

Abstract
Selectins are glycan-binding adhesion molecules which mediate the initial steps of leukocyte recognition of endothelium. Cytokines control numerous aspects of CD4 T helper differentiation, but how cytokines control induction of ligands for E- and P-selectin on T helper subsets remains poorly understood. Among 20 cytokines that affect T helper cell differentiation, we identified six, IL-12, IL-18, IL-27, IL-9, IL-25 and TGFβ1, that induce expression of selectin ligands on murine CD4 T cells above the low levels associated with TCR engagement. Collectively, these six cytokines could potentially account for selectin ligand expression on all of the currently defined non-sessile T helper lineages, including Th1, Th2, Th9, Th17 and Treg. Induction of selectin ligand expression by each of these six cytokines was almost completely inhibited by pharmacologic inhibition of p38 MAPK, but not other MAPKs, or by conditional genetic deletion of p38 alpha MAPK. Analysis of the expression of key glycosyltransferase genes revealed that p38 alpha signaling was selectively required for induction of Fut7 and Gcnt1, but not for induction of St3gal4 or St3gal6. Constitutively active MKK6, an immediate upstream activator of p38 MAPK, induced selectin ligand expression equivalent to that of cytokines, and this induction was completely dependent on expression of p38 alpha. Our results identify the repertoire of cytokines responsible for selectin ligand induction on CD4 T cells and provide a mechanistic link between T helper development and T cell migration.
INTRODUCTION
Ag-stimulated CD4 T cell differentiation leads to a number of functionally distinct cell fates that are determined primarily by cytokines in the extracellular milieu. Among six defined T helper (Th) cell lineages, Th1, Th2, Th17, and Th9 are defined by their signature cytokines IFNγ, IL-4, IL-17 and IL-9, respectively, and mediate both protective immunity to specific classes of pathogens as well as distinct types of inflammation (1–4). T follicular helper (Tfh) cells provide essential help for B cells in the germinal center (5,6), and regulatory T cells (Treg) are essential components of immunologic tolerance (7,8). Each of these six lineages also requires specific cytokines and corresponding sets of transcription factors to promote their development and preclude the development of alternative fates, with three of these (Th17, Th9 and Treg) sharing a requirement for signaling by TGFβ1 (9–11). With the exception of Tfh cells, which are mostly sessile and confined to germinal centers, cells of each of the other lineages require the ability to access peripheral tissues for their effector function. However, despite an abundance of information on transcription factors and cytokines which positively or negatively regulate these Th cell fate choices, mechanisms which control the ability of each Th cell subset to migrate to peripheral sites of inflammation remain largely undefined.
Selectins are a family of three glycan-binding adhesion molecules that mediate the initial steps of tethering and rolling on endothelium during migration to secondary lymphoid organs and sites of inflammation (12–14). E- and P-selectin (CD62E and CD62P) are inducibly expressed on endothelium at sites of inflammation and are critical for recruitment of all classes of leukocytes. Glycans that function as selectin ligands are absent from naive T cells and are inducibly expressed on CD4 cells as a function of both TCR engagement and the cytokine milieu (15,16). IL-12 is a potent inducer of selectin ligands on activated CD4 cells, whereas IL-4 either has no effect or actively inhibits selectin ligand expression (15–17). TGFβ1 is also a potent inducer of selectin ligand expression on CD4 T cells in both humans and mice (18,19). However, other cytokines that might play a role in regulation of selectin ligand expression have not been identified, and signal transduction pathways that underlie the ability of IL-12 or TGFβ1 to induce selectin ligands are incompletely understood.
We screened a large panel of cytokines known to regulate T cell function and identified a subset which induce selectin ligand expression. While highly disparate in their canonical signaling mechanisms, these six T helper-promoting cytokines each activate p38 MAP kinase and require p38 MAPK activity for selectin ligand induction. Moreover, p38 MAPK activity was mediated exclusively by the p38 alpha isoform, and was selectively required for expression of only a subset of relevant glycosyltransferases (GTases). These results identify cytokines capable of selectin ligand induction on all defined classes of T helper cells, and identify a common pathway through which they function.
MATERIALS & METHODS
Mice
WT C57BL/6 mice were originally obtained from Jackson Labs and were maintained in our colony under specific pathogen-free conditions. Mice with a “floxed” p38 alpha allele were kindly provided by Dr. Mercedes Rincon, University of Vermont, and were crossed with mice expressing the cre recombinase under the control of the CD4 locus, which were originally obtained from Jackson Labs and maintained in our colony. For all experiments involving mice with conditional deletion of p38 alpha, cre-negative littermates were used as controls. Mice of both genders were used, and were matched within an experiment, and no differences between genders were observed. All experiments were approved by the Northwestern University IACUC.
T cell activation and culture
Splenic CD4 T cells isolated from WT C57BL6 mice by positive selection using CD4 magnetic beads (Miltenyi) were activated for ~40 hrs by plate-bound anti-CD3/anti-CD28, and cultured with the indicated cytokines (see Fig 1 legend) as described (17,20), with or without pharmacologic inhibitors. Cytokines were added either at the initiation of culture, or following addition of pharmacologic inhibitors (or vehicle only; see figure legends) or retroviral transduction, as indicated in each figure legend. Ecotropic recombinant retrovirus was produced by transfecting Phoenix-Eco cells. Activated T cells were transduced by spinfection on day 2 of activation with supernatants containing recombinant retrovirus encoding eGFP plus either no cDNA, dominant negative (DN) p38 MAPK, or constitutively active (ca) MKK6, Rac1 or Rac2, as described (21). For all cultures, cells were harvested every 2 days, analyzed by FACS, and recultured in fresh media with (DN p38, inhibitors) or without (caMKK6, caRac1, caRac2) cytokines. Cytokines (see fig 1 legend for concentrations used) were all murine except TGFβ1 and were from Peprotech (IL-2, IL-4, IL-6, IL-7, IL-9, IL-12, IL-15, IL-21, IL-23); Ebioscience (IL-10, IFNγ, IL-27, TNFα); R&D Systems (TGFβ1, TSLP, IFNβ); Reprokine (IL-18, IL-25); or Axxora (IL-33).
Fig 1. Cytokines which induce selectin ligands on murine CD4 T cells.
