

Abstract
Transmissible tumors are those that have transcended the bounds of their incipient hosts by evolving the ability to infect another individual through direct transfer of cancer cells; thus becoming parasitic cancer clones. Coitus, biting, and scratching are transfer mechanisms for the two primary species studied, the domestic dog (Canis lupus familiaris) and the Tasmanian devil (Sarcophilus harrisii). Canine transmissible venereal tumors (CTVT) are likely thousands of years old, and have successfully travelled from host to host around the world, while the Tasmanian devil facial tumor disease (DFTD) is much younger and geographically localized. The dog tumor is not necessarily lethal, while the devil tumor has driven the population to near extinction. Transmissible tumors are uniform in that they have complex immunologic profiles, which allow them to escape immune detection by their hosts, sometimes for long periods of time. In this review we explore how transmissible tumors in CTVT, DFTD, and as well as the soft-shelled clam and Syrian hamster can advance studies tumor biology.
Keywords: cancer, infectious, transmissible, clonal, canine, devil
Types of TransmissibleTumors
Transmissible tumors are by definition spread directly by transfer of cells between individuals. They are clonal in origin, suggesting an ancient, singular event from which all modern tumors evolved. The most-well studied transmissible tumor is CTVT, dubbed the “oldest continuously propagated cell lineage” [1, 2]. Originally termed Sticker’s sarcoma, it has been observed for over 200 years and is spread primarily by coitus and oral contact [3, 4]. It was initially defined as a histocytic tumor by Novinski in 1876, who demonstrated transmissibility by rubbing an excised tumor from one dog onto the genital mucosa of another [5, 6]. CTVT tumors feature strong genetic identity with one another, but are markedly distinct from their extant, transient host [1, 2, 7, 8]. CTVT is endemic in more than 90 countries and believed to be the most widely disseminated tumor in existence [6, 9, 10–12].
DFTD is a much younger tumor, first diagnosed in 1996 in the Tasmanian devil, the largest marsupial carnivore [13, 14]. Originating in the northeast corner of Tasmania, DFTD has reached epidemic proportions. Between 1996 and 2006 it was observed in 41 locales, encompassing 50% of the island [13]. In contrast to CTVT, DFTD is not a venereal tumor, rather it is spread by biting, often during mating or feeding [15–17]. The tumor forms around the face and neck [18], ulcerating and causing death within six months of onset by asphyxiation or starvation [19]. This underscores a very real concern that current rates of infection may drive the population to extinction [13, 20, 21]. In the following, we compare and contrast available genomic data from these two transmissible tumors. We consider, specifically, how genomic advances have changed our view of these two tumors. Finally, we briefly discuss additional manifestations of transmissible tumors in two other species, the Syrian hamster and the softshelled clam.
Canine Transmissible Venereal Tumor
CTVT is a Parasitic Tumor
Several lines of evidence support CTVT as a naturally transmissible allograft. It can only be induced in a naïve individual by transplanting living tumor cells; and neither frozen, heated, desiccated, killed or filtered cells transmit the tumor [5, 22–24]. Also, karyotypes of tumors collected from different regions are more divergent than those from the same region, which are themselves highly similar, confirming cellular transfer as well as clonality [24, 25] (Box 1). Finally, electron microscopy has not detected oncogenic viral particles in tumor cells, further supporting this paradigm [26–28]. The tumor clone is demonstrably long-lived and stable [29], with experimental passaging for 40 generations (564 dogs) over 17 years producing no changes in histopathology [30].
Box 1. Key features of CTVT.
CTVT is a transmissible tumor believed to have originated hundreds or thousands of years ago.
Tumor is clonal, meaning that tumors from all infected canids share strong genetic identity.
Distinguishing somatic mutations, which drive tumor growth from those, found in the original or transient host is important for understanding how the tumor evades host immunity.
The tumor evades how immune detection by accumulated mutations in all pathways related to recognition of self versus nonself.
Genomic approaches including large catalogs of variation found in modern canids are critical for identifying somatic drivers of tumor growth.
The epidemiology of CTVT suggests no sex bias [12], or breed barrier [9, 10, 22]. Many canids including dogs, wolves, foxes and coyotes can be infected [24, 29]. However there is no evidence for transfer between distant species, as inoculation into rats, mice, hamsters, and cats has been unsuccessful [31].
The tumor generally remains localized to the external genitalia [9] (Figure 1). Histologic staining shows it to be characterized by large diffuse masses of compact round or polyhedral neoplastic cells [24]. Canid sexual intercourse entails a long (up to 30 minute) “tie” where male and female genitalia are in direct physical contact, often resulting in abraded tissue, likely contributing to the spectacular success of the tumor. Based on morphology and histologic staining patterns, CTVT was originally proposed to be histiocytic [24, 25], lymphatic, or reticuloendothelial [32]. Subsequent immunohistochemistry has since assigned it to macrophage origin [33, 34].
Figure 1.
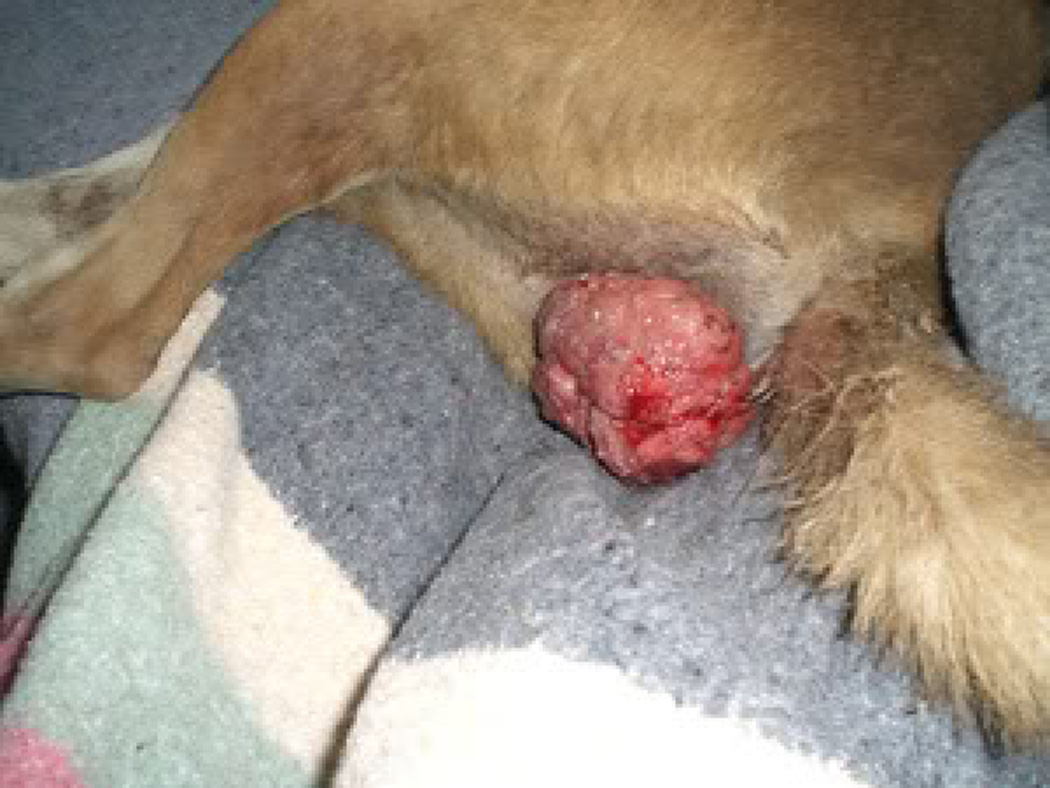
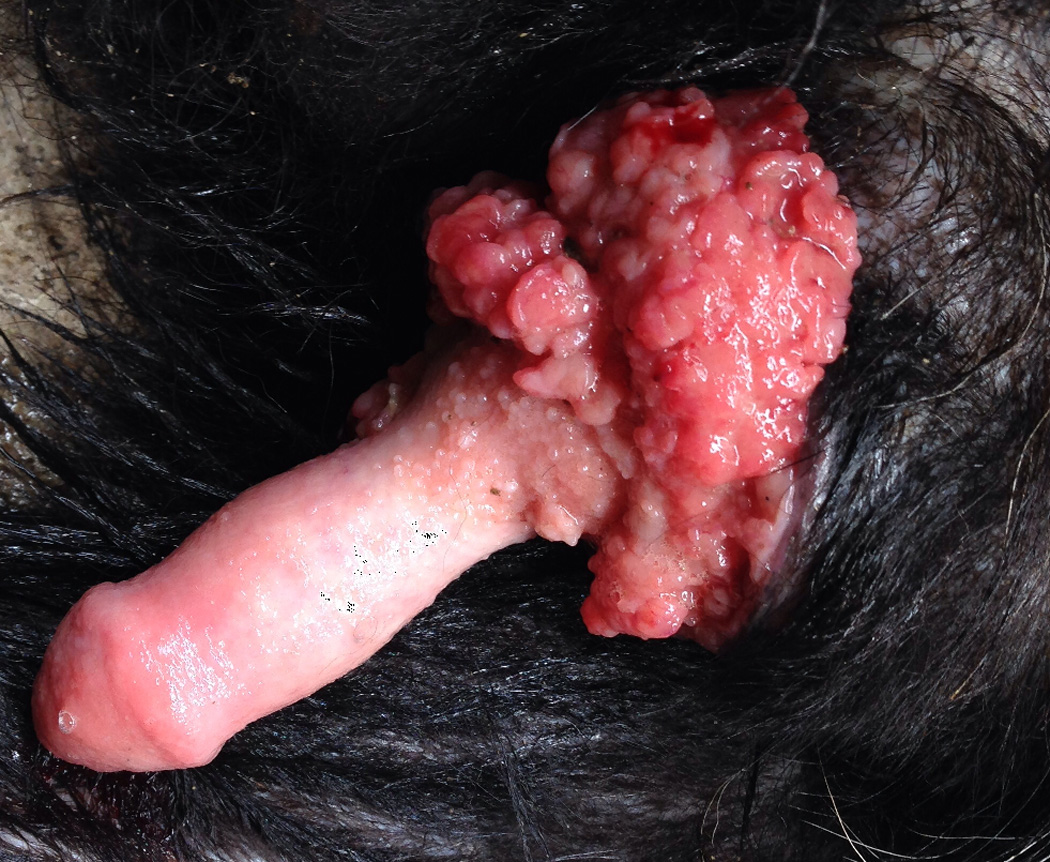
Photographs of external genitalia infected with CTVT in (A) Female and (B) Male.
