.

Completion of (±)-Dehaloperophoramidine (274)
4.2 Asymmetric Total Syntheses of Perophoramidine
To date, three asymmetric total syntheses of perophoramidine have been reported, the first of which was disclosed by Qin and coworkers. 50 During their studies towards the communesin alkaloids, Qin and coworkers were faced with the task of reducing an azide functionality in pentacycle 287 to the corresponding amine employing Lewis acidic conditions (Scheme 46). However, upon exposure of the starting material to the reaction conditions, the authors were surprised not to observe the amine product but instead isolated lactam 289 in 65-70% yield along with bromoindole 288. They posited that these products are obtained through a retro-Diels-Alder reaction forming diene 290, which would undergo capture with solvent. With this result in hand, the authors were interested in exploiting the reverse Diels-Alder process as a key transformation to access perophoramidine (10) in an asymmetric fashion. To induce stereoselectivity, an appropriate chiral auxiliary could be placed on the N-H of the δ-lactam.
Scheme 46.

Qin's Observed Retro-[4+2] Process
Diene precursor 296, was prepared starting from isatin 291 (Scheme 47). Grignard addition followed by TBS protection of the tertiary alcohol afforded oxindole 292 in excellent yield. Ozonolysis of the allyl moiety provided an aldehyde in 84%, which was condensed with the Ellman auxiliary 29351 and was reduced to deliver sulfinamide 294 in 85% yield over 2 steps. While other auxiliary groups were tested as well, the authors found that the Ellman auxiliary performed best both in terms of yield and d.r (vide infra). Sulfinamide 294 was treated with Boc2O under basic conditions to facilitate the lactam rearrangement (87%), and the resulting silyl lactam was treated with TBAF to cleave the TBS group, arriving at hydroxy δ-lactam 295 in 90%. Finally, the diene precursor 296 was accessed by conversion of the hydroxyl group to the unstable chloride by treatment with SOCl2.
Scheme 47.

Qin's Synthesis of [4+2] Precursor 296
With diene precursor 296 in hand, the key diastereoselective Diels-Alder reaction was explored. Reaction of indole 297 and the diene precursor 296 with 2 equiv of AgBF4 in DCM resulted in formation of the desired Diels-Alder product 299 in 74% yield and a 5.9:1 dr favoring the trans product corresponding to the relative stereochemistry observed in perophoramidine (10) (Scheme 48). Screening other silver salts and solvents revealed that employing AgClO4 in PhMe was optimal for the transformation and delivered the desired Diels-Alder product 299 in 77% yield and a 11:1 d.r. The yield of the transformation could be increased further to 88% by utilizing 3 equiv of the diene precursor. To rationalize the stereochemical outcome of the Diels-Alder process, the authors propose that the cycloaddition proceeds through exo transition state 298 with the trans, trans diene. This diene geometry is preferred to avoid unfavorable electronic repulsion between the lactam oxygen and aniline carbamate.
Scheme 48.

Completion of (+)-Perophoramidine (10)
With an optimized set of conditions to access pentacycle 299, the synthesis of perophoramidine (10) was elaborated. Chemoselective chlorination of the indoline ring was accomplished with the use of sodium hypochlorite, which also served to cleave the auxiliary on the δ-lactam. The dichloride product 300, was treated with PCC to oxidize the methyl group to the formyl group and this formyl compound was treated with Meerwein's salt to cleave the Boc group and convert the δ-lactam into imino ether 301. Cleavage of the phthalimide group was accomplished with MeNH2, and the resulting primary amine was cyclized by refluxing in chloroform to arrive at amidine 302 in 77% over 2 steps. Oxidation with MnO2 in DCM converted the aminal to bisamidine 303, and PPTS was used to equilibrate bisamidine 303 to the thermodynamically favored bisamidine 304. Finally, chemoselective methylation of 304 with NaHMDS and MeOTf in THF afforded (+)-perophoramidine (10) in 73%. The natural product was accessed in 17 steps (longest linear sequence) and ~11% overall yield.