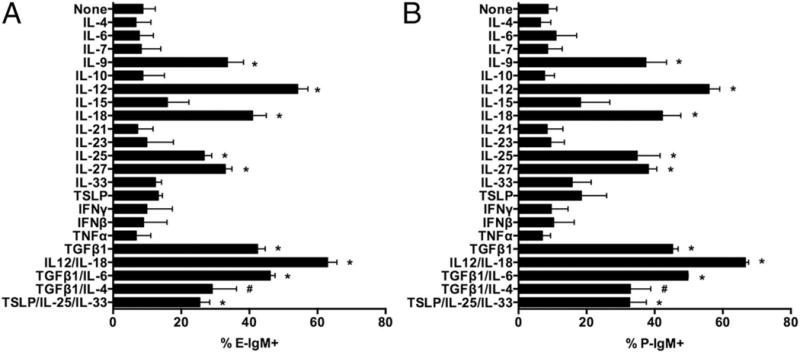
CD4+ T cells were activated with plate-bound anti-CD3/CD28 in the presence of the indicated cytokines as described in Materials & Methods. FACS analysis for selectin ligands was performed every 2 days; results are from day 8. IL-4 (10 ng/ml), IL-6 (20 ng/ml), IL-7 (10U/ml), IL-9 (20 ng/ml), IL-10 (20 ng/ml), IL-15 (10 ng/ml), IL-18 (50 ng/ml), IL-21 (20 ng/ml), IL-23 (20 ng/ml), IL-25 (25 ng/ml), IL-27 (100 ng/ml), IL-33 (10 ng/ml), TSLP (10 ng/ml), IFNγ (10 ng/ml), IFNβ (1000U/ml), TNFα (20 ng/ml), IL-12 (10 ng/ml), TGFβ1 (5 ng/ml). (A) E-selectin; (B) P-selectin. Values are mean ± SD for n=3. * p<0.01 vs IL-2 alone. # p<0.01 vs TGFβ1 alone.
Flow cytometry
Expression of E- and P-selectin ligands on T cells was determined by staining with E- or P-selectin/IgM chimeras followed by AF647-anti-human IgM, as described (17,19). We and others have previously shown that staining with these selectin chimeras closely and quantitatively tracks the ability of cells to roll on these selectins (15,17,18,20,22–24). Live/dead gating was with Sytox blue. Retrovirally transduced cells were identified by eGFP expression. Data was collected on a BD FACS Canto and analyzed using Flojo software.
Western blotting
Western blotting of whole cell lysates representing 1–2 × 106 cells for either total or phospho-p38 MAPK was as described (17).
qRT-PCR
Total RNA was isolated from CD4 cells on day 8 using Trizol, and 1 μg was reverse transcribed using the Superscript III system and random hexamers. cDNA was amplified and quantitated with SYBR green using primers specific for mouse St3gal4, St3gal6, Fut7 and Gcnt1. Data were normalized to the housekeeping gene Hprt, and further normalized by assigning the normalized value for each gene in the absence of cytokines or inhibitors to 1.
Statistical analysis
Comparison between groups was by Multiple t test within Prizm 6.
RESULTS
Identification of selectin ligand-inducing cytokines
We selected a panel of cytokines based on their ability to affect T helper differentiation, and tested them individually or in physiologically relevant combinations for their ability to induce selectin ligands above the low levels associated with T cell activation alone. We included IL-2 in all cultures, because inclusion of IL-2 does not alter the level of selectin ligands on viable CD4 T cells but does increase cell yield and viability (data not shown). This screen identified six T helper promoting cytokines exhibiting strong selectin ligand-inducing activity: IL-12, IL-18, IL-27, IL-9, IL-25, and TGFβ1 (Fig 1). No cytokines selectively induced only E- or P-selectin ligands. Selectin ligand expression on CD4 T cells cultured with TGFβ1 was not altered by inclusion of IL-6, and IL-6 alone had no significant selectin ligand-inducing activity, nor did other STAT3-activating cytokines, including IL-10 and IL-21. IL-4 inhibited selectin ligand induction by TGFβ1. Because IL-4 does not induce selectin ligand expression, how Th2 cells in vivo express selectin ligands (25,26) has been enigmatic. Our finding that IL-25, known to promote Th2 development (27), is a strong inducer of selectin ligands therefore suggests that IL-25 could be the major physiologic inducer of selectin ligands on Th2 cells in vivo. Although IL-9 is not known to promote the development of specific T helper lineages on its own, autocrine IL-9 could amplify selectin ligand expression on Th9 or Th2 cells. Other γc-activating cytokines, including IL-2, IL4, IL-7 and IL-15, had no significant effect. IL-21 and IL-23, both of which promote Th17 generation and/or expansion (28), were without effect. Notably, both IFNβ and IFNγ were without effect, despite being potent inducers of T-bet (29), which is critical for selectin ligand induction in response to IL-12 (20). For each of these cytokines, selectin ligands were expressed on a distinct subpopulation of cells (Fig 2A), and exhibited identical kinetics of expression, with the percentage of cells expressing selectin ligands increasing throughout the culture until at least day 10 (Fig 2B). Dose response curves (Fig 2C) showed that the doses used were near maximal. Together, these results identify a restricted subset of cytokines that collectively could account for selectin ligand induction on each of the currently defined non-sessile T helper lineages.
Fig 2. Selectin ligand expression in response to inflammatory cytokines.
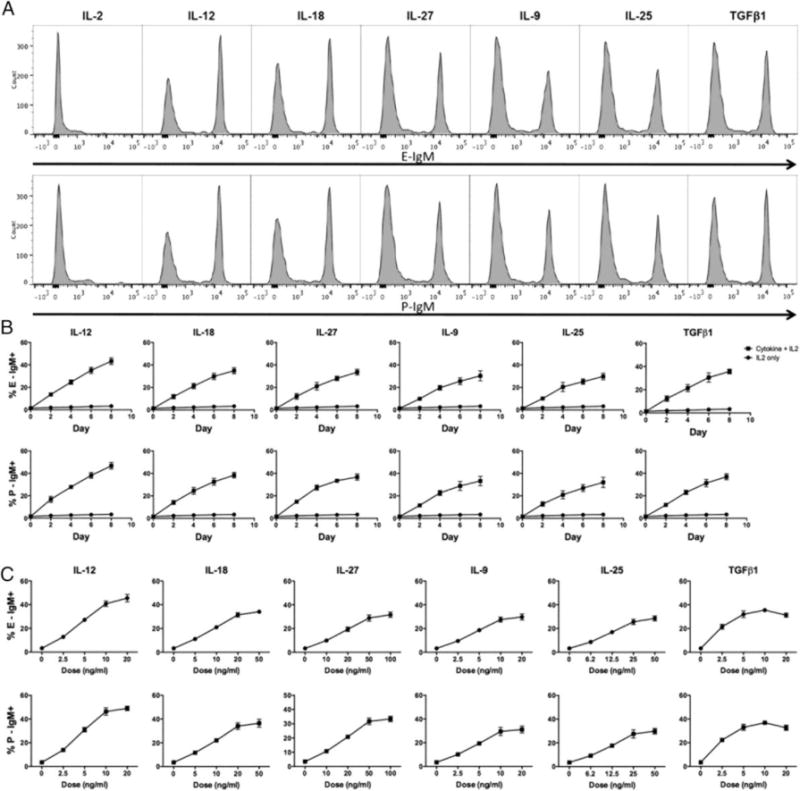
(A) Representative FACS plots of E- and P-selectin ligands on CD4 T cells cultured with each of the six cytokines identified as in Fig 1. (B) Kinetics of expression of selectin ligands in response to these six cytokines; doses used are as in fig 1. (C) Dose response curves for each cytokine; data are from day 8.