Tumor Dissemination
Early cytogenetic support for the clonality of all CTVT was demonstrated by extensive conservation of genomic imbalances across tumors (Figure 2) [26, 29, 35, 36]. Analysis of microsatellites, mitochondrial DNA (mtDNA) and the major histocompatility locus (MHC) in 40 dogs from five continents also showed that worldwide CTVT is derived from a single neoplastic clone [1]. Two major tumor clades were proposed, one in coyote and another encompassing gray wolf and domestic dog lineages. This data suggested that CTVT originated in the pre-domestication Canis lupus lineage [37]. These findings would later be refuted using modern genomic methods [2, 8].
Figure 2.

Copy number alterations in 5 CTVT cases (6A, 29M, 57C, 72C, 82K) assayed by aCGH (taken from [36]). Genomic copy number relative to domestic dog indicated as increase (green), decrease (red), and even (yellow) with the sixth column as consensus call.
Epidemiologic studies of CTVT, done using mtDNA, identify a close relationship among tumor haplotypes from Mexico, the U.S., Chile and Brazil. Asian haplotypes were more divergent, though most closely related to American haplotypes, suggesting that tumors in the U.S., although rare, may have originally disseminated from Asiatic lineages [38].
Recent data suggest that CTVT has likely existed on all continents (except Antarctica) for decades [11], or longer ([12] and refs therein). The tumor reservoir is largely stray and wild dogs in regions of indigenous populations [11, 12]. The disease is essentially eradicated in Canada, the United Kingdom, several additional European countries, and New Zealand due to high levels of veterinary surveillance. CTVT affects between one and 10% of dogs in Africa, Asia, and South and Central America, with the highest incidence in Belize (10–20%) [12].
CTVT Tumor Origins and Whole Genome Sequencing
Initially, the tumor was proposed to have originated 200–2500 years ago, but this was based on the absence of any molecular evidence [1]. In later microsatellite analysis, the estimate increased to 6000 years [37]. However, this method assesses only a small portion of the genome, and may not be well suited to estimate tumor evolution. Finally, using a controversial molecular clock approach in which a steady evolutionary rate based on human medulloblastoma was applied, the tumor age was proposed to be 11,000 years [2]. The true answer is unknown, but estimates between 6000 and 11,000 years are generally considered to be the most accurate. Two steps needed to correctly date the tumor include an accurate genome sequence/assembly not biased toward the modern boxer, and population level tumor sampling.
The above studies led to queries regarding the nature of the first dog to develop the tumor. Two distinct approaches have been taken to address this question. Murchison and colleagues [2] used principal components analysis (PCA) to analyze nearly 24,000 previously published canid genotypes [39, 40], together with DNA sequence generated from two tumors and their hosts (Brazil and Australia). They concluded that the founder dog likely belonged to the “ancient” group our research previously demonstrated includes huskies, malamutes, basenjis and several Asian breeds [40, 41]. Their initial sequence analysis was highly informative, providing no evidence that CTVT is undergoing strong positive selection and suggesting that few changes are now required for tumor survival [2]. Only a modest number of somatic variants differed between the two currently sequenced tumors, indicating derivation from a single source. Finally, they reported that 1.9 million SNVs found in both tumors were likely to be somatic, which is several orders of magnitude more than the average human tumor. The authors also showed that mutations in major known cancer genes, including CDKN2A, MYC, ERG, etc. are both characteristic and critical for CTVT survival [2].
We, in turn, addressed the founder question by identifying germline-specific variants within the CTVT sequence after comparison to whole genome sequence (WGS) from 186 modern canids [8]. Implementing maximum likelihood phylogenetic inference across 1.3 million founder-inherited alleles, we identified the founder as a post-domestication canid with significant identity to Arctic spitz breeds [8], as suggested previously [2]. Subsequent PCA with only these breeds clustered CTVT between Huskies and Malamutes, indicating that the founder was related to the ancestor of these two modern breeds [8].
Since our goals were to not only to more accurately identify SNVs, but also to find insertion/deletions (indels) and structural variations (SVs), our analysis relied on high-coverage WGS (mean 37.9×) from the 186 diverse dogs mentioned previously. This included American Kennel Club-recognized and international breeds, village dogs, and wild canids. Thus, we created the largest existing catalog of genome-wide canine variation to date, consisting of 28.01 million SNVs, 12.62 million indels, and 31,613 SVs, which includes over 99% of SNVs and indels, and over 95% of SVs found in modern canids [8]. Previous studies utilized only the canine dbSNP database, which contains only SNVs and encompasses less than one-third of the variants found in the average canid WGS (Figure 3) [8] (Outstanding Questions).
Figure 3.
