

Abstract
Structures of over 30 different G protein-coupled receptors (GPCRs) have advanced our understanding of cell signaling and provided a foundation for structure-guided drug design. This exciting progress required the development of three complimentary methods to facilitate GPCR crystallization, one of which is the thermostabilization of receptors by systematic mutagenesis. However, the reason why a particular mutation, or combination of mutations, stabilizes the receptor is not always evident from a static crystal structure. Molecular dynamics simulations have been used to identify and estimate the energetic factors that affect thermostability through comparing the dynamics of the thermostabilized receptors with structure-based models of the wild-type receptor. The data indicate that receptors are stabilized through a combination of factors, including an increase in receptor rigidity, a decrease in collective motion, reduced stress at specific residues, and the presence of ordered water molecules. Predicting thermostabilizing mutations computationally represents a major challenge for the field.
Keywords: thermostability, GPCR, dynamics, structure
The importance of GPCR thermostability
Over 120 structures of G protein-coupled receptors (GPCRs), with different ligands bound and in a variety of different conformations, have revolutionized our understanding of the architecture of GPCRs, the molecular mechanism of receptor activation and the molecular determinants for ligand efficacy [1–3]. Engineering GPCRs has proven essential to facilitate the formation of well-diffracting crystals and three complementary technologies have been developed [4]: (1) fusion of small soluble proteins such as T4 lysozyme (T4L) [5] and thermostabilized cytochrome b562 (BRIL) [6] into intracellular loop 3 (ICL3) or to the amino terminus of the receptors; (2) isolation of conformation-specific camelid or murine antibodies that lock the receptor in a specific state [7]; (3) conformational thermostabilization of the receptors by systematic mutagenesis [8]. In each of these strategies, the stability of the resulting GPCR-ligand complex is a key factor in determining whether crystals can be produced. The stability of receptors with T4L or BRIL fusions is usually dictated by the ligand, and it is often the case that receptor crystals can only be obtained using ligands with very high affinity (10 pM – 10 nM). This is also often the case with antibodies, but in at least one instance the camelid heavy chain antibody (nanobody) imparted sufficient stability to the receptor to allow the structure of the β2-adrenergic receptor to be determined bound to a native ligand, adrenaline [9]. In contrast to receptor fusions and antibody strategies, conformational thermostabilization specifically selects for improved thermostability of the detergent-solubilized receptor, and therefore crystallization of the receptor becomes largely independent of the ligand affinity, although it is still the case that no ligand-binding GPCR has had its structure determined in the absence of ligand. Engineering disulfide bridge in the receptor structure was one of the earlier strategies that was used to stabilize the rhodopsin structure [10].
In defining the first structure of a given GPCR, it is largely immaterial which methodology is used. Where structures of identical or similar receptors have been determined using either thermostabilized receptors or receptor-T4L fusions, the structures are virtually identical, within experimental error, and major differences are usually confined to the loop regions due to different crystal contacts [8]. However, if multiple structures of a receptor are required to understand ligand efficacy or for hit-to-lead optimization during drug development, there are significant advantages in the conformational thermostabilization strategy. For example, the thermostabilized β1-adrenergic receptor (β1AR) has been co-crystallized with 12 different ligands (inverse agonist, weak partial agonist, partial agonist, full agonist) [11–15] that has led to a detailed understanding of the molecular determinants of ligand efficacy and the initial molecular events upon agonist binding that increase the probability of the receptor becoming active and capable of binding a G protein. In the case of the thermostabilized adenosine A2A receptor, multiple structures of lead compounds bound to the receptor led to the development of preclinical candidates for the treatment of Parkinson’s disease [16]. The best compounds were based on a novel scaffold that accessed regions of the ligand binding pocket that were previously assumed to be involved only in agonist binding, although it is clear from the pharmacology and the structure that the ligand is an inverse agonist [17].
Thermostabilized GPCRs are highly desirable for both structure determination and drug development, but the thermostabilizing process is time-consuming and costly. Given the number of thermostabilized receptors engineered (Table 1) and our in-depth understanding of receptor structure, is it possible to perform thermostabilization of a receptor computationally? What is the reason why a particular mutation or combination of mutations has a stabilizing effect? The latter question was particularly perplexing, because it was usually not evident from a static crystal structure why mutations were thermostabilizing. Therefore a series of molecular dynamics (MD) simulations have been performed on thermostabilized GPCRs [18–20] to elucidate the energetic factors that affect the thermostability and dynamics of a mutant receptor. Here we discuss our understanding of how thermostabilization results in a thermostable receptor and the progress that has been made on predicting thermostabilizing mutations computationally. Although our emphasis here is on GPCRs, the methodology is equally applicable to other membrane proteins, and the structures of a thermostabilized transporter [21] and a thermostabilized ion channel [22] have been determined recently.
Table 1.