Cytokines induce selectin ligands on CD4 cells via p38 alpha MAPK
Induction of selectin ligands on human CD4 T cells in response to TGFβ1 is blocked by SB203580, a specific pyrimidazole inhibitor of p38 MAP kinase (18). IL-12, IL-18 and IL-27 are also known to activate p38 MAPK (30–32). We therefore asked whether p38 MAPK was a common pathway for selectin ligand induction by these six cytokines. CD4 T cells were activated by anti-CD3/CD28 overnight in the absence of cytokines to enhance cytokine responsiveness, rested for 6 hours, and then cultured with the six selectin ligand-inducing cytokines for 15 min. Each of the six cytokines strongly activated p38 MAPK, and for each cytokine this effect was dependent on prior T cell activation (Fig 3A).
Fig 3. Selective role of p38 MAPK in cytokine-induced selectin ligand expression.
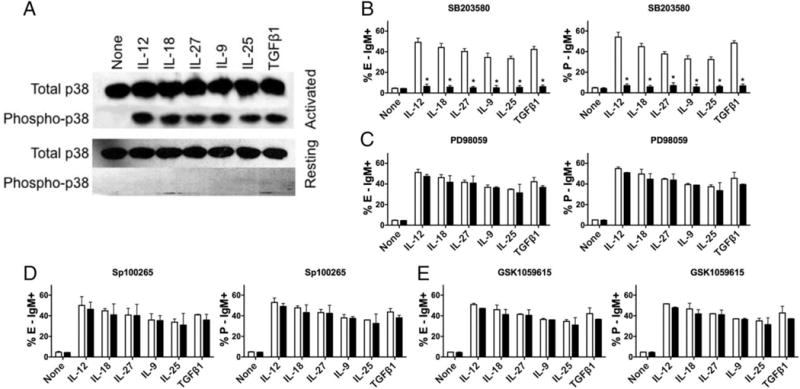
(A) CD4 T cells were either activated with anti-CD3/CD28 (top panels) or not (bottom), rested for 6 hours, exposed to the indicated cytokine for 15 min, and whole cell lysates were Western blotted for either total or phospho-p38 MAPK as indicated. Representative of 3 similar experiments. (B-E) CD4 T cells were activated by anti-CD3/CD28 without cytokine or inhibitor for 2 days, harvested, and cultured with cytokine plus (B) 5 μM SB203580; (C) 10 μM PD98059; (D) 5 μM Sp100265; or (E) 10 nM GSK1059615 (filled symbols) or vehicle (DMSO) only (open symbols), as described in Materials & Methods; data are from day 8. B-D, mean ± SD for 3 (B) or 2 (C-E) experiments. *p<0.01 vs no inhibitor.
We then activated CD4 T cells with anti-CD3/CD28 in the absence of cytokines, and then cultured them with each of the six cytokines in the presence or absence of specific inhibitors of different MAPK pathways. Selectin ligand induction in response to each of these six cytokines was nearly completely blocked by 5 μM SB203580 (Fig 3B), but was not affected by pharmacologic inhibition of either ERK MAPK activation by 10 μM PD98059 (Fig 3C) or JNK MAPK activation by 5 μM SP100265 (Fig 3D). No defects in T cell proliferation or viability were observed in cultures containing SB203580 (data not shown). Because this dose of SB203580 was reported to also affect the PI3K pathway (33), we directly tested whether pharmacologic inhibition of PI3K by GSK1059615 would have any effect on selectin ligand induction in response to these cytokines, and found that it did not (Fig 3E). In addition, retroviral overexpression of a DN p38 also blocked selectin ligand induction in response to all six cytokines (data not shown). These results indicate that the p38 MAPK pathway is selectively involved in selectin ligand induction in response to diverse inflammatory cytokines.
Four p38 isoforms, p38α, p38β, p38γ, and p38δ, have been identified, three of which (p38α, p38β, and p38δ) are expressed in CD4 T cells (34). Of the expressed isoforms, p38β is expressed at very low levels (34), and p38δ is insensitive to inhibition by SB203580 (35). Taken together, these considerations clearly implicate p38α as the primary isoform responsible for cytokine-induced selectin ligand expression. To test this, we analyzed CD4 T cells from mice with a conditional deletion of p38α driven by CD4-cre. Consistent with previous reports (36–38), these mice exhibit no detectable alterations in T cell development or in peripheral CD4 or CD8 T cell numbers (data not shown). CD4 T cells were isolated from cre+ and cre-negative littermates and analyzed as above for selectin ligand induction by these six cytokines. The results (Fig 4) show that conditional genetic inactivation of p38α in T cells abrogates the expression of selectin ligands in response to these six cytokines, phenocopying the results obtained with pharmacologic inhibition. Taken all together, our results offer strong evidence that p38α MAPK is required for selectin ligand induction on CD4 T cells in response to this panel of cytokines.
Fig 4. Selectin ligand induction in response to inflammatory cytokines requires p38 alpha.
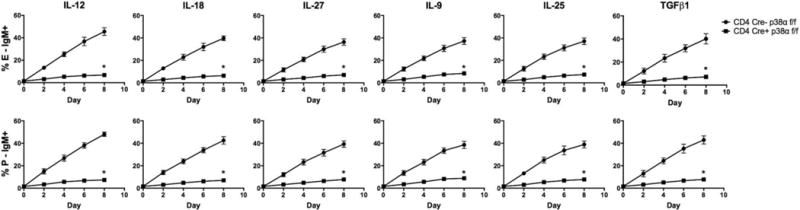
CD4 cells were isolated from p38α cKO mice or their cre-negative littermates and cultured with the six cytokines or IL-2 alone as above. FACS analysis was performed every two days. Values are mean ± SD for n=3. *p<0.01 vs cre-negative cells.
p38α MAPK is required for induction of Fut7 and Gcnt1
To identify mechanisms of p38 MAPK-dependent selectin ligand induction, we next examined expression of glycosyltransferases (GTases) known to be involved in selectin ligand biosynthesis in murine CD4 T cells (39–41). CD4 T cells activated and cultured with each of the six cytokines or IL-2 only, both with and without SB203580, were analyzed by qRT-PCR for expression of genes encoding Fut7, Gcnt1, St3gal4 and St3gal6. The results (Fig. 5) show that expression of each of these GTase genes was coordinately increased by these six cytokines, with the most potent induction in response to IL-12 and TGFβ1, roughly corresponding to the percentage of selectin ligand-positive cells generated in the presence of each cytokine (Fig 1). Inhibition of p38 MAPK by SB203580 strongly inhibited expression of Fut7 and Gcnt1, but had no significant effect on St3gal4 or St3gal6 (Fig 5). Essentially identical results were obtained using p38α-deficient CD4 T cells, where we also observed strong inhibition of Fut7 and Gcnt1 gene expression, but no significant effect on St3gal4 or St3gal6 (Fig 6). These results show that p38α MAPK signaling is selectively required for cytokine-driven upregulation of two key GTases in response to T helper-promoting inflammatory cytokines.
Fig 5. Selective inhibition of Fut7 and Gcnt1 by p38 MAPK inhibition.