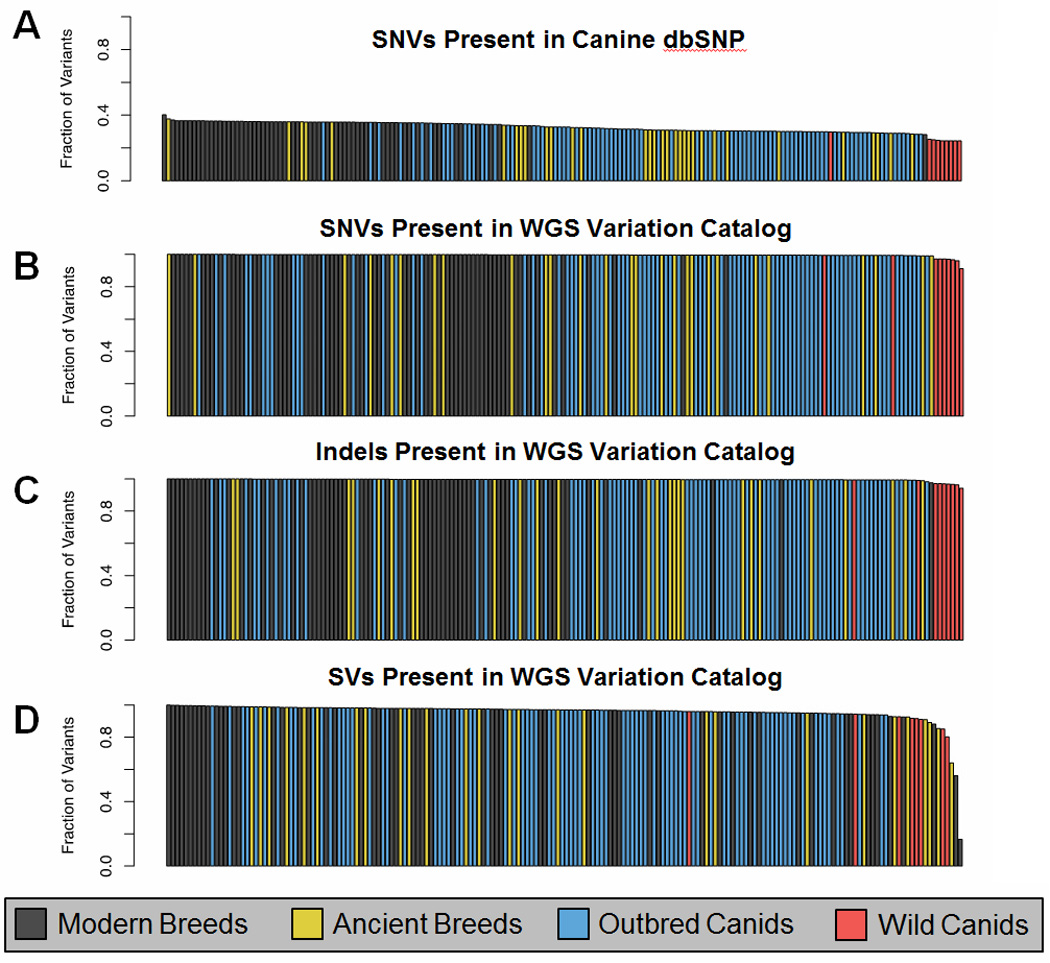
186 canid variant catalog included the two host dogs, 64 dogs representing 40 modern breeds, 27 dogs representing 12 ancient breeds, 86 outbred canids spanning four continents, and nine wild canids. (A) Fraction of SNVs found in canine dbSNP (mean 32.65%). Sequenced canids. (B–D) Fraction of SNVs, indels, and SVs found in a single canid that are present in at least one other canid in the catalog (means of 99.55%, 99.57%, and 95.63%, respectively) (Taken from [8]).
We compared our catalog to high quality variants identified in our analysis of the two published CTVT tumors [2]. We classified CTVT variants found in the catalog as putative founder alleles, and novel variants as likely somatic mutations [42]. We found that 75.6% of SNVs, 92.1% of indels, and 17.6% of SVs originated in the founder’s germline, with the remainder being strong candidates for somatic acquisition [8]. The mutation signature of putative founder alleles was indistinguishable from that of modern canids, while somatic variation was more consistent with human melanoma [43], a tumor induced by UV exposure. This supports the existing hypothesis that UV exposure was central to initial CTVT tumor formation [2]. (Text Box 1) [2]. Tumor genes found by this method which had deleterious nonsynonymous changes included those associated with cell adhesion, extracellular cohesion, cytoskeletal, cell membrane structures, apoptotic signal transduction and chromatin organization.
CTVT Host Immune Interactions
CTVT is unique in that host survival is high, though studies differ on the rate of spontaneous regression [24, 44–46] (Outstanding Questions). Although not considered metastatic, Das and Sahay report that up to 7% of dogs with CTVT demonstrate metastasis [47]. Strakova and Murchison put the number at 0–5%, dependent on geographic region [12]. The disturbing appearance of the tumor, which is often bright red, highly vascularized, ulcerated and necrotic (Figure 1), with an unsightly discharge [6], incentivizes owners to treat the tumor using chemotherapy. Tumors are generally responsive, and recede quickly [11, 48, 49]. As a result, life history studies of CTVT are rare.
CTVT does a remarkable job evading the host immune system for extended periods (Box 1). Murgia first proposed CTVT tumors down-regulated expression of MHC class I molecules and that class II was essentially absent [1]. Belov argued that genetic diversity at MHC genes could play a critical role in population susceptibility to contagious cancer, particularly if the species underwent genetic bottlenecks and/or lost MHC diversity [50]. This model does not fit the canine scenario since the tumor spreads between canine species, and dogs feature highly diverse HLA systems [51, 52]. We therefore investigated the precise nature of the somatic mutations.
Following variant filtering using our catalog, we found the two sequenced CTVT tumors shared nearly a million high-confidence somatic substitutions [8], an order of magnitude more than even the most mutated human cancers [43]. Functional annotation highlighted nearly 7000 of these. Since many are likely damaging, we used Gene Set Enrichment Analysis [53] to identify pathways with the most high-impact somatic disruptions (Figure 4). We found somatic mutations spanning all aspects of somatic cell participation in immune surveillance, including candidates in the self-antigen presentation pathway and each component of the transporter associated with antigen processing. Both tumors had deleterious mutations that disrupt both initiators and executors of apoptosis (Figure 3). Finally, mutations were observed in genes involved in maintenance of genome integrity. Though not unique to CTVT, the sheer numbers and redundancy of these mutations was surprising; indicating thousands of years of cryptic selective pressures. These evolutionary processes, CTVT’s exclusivity to canids, and the mechanisms leading to regression are intriguing questions that require further inquiry (Outstanding Questions).
Figure 4.
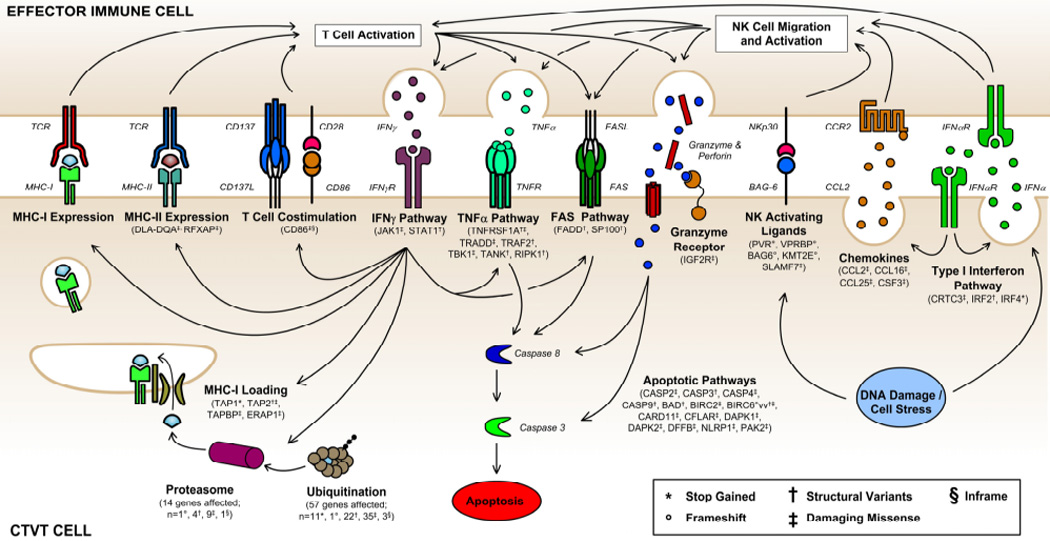
Somatic mutations shared by both CTVT tumors impact all aspects of pathways involved in immunosurveillance (Taken from [8]).
Tasmanian Devil Facial Tumor
Evolution and Recent History of DFTD
DFTD was first noted in the northeast portion of the island of Tasmania in 1996 [13] (Figure 5A). Like CTVT, DFTD is a clonal cell line transmitted as an allograft [16], but is restricted to Tasmania, the only natural devil habitat (Box 2) [54]. Since 1996, DFTD has spread rapidly across Tasmania, where it has decimated populations by nearly 90% in some regions [14, 20]. Indeed, estimates from 2004–2007 suggested that the disease was endemic across 70% of Tasmania [14, 20]. The rapid success of the disease may be due in part to the fact that devils are non-territorial and highly vagile, with home ranges up to 27 km2 [14]. Additionally, they are known to bite one another both during feeding (Figure 5A) and mating, facilitating transfer (Box 2) [15].