Wang and coworkers disclosed a catalytic asymmetric total synthesis of (+)-perophoramidine (10) employing a Ni-catalyzed addition of an indole into an aza-o-xylylene intermediate generated from a 3-bromooxindole (Scheme 49). 52 Using this methodology, the authors could forge both quaternary stereocenters in a single enantioselective transformation. Optimization studies on the coupling of indoles with 3-bromoxindole showed that the two substrates could be readily coupled in the presence of a nickel Lewis acid catalyst and chiral diamine ligand. In particular, the combination of Ni(OAc)2 and 3-OMePh-substituted diamine ligand 305 proved optimal to facilitate the transformation. To utilize this methodology in the total synthesis of (+)-perophoramidine (10), indole 306 was coupled with bromooxindole 307 under the optimized reaction conditions, affording compound 308 in 51% yield, a 12:1 dr and 90% ee.
Scheme 49.

Wang's Ni-Catalyzed Addition of Indoles to Bromo-Oxindoles
Next, the oxindole was sulfonated with NsCl, and the phthalimide group was cleaved with hydrazine triggering a transamidation sequence and providing pentacycle 309 in 84% over 2 steps (Scheme 50). The indoline ring was chlorinated employing NCS, and the nosyl group was cleaved from the aminal with thiophenol providing lactam 310. The lactam was sulfonated with NsCl and the azide was reduced facilitating another transamidation sequence. The resulting δ-lactam was methylated chemoselectively at the sulfonamide to deliver pentacyclic aminal 311 in 90% yield over 3 steps. Oxidation of the pentacyclic aminal 311 was accomplished with PCC, and the resulting amidine (312) was protected on both the lactam and amidine with Boc2O. Treatment of Boc-amidine 313 with LiAlH4 led to reduction of the amidine and cleavage of the Boc group present on the lactam. N–H lactam 314 was activated as the imino ether (315) in 87% yield by treatment with Me3OBF4. Finally, (+)-perophoramidine (10) was completed with cleavage of the nosyl group employing thiophenol, removal of the Boc group, and oxidation of the aminal to the amidine.
Scheme 50.

Completion of (+)-Perophoramidine (10)
Most recently, Trost and coworkers reported a catalytic asymmetric total synthesis of (−)-perophoramidine. 53 Unlike many other syntheses, which utilized a [4+2]-cycloaddition as their key step, the Trost group developed a route to this natural product utilizing a Mo-catalyzed asymmetric allylic alkylation (Mo-AAA) as the key enantiodetermining process. The synthesis commenced with the preparation of the two Mo-AAA coupling partners (Scheme 51). Allylic phosphate 317 was prepared from 4-bromo-2-fluorobenzaldehyde 316 in 5 steps, and oxindole 319 was prepared from tryptophol in 3 steps. Coupling of these components proceeded smoothly in the presence of a Mo(0) precatalyst and picolinic acid-derived ligand (S,S)-318. Upon basic workup, the oxindole product 320 was obtained in an excellent 89% isolated yield, 19:1 branched:linear ratio, 8:1 dr and 97% ee.
Scheme 51.

Mo-AAA Reaction of Oxindole 319 and allylic Phosphate 317.
With a robust means to construct one of the quaternary stereocenters in the target, assembly of the southern tetracyclic ring system was examined (Scheme 52). To perform a reductive cyclization, oxindole 320 was converted to the dimethylallyl carbonate 322. This intermediate was treated with LiEt3BH to chemoselectively reduce the oxindole to the N,O-aminal, which was treated with a Pd(0) source to remove the dimethylallyl group and concurrently cyclize aminal 323. This tetracyclic intermediate was treated with NCS to introduce the chlorine substituents in the final target providing dichloroaminal 324. To construct the northern 6-membered ring, the N–H dichloroaminal 324 was alkylated with PMB-Br with concomitant cleavage of the TBS group upon workup, and the primary alcohol intermediate was converted to azide 325 under Mitsunobu conditions. The olefin was cleaved oxidatively, oxidized, and converted to ester 326, and this intermediate was treated with PMe3 to reduce the azide and cyclize lactam 327.
Scheme 52.

Synthesis of Pentacylclic Lactam 327.