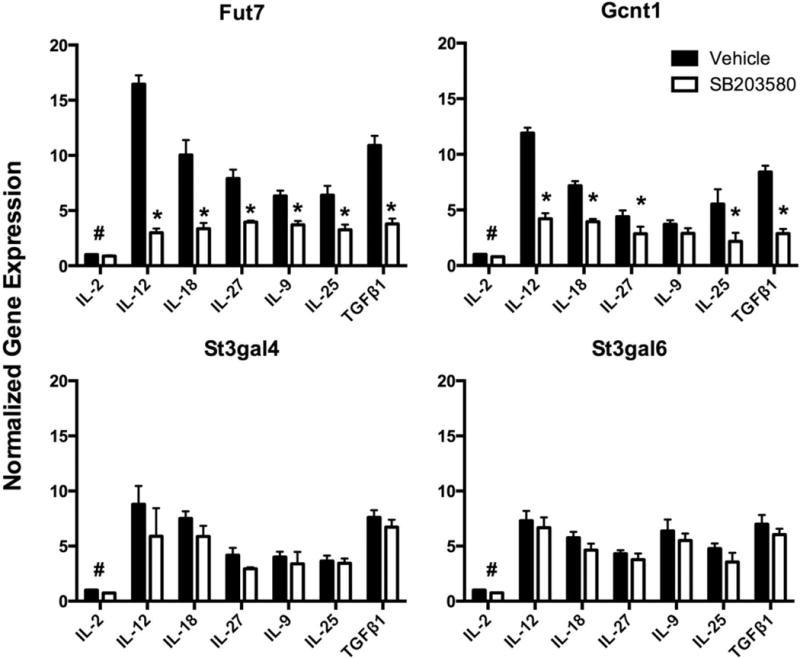
CD4 T cells were activated and cultured with cytokines or IL-2 only in the presence (open bars) or absence (closed bars) of 5 μM SB203580 as above, harvested at day 8, resuspended in Trizol, and 1 μg of total RNA was reverse transcribed. Expression of Fut7, Gcnt1, St3gal4, and St3gal6 was quantitated using SYBR green. Levels of mRNA for each enzyme in the absence of cytokines and inhibitors was assigned a value of 1. Values are mean ± SD for n=3. *p<0.01 vs IL-2 only group; #, p<0.01 for both groups different from all other groups.
Fig 6. Inhibition of Fut7 and Gcnt1 expression by genetic inactivation of Mapk14.
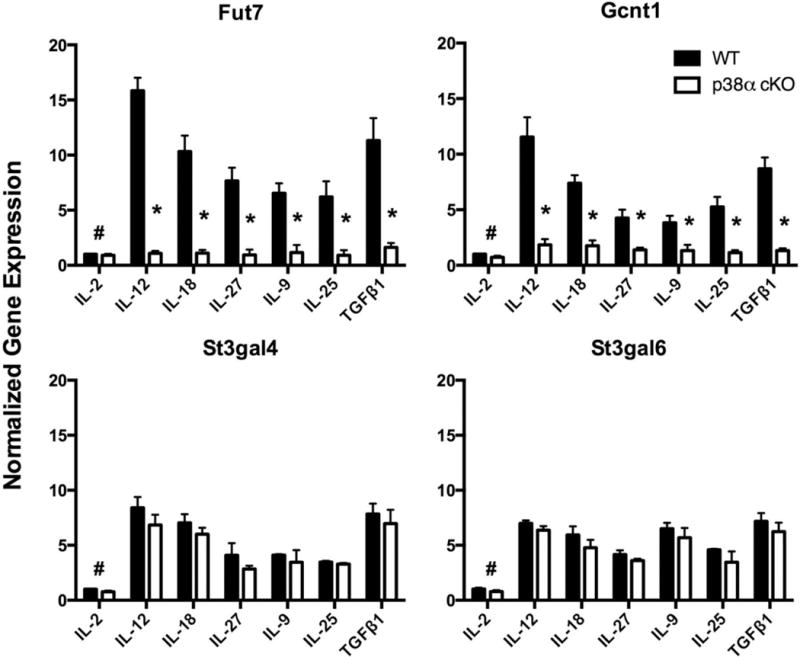
Identical setup as in fig 5, except that instead of cells being treated with SB203580, RNA was isolated on day 8 from the p38α MAPK-deficient CD4 and cre-negative control cells in fig 4. Values are mean ± SD for n=3. *p<0.01 vs IL-2 only group.
Constitutive activation of p38α is sufficient for induction of selectin ligands
Finally, to determine whether activation of p38 MAPK was sufficient for induction of selectin ligands, CD4 T cells were activated and transduced with RV expressing a constitutively active (ca) form of MKK6, the immediate upstream activator of p38 MAPK, and cultured in the absence of selectin ligand-inducing cytokines. We found that expression of caMKK6 induced levels of selectin ligands similar to those of cytokines (Fig 7). Similar results were found using caRac1 or caRac2, which activate MEKK4, the upstream activator of MKK6 (Fig 7). To ensure that this response was entirely dependent on p38α, these experiments were also carried out with p38α-deficient CD4 cells. Induction of selectin ligands by each of these upstream p38 activators was absent in cells deficient in p38α (Fig 7). Thus, activation of p38α is essential for induction of selectin ligands on CD4 T cells in response to these six inflammatory cytokines, and is sufficient for induction of selectin ligands in the absence of inducing cytokines.
Fig 7. Induction of selectin ligands in response to caMKK6 requires p38α.
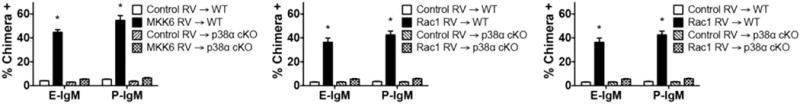
CD4+ T cells from p38α MAPK-deficient CD4 and cre-negative control cells were activated, transduced with RV expressing caMKK6, caRac1, caRac2, or no cDNA, and cultured with IL-2 only. Cells were analyzed by FACS for E-selectin ligands and P-selectin ligands as above; data are from day 8. Values are mean ± SD for n=3. *p<0.01 vs ctrl RV.
DISCUSSION
Although cytokines control numerous aspects of the immune response and host defense, how cytokines control leukocyte traffic, particularly T cell traffic, remains poorly understood. Similarly, it has been known for some time that specific cytokines or combinations of cytokines drive the differentiation of specific T helper subsets, but how this is integrated into the regulation of T cell migration has been largely unclear. Selectins are critically involved in control of T cell traffic, and expression of selectin ligands is critical for recruitment of multiple classes of inflammatory T helper cells to migrate to at least the skin and gut. Th1 cell migration to the skin during DTH (42,43) and to the gut (44), Th2 cell recruitment to the skin in atopic dermatitis (45,46), and Th17 cell migration to the gut (47,48), all require selectin ligand expression on inflammatory T cells, and Th9 cells are also skin-tropic and proinflammatory (49). Treg also require the ability to access peripheral tissues in order to downregulate immune responses (50). Thus, how selectin ligand expression is regulated on T helper cells of all classes is a key question in understanding the pathogenesis of a range of chronic inflammatory disorders.