Crystal structures determined of thermostabilized GPCRs
| Receptor | Mutant name |
Stabilized conformation |
Number of mutations |
G protein activation |
Thermo- stabilization methodology |
Ref |
|---|---|---|---|---|---|---|
| β1-adrenergic receptor (β1AR) | m23 | inactive | 6 | yes | SM* | [15] |
| Adenosine A2Areceptor (A2AR) | StaR2 | inactive | 8 | no | SM | [28] |
| GL31 | active-like | 4 | weak | SM | [29] | |
| Neurotensin receptor (NTSR1) | GW5 | active-like | 6 | no | SM | [30] |
| ELF | active-like | 3 | yes | SM | [27] | |
| TM86V | Agonist-bound, inactive | 11 | weak | directed evolution | [31] | |
| Chemokine receptor (CCR5) | - | inactive | 4 | no | Rational design | [32] |
| Free fatty acid receptor (FFA1R) | - | inactive | 4 | weak | SM | [33] |
| Corticotrophin release factor receptor (CRF1R) | - | inactive | 12 | ND* | SM | [34] |
| Metabotropic glutamate receptor (mGlu5) | - | inactive | 6 | ND | SM | [35] |
Scanning mutagenesis; ND, not determined
The effect of thermostabilization on GPCR pharmacology
To understand the results from the MD simulations discussed below, it is important to appreciate how the thermostabilizing mutations have altered the pharmacology of the receptor and also the conformation of the receptor observed in the crystal structure. GPCRs exist in multiple conformations, such as the inactive, active and other intermediate conformational states [2]. Inactive states are non-signaling receptor conformations which do not activate a G protein, for example, a receptor bound to a full inverse agonist, whereas active states are signaling GPCR conformations which, for example, catalyze nucleotide exchange at the Gα subunit of a heterotrimeric G protein. The β2-adrenergic receptor (β2AR) bound to the antagonist carazolol is regarded as an inactive state of the receptor [5], whilst β2AR bound to the full agonist BI167107 and a nucleotide-free heterotrimeric G protein is regarded as being in the fully active state [36]. However, in some structures of GPCRs bound to agonists, the receptor is described as being in an active-like state, because the receptor conformations have the majority of the characteristics of activated GPCRs, but due to the absence of a heterotrimeric G protein, the fully active conformation is not attained. This has been observed for the structures of A2AR and NTSR1 bound to agonists (Table 1). To add further complexity, different receptor parts such as the ligand binding pocket and the G protein interface may adopt different conformational states within the same GPCR [37, 38]. This is observed in the structure of NTSR1 evolved to be thermostable in the presence of agonist, where the intracellular half of the receptor represents the inactive state of the receptor and yet the extracellular portion of the receptor encompassing the ligand binding pocket is similar in conformation to the active-like state of the receptor [31].
Detailed MD simulations have been performed based on only four thermostabilized receptor structures, namely β1AR-m23, A2A-StaR2, A2A-GL31 and NTSR1-GW5 (Table 1 and Glossary). Each receptor was stabilized by a process of systematic alanine scanning mutagenesis coupled to a radioligand thermostability assay performed on detergent-solubilized receptor mutants. Single thermostabilizing mutations were then combined to make an optimally stable mutant that contained between 4–8 mutations (Table 1). β1AR-m23 and A2A-StaR2 were stabilized using the antagonists 3H-dihydroalprenolol and 3H-ZM241385, respectively, and it was found that the affinities of antagonists were similar to the wild-type receptor, but the affinities for agonists were considerably reduced [23, 25]. Thus it appeared that the thermostabilization process had altered the equilibrium between the inactive and active states so that the receptors were preferentially in the inactive state, later confirmed upon structure determination [15, 28]. In the case of β1AR-m23, the mutant can still couple to and activate G proteins although with reduced ability, whereas no coupling to G proteins was observed for A2A-StaR2. In contrast to β1AR-m23 and A2A-StaR2, the thermostabilization procedure for the engineering of A2A-GL31 and NTSR1-GW5 used the radiolabelled agonists 3H-NECA and 3H-neurotensin, respectively [28, 39]. Pharmacological evaluation of these thermostabilized receptors thus found the opposite to that observed for β1AR-m23 and A2A-StaR2 i.e. the affinities of agonists remained similar to the wild-type receptors whilst the affinities of antagonists were considerably weaker. The structures of both A2A-GL31 [28] and NTSR1-GW5 [30] showed that the receptors were in an active-like state, because rotamer changes of key amino acid side chains and helix movements in the core of the receptors had occurred, but the cytoplasmic end of TM6 partially occluded the G protein-binding site, whereas in the fully active state it would be expected to have moved a further 4–8 Å. Coupling to G proteins was impaired, with A2A-GL31 showing very weak ability to activate G proteins [39] whilst NTSR1-GW5 could not activate G proteins [30]. However, in the latter case, reversion of three out of the six of the thermostabilizing mutations restored the ability of the NTSR1 mutant to activate Gαq to almost the levels observed for the wild-type receptor [40].
Insights from Molecular Dynamics simulations of thermostable mutant GPCRs
It is possible to rationally engineer thermostability in a GPCR by, for example, creating a salt bridge in the extracellular loops of β1AR [41]. Thermostabilization can also be explained for the R68S mutation in thermostabilized β1AR through the removal of charge repulsion and the creation of a hydrogen bond [13]. The Y227A5.58 mutation in β1AR could also be rationalized, as it appears that the tyrosine is important in the stabilization of the activated state of the receptor [4]. However, many other mutations defy such a simplistic interpretation, which also ignores the fact that it is the free energy of the whole receptor that is important for stability, and a single mutation may have global consequences on the dynamics of the whole receptor, which are impossible to quantify from cursory inspection of a static crystal structure. Therefore MD simulations were performed on the thermostable mutants and the respective wild-type receptors, specifically the antagonist-bound inactive states of β1AR-m23 and A2A-StaR2, and the agonist-bound active-like states of NTSR1-GW5 and A2A-GL31. It must be noted that transitions from the inactive state to the active state of a GPCR cannot be simulated using brute force MD simulations possibly due to insurmountable energy barriers to go to the active state in the absence of the agonist and the G protein. Thus all the simulations discussed explore receptor conformations within an explicit lipid bilayer of either the inactive state or the active-like state, depending upon the starting structure used for the MD simulations.