With 5 of the 6 rings installed, the Boc group was cleaved thermally, and the aminal intermediate was oxidized to amidine 328 using PhI(OAc)2 (Scheme 52). Amidine 328 was treated with Meerwein's reagent to convert the lactam to imino ether 329, and this intermediate was allylated selectively on carbon employing allyl iodide in near quantitative yield and >20:1 dr. The C vs N regioselectivity of the iminoether allylic alkylation showed an interesting dependence on counterion wherein the lithium salt favoured N-alkylation but the potassium salt gave exclusively C-alkylation. To construct the final 5-membered ring, the olefin was converted to an aldehyde via ozonolysis, and this intermediate was converted to secondary amine 331 employing a Lewis-acid catalyzed reductive amination. Finally, the (−)-perophoramidine (10) was accessed by thermal cyclization of the amine onto the pendant imino ether in compound 331, followed by cleavage of the PMB group under acidic conditions.
4. Conclusions
The communesin alkaloids and perophoramidine (10) are intriguing alkaloids that have gained attention from the scientific community because of their unique structures and diverse biological activities. Elegant strategies have been developed to tackle these complex, densely functionalized natural products including inverse electron-demand hetero-Diels-Alder reactions, cyclopropanations, regio- and enantioselective allylic alkylations, as well as other novel approaches. The efficiency of these syntheses should enable the pursuit of structure activity relationships.
Scheme 53.

Completion of (−)-perophoraminde (10).
Acknowledgments
We thank the NSF (CHE-1145236) and the NIH (GM 033049) for their generous support of our programs. M.O. thanks The John Stauffer Memorial Fellowship and Stanford Graduate Fellowship for financial support.
Biography
 Barry M. Trost was born in Philadelphia, PA in 1941 and studied at the University of Pennsylvania (BA, 1962). He obtained his PhD in 1965 at MIT. He moved to the University of Wisconsin where he was made Professor in 1969 and subsequently Vilas Research Professor in 1982. He moved to Stanford University in 1987 and became Tamaki Professor of Humanities and Sciences in 1990. In addition to holding Visiting Professorships at several universities worldwide, he has been awarded numerous prizes. His interests span the entire field of organic synthesis, particularly in the development of novel methodology and strategy for total synthesis of bioactive complex molecules.
Barry M. Trost was born in Philadelphia, PA in 1941 and studied at the University of Pennsylvania (BA, 1962). He obtained his PhD in 1965 at MIT. He moved to the University of Wisconsin where he was made Professor in 1969 and subsequently Vilas Research Professor in 1982. He moved to Stanford University in 1987 and became Tamaki Professor of Humanities and Sciences in 1990. In addition to holding Visiting Professorships at several universities worldwide, he has been awarded numerous prizes. His interests span the entire field of organic synthesis, particularly in the development of novel methodology and strategy for total synthesis of bioactive complex molecules.
 Maksim Osipov was born in Lvov, Ukraine in 1985. After immigrating to the United States, he attended the University of Pittsburgh where he obtained his B.Sc. in chemistry. From there, Maksim attended Stanford University where he obtained his Ph.D. under the direction of Prof. Barry M. Trost developing asymmetric methods for the total synthesis of alkaloids. Maksim is currently a medicinal chemist at Flexus Biosciences where he developing drugs for cancer immunotherapy.
Maksim Osipov was born in Lvov, Ukraine in 1985. After immigrating to the United States, he attended the University of Pittsburgh where he obtained his B.Sc. in chemistry. From there, Maksim attended Stanford University where he obtained his Ph.D. under the direction of Prof. Barry M. Trost developing asymmetric methods for the total synthesis of alkaloids. Maksim is currently a medicinal chemist at Flexus Biosciences where he developing drugs for cancer immunotherapy.
