

Abstract
Orofacial clefts (OFCs) represent a large fraction of human birth defects and are one of the most common phenotypes affected by large copy number variants (CNVs). Due to the limited number of CNV patients in individual centers, CNV analyses of a large number of OFC patients are challenging. The present study analyzed 249 genomic deletions and 226 duplications from a cohort of 312 OFC patients reported in two publicly accessible databases of chromosome imbalance and phenotype in humans, DECIPHER and ECARUCA. Genomic regions deleted or duplicated in multiple patients were identified, and genes in these overlapping CNVs were prioritized based on the number of genes encompassed by the region and gene expression in embryonic mouse palate. Our analyses of these overlapping CNVs identified two genes known to be causative for human OFCs, SATB2 and MEIS2, and 12 genes (DGCR6, FGF2, FRZB, LETM1, MAPK3, SPRY1, THBS1, TSHZ1, TTC28, TULP4, WHSC1, WHSC2) that are associated with OFC or orofacial development. Additionally, we report 34 deleted and 24 duplicated genes that have not previously been associated with OFCs but are associated with the BMP, MAPK and RAC1 pathways. Statistical analyses show that the high number of overlapping CNVs is not due to random occurrence. The identified genes are not located in highly variable genomic regions in healthy populations and are significantly enriched for genes that are involved in orofacial development. In summary, we report a CNV analysis pipeline of a large cohort of OFC patients and identify novel candidate OFC genes.
Electronic supplementary material
The online version of this article (doi:10.1007/s00439-015-1606-x) contains supplementary material, which is available to authorized users.
Introduction
Orofacial clefts (OFCs) are characterized by orofacial dysmorphism that may extend from the oral cavity to the whole face, involving also the eyes and ears in the most severe cases. OFCs represent the most common craniofacial malformations and a large fraction of all human birth defects. Collectively, the prevalence of OFCs varies between 1.5 and 25 per 10,000 births worldwide (Mossey and Castilla 2003) depending on geographical location, ethnicity and even socioeconomic status (Murthy and Bhaskar 2009). Although OFCs are repairable surgically with only rare exceptions, such as oblique facial cleft, these defects lead to a wide spectrum of lifelong complications, including feeding difficulties, velopharyngeal insufficiency, speech and acoustic impairment, orthodontic problems, psychosocial issues and economic burden due to medical and behavioral interventions, which greatly affect the quality of life (Mossey et al. 2009; Strauss and Cassell 2009; Wehby and Cassell 2010; Shkoukani et al. 2013). Due to the complex multidisciplinary interventions required to treat the lifelong morbidity associated with OFCs, the costs for these disorders have been estimated to be greater than $100,000 per individual (http://www.cdc.gov). Both the frequency and significant healthcare burdens imposed by OFCs emphasize the need to identify the genetic causes and the molecular and cellular mechanisms of these disorders, which will enable the ultimate aim of improving diagnosis, counseling, care and treatment for affected individuals.
Traditionally, OFCs are classified by phenotypes, based on the severity and the anatomical regions involved. The most common OFC phenotypes are cleft palate (CP), cleft lip (CL) and cleft lip and palate (CLP). However, rare OFCs have also been observed in humans, which may affect other oral structures, such as mandible or lower lip, or even the whole face, including nose, cheeks, eyes and forehead into the hairline. OFCs are generally considered to emerge from disruptions of distinct morphogenic processes which occur at different stages of embryological development (Shkoukani et al. 2013). OFCs are defined as complex multifactorial polygenic traits arising from many etiologies, including single-gene mutations, chromosome aberrations, intrauterine environment anomalies, improper maternal nutrient intake (deficiency of folic acid or zinc, excess of retinoic acid), exposure to teratogens (maternal smoking, alcohol or drug consumption, chemical pollutants), stress, infections, and even parental age and weight during pregnancy seem to contribute to the pathology (Dravet et al. 1992; Abrishamchian et al. 1994; Derijcke et al. 1996; Munger et al. 1996; Abel 1998; Hernández-Diaz et al. 2000; Little et al. 2004; Canfield et al. 2005; Jugessur and Murray 2005; Tamura et al. 2005; Villamor et al. 2008; Mossey et al. 2009; Murthy et al. 2009). The genetic component of OFC etiology is relevant, as demonstrated by the tenfold increased risk that has been observed in monozygotic (40 %) vs dizygotic twins (4.2 %) (Wyszynski et al. 1996).