Comparison of the energetics of thermostable mutants to the wild-type receptors
Analysis of the enthalpic and entropic contributions (see Glossary) to the free energy of stabilization for the four thermostable mutants (β1AR-m23, A2A-StaR2, A2A-GL31 and NTSR1-GW5) suggest that a significant proportion of the stabilization comes from the enthalpic component of the free energy [18, 19] except for β1AR-m23, where entropic contributions stabilize the receptor as much as enthalpic contributions [20]. In the case of β1AR-m23, the flexibility of the side chains (but not the main chain) of the amino acids in the transmembrane (TM) domain increase in β1AR-m23 compared to the wild-type receptor. Analysis of the structural basis for the enthalpic stabilization of the thermostable mutants showed an increase in the number and strength of inter-helical hydrogen bonds and van der Waals packing interactions. MD simulations of the thermostable mutants showed rearrangement of side chain conformations in residues neighboring the mutations leading to formation of new inter-helical hydrogen bonds and favorable van der Waals packing interactions. Thermostable mutations also led to improved protein-lipid interactions, with the lipid forming a tight layer of annular lipids around the surface of the protein contributing to favorable hydrophobic matching interactions [18]. Note that in the thermostability assays performed on detergent-solubilized GPCRs, there will be considerable amounts of lipid present (see discussion in reference 51). MD simulations also revealed that the interhelical packing interactions dominate in the intracellular region of the receptors compared to other regions in the thermostable mutants. The decreased receptor flexibility at the intracellular receptor surface may in part explain why the active-like A2A-GL31 and NTSR1-GW5 mutants show a dramatic reduction in their ability to catalyze nucleotide exchange at the G protein, because flexibility of the intracellular TM regions may be a key prerequisite to activate G proteins or arrestins.
For the A2A-GL31 and NTSR1-GW5 mutants thermostabilized in an active-like conformation, it is essential to have an agonist bound to achieve thermostability [23, 26], presumably because in the absence of agonist the receptor is in the inactive state and the thermostabilizing mutations are specific for the active-like state. Thus, in the absence of agonist, these thermostabilized receptors are far less stable than the wild-type receptor after detergent solubilization. This was investigated by MD simulations which showed that the binding of the agonist, adenosine, stabilizes the active-like mutant A2A-GL31 by 19 kcal/mol compared to the wild-type A2AR [19]. In contrast, receptors stabilized in the inactive state are also extremely stable in the absence of ligand, presumably because the ligand-free receptor is very similar to the inactive state.
Comparison of the dynamics of thermostable mutants to the wild-type receptors
MD simulations suggest that the thermostable mutants are less flexible than their wildtype counterparts. Analysis of the collective motion in the inactive β1AR-m23 and A2A–StaR2 without ligands showed that the dominant motions in both receptors are a global pulsating motion of the opening and closing of the extracellular ligand binding site. This movement is restricted when an inverse agonist is bound to the respective thermostable mutants compared to the wild-type receptors [19, 20]. Collective motion analysis of the dynamics of the active-like mutants A2A-GL31 and NTSR1-GW5 shows an outward movement of the intracellular parts of TM5 and TM6 with respect to TM3, as seen for example in the crystal structure of the β2AR in complex with the Gs protein [36]. However, A2A-GL31 and NTSR1-GW5 and other thermostable mutants we have simulated show less movement in the intracellular regions of TM5 and TM6 with respect to TM3, compared to their respective wild-type receptors, as shown schematically in Fig. 1. This may explain why these thermostable mutants only poorly activate the G proteins compared to their wild-type receptors and is consistent with the increased rigidity of the intracellular regions as described above [23, 30]. Comparison of the dynamics of the intracellular parts of TM5 and TM6 of the antagonist bound inactive mutant A2A-StaR2 to the active-like mutant A2A-GL31, shows that the flexibility of TM6 with respect to TM3 is more restricted in the antagonist bound A2A-StaR2 than in the agonist bound A2A-GL31 mutant. This is understandable since the tight interaction between TM3 and TM6 will improve the stabilization of the inactive state of the receptor.
Figure 1.
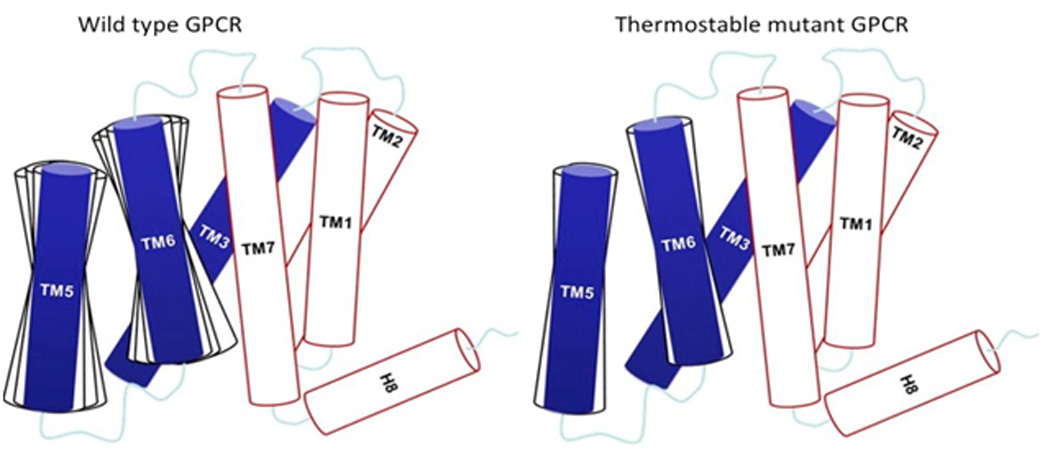
Schematic representation of the dynamics of the thermostable mutant GPCR compared to its wild-type receptor. This schematic is based on the analysis of the extensive MD simulation results on four thermostabilized GPCRs. TM5 and TM6 are more dynamic in the wild-type receptor compared to their thermostable mutant counterpart.