In this report, we identify a small group of inflammatory cytokines, IL-12, IL-18, IL-27, IL-9, IL-25, and TGFβ1, most of which have defined roles in the differentiation of specific T helper subsets, that induce selectin ligands on activated CD4 T cells. We further show that induction of selectin ligands by this group of cytokines requires p38 MAPK activity, mediated specifically by the p38 alpha isoform, despite otherwise distinct signaling mechanisms. Our findings identify a common signaling mechanism underlying expression of selectin ligands on distinct classes of CD4 T cells, and provide a foundation for further dissecting molecular mechanisms that coordinate regulation of T helper differentiation and T helper migration.
Although MAPK p38β is expressed at much lower levels than p38α in CD4 T cells (34), more recent research has shown that p38β represents as much as one third of the active, i.e. phosphorylated species following activation specifically through the TCR (51). Whether this degree of relative phosphorylation of p38β is also induced by cytokines in unknown. As mentioned above, p38β activity is also inhibited by SB203580. However, our results using p38α conditional knockout mice show clearly that p38α accounts for essentially all of the activity in response to cytokines, indicating that p38β plays little or no role in these responses. Data showing that caMKK6 or caRac1/2 can induce selectin ligands also suggest that cytokines trigger primarily the classical pathway of p38 MAPK activation.
At least four glycosyltransferases contribute to selectin ligand formation: FucT-VII, C2GlcNAcT-I, ST3Gal-IV and ST3Gal-VI (39–41,52,53). Although expression of the genes encoding each of these enzymes was upregulated by these six cytokines, we found that only Fut7 and Gcnt1 induction are dependent on p38 MAPK signaling. This finding implies that these cytokines trigger additional, p38α MAPK-independent signaling pathways that are responsible for upregulation of St3gal4 and St3gal6, which will be important to identify. Our results implicate Fut7 and Gcnt1 as targets of a p38 alpha MAPK-dependent genetic program that controls T cell migration in diverse settings of inflammation.
It is possible that our results could be explained in part by a requirement for p38α MAPK in expression of cell surface receptors for the selectin ligand-inducing cytokines we have identified. Little is known about signaling pathways that control expression of most of these cytokine receptors. However, inhibition of p38 activity does not affect the ability of IL-12 to trigger STAT4 phosphorylation (30), and receptors for TGFβ1 are constitutively expressed on T cells. In addition, caMKK6 induced high levels of selectin ligand expression which are absolutely dependent on p38α MAPK. These findings make it unlikely that inhibition of receptor expression is a common mechanism underlying our findings.
IL-12 promotes Th1 development, and both IL-18 and IL-27 can also induce or augment Th1 development (31,32,54,55). We and others have previously shown that IL-12 potently induces selectin ligands (15–17), and we show here that IL-18 and IL-27 also do so. Th1 development and selectin ligand induction by IL-12 requires both Stat4 and T-bet (17,20,56,57). Like IL-12, IL-18 and IL-27 also induce T-bet (58,59), which may be required for selectin ligand induction by these cytokines, and induction of T-bet by these cytokines appears to require p38 MAPK signaling (32,60). Also like IL-12, both IL-18 and IL-27 are products of APC, suggesting multiple ways through which APC can regulate selectin ligand expression in CD4 T cells. Recent evidence indicates that IL-27 may be additionally important for modulation of inflammation in part via its ability to induce IL-10 (61), suggesting a mechanism for maintenance of T cell recruitment during the resolution phase of an inflammatory response.
The downstream targets that link p38 MAPK signaling to induction of Fut7 and Gcnt1 in T cells are presently unclear, and may not be identical for each of these cytokines. A number of widely expressed transcription factors have been identified as direct targets of p38 MAPK, including ATF-2, CREB, CHOP, MEF2C, Runx, and STATs (62,63). IL-12 induces phosphorylation of STAT4 serine 721 by p38 MAPK, and this enhances transcriptional potential for a subset of STAT4 gene targets (64), which may include Gcnt1, since STAT4 is essential for IL-12-induced expression of Gcnt1 (17). IL-9 and IL-27 activate STAT1, STAT3 and/or STAT5 in various cell types, including T cells, suggesting that one or more of these STAT proteins could function downstream of IL-9 and IL-27 in selectin ligand induction, analogous to STAT4 in IL-12-induced responses. Consistent with this, p38 MAPK phosphorylates serine 727 in both STAT1 and STAT3, which is homologous to serine 721 in STAT4. These serine residues are located in a highly conserved region of the transactivation domains of STAT1, STAT3 and STAT4, and p38-mediated phosphorylation of serine 727 in STAT1 and STAT3 also enhances their transcriptional activity (65,66).
Distinct mechanisms involving other p38 targets seem likely at least for TGFβ1 and IL-25. TGFβ1 signaling utilizes multiple MAPK pathways in addition to Smad transcription factors, and both p38 and ERK MAPK have been shown to phosphorylate Smad proteins (67), modulating their activity. Determining whether phosphorylation of Smad proteins by p38 alpha MAPK is involved in selectin ligand induction, or whether the requirement for p38 MAPK signaling is independent of Smad transcriptional activity, is a key question going forward. The E3 ubiquitin ligase Act1/CIKS is an adaptor protein essential for at least some signaling through the IL-25R and related family members (68). Act1 couples the IL25R to multiple downstream effectors, including Traf6 (69,70). It will be important to determine whether Act1, or Traf6, is required for p38 MAPK activation by IL-25. It will also be important to determine how the IL9R couples to p38 activation.
In summary, we have shown that a diverse group of inflammatory cytokines can potentially collectively account for expression of selectin ligands on all defined murine T helper subsets, and that selectin ligand induction in response to these functionally distinct cytokines requires p38α MAPK. The undoubtedly diverse mechanisms underlying the requirement for p38α MAPK in GTase gene induction in response to these inflammatory cytokines will require further investigation. Our results identify p38α MAPK as a common and essential pathway through which a select group of cytokines responsible for promoting the development of diverse T helper lineages can couple differentiation of specific T helper lineages to expression of key homing molecules required for effector function in peripheral tissues.
Acknowledgments
The authors thank numerous colleagues for provision of cytokines used in this study.
Footnotes
Supported in part by the Chicago Biomedical Consortium with support from the Searle Funds at The Chicago Community Trust (GSK); and by Public Health Service grants AI045515, AI057459, and AI095282 (MHK). OA was supported by PHS F31 Al100542. Support provided by the HB Wells Center was in part from the Riley Children’s Foundation.