OFCs can be categorized into syndromic and non-syndromic forms, according to the presence or absence of other cognitive or structural anomalies occurring outside the cleft area in the affected individuals (Cobourne 2004). At least 275 syndromes, whose primary features include OFCs, have been identified but the genetic causes are known only for 75 % of them (Leslie and Marazita 2013). Classical genetic studies of syndromic forms of OFCs have identified causative mutations in genes including IRF6 [Van der Woude syndrome (OMIM #119300), popliteal pterygium syndrome (OMIM #119500)], MSX1 [cleft-associated tooth agenesis (OMIM #106600)] and TP63 [ankyloblepharon-ectodermal dysplasia-clefting (OMIM #106260), ectrodactyly-ectodermal dysplasia-clefting (OMIM #225060)], through linkage analysis, candidate gene approaches and confirmation in studies using animal models (Celli et al. 1999; van den Boogaard et al. 2000; McGrath et al. 2001; Kondo et al. 2002; Dixon et al. 2011; Leslie and Marazita 2013). With the fast development of Next-Generation Sequencing technology, whole exome-sequencing studies have identified several causative genes in syndromic forms of OFCs such as MLL2 [Kabuki syndrome (OMIM #147920)], DHODH [Miller syndrome (OMIM #263750)] and RIPK4 [Bartsocas-Papas syndrome (OMIM #263650)] (Ng et al. 2010a, b; Kalay et al. 2012; Mitchell et al. 2012; Setó-Salvia and Stanier 2014).
The identification of causative sequence variants and the associated genes for non-syndromic OFCs remains challenging, as such cases are often sporadic. Several genes involved in syndromic forms of OFCs have been implicated in non-syndromic OFCs, such as IRF6 and MSX1 (Lidral et al. 1998; Van den Boogaard et al. 2000; Jezewski et al. 2003; Suzuki et al. 2004; Vieira et al. 2004; Zucchero et al. 2004; Rahimov et al. 2008; Birnbaum et al. 2009). Genome-wide association studies (GWAS) are often performed to investigate genes and loci contributing to the risk of OFC by statistical analyses. Among genes within ORF loci identified by GWAS, IRF6 has been demonstrated by various models as the causative gene, whereas the role of other genes, such as ABCA4 and MAFB, remains to be assessed (Beaty et al. 2010).
Large structural alterations of the genome, including deletions and duplications of genomic regions termed copy number variations (CNVs), have been studied in OFC patients using classical genetic analyses such as FISH, CGH arrays or, more recently, SNP arrays (FitzPatrick et al. 2003; Mulatinho et al. 2008; Barber et al. 2013; Izzo et al. 2013). Some of the identified genes including SUMO1, CLPTM1L and BMP2 have also been validated in animal models (Shi et al. 2009; Sahoo et al. 2011; Williams et al. 2012). However, due to the relatively low number of OFC patients exhibiting CNVs that are available in individual research centers, a systematic CNV study of a large number of OFC patients has not yet been performed to investigate the etiology of OFCs. Recently, new consortia have been organized to create comprehensive databases of clinical case data by combining the resources from different medical centers worldwide. One of these databases is DECIPHER (Database of Genomic Variation and Phenotype in Humans using Ensembl Resources, https://decipher.sanger.ac.uk/), a database of chromosomal imbalance and phenotype in human with information of more than 25,000 patients, contributed by a consortium of clinical genetic centers and diagnostic laboratories from thirty different countries (Bragin et al. 2014). Similarly, ECARUCA (European Cytogeneticists Association Register of Unbalanced Chromosome Aberrations, http://www.ecaruca.net) is another database that provides both clinical and molecular details on unbalanced chromosomal aberrations, recording over 4800 clinical cases so far (Vulto-van Silfhout et al. 2013). These publicly accessible databases provide rich resources to systematically study genetic mechanisms in a large number of CNV patients with OFCs.
Here, we report a comprehensive bioinformatics and statistical analysis to identify candidate causative genes involved in OFCs by analyzing common CNV regions shared by OFC patients retrieved from DECIPHER and ECARUCA databases. The analysis pipeline includes retrieving patient and CNV data from databases, the identification of overlapping genomic regions and the prioritization of candidate genes in the genomic regions followed by statistical analyses. This study identifies two previously known OFC genes and several novel candidate OFC genes.