To understand the cause behind the reduced flexibility in the thermostable mutants compared to the wild-type receptors, we calculated the net force on each residue (which we define as stress) in the thermostable mutants β1AR-m23, A2A-StaR2, A2A-GL31 and NTSR1-GW5, and compared it to that of the respective wild-type receptors [18–20, 42]. The reasoning behind this calculation is that the net force or stress on any object would lead to movement of that object and the GPCR needs to be flexible to bind and activate the G protein. This flexibility could come from the stress on residues that are required for activation of the receptor. We found that the calculated stress on each residue in the thermostable mutants is always lower than the corresponding stress in the wild-type receptors [18, 19, 42]. This reduced stress per residue in the thermostable mutants leads to lower flexibility of the receptor in a given conformational state stabilized by the bound ligand. However, the reduction of stress also leads to an attenuating effect on the activity of the thermostable receptor mutants in the presence of agonist and G protein. Therefore a balance between reduction of stress and gain in thermostability are important factors in predicting thermostable mutations for GPCRs. Note that the recently described active-like NTSR1-ELF mutant [40] is able to activate G protein, in contrast to the active-like NTSR1-GW5. Thus NTSR1-ELF will offer the unique opportunity to compare the dynamics of those two NTSR1 variants in view of their ability to stimulate nucleotide exchange at the G protein.
Role of water in thermostabilization of GPCRs
Analysis of high-resolution crystal structures of GPCRs along with the waters present in the crystal structures have shown co-localization of water molecules in the vicinity of highly conserved amino acid motifs [43]. In addition, the results from MD simulations on several GPCRs have led to the speculation that water molecules play a functional role in the activation of GPCRs [44–48]. Analysis of the energetics of water molecules in the ligand binding sites has also been used to guide structure-based drug design [49]. We have used the MD simulation trajectories to analyze the role of waters in the thermostabilization effect of GPCRs, but not in receptor activation. As shown for NTSR1, the interhelical hydrogen bonds that are specific to the thermostable mutant and not present in the wild-type receptor are water mediated (Fig. 2). The number of water molecules in the first solvent shell close to the receptor in the inside of the seven TM barrel is higher in the thermostable mutant NTSR1-GW5 compared to the wild-type NTSR1. The water molecules form tight hydrogen bonded water clusters that strengthen the interhelical interactions (Fig. 2). These water clusters resemble the tightly bound “ice-like” water clusters [50, 51] that have been observed in thermophilic proteins. We speculate that these clusters of waters could also be used to absorb the thermal energy when heating the thermostable mutants without disrupting the three-dimensional fold of the protein to preserve its integrity.
Figure 2.
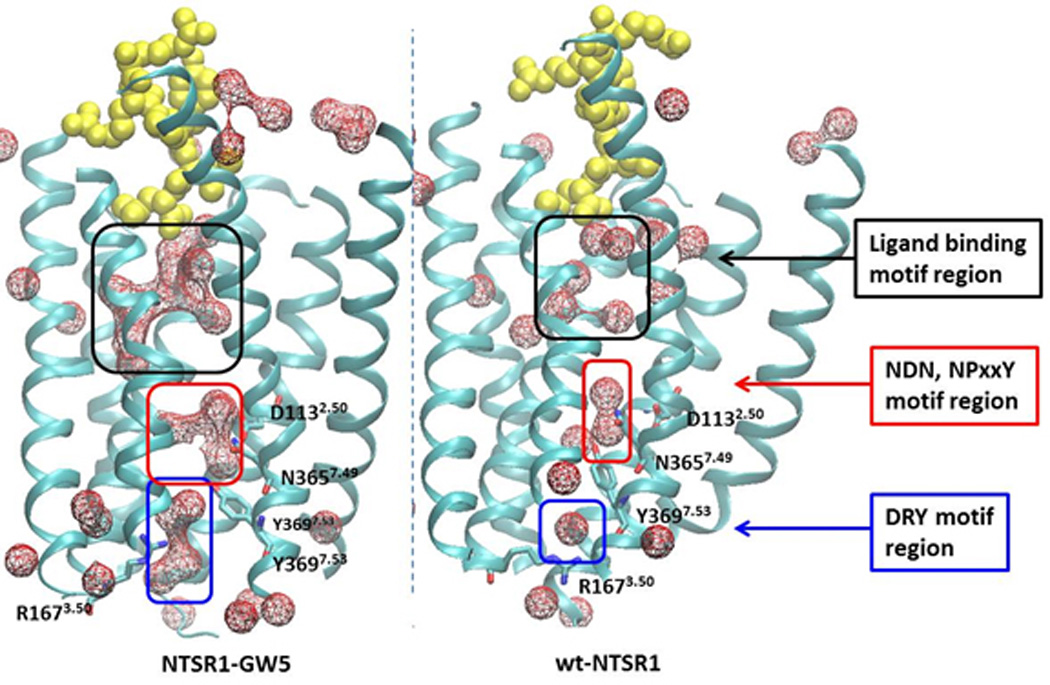
Role of water in thermostabilization of GPCRs. Clusters of tightly bound water molecules, shown as mesh in the figure, accumulate between the transmembrane helices of the thermostable mutant NTSR1-GW5 to stabilize the receptor, compared to the wild-type.