References
- 1.Zhu J, Paul WE. Peripheral CD4+ T-cell differentiation regulated by networks of cytokines and transcription factors. Immunol Rev. 2010;238:247–262. doi: 10.1111/j.1600-065X.2010.00951.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abbas AK, Murphy KM, Sher A. Functional diversity of helper T lymphocytes. Nature. 1996;383:787–793. doi: 10.1038/383787a0. [DOI] [PubMed] [Google Scholar]
- 3.Iwakura Y, Ishigame H, Saijo S, Nakae S. Functional specialization of interleukin-17 family members. Immunity. 2011;34:149–162. doi: 10.1016/j.immuni.2011.02.012. [DOI] [PubMed] [Google Scholar]
- 4.Kaplan MH. Th9 cells: differentiation and disease. Immunol Rev. 2013;252:104–115. doi: 10.1111/imr.12028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nurieva RI, Chung Y, Hwang D, Yang XO, Kang HS, Ma L, Wang YH, Watowich SS, Jetten AM, Tian Q, Dong C. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity. 2008;29:138–149. doi: 10.1016/j.immuni.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chtanova T, Tangye SG, Newton R, Frank N, Hodge MR, Rolph MS, Mackay CR. T follicular helper cells express a distinctive transcriptional profile, reflecting their role as non-Th1/Th2 effector cells that provide help for B cells. J Immunol. 2004;173:68–78. doi: 10.4049/jimmunol.173.1.68. [DOI] [PubMed] [Google Scholar]
- 7.Kim JM, Rasmussen JP, Rudensky AY. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol. 2007;8:191–197. doi: 10.1038/ni1428. [DOI] [PubMed] [Google Scholar]
- 8.Rubtsov YP, Rudensky AY. TGFbeta signalling in control of T-cell-mediated self-reactivity. Nat Rev Immunol. 2007;7:443–453. doi: 10.1038/nri2095. [DOI] [PubMed] [Google Scholar]
- 9.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 10.Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 11.Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med. 2005;201:1061–1067. doi: 10.1084/jem.20042276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Butcher EC. Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell. 1991;67:1033–1036. doi: 10.1016/0092-8674(91)90279-8. [DOI] [PubMed] [Google Scholar]
- 13.Kansas GS. Selectins and their ligands: current concepts and controversies. Blood. 1996;88:3259–3286. [PubMed] [Google Scholar]
- 14.Ley K, Kansas GS. Selectins in T cell recruitment to non-lymphoid tissues and sites of inflammation. Nat Rev Immunol. 2004;4:325–335. doi: 10.1038/nri1351. [DOI] [PubMed] [Google Scholar]
- 15.Wagers AJ, Waters CM, Stoolman LM, Kansas GS. IL-12 and IL-4 control T cell adhesion to endothelial selectins through opposite effects on FucT-VII gene expression. J Exp Med. 1998;188:2225–2231. doi: 10.1084/jem.188.12.2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Austrup F, Vestweber D, Borges E, Lohning M, Brauer R, Herz U, Renz H, Hallmann R, Scheffold A, Radbruch A, Hamann A. P- and E-selectin mediate recruitment of T helper 1 but not T helper 2 cells into inflamed tissues. Nature. 1997;385:81–83. doi: 10.1038/385081a0. [DOI] [PubMed] [Google Scholar]
- 17.White SJ, Underhill GH, Kaplan MH, Kansas GS. Cutting Edge: Differential requirements for Stat4 in expression of glycosyltransferases responsible for selectin ligand formation in Th1 cells. J Immunol. 2001;167:628–631. doi: 10.4049/jimmunol.167.2.628. [DOI] [PubMed] [Google Scholar]
- 18.Wagers AJ, Kansas GS. Potent induction of FucT-VII by TGF-β1 through a p38 MAP kinase-dependent pathway. J Immunol. 2000;165:5011–5016. doi: 10.4049/jimmunol.165.9.5011. [DOI] [PubMed] [Google Scholar]
- 19.Ebel ME, Kansas GS. Defining the functional boundaries of the murine alpha1,3-fucosyltransferase Fut7 reveals a remarkably compact locus. J Biol Chem. 2014;289:6341–6349. doi: 10.1074/jbc.M113.511790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Underhill GH, Zisoulis DG, Kolli KP, Ellies LG, Marth JD, Kansas GS. A crucial role for T-bet in selectin ligand expression in T helper 1 (Th1) cells. Blood. 2005;106:3867–3873. doi: 10.1182/blood-2005-03-0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zisoulis DG, Kansas GS. H-Ras and PI3K cooperate to induce FucT-VII expression in Jurkat T cells. J Biol Chem. 2004;279:39495–39505. doi: 10.1074/jbc.M407904200. [DOI] [PubMed] [Google Scholar]
- 22.Barry SM, Zisoulis DG, Neal JW, Clipstone NA, Kansas GS. Induction of FucT-VII by the Ras/MAP kinase cascade in Jurkat T cells. Blood. 2003;102:1771–1778. doi: 10.1182/blood-2002-11-3551. [DOI] [PubMed] [Google Scholar]
- 23.Knibbs RN, Craig RA, Maly P, Smith PL, Wolber FM, Faulkner NE, Lowe JB, Stoolman LM. a1,3-fucosyltransferase VII dependent synthesis of P- and E-selectin ligands on cultured T lymphoblasts. J Immunol. 1998;161:6305–6315. [PubMed] [Google Scholar]
- 24.Knibbs RN, Craig RA, Natsuka S, Chang A, Cameron M, Lowe JB, Stoolman LM. The fucosyltransferase FucT-VII regulates E-selectin ligand synthesis in human T cells. J Cell Biol. 1996;133:911–920. doi: 10.1083/jcb.133.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kretschmer U, Bonhagen K, Debes GF, Mittrucker HW, Erb KJ, Liesenfeld O, Zaiss D, Kamradt T, Syrbe U, Hamann A. Expression of selectin ligands on murine effector and IL-10-producing CD4+ T cells from non-infected and infected tissues. Eur J Immunol. 2004;34:3070–3081. doi: 10.1002/eji.200424972. [DOI] [PubMed] [Google Scholar]
- 26.Syrbe U, Hoffmann U, Schlawe K, Liesenfeld O, Erb K, Hamann A. Microenvironment-dependent requirement of STAT4 for the induction of P-selectin ligands and effector cytokines on CD4+ T cells in healthy and parasite-infected mice. J Immunol. 2006;177:7673–7679. doi: 10.4049/jimmunol.177.11.7673. [DOI] [PubMed] [Google Scholar]
- 27.Angkasekwinai P, Park H, Wang YH, Wang YH, Chang SH, Corry DB, Liu YJ, Zhu Z, Dong C. Interleukin 25 promotes the initiation of proallergic type 2 responses. J Exp Med. 2007;204:1509–1517. doi: 10.1084/jem.20061675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zuniga LA, Jain R, Haines C, Cua DJ. Th17 cell development: from the cradle to the grave. Immunol Rev. 2013;252:78–88. doi: 10.1111/imr.12036. [DOI] [PubMed] [Google Scholar]