Materials and methods
Data collection
The patients included in our study were retrieved from two publicly accessible web-based databases of genomic variants and chromosomal aberrations in humans, DECIPHER and ECARUCA, reported till July 2014. Two main criteria were used to select patients: the presence of OFCs, alone or in combination with other phenotypes, and the availability of the CNV location coordinates. Firstly, the terms ‘cleft’ and ‘bifid uvula’ were used to perform the search in the databases, and subsequently, for each identified patient the phenotypes were checked to exclude cases of cleft that do not involve the oral region (e.g., eyelid cleft). To be able to obtain a large number of cases, we decided to include patients with syndromic and non-syndromic OFCs. After selecting the relevant patients, further patient details (ID number, OFCs and other phenotypes, presence of overlapping syndromes) and CNV information (CNV type, size, genomic location in GRCh37/hg19) from ECARUCA and DECIPHER were collected (Supplementary Table 1).
Identification of overlapping CNV regions
Several Linux BEDtools were used sequentially to identify the deleted or duplicated genomic regions shared among OFC patients (Quinlan and Hall 2010). After sorting based on the genomic locations of CNVs, genomeCoverageBed was run to define the common genomic regions shared by patients’ CNVs, named overlapping regions. For each overlapping region the common genomic sequence (chr:start–end) and the total number of overlaps were analyzed. Subsequently, the BEDtools intersectBed and groupbyBed with the option ‘collapse’ were used in combination to join the BED files of common CNV regions with a list containing patients’ IDs for retrieving the patients who shared deleted or duplicated regions.
For randomization, the shuffleBed command was run repeatedly on the same total number of regions with the same sizes as the corresponding actual deletion and duplication lists to obtain 1000 random permutations. For the random permutations, assembly gaps (telomeres and short arm of chr13, 14, 15, 21, 22), alternative haplotype sequences (e.g., chr6_ssto_hap7) and random contigs (e.g. chr4_gl000193_random) as reported in the UCSC Genome Browser archive (http://hgdownload.soe.ucsc.edu/goldenPath/hg19/database/) were excluded. The same pipeline that was used to identify common deletions and duplications of patients was applied to identify the overlapping regions in each randomized CNV list for the two sets of 1000 randomized lists, termed as randomized overlapping region lists. In each randomized overlapping region list, the mean of the number of the overlaps was calculated, obtaining 1000 means per set, and subsequently the overall mean of the set was calculated based on the 1000 means. To visualize the data, the overall mean and the overall standard deviation were computed with R statistical program (http://www.r-project.org/), and then used to calculate the z score of each randomized list given by:
| 1 |
where μi represents the mean of overlaps of a specific randomized list (i), indicates the overall mean (average of all list means) while σ states the overall standard deviation. Formula (1) refers to a specific randomized list (i), but it was applied to all 1000 elements of the randomization set (R). Deletions and duplications were processed separately.
The Shapiro–Wilk test showed that the obtained distribution of the z score was not a normal distribution in both deletion and duplication sets, and therefore exact p values could not be computed. Instead, empirical p values based on counting the number of randomization scores that matched or exceeded the real scores were used. The z scores based on the list of overlapping deletions and of overlapping duplications derived from patients’ CNVs were calculated, using the same formula (1) that was applied to the randomized lists.
Gene retrieval, prioritization and OFC gene enrichment analysis
The UCSC Table Browser (https://genome.ucsc.edu/cgi-bin/hgTables) was used to generate a list of encompassed RefSeq genes for each overlapping region, including not only protein-coding genes but also pseudogenes, miRNAs and long non-coding RNAs (lncRNAs). Gene prioritization was performed based on three criteria: the number of overlapping CNVs (≥2), the number of genes in the overlapping regions (≤5, Supplementary Figure 1) and gene expression levels in mouse embryonic palate detected in an RNA-Seq analysis (nRPK ≥ 59.00, Supplementary Figure 2).
To test whether the prioritized gene list is enriched for known OFC genes, a panel of 126 OFC genes that have been shown to be involved in OFCs or craniofacial development was assembled based on the existing literature, hereafter referred to as OFC-associated genes (OFC-AGs) (Supplementary Table 2). Subsequently, the fold enrichment of the proportion of these OFC-AGs in the prioritized gene list was calculated relative to the proportion of OFC-AGs in all genes retrieved from the deleted or duplicated CNVs. The hypergeometric test was used to evaluate the significance of the enrichment.