Figure 5.


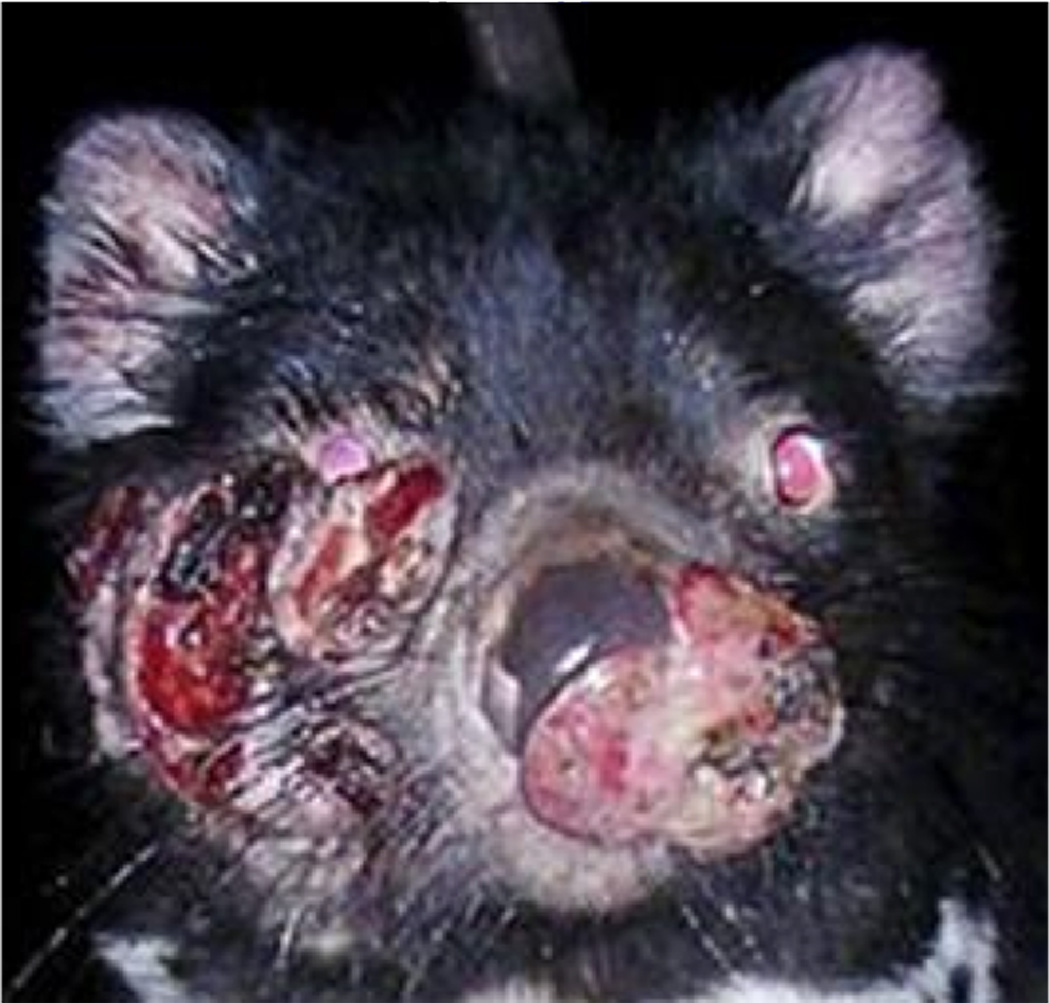
Tasmanian devils harbor DFTD. (A) Devils bite and gnaw as a matter of fighting and of sociality. (B) Normal devil facial features. (C) Severely affected devil with DFTD
Box 2. Distinguishing features of DFTD.
The Tasmanian Devil tumor (DFTD) developed much more recently then CTVT and was first described in Tasmania in 1996, where it is restricted to that island.
Found only in Tasmanian Devils, the tumor is lethal, unlike CTVT and is spread by biting and scratching; DFTD has brought the species to the brink of extinction.
DFTD is comprised of Schwann like cells, suggesting a neural crest origin for the tumor.
Key Immune features allow the tumor to escape host surveillance including differential expression of key miRNAs and epigenetic down-regulation of Class I MHC molecules.
Tumor Origins
Compared to the normal appearance of devils (Figure 5B), DFTD tumors are disfiguring, initially appearing on the face and mouth (Figure 5C). About 65% undergo true metastasis to internal organs, particularly the lungs [15, 18]. The cells are round and characterized as undifferentiated, soft neoplasms [18]. They are composed of infiltrative masses contained within a pseudocapsule, which is itself supported by fibrovascular stroma. Tumors are both ulcerated and exudative [18] (Figure 5C).
Clonality of DFTD has been established several times, initially by karyotypic data showing that tumors from different individuals contain the same complex chromosomal rearrangements [16, 55]. This was followed by microsatellite and MHC analysis indicating a lack of diversity across tumors, consistent with clonal transmissibility [15]. This was verified by Murchison who showed that DFTD tumors shared similar microsatellite genotypes across all loci tested, regardless of location, sex, or age of devil [56].
DFTD Cell Type of Origin
The ancestral cell type of DFTD was originally categorized as neuroendocrine because tumors express diagnostic markers from neuron-specific enolase, synaptophysin, and vimentin [57]. More recently, hierarchical clustering showed high concordance between tumor cells and those associated with the peripheral nervous system. Subsequent antibody staining indicated that tumor cells produce a Schwann cell-specific protein, periaxin [56]. Schwann and neuroendocrine cells are both derived from the neural crest and overlap in gene expression, suggesting that DFTD is derived from a precursor neural crest cell (Box 2). The Schwann cell marker periaxin is considered a sensitive and specific diagnostic for DFTD tumors [58].
Genetic Variation and Tumor Success
How could the DFTD tumor have arisen so quickly? Overall, the species possesses limited genetic diversity and low heterozygosity (14, 60). Assessing devils from six geographic regions using microsatellites revealed similar genetic profiles and no signatures of bottlenecks or recent expansions. Heterozygosity was estimated to range from only 0.39–0.47 while allelic diversity was 2.7–3.3; only 2/3 to 1/2 of that observed in other Australian marsupials. Such low diversity is likely related to restricted habitat, founder effects, disease burden, as well as natural fluxation in the population [13, 59]. In addition, long-term low population density over time, as the devils have experienced, leads to popular sire effects, which further reduces diversity. In 2011, two Tasmanian devils and their tumors from distant parts of Tasmania were sequenced, and a de novo assembly performed [60]. Analysis of these data combined with mtDNA of extant and museum specimens, again suggested low diversity in contemporary populations. This perfect storm of events, which reduced both population size and genetic diversity, set the stage for the current high magnitude DFTD outbreak.
Murchison et al. also sequenced two Tasmanian devils and two DFTD cell lines, reporting 15,000–17,000 somatic mutations in each DFTD, again substantially higher than most human cancers [61]. Using mtDNA from 104 tumors, phylogenetic inference demonstrated that DFTD clearly derived from a single devil, likely located on the east coat of the island. To investigate subsequent divergence from this ancestor, the authors collected DFTD from 69 devils and genotyped them for 16 likely somatic variants, each of which was present in one or the other sequenced tumor, but not both. Two distinct lineages exist; one from Forestier Peninsula, and a divergent subpopulation that bloomed between 2007–2010. It is this subpopulation and its derived subclones that now populate most of the island.
Tasmanian Devil MHC
When considering spread of any transmissible tumor epidemic, one can imagine two ways in which MHC fails the host. First, the tumor can escape the immune system by modulating expression of MHC genes. DFTD does not display functional MHC class I molecules, in vivo or in vitro, and thus avoids recognition by host CD8+ T cells. This has been demonstrated to be the result of coordinated epigenetic down-regulation of multiple aspects within the antigen presenting system, likely during the initial neoplastic transformation [62]. Alternatively, the host (devils) may simply lack enough genetic diversity to recognize the tumor as non-self (Box 2). Clearly, the former is at work with CTVT [8]. However, both may be important for DFTD. Analysis of Tasmanian devil MHC also suggests low genetic diversity in the devil population [15]. Since the tumor is an allograft, it should be recognized and rejected by the host immune system. However, Siddle and colleagues collected lymphocytes from 30 eastern Tasmanian devils, and tested pools against each other as well as lymphocytes isolated from other parts of the island. Despite differing origins, no mixed lymphocyte responses were detected. This very striking result highlights the lack of MHC diversity in the population, i.e. the devil immune system is incapable of recognizing foreign lymphocytes as non-self, much less as tumor [15].