References
- 1.Numata A, Takahashi C, Ito Y, Takada T, Kawai K, Usami Y, Matsumura E, Imachi M. Tetrahedron Lett. 1993;34:2355–2358. [Google Scholar]
- 2.a Ratnayake AS, Yoshida WY, Mooberry SL, Hemscheidt TK. J. Org. Chem. 2001;66:8717–8721. doi: 10.1021/jo010335e. [DOI] [PubMed] [Google Scholar]; b Ratnayake AS, Yoshida WY, Mooberry SL, Hemscheidt TK. J. Org. Chem. 2003;68:1640. doi: 10.1021/jo010335e. retraction. [DOI] [PubMed] [Google Scholar]
- 3.Jadulco R, Edrada RA, Ebel R, Berg A, Schaumann K, Wray V. J. Nat. Prod. 2004;67:78–81. doi: 10.1021/np030271y. [DOI] [PubMed] [Google Scholar]
- 4.Hayashi H, Matsumoto H, Akiyama K. Biosci. Biotechnol. Biohem. 2004;68:753–756. doi: 10.1271/bbb.68.753. [DOI] [PubMed] [Google Scholar]
- 5.Dalsgaard PW, Blunt JW, Munro MHG, Frisvad JC, Christophersen C. J. Nat. Prod. 2005;68:258–261. doi: 10.1021/np049646l. [DOI] [PubMed] [Google Scholar]
- 6.May JA, Stoltz BM. Tetrahedron. 2006;62:5262–5271. [Google Scholar]
- 7.a Steven A, Overman LE. Angew. Chem. Int. Ed. 2007;46:5488–5508. doi: 10.1002/anie.200700612. [DOI] [PubMed] [Google Scholar]; b Schmidt MA, Movassaghi M. Synlett. 2008:313–324. [Google Scholar]
- 8.Eccles GR. Proc. Am. Pharm. Assoc. 1888;84:382. [Google Scholar]
- 9.Woodward RB, Yand N, Katz TJ. Proc. Chem. Soc. London. 1960:76. [Google Scholar]
- 10.Robinson R, Teuber HJ. Chem. Ind. 1954:783. [Google Scholar]
- 11.a Hamor TA, Robertson JM, Shrivastave HN, Silverton JV. Proc. Chem. Soc. London. 1960:78. [Google Scholar]; b Hamor TA, Robertson JM. J. Chem. Soc. 1962:194. [Google Scholar]
- 12.Hodson HF, Robinson B, Smith GF. Proc. Chem. Soc. London. 1961:465–466. [Google Scholar]; b Saxton JE, Bardsley WG, Smith GF. Proc. Chem. Soc. London. 1962:148. [Google Scholar]; c Nakano T, Martín A. Planta Med. 1976;30:186–188. doi: 10.1055/s-0028-1097715. [DOI] [PubMed] [Google Scholar]; d Ripperger H. Pharmazie. 1982;37:867. [Google Scholar]; e Lajis NH, Mahmud Z, Toia RF. Planta Med. 1993;59:383–384. doi: 10.1055/s-2006-959709. [DOI] [PubMed] [Google Scholar]; f Duke RK, Allan RD, Johnston GAR, Mewett KN, Mitrovic AD, Duke CC, Hambley TW. J. Nat. Prod. 1995;58:1200–1208. doi: 10.1021/np50122a007. [DOI] [PubMed] [Google Scholar]; g Verotta L, Pilati T, Tatò M, Elisabetsky E, Amador TA, Nunes DS. J. Nat. Prod. 1998;61:392–396. doi: 10.1021/np9701642. [DOI] [PubMed] [Google Scholar]
- 13.Tokuyama T, Daly JW. Tetrahedron. 1983;39:41–47. [Google Scholar]
- 14.a Grant IJ, Hamor TA, Robertson JM, Sim GA. Proc. Chem. Soc. London. 1962:148. [Google Scholar]; b Grant IJ, Hamor TA, Robertson JM, Sim GA. J. Chem. Soc. 1965:5678. [Google Scholar]
- 15.Adjibade Y, Weniger B, Quirion JC, Kuballa B, Cabalion P, Anton R, Phytochemistry R. 1992;31:317–319. [Google Scholar]
- 16.Verotta L, Pilati T, Tatò M, Elisabetsky E, Amador TA, Nunes DS. J. Nat. Prod. 1998;61:392–396. doi: 10.1021/np9701642. [DOI] [PubMed] [Google Scholar]