Phenotype mapping
In addition to OFCs, other phenotypes of patients who have deletions or duplications encompassing one or more candidate genes were classified based on the phenotypic feature hierarchy of the Human Phenotype Ontology (HPO) (http://www.human-phenotype-ontology.org/) (Robinson and Mundlos 2010). HPO terms (phenotypic features) annotated to the patients were mapped to the top level of the term hierarchy, which consists of 23 broad phenotypic categories (e.g., abnormality of the nervous system) (Supplementary Table 3). This allows a coarse-grained characterization and comparison of patient phenotypes.
Generation of RNA-Seq data
All animal experiments were approved by the University of Manchester Ethical Review Committee and performed in accordance with the Animals (Scientific Procedures) Act, 1986, United Kingdom. Matings were established between male and female CD1 mice, the morning of the vaginal plug being counted as E0.5. Microdissected facial processes from E10 and E11 and palatal shelves from E12, E13 and E14 embryos were collected and pooled according to their stages to obtain sufficient amount, and RNA was isolated using the Qiagen RNeasy kit. RNA-Seq libraries were generated using the SOLiDTM Total RNA-Seq Kit. Samples were run on SOLiDTM v4 for single-end 50 bp reads. Poor reads were filtered from the data with SOLiD Preprocess Filter. Reads were mapped to the mouse genome (mm9, NCBI Build 37) using Bowtie 0.12.7 (http://bowtie.cbcb.umd.edu) (Langmead et al. 2009) and assigned to RefSeq transcripts with Partek Genomics Solution (version 6.5, Copyright 2009, Partek Inc., St. Charles, MO, USA). Transcript reads were normalized and differential expression analyzed with DESeq2 (Love et al. 2014). The normalized counts from DESeq2 analysis were then converted to normalized expression value nRPK, by dividing normalized counts by the size (kilobases) of the specific isoform transcript length. A mean of 59.00 nRPK was obtained from expression of all genes and an expression level of ≥59.00 nRPK was set as the cutoff for the candidate gene prioritization (Supplementary Figure 2). The RNA-Seq data are available from ArrayExpress: E-MTAB-3157.
Analysis of genomic variability score
The DGV (Database of Genomic Variants, http://dgv.tcag.ca/) (MacDonald et al. 2014) dataset was retrieved using the UCSC Table Browser. The human genome (GRCh37/hg19) was divided into windows of fixed size, 1 Mb, using the tool windowBed (Quinlan and Hall 2010), and the numbers of CNVs (observed gains and losses) were summed to obtain the total number of variants in each region. Next, the overall number of structural variants per window was determined using the intersectBed and groupbyBed commands in combination. The windows encompassing assembly gaps (telomeres and short arm of chr13, 14, 15, 21, 22), alternative haplotype sequences (e.g., chr6_ssto_hap7) and random contigs (e.g., chr4_gl000193_random) reported in the UCSC Genome Browser archive (http://hgdownload.soe.ucsc.edu/goldenPath/hg19/database/) were removed to avoid bias. To approximate a normal-like distribution, the logarithmic conversion was applied to the variant counts of the resulting genomic windows. The overall mean (μ) of all the count logarithms was computed as well as the overall standard deviation (σ) with R statistical program, and subsequently used to calculate the z score as follows:
| 2 |
| 3 |
| 4 |
where log10(ci) represents the base-10 log of the count value of structural variants (c) in a specific window (i) according to DGV, n indicates the total number of windows generated from the whole genome (GRCh37/hg19), μ is the overall mean (average of the base-10 count logs of all windows) while σ is the overall standard deviation. Formula (4) was applied to all the elements of the window set (W). In this case, the z score was considered as a measure of genome variability.
The genomic windows were intersected with deleted and duplicated regions shared by at least two OFC patients. In case a deleted or duplicated region overlapped multiple windows, the mean of the counts was calculated and then used to determine a single z score per region. The variability of known OFC genes and potential candidate OFC genes identified in this work and the variability of OFC-AGs from the literature (Supplementary Table 2) were also evaluated by intersecting their genomic locations with the list of windows. The gene locations were retrieved from the Ensembl database (http://www.ensembl.org/index.html) setting the consistent assembly GRCh37/hg19.
Results
Patient cohort
A total of 312 unrelated patients presenting different forms of OFCs (including both syndromic and non-syndromic forms) were analyzed in this study, including 295 retrieved from DECIPHER and 17 from ECARUCA in July 2014 (Table 1). All 312 patients appear to be unique, as the genomic locations of CNVs in these patients are all different (Supplementary Table 1). The most common OFC phenotype in this cohort is CP with 197 patients affected. In addition, there are 41 CLP patients, 30 CL patients, 30 bifid uvula patients and a small number of patients exhibiting minor cleft phenotypes, such as mandible cleft, alveolar ridge cleft and facial cleft (Table 1). In case a minor cleft type was present in combination with main OFC phenotypes, the patient was ascribed to the main OFC group.