DFTD and Possible Extinction
Due to the severity of the DFTD epidemic, major concerns exist that this unusual metatherian mammal will become extinct [13, 20, 21] in perhaps as few as 35 years [21]. As devils are unable to recognize the clonal facial tumors as foreign and mount an effective immune response, it has been proposed that the only effective way to deal with the epidemic is to isolate unaffected animals while developing a rational plan for culling affected individuals from the population [15]. Recently, the development of a potential vaccine for DFTD has provided new hope [63]. Whether these or other actions are taken, it is imperative that they be taken rigorously and soon.
Syrian Hamsters
In contrast to the single origin of CTVT and DFTD, transmissible tumors with multiple, independent origins occur within the golden Syrian hamster (Mesocricetus auratus) (Figure 6A). Numerous, spontaneously occurring sarcoma lineages have been discovered in different experimental populations originating from the same founder [64]. These tumors are reportedly aggressive, highly transmissible and generally fatal.
Figure 6.


Two additional species that harbor transmissible tumor “models” (A) Syrian hamster. (B) Soft-shelled clam.
Subsequent research shows that the sarcomas naturally spread within a laboratory population without inoculation, presumably by hamsters gnawing at tumors and cannibalism [65]. Surprisingly, when present in the bloodstream of experimentally inoculated hamsters, tumor cells can also be transmitted between colony members via mosquito bite, with metastases forming within days of vector transfer [66]. Initial reports argue strongly that there is no evidence for viral induction of tumorgenesis associated with original reports of the hamster disease, and tumor cells reportedly possessed a stable karyotype through numerous passages [67], including one spanning 11 years and 288 passages. In every experimental case throughout these passages, inoculation of the tumor produced metastases that spread freely and invariably proved fatal.
In 1975 Ambrose and Coggin [68] published a report describing an epidemic of lymphomas that arose spontaneously on three occasions at an animal facility housing both random bred and inbred Syrian hamsters. This story is distinct from those described above in that tumor formation in these hamsters was not apparently dependent on the ingestion of the tumor by healthy individuals, scratching, biting or sexual contact, etc. Rather, the tumor appeared in the colony each time spontaneously [68]. It occurred in a facility that had noted no history of natural appearance of lymphomas in hamsters. It was, however, similar to the above story of golden Syrian hamsters in that once in the colony it spread quickly. The addition of healthy new hamsters to the diseased part of the colony resulted in those animals contracting the same tumor type as the original index case. New animals housed with hamsters that had no evidence of disease remained healthy for the one-year duration of the experiment. Affected individuals experienced metastases, often to distant organs, and death. Despite efforts to clean the affected rooms, the epizootic appeared repeatedly in newly introduced animals.
The authors gave considerable thought as to the cause of the initial infection. They found no evidence for retroviridae or oncogenic viruses in the tumors or hosts. Whatever the causative agent, it was resistant to UV inactivation, and antiviral cleaning agents. The investigators argued initially that procedures undertaken in the laboratory had no likely relationship with tumor formation. However the treatments animals experienced included injections with the polyoma virus Simian Virus 40 (SV40), now known to be a DNA tumor virus, as well as various adenoviruses. The tumor extracts were negative for STV0 T antigen, the hallmark of viral infection. Additionally, a follow-up publication utilizing cell-free extracts showed the presence of a naturally transmitting, oncogenic, naked DNA viroid-like agent, the origins of which were unclear [69]. These studies did not extend beyond the report of the viroid like agent, raising the question as to its role in initiation and transmission. Further, the relationship between these studies and those done in the 1970s and 80s examining the transmissible Golden Syrian hamster sarcoma are ambiguous. Thus, it may be that all Syrian hamster transmissible tumors are virally induced; while it is unclear if virus or what elements of the virus are needed for transmissibility.
In any event, over time it is likely that other examples of transmissible mammalian tumors will appear in the literature, perhaps revealing as yet unidentified modes of transmission, such as lactation. Careful studies are needed to assess the potential role of viruses, prions, or other vectors in initiation and transmission of such tumor events.
Other Potential Transmissible Tumors
Soft Shelled Clams
Dogs, Tasmanian devils and Syrian hamsters provide the only current examples of transmissible tumors among mammals. But to fully explore the biology of transmissible tumors it is essential to consider non-mammalian species. Recent studies suggest a possible candidate has emerged from the Phylum Mollusca (e.g. snails, clams, oysters, mussels) (Figure 6B). The soft shell clam (Mya arenaria) is distributed along the east coast of North America, and since the late 1960’s has been the subject of numerous reports of a disseminated leukemia-like neoplasia [70–71].
Clams have an open circulatory system comprised of hemolymph, which surrounds and bathes body organs. Hemolymph contains elements similar to mammalian blood, including respiratory proteins and hemocytes, which are the primary line of defense against foreign matter, analogous to mammalian phagocytes. The clam neoplasia is characterized by the hemolymph containing anywhere from 1% to 100% mitotic hemocytes [72]. Neoplastic hemocytes show loss of phagocytic ability and promotion of dissemination into tissues [73]. During disease progression, amitotic hemocytes are replaced by proliferating leukemia cells that invade all tissues, leading to death [74].
The strongest evidence for a transmissible neoplasia in clams comes from studies of unfiltered hemolymph and lysed hemocytes that induce cancer in healthy individuals [75]. Subsequent studies were consistent with a retrovirus or retrotransposon etiology [76].
Studies from the 1990s reported reverse transcriptase (RT) activity in the tissues of affected clams [77]. Recently, Arriagada and colleagues detected two to 10-fold higher RT levels in cell-free hemolymph and 20–50× higher in cultured hemocytes from neoplastic versus healthy clams [74]. Examination of the hemolymphatic transcriptome identified the presence of retroviral protease, RT and integrase. In combination, these represent a complete copy of a previously uncharacterized retroelement insertion from the gypsy family. Dubbed ‘Steamer’, this element shows a significantly elevated DNA copy number in diseased cells. Analysis of Steamer integration sites, mt-DNA, SNVs, and microsatellites, demonstrates that the genotypes of neoplastic cells collected from multiple locations including New York, Maine, and Prince Edward Island are nearly identical to one another, and all cases differed significantly from their transient hosts [78]. The authors conclude that the cancer is spreading as a clonal, transmissible cell derived from a single original clam.
While the above data support the transmissible tumor hypothesis, no conclusive evidence has identified the mechanism of neoplastic transfer. Clams are primarily sessile organisms that do not normally come into contact with each other at any point in their life cycle. They are external fertilizers and broadcast spawners, releasing eggs and sperm into the surrounding seawater. The resulting “embryos” eventually settle out of the water column and undertake a benthic existence with little or no movement for the rest of their lives. Clearly, additional studies are needed before we can conclude that soft-shell clams or other mollusk species truly have transmissible tumors [79].
Concluding Remarks and Future Perspectives
While many types of cancer have been described in birds, amphibians, reptiles and especially fish, to date there have been no reports of a transmissible tumor among any of these non-mammalian vertebrates (Figure 7, Key Figure). Given that these four groups collectively represent about 55,000 described species with incredibly rich and diverse behavioral repertoires, compared to ~5,500 species of mammals, it is not unreasonable to expect that examples of transmissible tumors will ultimately be reported. In fact, given the efficacy of evolutionary pressures in generating life to fill biological niches, it would be unreasonable to expect that only four species on the planet be burdened with transmissible tumors. The path to transmissibility is already less obscured in that convergent evolution exists between CTVT and DFTD. Presentation of Class I MHC molecules is ablated by redundant somatic mutation in the former, and by coordinated epigenetic down-regulation in the latter, leading to host immune avoidance. This clearly underscores the importance of this early mechanism in overcoming an essential hurdle for these tumors. The prolonged timeframe of CTVT evolution may have begun with a similar epigenetic pathway as is seen in modern DFTD.