- 17.Peterson EA, Overman LE. PNAS. 2004;101:11943–11948. doi: 10.1073/pnas.0402416101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a Noole A, Sucman NS, Kabeshov MA, Kanger T, Macaev FZ, Malkov AV. Chem. Eur. J. 2012;18:14929–14933. doi: 10.1002/chem.201203099. [DOI] [PubMed] [Google Scholar]; b Mitsunuma H, Shibasaki M, Kanai M, Matsunaga S. Angew. Chem. Int. Ed. 2012;51:5217–5221. doi: 10.1002/anie.201201132. [DOI] [PubMed] [Google Scholar]; c Trost BM, Malhotra S, Chan WH. J. Am. Chem. Soc. 2011;133:7328–7331. doi: 10.1021/ja2020873. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Lemieux RM, Meyers AI. J. Am. Chem. Soc. 1998;120:5453–5457. [Google Scholar]
- 19.a Siengalewicz P, Gaich T, Mulzer J. Angew. Chem. Int. Ed. 2008;47:8170–8176. doi: 10.1002/anie.200801735. [DOI] [PubMed] [Google Scholar]; b Zuo Z, Ma C. Isr. J. Chem. 2011;51:434–441. [Google Scholar]
- 20.May JA, Zeidan RK, Stoltz BM. Tetrahedron Lett. 2003;44:1203–1205. [Google Scholar]
- 21.Crawley SL, Funk RL. Org. Lett. 2003;5:3169–3171. doi: 10.1021/ol034407v. [DOI] [PubMed] [Google Scholar]
- 22.Crawley SL, Funk RL. Org. Lett. 2006;8:3995–3998. doi: 10.1021/ol061461d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.a Ploux O, Caruso M, Chassaing G, Marquet A. J. Org. Chem. 1988;53:3154–3158. For similar aziridine syntheses, see. [Google Scholar]; b Tokés A, Likei G, Janzsó G. Synth. Commun. 1990;20:1905–1913. [Google Scholar]; c Davoli P, Forni A, Moretti I, Prati F, Torre G. Tetrahedron. 2001;57:1801–1812. [Google Scholar]; d Comstock LR, Rajski SR. Tetrahedron. 2002;58:6019–6026. [Google Scholar]
- 24.Artman GD, III, Weinreb SM. Org. Lett. 2003;5:1523–1526. doi: 10.1021/ol034314d. [DOI] [PubMed] [Google Scholar]
- 25.Fürstner A, Weintritt H. J. Am. Chem. Soc. 1998;120:2817–2825. [Google Scholar]
- 26.Seo JH, Artman GD, III, Weinreb SM. J. Org. Chem. 2006;71:8891–8900. doi: 10.1021/jo061660a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.a Boal BW, Schammel AW, Garg NK. Org. Lett. 2009;11:3458–3461. doi: 10.1021/ol901383j. [DOI] [PubMed] [Google Scholar]; b Schammel AW, Boal BW, Zu L, Mesganaw T, Garg NK. Tetrahedron. 2010;66:4687–4695. doi: 10.1016/j.tet.2010.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.a Zu L, Boal BW, Garg NK. J. Am. Chem. Soc. 2011;133:8877–8879. doi: 10.1021/ja203227q. [DOI] [PubMed] [Google Scholar]; b Schammel AW, Chiou G, Garg NK. J. Org. Chem. 2012;77:725–728. doi: 10.1021/jo202078z. [DOI] [PubMed] [Google Scholar]
- 29.Schammel AW, Chiou G, Garg NK. Org. Lett. 2012;14:4556–4559. doi: 10.1021/ol302023q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Robertson FJ, Kenimer BD, Wu J. Tetrahedron. 2011;67:4327–4332. [Google Scholar]
- 31.George JH, Adlington RM. Synlett. 2008:2093–2096. [Google Scholar]
- 32.a Danielsson J, Somfai J. Org. Lett. 2014;16:784–787. doi: 10.1021/ol403516s. [DOI] [PubMed] [Google Scholar]; b Sanzone A, Somfai J. Eur. J. Org. Chem. 2015;16:3441–3449. [Google Scholar]
- 33.Ishida T, Tsukano C, Takemoto Y. Chem. Lett. 2013;41:44–46. [Google Scholar]
- 34.Ishida T, Takemoto Y, Tetrahedron Y. 2013;69:4517–4523. [Google Scholar]