Table 1.
Phenotypes in selected OFC patients from DECIPHER and ECARUCA
| Phenotypes | Number of patients | |
|---|---|---|
| Cleft lip (CL) | 24 | CL patients in total 30 |
| CL + Alveolar ridge cleft | 3 | |
| CL + Cleft mandible | 2 | |
| CL + Cleft lower lip | 1 | |
| Cleft lip and palate (CLP) | 38 | CLP patients in total 41 |
| CLP + Bifid uvula | 1 | |
| CLP + Cleft mandible | 2 | |
| Cleft palate (CP) | 186 | CP patients in total 197 |
| CP + Bifid uvula | 8 | |
| CP + Facial cleft | 1 | |
| CP + Alveolar ridge cleft | 1 | |
| CP + Cleft lower lip | 1 | |
| Bifid uvula | 30 | |
| Oral cleft (unspecified) | 10 | |
| Alveolar ridge cleft | 2 | |
| Cleft lower lip | 1 | |
| Facial cleft | 1 | |
| Total | 312 |
Identification of overlapping CNV regions in OFC patients
To identify the genomic regions that likely contain OFC genes, we identified the genomic CNV regions that are shared by multiple patients. From the cohort of 312 OFC patients with CNVs, 249 genomic deletions and 226 genomic duplications (Supplementary Table 1) were retrieved and analyzed to determine the overlap. Altogether, 146 deletions and 109 duplications that are shared by two or more patients were identified, and these regions are referred as overlapping CNVs hereafter (Table 2). One region of 0.48 Mb located on chr2 is shared among eight OFC patients with deletions, and two regions of 1.8 Kb and 0.5 Kb both on chr22 are shared among eight duplications (Fig. 1). To assess whether the degree of OFC CNV overlap occurs by chance, we performed a randomization analysis (Quinlan and Hall 2010). For the 1000 randomizations based on the deletion list, the maximum overlap number found in the set of randomized regions was 6, and the distribution was uniformly shifted towards lower overlap numbers than that of the OFC deletion list (Fig. 2a). To verify whether this shift is statistically significant, we calculated the z scores of the overlap numbers from the deletion list and those from the randomizations. The z score of the deletion list is 13.91, markedly higher than those of the randomizations, which are all included in the range −2.55 ≤ z ≤ +4.11 (Fig. 2c). As the z scores of randomized overlaps do not follow a normal distribution (p = 1.38 × 10−6, Shapiro–Wilk normality test), we used the conservative empirical p value of p ≤ 0.001. Similarly, the distribution of randomized overlapping regions based on the duplication list also appeared shifted toward lower numbers, with a maximum overlap number of 6 and a z score range of −2.69 ≤ z ≤ +4.36 (Fig. 2b). In comparison, the z score of the mean overlap number characterizing the OFC duplication list was 11.58, higher than those of the randomizations, with a significant empirical p value of p ≤ 0.001, as the randomization overlap numbers are not normally distributed (p = 1.98 × 10−8, Shapiro–Wilk normality test).
Table 2.
Overview of identified overlapping deletions and duplications
| Num. of overlaps | Deletions | Duplications | ||
|---|---|---|---|---|
| Num. of regions | Length average (bp) | Num. of regions | Length average (bp) | |
| 1a | 198 | 2242419.58 | 198 | 2078953.58 |
| 2 | 73 | 1808210.63 | 71 | 1747913.48 |
| 3 | 37 | 974799.30 | 23 | 471465.30 |
| 4 | 20 | 1076381.95 | 6 | 663154.00 |
| 5 | 8 | 1186606.63 | 2 | 330141.50 |
| 6 | 4 | 514565.50 | 2 | 515747.00 |
| 7 | 3 | 2487712.67 | 3 | 283718.67 |
| 8 | 1 | 484236.00 | 2 | 1177.50 |
aRegions with one overlap represent CNVs that are present in only one patient and are excluded in further analyses
Fig. 1.