Figure 7.
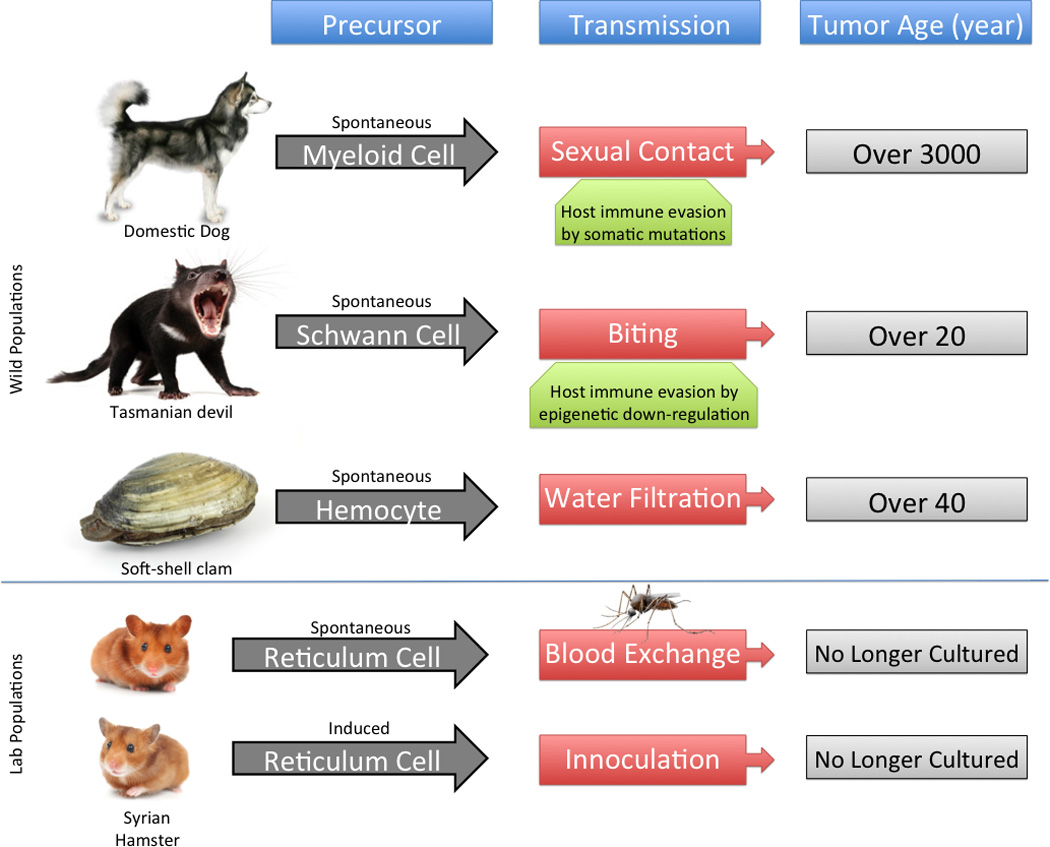
Summarization of transmissible tumors, including their original cell type, mode of transmission, potential mechanisms for host evasion, and approximate age.
Cancer is a disease of normal cells whose progeny have broken the bonds of normal function. In the case of transmissible tumors, even the known paradigm of cancer is broken. Because they are directly conferrable between hosts and therefore perpetuate for dozens to thousands of years, they represent a biological curiosity of the greatest magnitude that intrigues and concerns (Figure 7, Key Figure). It remains to be determined how these tumors originated, and why such events are so rare. Further, we need to better understand how dogs overcome the tumor and sometimes survive. We also need a clearer understanding of the epidemiology of each tumor, with data about speed of transmission and susceptible hosts. Thus, considerable work remains to understand the biology behind the three known mammalian transmissible tumors, and to understand how the mollusk tumors are transmitted. Such data will not only help predict if additional tumors are likely, and in what types of species, but will also inform on the nature of the requisite mechanisms of clonal transmissibility, providing a fundamental understanding of the functions underpinning the basic biology of evolving tumors.
Trends Box: Transmissible Tumors.
Recent whole genome sequencing of CTVT has allowed the first detailed glimpse into mechanisms allowing transmissibility.
Evaluation of CTVT against 186 canine whole genomes drastically increased the ability to distinguish between somatic and germline variants, leading to accurate classification of SNVs, indels, and SVs.
This evaluation as well as a more accurate depiction of tumor evolution and a better understanding the underlying immunology which facilitates the characteristic transmissibility of transmissible tumors
DFTD shows evidence for convergent evolution with the much older CTVT within class I MHC molecule presentation, indicating an essential hurdle for host immune evasion.
Additional transmissible tumor models in the Syrian hamster and soft-shelled clam may further highlight commonalities and divergences between tumor transmissibility mechanisms.
Outstanding Questions Box 1: Origins of transmissible tumors.
What factors have lead to the stunning success of CTVT as a parasitic cancer?
Could such factors be easily replicated in other species?
Are mammals particularly suited to transmissible cancers?
Why do some CTVT tumors resolve and others do not?
Acknowledgements
Thank you to Brennan Decker, Drs. Danielle Karyadi, and Heidi Parker for helpful comments on this manuscript. EAO and BWD are supported by the Intramural Program of the National Human Genome Research Institute.
Glossary
- aCGH
microarray comparative genomic hybridization
- CTVT
Canine venereal transmissible tumor
- dbSNP
database of single nucleotide variation
- DFTD
Tasmanian Devil Facial Tumor
- DLA
Dog leucocyte antigen locus
- DNA
Deoxyribonucleic acid
- dbSNP
database of single nucleotide polymorphisms
- HLA
Human leucocyte antigen locus
- Indel
Insertion/deletion
- LINE
Long interspersed nuclear element
- PCA
Principal components analysis
- RNA
ribonucleic acid
- SINE
short interspersed nuclear element
- SNP
Single nucleotide polymorphism
- SNV
Single nucleotide variant
- SV
Structural variant
- WGS
Whole genome sequence
- WSINE
Wallaby short interspersed nuclear element
- U.S.