Structural conservation of thermostable amino acid residue positions in GPCRs
To determine whether thermostable positions are transferable across receptors, we have analyzed the measured thermostability of all the single point mutants used to initially thermostabilize each receptor [8]. The experimental procedure for the isolation of thermostabilizing mutations started with alanine scanning mutagenesis where every residue was mutated to Ala (except for Ala residues that were mutated to Leu). Each mutant was then expressed and its thermostability was determined using a radiolabeled ligand, relative to the wild-type receptor [23, 25, 39]. Fig. 3 shows the measured thermostability data for single point mutants shown on the respective crystal structures of the thermostable mutants β1AR-m23, A2A-StaR2, A2A-GL31, and NTSR1-GW5, bound to their respective ligands. The position of the mutated amino acid residues that show significant thermostability are not conserved either in their positions within the three dimensional structures or the identity of the amino acid residue. Instead they are distributed throughout the receptor structures differently for each receptor. This shows that thermostability may not be transferable and it could stem from subtle changes upon mutation in some global property of the receptor structure. Experimental work supports this contention, as the six thermostabilizing mutations in turkey β1AR-m23 could be transferred to human β1AR and human β2AR [52], but the gain in thermostability was considerably less for human β2AR than human β1AR.
Figure 3.
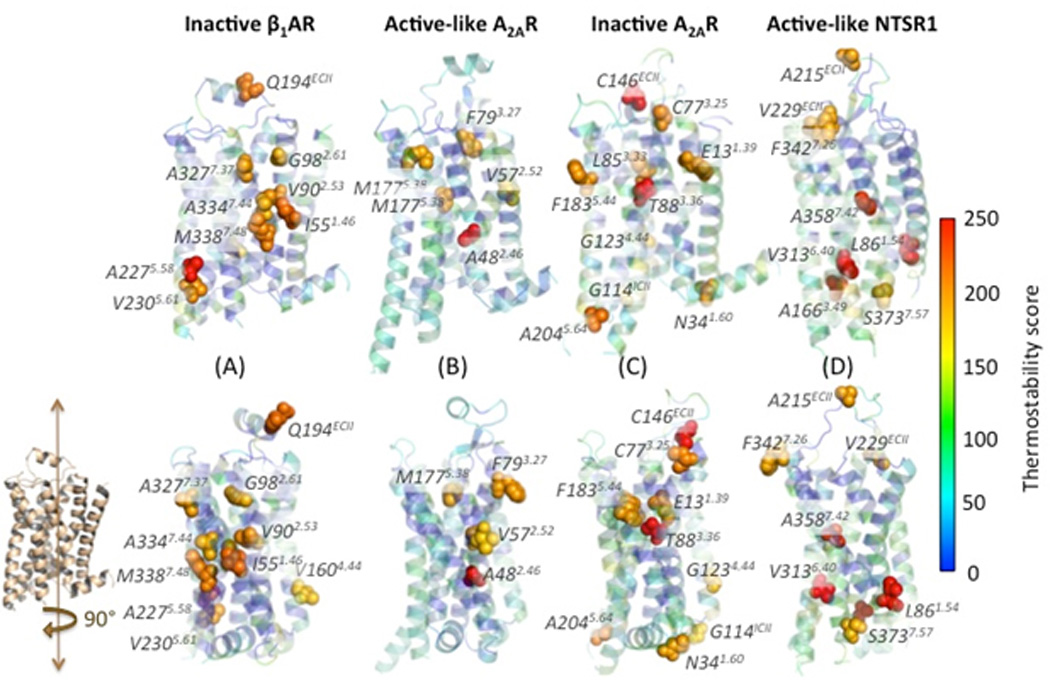
Experimentally measured thermostability scores ranging from blue to red (low to high) shown overlaid on the crystal structures of the thermostable mutants β1AR-m23, A2A-StaR2, A2A-GL31 and NTSR1-GW5. Thermostability was measured using the following radioligands: the antagonist 3H-dihydroalprenolol for turkey β1AR in the inactive state, the antagonist 3H-ZM241385 for A2AR in the inactive state, the agonist 3H-NECA for the A2AR in the active-like state, and the agonist 3H-neurotensin for NTSR1 in the active-like state. Each structure is shown in two mutually orthogonal views. The residues with high thermostability are marked in spheres and labeled in each receptor.
Predicting thermostable mutations
Computational prediction of thermostabilizing mutations is a challenging process since the thermostability could arise from thermodynamic stability or increasing the kinetic barrier to receptor unfolding, intermeshed with subtle changes in other global properties of the receptor as described above. In one method to improve thermostability, Chen et al. exploited the void space present in the structure of β1AR-m23 combined with sequence conservation information to computationally design single, double and triple mutants using RosettaMembrane [53] that would fill up the void space and improve packing interactions between amino acid residues in the receptor. These predicted mutants of β1AR were later tested using thermostability assays under saturating concentrations of the antagonist and, in the best cases, were found to improve thermostability of the receptor significantly [54]. In a different approach, we developed a systematic rapid screening computational method, LITiConDesign for predicting single point alanine mutations to thermostabilize GPCRs [55, 42]. It involves generating an ensemble of conformations using the conformational sampling method called LITiCon [55–57]. The computational thermostability score calculated for each mutant in LITiConDesign is based on the enthalpy difference between the mutant and the wild-type, as well as other properties such as stress on each residue and the extent of involvement of each residue in allosteric communication of the ligand binding site to the G protein interaction surface [38, 42]. Mutating residues with high stress or residues that are involved in allosteric communication between the ligand binding region and G protein coupling region leads to poor thermostability. Starting from homology models of GPCRs LITiConDesign identified the most stable thermostable mutants in the top 35% of the predicted mutants. This reduces the number of experiments by two-thirds and is a good starting point especially for orphan GPCRs with very little structural information. These methods lay the groundwork for future advancements as more experimental data emerge on thermostable mutations.