Genomic regions overlapped by eight genomic deletions and by eight genomic duplications in OFC patients. The coordinates of the start and the end of the CNVs are indicated inside the red/blue bars, if the length of the CNVs are longer than what are shown in the screenshot. The encompassed RefSeq genes and the chromosome ideogram are indicated under the bars. a. UCSC Genome Browser screenshot (assembly GRCh37/hg19) of the genomic region on chr2 (0.48 Mb, dashed box) overlapped by eight genomic deletions (red bars) in OFC patients. b. UCSC Genome Browser screenshot (assembly GRCh37/hg19) of the two genomic regions located on chr22 (1.8 Kb and 1.5 Kb, dashed boxes) overlapped by eight genomic duplications (blue bars) in eight OFC patients
Fig. 2.

Distribution of overlapping frequencies of CNVs from OFC patients and randomizations. a Distributions of the number of overlapping regions of 249 genomic deletions among OFC patients (solid line), in comparison to the average number (μ, dashed line) of the overlapping regions in 1000 randomizations. σ, standard deviation. μ ± 2σ, dotted lines. Y-axis: number of regions; X-axis: number of overlaps. b Distribution of the number of overlapping regions of 226 genomic duplications among OFC patients (solid line), in comparison to the average number (μ, dashed line) of overlapping regions in 1000 randomizations. σ, standard deviation. μ ± 2σ, dotted lines. Y -axis: number of regions; X-axis: number of overlaps. c The z scores of the mean of the overlap frequencies of genomic deletions (red dot, z = 13.91), and duplications (blue dot, z = 11.58) are highlighted by dashed lines. Kernel density plots on the left side show z score distributions of the means of the overlap frequencies from 1000 randomizations based on genomic deletions (red solid curve) and on genomic duplications (blue solid curve). Y-axis: density, X-axis: z score (based on the overall mean)
Candidate gene identification in overlapping CNVs among OFC patients
To identify candidate OFC genes in the CNVs, we searched for genes that are shared by multiple patients and applied a prioritization pipeline. In total, 5809 and 5941 RefSeq genes were retrieved from 249 genomic deletions and 226 genomic duplications, respectively, including protein-coding genes, pseudogenes, miRNAs and long non-coding RNAs. After the gene retrieval, several prioritization criteria were used to identify potential causative genes. Firstly, we excluded genes that are deleted or duplicated in only one patient, resulting in 1651 deleted genes and 1887 duplicated genes. Secondly, the number of genes in the overlapping regions was assessed (Supplementary Figure 1). Among these CNVs, one deleted region covers more than 84 genes and 19 deleted regions contain only one gene. For duplications, one region covers more than 200 genes and in 23 regions only one gene was duplicated. In general, the number of deletions or duplications decreases when the number of their encompassing genes increases. For deletions, there is no clear inflection point at which this number shows a sharp change. Both three and six genes are associated with rather major changes (Supplementary Figure 1a), and deletions with more than six genes fluctuate in frequency at low levels. For duplications on the other hand, there is a clear inflection point at five genes (Supplementary Figure 1b). As the contribution to causality of each gene in regions with a large number of deleted or duplicated genes is difficult to assess, we decided to take a cutoff of the number of genes in the region (≤5 genes, Supplementary Figure 1), and therefore the genes present in regions that contained five genes or fewer were selected. We chose this threshold as it corresponds to a clear inflection point for the duplications and lies between two changing points for the deletions (Supplementary Figure 1). This prioritization step gave rise to 117 deleted and 88 duplicated genes present in the common CNV regions shared by OFC patients (Supplementary Table 4). Subsequently, gene expression in mouse embryonic palatal shelves detected by an RNA-Seq analysis was used as the last prioritization step. To select genes with relatively high expression in developing mouse palates, the distribution of expressed genes (nRPK, normalized reads per kilobase, >0) detected at all analyzed stages (E10-14) was plotted to identify the mean expression level of all genes, which lies at nRPK = 59.00 (Supplementary Figure 2). Finally, the candidate genes with an expression level higher than 59.00 nRPK at any of the five stages (Supplementary Table 4) were selected as potential candidate OFC genes, resulting in 45 genes in deleted CNVs and 27 in duplicated CNVs (Supplementary Table 5).