United States
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Murgia C, et al. Clonal origin and evolution of a transmissible cancer. Cell. 2006;126:477–487. doi: 10.1016/j.cell.2006.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murchison EP, et al. Transmissible [corrected] dog cancer genome reveals the origin and history of an ancient cell lineage. Science. 2014;343:437–440. doi: 10.1126/science.1247167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blaine DP. A domestic treartise on the diseases in dogs and horses. T Boosey; 1810. [Google Scholar]
- 4.Marcos R, et al. Cutaneous transmissible venereal tumor without genital involvement in a prepubertal female dog. Vet. Clin. Pathol. 2006;35:106–109. doi: 10.1111/j.1939-165x.2006.tb00097.x. [DOI] [PubMed] [Google Scholar]
- 5.Nowinsky M. Zur Frage ueber die Impfung der krebsigen Geschwuelste. Zentralbl Med Wissensch. 1876;14:790–791. [Google Scholar]
- 6.Das U. Review of canine transmissible venereal sarcoma. Vet. Res. Commun. 2000;24:545–556. doi: 10.1023/a:1006491918910. [DOI] [PubMed] [Google Scholar]
- 7.Katzir N, et al. Common origin of transmissible venereal tumors (TVT) in dogs. Oncogene. 1987;1:445–448. [PubMed] [Google Scholar]
- 8.Decker B, Davis BW, Rimbault M, Long AH, Karlins E, Jagannathan V, Reiman R, Parker HG, Drogemuller C, Corneveaus JJ, Chapman ES, Trent JM, Leeb T, Huentelman MJ, Wayne RK, Karyadi DM, Ostrander EA. Comparison against 186 canid whole genome sequences reveals survival strategies of an ancient clonally transmissible canine tumor. Genome Res. 2015 doi: 10.1101/gr.190314.115. (In Press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ndiritu CG, et al. Extragenitally located transmissible venereal tumor in dogs. Mod. Vet. Pract. 1977;58:940–946. [PubMed] [Google Scholar]
- 10.Murchison EP. Clonally transmissible cancers in dogs and Tasmanian devils. Oncogene. 2008;27(Suppl 2):S19–S30. doi: 10.1038/onc.2009.350. [DOI] [PubMed] [Google Scholar]
- 11.Ganguly B, et al. Canine transmissible venereal tumour: a review. Vet. Comp. Oncol. 2013 doi: 10.1111/vco.12060. [DOI] [PubMed] [Google Scholar]
- 12.Strakova A, et al. The changing global distribution and prevalence of canine transmissible venereal tumour. BMC Vet. Res. 2014;10:168. doi: 10.1186/s12917-014-0168-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hawkins CE, Baars C, Hesterman H, Hocking GJ, Jones ME, Lazenby B, Mann D, Mooney N, Pemberton D, Pyecroft S, et al. Emerging disease and population decline of an island endemic, the Tasmanian devil Sarcophilus harrisii. Biol. Conserv. 2006;131:307–324. [Google Scholar]
- 14.Jones ME, et al. Genetic diversity and population structure of Tasmanian devils, the largest marsupial carnivore. Mol. Ecol. 2004;13:2197–2209. doi: 10.1111/j.1365-294X.2004.02239.x. [DOI] [PubMed] [Google Scholar]
- 15.Siddle HV, et al. Transmission of a fatal clonal tumor by biting occurs due to depleted MHC diversity in a threatened carnivorous marsupial. Proc. Natl. Acad. Sci. U.S.A. 2007;104:16221–16226. doi: 10.1073/pnas.0704580104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pearse AM, et al. Allograft theory: transmission of devil facial-tumour disease. Nature. 2006;439:549. doi: 10.1038/439549a. [DOI] [PubMed] [Google Scholar]
- 17.Hamede RK, et al. Contact networks in a wild Tasmanian devil (Sarcophilus harrisii) population: using social network analysis to reveal seasonal variability in social behaviour and its implications for transmission of devil facial tumour disease. Ecol. Lett. 2009;12:1147–1157. doi: 10.1111/j.1461-0248.2009.01370.x. [DOI] [PubMed] [Google Scholar]
- 18.Loh R. The pathology of devil facial tumor disease (DFTD) in Tasmanian Devils (Sarcophilus harrisii) Vet. Pathol. 2006;43:890–895. doi: 10.1354/vp.43-6-890. [DOI] [PubMed] [Google Scholar]
- 19.Pyecroft SB, et al. Towards a case definition for devil facial tumour disease: What is it? EcoHealth. 2007;4:318–325. [Google Scholar]
- 20.Lachish S, et al. Demography, disease and the devil: life-history changes in a disease-affected population of Tasmanian devils (Sarcophilus harrisii) J. Anim. Ecol. 2009;78:427–436. doi: 10.1111/j.1365-2656.2008.01494.x. [DOI] [PubMed] [Google Scholar]
- 21.McCallum H, et al. Transmission dynamics of Tasmanian devil facial tumor disease may lead to disease-induced extinction. Ecology. 2009;90:3379–3392. doi: 10.1890/08-1763.1. [DOI] [PubMed] [Google Scholar]
- 22.Cohen D. The canine transmissible venereal tumor: a unique result of tumor progression. Adv. Cancer Res. 1985;43:75–112. doi: 10.1016/s0065-230x(08)60943-4. [DOI] [PubMed] [Google Scholar]
- 23.Rust JH. Transmissible lymphosarcoma in the dog. J. Am. Vet. Med. Assoc. 1949;114:10–14. [PubMed] [Google Scholar]
- 24.Mukaratirwa S, et al. Canine transmissible venereal tumour: cytogenetic origin, immunophenotype, and immunobiology. A review. Vet. Q. 2003;25:101–111. doi: 10.1080/01652176.2003.9695151. [DOI] [PubMed] [Google Scholar]
- 25.Mozos E, et al. Immunohistochemical characterization of canine transmissible venereal tumor. Vet. Pathol. 1996;33:257–263. doi: 10.1177/030098589603300301. [DOI] [PubMed] [Google Scholar]
- 26.Murray M, et al. A study of the cytology and karyotype of the canine transmissible venereal tumour. Res. Vet. Sci. 1969;10:565–568. [PubMed] [Google Scholar]
- 27.Moulton JD. Toumors of Domestic Animmals. University of California Press; 1990. [Google Scholar]
- 28.Cockrill JM, et al. Ultrastructural characteristics of canine transmissible venereal tumor at various stages of growth and regression. Am. J. Vet. Res. 1975;36:677–681. [PubMed] [Google Scholar]
- 29.Adams EW, et al. Cytogenetic observations on the canine venereal tumor in long-term culture. Cornell Vet. 1981;71:336–346. [PubMed] [Google Scholar]
- 30.Karlson AG, et al. The transmissible venereal tumor of dogs: observations on forty generations of experimental transfers. Ann. N. Y. Acad. Sci. 1952;54:1197–1213. doi: 10.1111/j.1749-6632.1952.tb39989.x. [DOI] [PubMed] [Google Scholar]
- 31.Cockrill JM, et al. Transmission of transmissible venereal tumor of the dog to the coyote. Am. J. Vet. Res. 1979;40:409–410. [PubMed] [Google Scholar]
- 32.Gimeno EJ, et al. Intermediate filament expression and lectin histochemical features of canine transmissible venereal tumour. APMIS. 1995;103:645–650. doi: 10.1111/j.1699-0463.1995.tb01417.x. [DOI] [PubMed] [Google Scholar]
- 33.Sandusky GE, et al. Diagnostic immunohistochemistry of canine round cell tumors. Vet. Pathol. 1987;24:495–499. doi: 10.1177/030098588702400604. [DOI] [PubMed] [Google Scholar]
- 34.Albanese F, et al. Primary cutaneous extragenital canine transmissible venereal tumour with Leishmania-laden neoplastic cells: a further suggestion of histiocytic origin? Vet. Dermatol. 2002;13:243–246. doi: 10.1046/j.1365-3164.2002.00301.x. [DOI] [PubMed] [Google Scholar]
- 35.Makino S. Some epidemiologic aspects of venereal tumors of dogs as revealed by chromosome and DNA studies. Ann. N. Y. Acad. Sci. 1963;108:1106–1122. doi: 10.1111/j.1749-6632.1963.tb13438.x. [DOI] [PubMed] [Google Scholar]
- 36.Thomas R, et al. Extensive conservation of genomic imbalances in canine transmissible venereal tumors (CTVT) detected by microarray-based CGH analysis. Chrom. Res. 2009;17:927–934. doi: 10.1007/s10577-009-9080-8. [DOI] [PubMed] [Google Scholar]
- 37.Rebbeck CA, et al. Origins and evolution of a transmissible cancer. Evolution. 2009;63:2340–2349. doi: 10.1111/j.1558-5646.2009.00724.x. [DOI] [PubMed] [Google Scholar]
- 38.Bautista-Gomez LG, et al. Analysis of canine transmissible veneral tumor genotypes using the D-loop region of mitochondrial DNA. Genes Genet. Syst. 2011;86:351–355. doi: 10.1266/ggs.86.351. [DOI] [PubMed] [Google Scholar]