Concluding Remarks
MD simulations have given us insights into the dynamics and energetics of the thermostable mutants in comparison to their wild-type counterparts and have provided us an understanding of how the thermostable mutants are stabilized through the formation of water-mediated interhelical hydrogen bonds and van der Waals interactions. There are many challenges in understanding the pharmacology and conformational dynamics of GPCRs especially in view of the recent findings of biased signaling properties. Computational methods such as molecular dynamics simulations in combination with biochemical and biophysical assays investigating the pharmacological properties and structural dynamics of GPCRs, will provide detailed mechanistic insight into the subtleties of GPCR signaling. The first step towards addressing the signaling specificity of GPCRs is to stabilize them in complex with various G proteins, kinases, or arrestin molecules. However, there are significant experimental challenges arising from the dynamic conformational flexibility exhibited by GPCR-transducer complexes. As a result, the experiments are tedious and time-consuming, and have relied largely on educated “trial and error” experimentation. Progress in understanding signaling properties of GPCRs can be accelerated if we can provide a foundation for experiments that will have a higher probability of success. As enumerated in the Outstanding Questions box, a key challenge is to enhance the probability of predicting thermostabilizing mutations in receptors that have not been crystallized and are unrelated to receptors whose structures are known. Only a single structure of a GPCR bound to a G protein has been obtained, and similarly there is only a single structure of a GPCR bound to arrestin. Ideally many more structures need to be obtained to increase out understanding of G protein and arrestin coupling, but the extreme difficulty in preparing the complexes and obtaining welldiffracting crystals suggests that computational techniques could be well placed to facilitate structure determination by predicting potential avenues for thermostabilization.
Can we develop a reliable and time-efficient computational method that will provide a foundation for concurrent experiments to stabilize and purify GPCR-transducer complexes?
Another challenge is to solve the crystal structures of unrelated GPCRs covering more of their phylogenetic tree. Progress towards a complete picture of the breadth of GPCR biology depends crucially on generating highly diffracting crystals. The emerging question facing up to this challenge is: Can we predict accurately, thermostabilizing mutations in GPCRs when the structure has not been determined?
Is it possible to develop brute force multiscale molecular dynamics methods that do not bias simulations to study the large scale conformation dynamics of GPCRs to provide insight into signaling mechanisms?
Acknowledgements
N.V. thanks Dr. Sangbae Lee, Dr. Supriyo Bhattacharya and Mr. Benjamin Levy for their help with the preparation of some of the figures and references. Funding for this work was provided by NIH-RO1GM097261. CGT is funded by the Medical Research Council (MRC U105197215). The research of RG is supported by the Intramural Research Program of the National Institute of Neurological Disorders and Stroke, the National Institutes of Health.
Glossary
- Active-like conformation
Agonist-occupied receptor conformations with characteristics of activated GPCRs in the absence of a heterotrimeric G protein. An active-like conformation can be considered as an intermediate state approaching the active state.
- Active conformation
Signaling GPCR conformations that, for example, catalyze nucleotide exchange at the Gα subunit of a heterotrimeric G protein.
- A2A-GL31
Thermostabilized active-like adenosine A2A receptor with the mutations L48A2.46, A54L2.52, T65A2.63, Q89A3.37 [23]
- A2A-Star2
Thermostabilized inactive adenosine A2A receptor with the mutations A54L2.52, T88A3.36, R107A3.55, K122A4.43, L202A5.63, L235A6.37, V239A6.41, S277A7.42 [24].
- β1AR-m23
Thermostabilized inactive β1-adrenergic receptor with the mutations R68S1.59, M90V2.53, Y227A5.58, A282L6.27, F327A7.37, F338M7.48 [25].
- Conformational thermostabilization
Technique that results in a thermostabilized receptor that has the equilibrium between the inactive and active state perturbed by that process, leading to an increase in the stability of the receptor locked into a particular conformation. Thermostabilized receptor mutants can be identified by the systematic alanine scanning mutagenesis approach or by directed evolution.
- Directed evolution
High-throughput approach by error prone PCR and FACS-based selection of receptor variants with increased stability in detergent and/or higher expression.
- Inactive conformation
Non-signaling receptor conformations, which do not activate a G protein, for example, in the presence of a full inverse agonist.
- NTSR1-GW5
Thermostabilized active-like neurotensin receptor with the mutations A86L1.54, E166A3.49, G215AECL2, L310A6.37, F358A7.42, V360A7.44 [26]
- NTSR1-ELF
Thermostabilized active-like neurotensin receptor with the mutations A86L1.54, G215AECL2, V360A7.44 [27]
- Rational design
Mutations are based on computational predictions using crystal structures, followed by experimental verification of the mutants.
- Systematic alanine scanning approach
Receptors containing single point mutations are probed for stability by exposure to heat; receptor stability is further improved by iterative combination of single thermostabilizing mutations. If the heat step is conducted in the presence of a particular ligand such as an inverse agonist or agonist, receptor mutants are expected to be stabilized in inactive states or active-like conformations, respectively.