Among genes in deleted CNVs, two of the genes, SATB2 and MEIS2, which are deleted in eight and five OFC patients (Table 3; Supplementary Tables 2, 5, 6), respectively, have been reported as causative for CP and CLP in several human and animal studies (FitzPatrick et al. 2003; Beaty et al. 2006; Britanova et al. 2006; Dobreva et al. 2006; Erdogan et al. 2007; Leoyklang et al. 2007; Crowley et al. 2010; Johansson et al. 2014; Rainger et al. 2014; Louw et al. 2015). Six additional genes (THBS1, TSHZ1, TTC28, WHSC1, WHSC2 and LETM1) encompassed by deleted CNVs have been proposed as the potentially causative genes in critical genomic regions for OFC syndromes (Table 3; Supplementary Tables 2, 5) (Wright et al. 1997, 1999; Stec et al. 1998; Zollino et al. 2000, 2003; Schlickum et al. 2004; Nishiwaki et al. 2006; Coré et al. 2007; Maas et al. 2008; Dostal et al. 2009; Heinonen and Maki 2009; Davidson et al. 2012; Shimizu et al. 2014; Liu et al. 2015). Furthermore, three candidates in deleted CNVs, FGF2, FRZB and SPRY1, have been shown to contribute to orofacial development in animal models and to be involved with signaling pathways whose disruption leads to OFCs in human (Table 3, Supplementary Tables 2, 5) (Hoang et al. 1996; Lin et al. 1997; Hoang et al. 1998; Mansukhani et al. 2000; Moore et al. 2002; Ignelzi et al. 2003; Sasaki et al. 2006; Szabo-Roger et al. 2008; Dickinson et al. 2009; Porntaveetu et al. 2010; Yang et al. 2010; Kamel et al. 2013). We further identified 34 novel candidates which have not previously been associated with OFCs (Supplementary Table 5). Among 27 genes in duplicated CNVs, three of them, DGCR6, MAPK3 and TULP4 (Table 3; Supplementary Tables 2, 5), have been previously associated with orofacial development or proposed as causative for OFC syndromes (Demczuk et al. 1996; Lindsay and Baldini 1997; Yamamoto et al. 2003; Singh et al. 2007; Nakamura et al. 2009; Das Chakraborty et al. 2012; Vieira et al. 2015), while the remaining 24 genes are novel candidates (Supplementary Table 5).
Table 3.
Genes that are identified in overlapping CNVs and have been associated with OFCs and orofacial development
| Gene | Overlapping CNVs | Syndromic and non-syndromic OFCs | References |
|---|---|---|---|
| SATB2 | 8 Deletions | Glass Syndrome (OMIM#612313). Isolated CP | FitzPatrick et al. (2003), Beaty et al. (2006), Britanova et al. (2006), Dobreva et al. (2006), Leoyklang et al. (2007), Rainger et al. (2014) |
| MEIS2 | 5 Deletions | CP associated with cardiac defects and with 15q14 deletion syndrome | Erdogan et al. (2007), Crowley et al. (2010), Johansson et al. (2014), Louw et al. (2015) |
| FGF2 | 3 Deletions | Involved in craniofacial osteogenesis and suture homeostasis. Ligand of FGFR1 and FGFR2 (known OFC causative genes) | Britto et al. (2002), Sasaki et al. (2006), Szabo-Rogers et al. (2008), Nikopensius et al. (2010), Porntaveetus et al. (2010), Wang et al. (2013) |
| FRZB | 2 Deletions | Involved in primary ossification of craniofacial regions via WNT pathway (mouse, zebrafish, xenopus) | Hoang et al. (1996, 1998), Lin et al. (1997). Dickinson and Sive (2009), Kamel et al. (2013) |
|
LETM1
WHSC1 WHSC2 (NELFA) |
3 Deletions | Candidates for Wolf–Hirschhorn syndrome [OMIM #194190] | Wright et al. (1997, 1999), Stec et al. (1998), Zollino et al. (2000, 2003), Schlickum et al. (2004), Maas et al. (2008), Shimizu et al. (2014), Liu et al. (2015) |
| 3 Deletions | |||
| 3 Deletions | |||
| SPRY1 | 3 Deletions | CP in transgenic mice. Primary paralog of SPRY2 [Ensembl] | Yang et al. (2010) |
| THBS1 | 2 Deletions | Craniofacial defects in KO mice. Candidate for 18q deletion syndrome [OMIM#601808] | Nishiwaki et al. (2006), Heinonen and Maki (2009) |
| TSHZ1 | 6 Deletions | Role in Peters-plus syndrome [OMIM#261540] | Coré et al. (2007), Dostal et al. (2009) |
| TTC28 | 3 Deletions | Likely gene responsible for Pierre-Robin sequence (including CP) in a case report | Davidson et al. (2012) |
| DGCR6 | 5 Duplications | Candidate for DiGeorge syndrome [OMIM*601279] | Demczuk et al. (1996), Lindsay and Baldini (1997), Das Chakraborty et al. (2012) |