- 29.Zhu J, Jankovic D, Oler AJ, Wei G, Sharma S, Hu G, Guo L, Yagi R, Yamane H, Punkosdy G, Feigenbaum L, Zhao K, Paul WE. The transcription factor T-bet is induced by multiple pathways and prevents an endogenous Th2 cell program during Th1 cell responses. Immunity. 2012;37:660–673. doi: 10.1016/j.immuni.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang S, Kaplan MH. The p38 mitogen-activated protein kinase is required for IL-12-induced interferon gamma expression. J Immunol. 2000;165:1374–1380. doi: 10.4049/jimmunol.165.3.1374. [DOI] [PubMed] [Google Scholar]
- 31.Yang J, Zhu H, Murphy TL, Ouyang W, Murphy KM. IL-18-stimulated GADD45 beta required in cytokine-induced, but not TCR-induced, IFN-gamma production. Nat Immunol. 2001;2:157–164. doi: 10.1038/84264. [DOI] [PubMed] [Google Scholar]
- 32.Owaki T, Asakawa M, Fukai F, Mizuguchi J, Yoshimoto T. IL-27 induces Th1 differentiation via p38 MAPK/T-bet- and intercellular adhesion molecule-1/LFA-1/ERK1/2-dependent pathways. J Immunol. 2006;177:7579–7587. doi: 10.4049/jimmunol.177.11.7579. [DOI] [PubMed] [Google Scholar]
- 33.Lali FV, Hunt AE, Turner SJ, Foxwell BMJ. The pyridinyl imidazole inhibitor SB203580 blocks phosphoinositide-dependent protein kinase activity, protein kinase B phosphorylation, and retinoblastoma hyperphosphorylation in interleukin 2-stimulated T cells independently of p38 mitogen-activated protein kinase. J Biol Chem. 2000;275:7395–7402. doi: 10.1074/jbc.275.10.7395. [DOI] [PubMed] [Google Scholar]
- 34.Hale KK, Trollinger D, Rihanek M, Manthey CL. Differential expression and activation of p38 mitogen-activated protein kinase alpha, beta, gamma, and delta in inflammatory cell lineages. J Immunol. 1999;162:4246–4252. [PubMed] [Google Scholar]
- 35.Kumar S, McDonnell PC, Gum RJ, Hand AT, Lee JC, Young PR. Novel homologues of CSBP/p38 MAP kinase: activation, substrate specificity and sensitivity to inhibition by pyridinyl imidazoles. Biochem Biophys Res Comm. 1997;235:533–538. doi: 10.1006/bbrc.1997.6849. [DOI] [PubMed] [Google Scholar]
- 36.Kim JM, White JM, Shaw AS, Sleckman BP. MAPK p38 alpha is dispensable for lymphocyte development and proliferation. J Immunol. 2005;174:1239–1244. doi: 10.4049/jimmunol.174.3.1239. [DOI] [PubMed] [Google Scholar]
- 37.Berenson LS, Yang J, Sleckman BP, Murphy TL, Murphy KM. Selective requirement of p38alpha MAPK in cytokine-dependent, but not antigen receptor-dependent, Th1 responses. J Immunol. 2006;176:4616–4621. doi: 10.4049/jimmunol.176.8.4616. [DOI] [PubMed] [Google Scholar]
- 38.Hu P, Nebreda AR, Liu Y, Carlesso N, Kaplan M, Kapur R. p38alpha protein negatively regulates T helper type 2 responses by orchestrating multiple T cell receptor-associated signals. J Biol Chem. 2012;287:33215–33226. doi: 10.1074/jbc.M112.355594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maly P, Thall AD, Petryniak B, Rogers CE, Smith PL, Marks RM, Kelly RJ, Gersten KM, Cheng G, Saunders TL, Camper SA, Camphausen RT, Sullivan FX, Isogai Y, Hindsgaul O, von Andrian UH, Lowe JB. The α (1,3) fucosyltransferase FucT-VII controls leukocyte trafficking through an essential role in L-, E-, and P-selectin ligand biosynthesis. Cell. 1996;86:643–653. doi: 10.1016/s0092-8674(00)80137-3. [DOI] [PubMed] [Google Scholar]
- 40.Ellies LG, Tsuboi S, Petryniak B, Lowe JB, Fukuda M, Marth JD. Core 2 oligosaccharide biosynthesis distinguishes between selectin ligands essential for leukocyte homing and inflammation. Immunity. 1998;9:881–890. doi: 10.1016/s1074-7613(00)80653-6. [DOI] [PubMed] [Google Scholar]
- 41.Yang WH, Nussbaum C, Grewal PK, Marth JD, Sperandio M. Coordinated roles of ST3Gal-VI and ST3Gal-IV sialyltransferases in the synthesis of selectin ligands. Blood. 2012;120:1015–1026. doi: 10.1182/blood-2012-04-424366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Borges E, Tietz W, Steegmaier M, Moll T, Hallmann R, Hamann A, Vestweber D. P-selectin glycoprotein ligand-1 (PSGL-1) on T helper 1 but not on T helper 2 cells binds to P-selectin and supports migration into inflamed skin. J Exp Med. 1997;185:573–578. doi: 10.1084/jem.185.3.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Staite ND, Justen JM, Sly LM, Beaudet AL, Bullard DC. Inhibition of delayed-type contact hypersensitivity in mice deficient in both E-selectin and P-selectin. Blood. 1996;88:2973–2979. [PubMed] [Google Scholar]
- 44.Haddad W, Cooper CJ, Zhang Z, Brown JB, Zhu Y, Issekutz A, Fuss I, Lee HO, Kansas GS, Barrett TA. P-selectin and P-selectin glycoprotein ligand-1 are major determinants for Th1 cell recruitment to nonlymphoid effector sites in the intestinal lamina propria. J Exp Med. 2003;198:369–377. doi: 10.1084/jem.20020691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Santamaria Babi LF, Picker LJ, Perez Soler MT, Drzimalla K, Flohr P, Blaser K, Hauser C. Circulating allergen-reactive T cells from patients with atopic dermatitis and allergic contact dermatitis express the skin-selective homing receptor, the cutaneous lymphocyte-associated antigen. J Exp Med. 1995;181:1935–1940. doi: 10.1084/jem.181.5.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Biedermann T, Schwarzler C, Lametschwandtner G, Thoma G, Carballido-Perrig N, Kund J, de Vries JE, Rot A, Carballido JM. Targeting CLA/E-selectin interactions prevents CCR4-mediated recruitment of human Th2 memory cells to human skin in vivo. Eur J Immunol. 2002;32:3171–3180. doi: 10.1002/1521-4141(200211)32:11<3171::AID-IMMU3171>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 47.Angiari S, Donnarumma T, Rossi B, Dusi S, Pietronigro E, Zenaro E, Della Bianca V, Toffali L, Piacentino G, Budui S, Rennert P, Xiao S, Laudanna C, Casasnovas JM, Kuchroo VK, Constantin G. TIM-1 glycoprotein binds the adhesion receptor P-selectin and mediates T cell trafficking during inflammation and autoimmunity. Immunity. 2014;40:542–553. doi: 10.1016/j.immuni.