- 35.a Han S-J, Vogt F, Krishnan S, May JA, Gatti M, Virgil SC, Stoltz BM. Org. Lett. 2014;16:3316–3319. doi: 10.1021/ol5013263. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Han S-J, Vogt F, May JA, Krishnan S, Gatti M, Virgil SC, Stoltz BM. J. Org. Chem. 2015;80:528–547. doi: 10.1021/jo502534g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang D, Song H, Qin Y. Acc. Chem. Res. 2011;44:447–457. doi: 10.1021/ar200004w. [DOI] [PubMed] [Google Scholar]
- 37.Yang J, Song H, Xiao X, Wang J, Qin Y. Org. Lett. 2006;8:2187–2190. doi: 10.1021/ol0607138. [DOI] [PubMed] [Google Scholar]
- 38.Yang J, Wu H, Shen L, Qin Y. J. Am. Chem. Soc. 2007;129:13794–13795. doi: 10.1021/ja075705g. [DOI] [PubMed] [Google Scholar]
- 39.a Liu P, Seo JH, Weinreb SM. Angew. Chem. Int. Ed. 2010;49:2000–2003. doi: 10.1002/anie.200906818. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Seo JH, Liu P, Weinreb SM. J. Org. Chem. 2010;75:2667–2680. doi: 10.1021/jo100339k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Belmar J, Funk RL. J. Am. Chem. Soc. 2012;134:16941–16943. doi: 10.1021/ja307277w. [DOI] [PubMed] [Google Scholar]
- 41.a Fuchs JR, Funk RL. J. Am. Chem. Soc. 2004;126:5068–5069. doi: 10.1021/ja049569g. [DOI] [PubMed] [Google Scholar]; b Fuchs JR, Funk RL. Org. Lett. 2005;7:677–680. doi: 10.1021/ol047532v. [DOI] [PubMed] [Google Scholar]
- 42.Sakaitani M, Ohfune Y. Tetrahedron Lett. 1985;26:5543–5546. [Google Scholar]
- 43.Zuo Z, Xie W, Ma D. J. Am. Chem. Soc. 2010;132:13226–13228. doi: 10.1021/ja106739g. [DOI] [PubMed] [Google Scholar]
- 44.Ennis MD, Hoffman RL, Ghazal NB, Old DW, Mooney PA. J. Org. Chem. 1996;61:5813–5817. [Google Scholar]
- 45.Zuo Z, Ma D. Angew. Chem. Int. Ed. 2011;50:12008–12011. doi: 10.1002/anie.201106205. [DOI] [PubMed] [Google Scholar]
- 46.Fuchs JR, Funk RL. J. Am. Chem. Soc. 2004;126:5068–5069. doi: 10.1021/ja049569g. [DOI] [PubMed] [Google Scholar]
- 47.Sabahi A, Novikov A, Rainier JD. Angew. Chem. Int. Ed. 2006;45:4317–4320. doi: 10.1002/anie.200601278. [DOI] [PubMed] [Google Scholar]
- 48.a Rainier JD, Kennedy AR. J. Org. Chem. 2000;65:6213–6216. doi: 10.1021/jo000831n. [DOI] [PubMed] [Google Scholar]; b Rainier JD, Kennedy AR, Chase E. Tetrahedron Lett. 1999;40:6325–6327. [Google Scholar]; c Novikov AN, Kennedy AR, Rainier JD. J. Org. Chem. 2003;68:993–996. doi: 10.1021/jo026582f. [DOI] [PubMed] [Google Scholar]; d Novikov AN, Sabahi A, Nyong AM, Rainier JD. Tetrahedron Asymm. 2003;14:911–915. [Google Scholar]
- 49.Ishida T, Ikota H, Kurahashi K, Tsukano C, Takemoto Y. Angew. Chem. Int. Ed. 2013;39:10204–10207. doi: 10.1002/anie.201305581. [DOI] [PubMed] [Google Scholar]
- 50.Wu H, Xue F, Xiao X, Qin Y. J. Am. Chem. Soc. 2010;132:14052–14054. doi: 10.1021/ja1070043. [DOI] [PubMed] [Google Scholar]
- 51.a Ellman JA, Owens TD, Tang TP. Acc. Chem. Res. 2002;35:984–995. doi: 10.1021/ar020066u. [DOI] [PubMed] [Google Scholar]; b Robak MT, Herbage MA, Ellman JA. Chem. Rev. 2010;110:3600–3740. doi: 10.1021/cr900382t. [DOI] [PubMed] [Google Scholar]
- 52.Zhang H, Hong L, Kang H, Wang J R. Am. Chem. Soc. 2013;135:14098–14101. doi: 10.1021/ja408336v. [DOI] [PubMed] [Google Scholar]
- 53.Trost BM, Osipov M, Kruger S, Zhang Y. Y. Chem. Sci. 2015;6:349–353. doi: 10.1039/c4sc01826e. [DOI] [PMC free article] [PubMed] [Google Scholar]