- Enthalpy and Entropic contributions
The thermostability score for each single point mutant for a GPCR is calculated as the sum of two terms in the free energy, namely the enthalpy and entropy terms. Enthalpic contribution to the thermostability score comes from the interhelical hydrogen bonds or packing interactions formed or broken in the mutant. The entropic contribution comes from the increased or decreased flexibility in the main chain or side chain atoms of the amino acids.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Katritch V, et al. Structure-Function of the G Protein-Coupled Receptor Superfamily. Annu. Rev. Pharmacool. Toxicol. 2013;53:531–556. doi: 10.1146/annurev-pharmtox-032112-135923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenbaum DM, et al. The structure and function of G-protein-coupled receptors. Nature. 2009;459:356–363. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Venkatakrishnan AJ, et al. Molecular signatures of G-protein-coupled receptors. Nature. 2013;494:185–194. doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- 4.Tate CG, Schertler GFX. Engineering G protein-coupled receptors to facilitate their structure determination. Curr. Opin. Struct. Biol. 2009;19:386–395. doi: 10.1016/j.sbi.2009.07.004. [DOI] [PubMed] [Google Scholar]
- 5.Rosenbaum DM, et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 6.Chun E, et al. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure. 2012;20:967–976. doi: 10.1016/j.str.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steyaert J, Kobilka BK. Nanobody stabilization of G protein-coupled receptor conformational states. Curr. Opin. Struct. Biol. 2011;21:567–572. doi: 10.1016/j.sbi.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tate CG. A crystal clear solution for determining G-protein-coupled receptor structures. Trends Biochem. Sci. 2012;37:343–352. doi: 10.1016/j.tibs.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 9.Ring AM, et al. Adrenaline-activated structure of beta2-adrenoceptor stabilized by an engineered nanobody. Nature. 2013;502:575–579. doi: 10.1038/nature12572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Standfuss J, et al. Crystal structure of a thermally stable rhodopsin mutant. J. Mol. Biol. 2007;372:1179–1188. doi: 10.1016/j.jmb.2007.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moukhametzianov R, et al. Two distinct conformations of helix 6 observed in antagonist-bound structures of a beta(1)-adrenergic receptor. Proc. Natl. Acad. Sci. USA. 2011;108:8228–8232. doi: 10.1073/pnas.1100185108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Christopher JA, et al. Biophysical Fragment Screening of the beta(1)- Adrenergic Receptor: Identification of High Affinity Arylpiperazine Leads Using Structure-Based Drug Design. J. Med. Chem. 2013;56:3446–3455. doi: 10.1021/jm400140q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Warne T, et al. Crystal structures of a stabilized beta1-adrenoceptor bound to the biased agonists bucindolol and carvedilol. Structure. 2012;20:841–849. doi: 10.1016/j.str.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Warne T, et al. The structural basis for agonist and partial agonist action on a beta(1)-adrenergic receptor. Nature. 2011;469:241–244. doi: 10.1038/nature09746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warne T, et al. Structure of a beta1-adrenergic G-protein-coupled receptor. Nature. 2008;454:486–491. doi: 10.1038/nature07101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Langmead CJ, et al. Identification of Novel Adenosine A(2A) Receptor Antagonists by Virtual Screening. J. Med. Chem. 2012;55:1904–1909. doi: 10.1021/jm201455y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Congreve M, et al. Discovery of 1,2,4-Triazine Derivatives as Adenosine A(2A) Antagonists using Structure Based Drug Design. J. Med. Chem. 2012;55:1898–1903. doi: 10.1021/jm201376w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee S, et al. Structural Dynamics and Thermostabilization of Neurotensin Receptor 1. J. Phys. Chem. B. 2015;119:4917–4928. doi: 10.1021/jp510735f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee S, et al. Dynamic Behavior of the Active and Inactive State of Adenosine A2A Receptors. J. Phys. Chem. B. 2014;118(12):3355–3365. doi: 10.1021/jp411618h. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Niesen MJM, et al. Thermostabilization of the β1-Adrenergic Receptor Correlates with Increased Entropy of the Inactive State. J. Phys. Chem. B. 2013;117:7283–7291. doi: 10.1021/jp403207c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Penmatsa A, et al. X-ray structure of dopamine transporter elucidates antidepressant mechanism. Nature. 2013;503:85–90. doi: 10.1038/nature12533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen L, et al. X-ray structures of AMPA receptor-cone snail toxin complexes illuminate activation mechanism. Science. 2014;345:1021–1026. doi: 10.1126/science.1258409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lebon G, et al. Thermostabilisation of an Agonist-Bound Conformation of the Human Adenosine A(2A) Receptor. J. Mol. Biol. 2011;409:298–310. doi: 10.1016/j.jmb.2011.03.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Robertson N, et al. The properties of thermostabilised G protein-coupled receptors (StaRs) and their use in drug discovery. Neuropharmacology. 2011;60:36–44. doi: 10.1016/j.neuropharm.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 25.Serrano-Vega MJ, et al. Conformational thermostabilization of the beta 1-adrenergic receptor in a detergent-resistant form. Proc. Natl. Acad. Sci. USA. 2008;105:877–882. doi: 10.1073/pnas.0711253105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shibata Y, et al. Optimising the combination of thermostabilising mutations in the neurotensin receptor for structure determination. Biochim. Biophys. Acta. 2013;1828:1293–1301. doi: 10.1016/j.bbamem.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krumm BE, et al. Structural prerequisites for G-protein activation by the neurotensin receptor. Nat. Comm. 2015;6 doi: 10.1038/ncomms8895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dore AS, et al. Structure of the adenosine A(2A) receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure. 2011;19:1283–1293. doi: 10.1016/j.str.