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brown JB, Cheresh P, Zhang Z, Ryu H, Managlia E, Barrett TA. P-selectin glycoprotein ligand-1 is needed for sequential recruitment of T-helper 1 (Th1) and local generation of Th17 T cells in dextran sodium sulfate (DSS) colitis. Inflamm bowel Dis. 2012;18:323–332. doi: 10.1002/ibd.21779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schlapbach C, Gehad A, Yang C, Watanabe R, Guenova E, Teague JE, Campbell L, Yawalkar N, Kupper TS, Clark RA. Human TH9 cells are skin-tropic and have autocrine and paracrine proinflammatory capacity. Science Trans Med. 2014;6:219ra218. doi: 10.1126/scitranslmed.3007828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Siegmund K, Feuerer M, Siewert C, Ghani S, Haubold U, Dankof A, Krenn V, Schon MP, Scheffold A, Lowe JB, Hamann A, Syrbe U, Huehn J. Migration matters: regulatory T-cell compartmentalization determines suppressive activity in vivo. Blood. 2005;106:3097–3104. doi: 10.1182/blood-2005-05-1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jirmanova L, Giardino Torchia ML, Sarma ND, Mittelstadt PR, Ashwell JD. Lack of the T cell-specific alternative p38 activation pathway reduces autoimmunity and inflammation. Blood. 2011;118:3280–3289. doi: 10.1182/blood-2011-01-333039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Snapp KR, Heitzig CE, Ellies LG, Marth JD, Kansas GS. Differential requirements for the O-linked branching enzyme core 2 β1-6-N-glucosaminyltransferase (C2GlcNAcT-I) in biosynthesis of ligands for E-selectin and P-selectin. Blood. 2001;97:3806–3812. doi: 10.1182/blood.v97.12.3806. [DOI] [PubMed] [Google Scholar]
- 53.Ellies LG, Sperandio M, Underhill GH, Yousif J, Smith M, Priatel JJ, Kansas GS, Ley K, Marth JD. Sialyltransferase specificity in selectin ligand formation. Blood. 2002;100:3618–3625. doi: 10.1182/blood-2002-04-1007. [DOI] [PubMed] [Google Scholar]
- 54.Okamura H, Tsutsi H, Komatsu T, Yutsudo M, Hakura A, Tanimoto T, Torigoe K, Okura T, Nukada Y, Hattori K, et al. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature. 1995;378:88–91. doi: 10.1038/378088a0. [DOI] [PubMed] [Google Scholar]
- 55.Chen Q, Ghilardi N, Wang H, Baker T, Xie MH, Gurney A, Grewal IS, de Sauvage FJ. Development of Th1-type immune responses requires the type I cytokine receptor TCCR. Nature. 2000;407:916–920. doi: 10.1038/35038103. [DOI] [PubMed] [Google Scholar]
- 56.Kaplan MH, Sun YL, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature. 1996;382:174–177. doi: 10.1038/382174a0. [DOI] [PubMed] [Google Scholar]
- 57.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 58.Bachmann M, Dragoi C, Poleganov MA, Pfeilschifter J, Muhl H. Interleukin-18 directly activates T-bet expression and function via p38 mitogen-activated protein kinase and nuclear factor-kappaB in acute myeloid leukemia-derived predendritic KG-1 cells. Mol Can Therap. 2007;6:723–731. doi: 10.1158/1535-7163.MCT-06-0505. [DOI] [PubMed] [Google Scholar]
- 59.Kamiya S, Owaki T, Morishima N, Fukai F, Mizuguchi J, Yoshimoto T. An indispensable role for STAT1 in IL-27-induced T-bet expression but not proliferation of naive CD4+ T cells. J Immunol. 2004;173:3871–3877. doi: 10.4049/jimmunol.173.6.3871. [DOI] [PubMed] [Google Scholar]
- 60.Jones DC, Ding X, Zhang TY, Daynes RA. Peroxisome proliferator-activated receptor alpha negatively regulates T-bet transcription through suppression of p38 mitogen-activated protein kinase activation. J Immunol. 2003;171:196–203. doi: 10.4049/jimmunol.171.1.196. [DOI] [PubMed] [Google Scholar]
- 61.Awasthi A, Carrier Y, Peron JP, Bettelli E, Kamanaka M, Flavell RA, Kuchroo VK, Oukka M, Weiner HL. A dominant function for interleukin 27 in generating interleukin 10-producing anti-inflammatory T cells. Nat Immunol. 2007;8:1380–1389. doi: 10.1038/ni1541. [DOI] [PubMed] [Google Scholar]
- 62.Dong C, Davis RJ, Flavell RA. MAP kinases in the immune response. Ann Rev Immunol. 2002;20:55–72. doi: 10.1146/annurev.immunol.20.091301.131133. [DOI] [PubMed] [Google Scholar]
- 63.Collins A, Littman DR, Taniuchi I. RUNX proteins in transcription factor networks that regulate T-cell lineage choice. Nat Rev Immunol. 2009;9:106–115. doi: 10.1038/nri2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Visconti R, Gadina M, Chiarello M, Chen EH, Stancato LF, Gutkind JS, O’Shea JJ. Importance of the MKK6/p38 pathway for interleukin-12-induced STAT4 serine phosphorylation and transcriptional activity. Blood. 2000;96:1844–1852. [PubMed] [Google Scholar]
- 65.Goh KC, Haque SJ, Williams BR. p38 MAP kinase is required for STAT1 serine phosphorylation and transcriptional activation induced by interferons. EMBO J. 1999;18:5601–5608. doi: 10.1093/emboj/18.20.5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Turkson J, Bowman T, Adnane J, Zhang Y, Djeu JY, Sekharam M, Frank DA, Holzman LB, Wu J, Sebti S, Jove R. Requirement for Ras/Rac1-mediated p38 and c-Jun N-terminal kinase signaling in Stat3 transcriptional activity induced by the Src oncoprotein. Mol Cell Biol. 1999;19:7519–7528. doi: 10.1128/mcb.19.11.7519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kamato D, Burch ML, Piva TJ, Rezaei HB, Rostam MA, Xu S, Zheng W, Little PJ, Osman N. Transforming growth factor-beta signalling: role and consequences of Smad linker region phosphorylation. Cell Signal. 2013;25:2017–2024. doi: 10.1016/j.cellsig.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 68.Gu C, Wu L, Li X. IL-17 family: cytokines, receptors and signaling. Cytokine. 2013;64:477–485. doi: 10.1016/j.cyto.2013.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Claudio E, Sonder SU, Saret S, Carvalho G, Ramalingam TR, Wynn TA, Chariot A, Garcia-Perganeda A, Leonardi A, Paun A, Chen A, Ren NY, Wang H, Siebenlist U. The adaptor protein CIKS/Act1 is essential for IL-25-mediated allergic airway inflammation. J Immunol. 2009;182:1617–1630. doi: 10.4049/jimmunol.182.3.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Maezawa Y, Nakajima H, Suzuki K, Tamachi T, Ikeda K, Inoue J, Saito Y, Iwamoto I. Involvement of TNF receptor-associated factor 6 in IL-25 receptor signaling. J Immunol. 2006;176:1013–1018. doi: 10.4049/jimmunol.176.2.1013. [DOI] [PubMed] [Google Scholar]