- 39.vonHoldt BM, et al. A genome-wide perspective on the evolutionary history of enigmatic wolf-like canids. Genome Res. 2011;21:1294–1305. doi: 10.1101/gr.116301.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vonholdt BM, et al. Genome-wide SNP and haplotype analyses reveal a rich history underlying dog domestication. Nature. 2010;464:898–902. doi: 10.1038/nature08837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parker HG, et al. Genetic structure of the purebred domestic dog. Science. 2004;304:1160–1164. doi: 10.1126/science.1097406. [DOI] [PubMed] [Google Scholar]
- 42.Kumar A, et al. Exome sequencing identifies a spectrum of mutation frequencies in advanced and lethal prostate cancers. Proc. Natl. Acad. Sci. U.S.A. 2011;108:17087–17092. doi: 10.1073/pnas.1108745108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alexandrov LB, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liao KW, et al. Identification of canine transmissible venereal tumor cells using in situ polymerase chain reaction and the stable sequence of the long interspersed nuclear element. J. Vet. Diagn. Invest. 2003;15:399–406. doi: 10.1177/104063870301500501. [DOI] [PubMed] [Google Scholar]
- 45.Yang TJ. Immunobiology of a spontaneously regressive tumor, the canine transmissible venereal sarcoma (review) Anticancer Res. 1988;8:93–95. [PubMed] [Google Scholar]
- 46.Bellingham Smith G, Washbourn JW. Infective venereal tumours in dog. J. Pathol. Bateriol. 1898;5:99–111. [Google Scholar]
- 47.Dass LL, et al. Surgical treatment of canine transmisible venereal tumor--a retrospective study. Indian Vet. J. 1989;66:255–258. [Google Scholar]
- 48.Amber EI, et al. Single-drug chemotherapy of canine transmissible venereal tumor with cyclophosphamide, methotrexate, or vincristine. J. Vet. Intern. Med. 1990;4:144–147. doi: 10.1111/j.1939-1676.1990.tb00887.x. [DOI] [PubMed] [Google Scholar]
- 49.Singh J, et al. Clinico-pathological studies on the effect of different anti-neoplastic chemotherapy regimens on transmissible venereal tumours in dogs. Vet. Res. Commun. 1996;20:71–81. doi: 10.1007/BF00346579. [DOI] [PubMed] [Google Scholar]
- 50.Belov K. The role of the Major Histocompatibility Complex in the spread of contagious cancers. Mamm. Genome. 2011;22:83–90. doi: 10.1007/s00335-010-9294-2. [DOI] [PubMed] [Google Scholar]
- 51.Wagner JL, et al. Molecular analysis of the DLA-DR region. Tissue Antigens. 1996;48:549–553. doi: 10.1111/j.1399-0039.1996.tb02668.x. [DOI] [PubMed] [Google Scholar]
- 52.Wagner JL, et al. Molecular analysis and polymorphism of the DLA-DQA gene. Tissue Antigens. 1996;48:199–204. doi: 10.1111/j.1399-0039.1996.tb02629.x. [DOI] [PubMed] [Google Scholar]
- 53.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gill ED. The Australian Aborigines and the Tasmanian devil. Mankind. 1971;8:59–60. [Google Scholar]
- 55.Deakin JE, et al. Genomic restructuring in the Tasmanian devil facial tumour: chromosome painting and gene mapping provide clues to evolution of a transmissible tumour. PLoS Genet. 2012;8:e1002483. doi: 10.1371/journal.pgen.1002483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murchison EP, et al. The Tasmanian devil transcriptome reveals Schwann cell origins of a clonally transmissible cancer. Science. 2010;327:84–87. doi: 10.1126/science.1180616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Loh R. The immunohistochemical characterization of devil facial tumor disease (DFTD) in the Tasmanian Devil (Sarcophilus harrisii) Vet. Pathol. 2006;43:896–903. doi: 10.1354/vp.43-6-896. [DOI] [PubMed] [Google Scholar]
- 58.Tovar C, et al. Tumor-specific diagnostic marker for transmissible facial tumors of Tasmanian devils: immunohistochemistry studies. Vet. Pathol. 2011;48:1195–1203. doi: 10.1177/0300985811400447. [DOI] [PubMed] [Google Scholar]
- 59.Guiler ER. Observations on theTasmanian devil, Sarcophilus harrisii (Marsupialia: Dasyuridae)l. II. Reproduction Breeding and growth of pouch young. Aust. J. Zool. 1978;18:49–62. [Google Scholar]
- 60.Miller W, et al. Genetic diversity and population structure of the endangered marsupial Sarcophilus harrisii (Tasmanian devil) Proc. Natl. Acad. Sci. U.S.A. 2011;108:12348–12353. doi: 10.1073/pnas.1102838108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Murchison EP, et al. Genome sequencing and analysis of the Tasmanian devil and its transmissible cancer. Cell. 2012;148:780–791. doi: 10.1016/j.cell.2011.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Siddle HV, et al. Reversible epigenetic down-regulation of MHC molecules by devil facial tumour disease illustrates immune escape by a contagious cancer. Proc. Natl. Acad. Sci. U.S.A. 2013;110:5103–5108. doi: 10.1073/pnas.1219920110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kreiss A, et al. Evidence for induction of humoral and cytotoxic immune responses against devil facial tumor disease cells in Tasmanian devils (Sarcophilus harrisii) immunized with killed cell preparations. Vaccine. 2015;33:3016–3025. doi: 10.1016/j.vaccine.2015.01.039. [DOI] [PubMed] [Google Scholar]
- 64.Ashbel R. Spontaneous transmissible tumors in the Syrian hamsters. Nature. 1945;155:607. [Google Scholar]
- 65.Brindley DC, Banfield WG. A contagious tumor of the hamster. J. Natl. Cancer Inst. 1961;26:949–957. [Google Scholar]
- 66.Banfield WG, et al. Mosquito transmission of a reticulum cell sarcoma of hamsters. Science. 1965;148:1239–1240. doi: 10.1126/science.148.3674.1239. [DOI] [PubMed] [Google Scholar]
- 67.Cooper HL, MacKay CM, Banfield WG. Chromosome studies of a contagious reticulum cell sarcoma of the Syrian hamster. J. Natl. Cancer Inst. 1964;33:691–706. [PubMed] [Google Scholar]
- 68.Ambrose KR, Coggin JH., Jr An epizootic in hamsters of lymphomas of undetermined origin and mode of transmission. J. Natl. Cancer Inst. 1975;54:877–880. [PubMed] [Google Scholar]
- 69.Coggin JH, et al. Unusual filterable oncogenic agent isolated from hoizontally traqnsmitted syrian hamster lymphomas. Nature. 1981;290:336–338. doi: 10.1038/290336a0. [DOI] [PubMed] [Google Scholar]
- 70.Brown RS, et al. Prevalence of neoplasia in 10 New England populations of the soft-shell clam (Mya arenaria) Ann. N. Y. Acad. Sci. 1978;298:522–534. doi: 10.1111/j.1749-6632.1977.tb19287.x. [DOI] [PubMed] [Google Scholar]
- 71.Frierman EM, et al. Occurrence of hematopoietic neoplasms in Virginia oysters (Crassostrea virginica) J. Natl. Cancer Inst. 1976;56:319–324. doi: 10.1093/jnci/56.2.319. [DOI] [PubMed] [Google Scholar]
- 72.Böttger SA, Amarosa EJ, Geoghegan P, Walker CW. Chronic natural occurrence of disseminated neoplasia in select populations of the soft-shell clam, Mya arenaria, in New England. Northestern Nat. 2013:430–440. [Google Scholar]
- 73.Rodrick GE, et al. Microscopical studies of the hemocytes of bivalves and their phagocytic interaction with selected bacteria. Helgolander Meeresuntersuchungen. 1984;37:167–176. [Google Scholar]
- 74.Arriagada G, et al. Activation of transcription and retrotransposition of a novel retroelement, Steamer, in neoplastic hemocytes of the mollusk Mya arenaria. Proc. Natl. Acad. Sci. U.S.A. 2014;111:14175–14180. doi: 10.1073/pnas.1409945111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Walker C, et al. Mass culture and characterization of tumor cells from a naturally occurring invertebrate cancer model: applications for human and animal disease and environmental health. Biol. Bull. 2009;216:23–39. doi: 10.1086/BBLv216n1p23. [DOI] [PubMed] [Google Scholar]
- 76.Oprandy JJ, et al. 5-bromodeoxyuridine induction of hematopoietic neoplasia and retrovirus activation in the soft-shell clam, Mya arenaria. J. Invertebr. Pathol. 1983;42:196–206. doi: 10.1016/0022-2011(83)90062-9. [DOI] [PubMed] [Google Scholar]
- 77.House ML, et al. Soft shell clams Mya arenaria with disseminated neoplasia demonstrate reverse transcriptase activity. Dis. Aquat. Organ. 1998;34:187–192. doi: 10.3354/dao034187. [DOI] [PubMed] [Google Scholar]
- 78.Metzger MJ, et al. Horizontal transmission of clonal cancer cells causes leukemia in soft-shell clams. Cell. 2015;161:255–263. doi: 10.1016/j.cell.2015.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Barber BJ. Neoplastic diseases of commercially important marine bivalves. Aqua Liv. Resour. 2004;17:449–466. [Google Scholar]