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lebon G, et al. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature. 2011;474:521–525. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.White JF, et al. Structure of the agonist-bound neurotensin receptor. Nature. 2012;490:508–513. doi: 10.1038/nature11558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Egloff P, et al. Structure of signaling-competent neurotensin receptor 1 obtained by directed evolution in Escherichia coli. Proc. Natl. Acad. Sci. USA. 2014;111:E655–E662. doi: 10.1073/pnas.1317903111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tan Q, et al. Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science. 2013;341:1387–1390. doi: 10.1126/science.1241475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Srivastava A, et al. High-resolution structure of the human GPR40 receptor bound to allosteric agonist TAK-875. Nature. 2014;513:124–127. doi: 10.1038/nature13494. [DOI] [PubMed] [Google Scholar]
- 34.Hollenstein K, et al. Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature. 2013;499:438–443. doi: 10.1038/nature12357. [DOI] [PubMed] [Google Scholar]
- 35.Dore AS, et al. Structure of class C GPCR metabotropic glutamate receptor 5 transmembrane domain. Nature. 2014;511:557–562. doi: 10.1038/nature13396. [DOI] [PubMed] [Google Scholar]
- 36.Rasmussen SGF, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nygaard R, et al. The dynamic process of beta(2)-adrenergic receptor activation. Cell. 2013;152:532–542. doi: 10.1016/j.cell.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bhattacharya S, Vaidehi N. Differences in allosteric communication pipelines in the inactive and active states of a GPCR. BioPhys. J. 2014;107:422–434. doi: 10.1016/j.bpj.2014.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shibata Y, et al. Thermostabilization of the Neurotensin Receptor NTS1. J. Mol. Biol. 2009;390:262–277. doi: 10.1016/j.jmb.2009.04.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krumm BE, et al. Structural prerequisites for G protein activation by the neurotensin receptor. Nat. Comm. 2015;6:7895–7904. doi: 10.1038/ncomms8895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Miller JL, Tate CG. Engineering an ultra-thermostable beta(1)-adrenoceptor. J. Mol. Biol. 2011;413:628–638. doi: 10.1016/j.jmb.2011.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bhattacharya S, et al. Rapid Computational Prediction of Thermostabilizing Mutations for G Protein-Coupled Receptors. J. Chem. Theory. Comput. 2014;10:5149–5160. doi: 10.1021/ct500616v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hulme EC. GPCR activation: a mutagenic spotlight on crystal structures. Trends Pharmacol. Sci. 2013;34:67–84. doi: 10.1016/j.tips.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 44.Bhattacharya S, et al. Agonist-induced conformational changes in bovine rhodopsin: Insight into activation of G-protein-coupled receptors. J. Mol. Biol. 2008;382:539–555. doi: 10.1016/j.jmb.2008.06.084. [DOI] [PubMed] [Google Scholar]
- 45.Yuan S, et al. Activation of G-protein-coupled receptors correlates with the formation of a continuous internal water pathway. Nat. Comm. 2014;5:4733–4742. doi: 10.1038/ncomms5733. [DOI] [PubMed] [Google Scholar]
- 46.Grossfield A, et al. Internal hydration increases during activation of the G-protein-coupled receptor rhodopsin. J. Mol. Biol. 2008;381:478–486. doi: 10.1016/j.jmb.2008.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Angel TE, et al. Conserved waters mediate structural and functional activation of family A (rhodopsin-like) G protein-coupled receptors. Proc. Natl. Acad. Sci. USA. 2009;106:8555–8560. doi: 10.1073/pnas.0903545106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gerwert K, et al. The role of protein-bound water molecules in microbial rhodopsins. Bba-Bioenergetics. 2014;1837:606–613. doi: 10.1016/j.bbabio.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 49.Mason JS, et al. New insights from structural biology into the druggability of G protein-coupled receptors. Trends Pharmacol. Sci. 2012;33:249–260. doi: 10.1016/j.tips.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 50.Prabhu N, Sharp K. Protein-solvent interactions. Chem. Rev. 2006;106:1616–1623. doi: 10.1021/cr040437f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun T. An antifreeze protein folds with an interior network of more than 400 semi-clathrate waters (vol 343, pg 795, 2014) Science. 2014;343:969–969. doi: 10.1126/science.1247407. [DOI] [PubMed] [Google Scholar]
- 52.Serrano-Vega MJ, Tate CG. Transferability of thermostabilizing mutations between beta-adrenergic receptors. Mol. Membr. Biol. 2009;26:385–396. doi: 10.3109/09687680903208239. [DOI] [PubMed] [Google Scholar]
- 53.Barth P, et al. Toward high-resolution prediction and design of transmembrane helical protein structures. Proc Natl Acad Sci USA. 2007;104:15682–15687. doi: 10.1073/pnas.0702515104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen KYM, et al. Naturally evolved G protein-coupled receptors adopt metastable conformations. Proc. Natl. Acad. Sci. USA. 2012;109:13284–13289. doi: 10.1073/pnas.1205512109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Balaraman GS, et al. Structural insights into conformational stability of wild-type and mutant beta1-adrenergic receptor. Biophys. J. 2010;99:568–577. doi: 10.1016/j.bpj.2010.04.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bhattacharya S, et al. Ligand-stabilized conformational states of human beta(2) adrenergic receptor: Insight into G-protein-coupled receptor activation. Biophys. J. 2008;94:2027–2042. doi: 10.1529/biophysj.107.117648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bhattacharya S, Vaidehi N. Computational Mapping of the Conformational Transitions in Agonist Selective Pathways of a G-Protein Coupled Receptor. J. Am. Chem. Soc. 2010;132:5205–5214. doi: 10.1021/ja910700y. [DOI] [PubMed] [Google Scholar]