| MAPK3 (ERK1) | 2 Duplications | MAPK pathway involved in craniofacial development. Interactor of ERK2, whose disruption leads to OFCs in animal models | Yamamoto et al. (2003), Singh et al. (2007), Nakamura et al. (2009), O’Brien et al. (2015) |
| TULP4 | 2 Duplications | Statistically significant association in a population-based study | Vieira et al. (2015) |
To further test whether the genes identified within CNVs have an enrichment of known OFC genes, we selected a panel of 126 genes, which we termed OFC-associated genes (OFC-AGs), based on the following criteria: (i) genes harboring mutations causing syndromic and non-syndromic OFCs, or (ii) genes located near or within OFC GWAS loci and CNVs, or (iii) genes expressed in lip or palate primordia during development in animal models (Supplementary Table 2). In this panel, 50 genes are shown to be associated with non-syndromic OFCs, 58 are involved in the pathogenesis of syndromes whose features include OFCs, and 18 genes are implicated in both cases. Eleven OFC-AGs are present among the 45 deleted candidates and three among 27 duplicated candidates. This represent a statistically significant 29-fold enrichment (hypergeometric test, p = 7.2 × 10−16) of OFC-AGs in deleted CNV genes and a 22-fold enrichment (hypergeometric test, p = 8.6 × 10−6) in duplicated CNV genes (Table 4). Therefore, our data show that the prioritized gene list identified from overlapping CNVs contains a significant number of known OFC-AGs (Table 3).
Table 4.
Enrichment of OFC-associated genes (OFC-AGs) in candidate OFC genes identified by CNV analysis
| Deletions | Duplications | |||
|---|---|---|---|---|
| No. of total genes | No. of OFC-AGsa | No. of total genes | No. of OFC-AGs | |
| All genes without prioritization | 5809 | 49 | 5941 | 30 |
| Prioritized genes | 45 | 11 | 27 | 3 |
| Fold enrichment | 28.98 | 22.00 | ||
| P value (Hypergeometric test) | 7.2 × 10−16 | 8.6 × 10−6 | ||
aOFC-AGs: OFC-associated genes. Genes that have been associated with OFC or orofacial development based on extensive literature search (Supplementary Table 2)
One interesting question is whether the identified candidate genes are located in highly polymorphic genomic regions, named hypervariable regions (HVRs), in individuals without OFCs. To address this question, we first examined the variability in the whole human genome based on 2,135,523 structural variants from healthy individuals reported in the DGV database (January 2015) (MacDonald et al. 2014) by partitioning the genome to fixed windows and calculating the z score of the CNV counts within each window. A total of 2876 1 Mb windows with DGV variant counts were generated and the z scores in the healthy population resulted in a range from −5.80 to +3.65 (not normally distributed, Shapiro–Wilk normality test, p < 2.2 × 10−16) (Fig. 3a). In addition, we assessed the variability of the 126 OFC-AGs from the literature (Supplementary Table 2), and found that they are located within the variability range of the normal population, between −2.84 and +2.30. Next, the z scores of the genomic windows encompassing CNVs shared by OFC patients were calculated. The variability distributions of windows encompassing deletions and duplications are in both cases slightly shifted towards increased variability. Nevertheless, they are located within the variability range of the healthy population, from −2.92 to +3.02 for deleted CNVs and from −2.30 to +3.05 for duplicated CNVs but not in HVRs (Fig. 3a). These data show that the OFC CNVs are not located in HVRs in healthy individuals. Next, we focused on the windows containing the identified candidates, 45 deleted genes and 27 duplicated genes, to evaluate the location of those windows in the z score distribution. For two deleted (USP14 and ZMYND11) and four duplicated (RIC8A, PSMD13, SIRT3 and YES1) novel candidates, the variability score was not assessable due to the exclusion of the windows encompassing telomeric sequences. Importantly, the z scores of the other potential OFC candidates lie within the variability range of the normal population, varying between −2.05 (FAT4) and +2.21 (FRG1) for deleted CNV genes, and between −0.67 (FIRRE) to +1.72 (DGRC6) (Fig. 3) for duplicated CNV genes, similar to those of the range of the OFC-AGs.
Fig. 3.
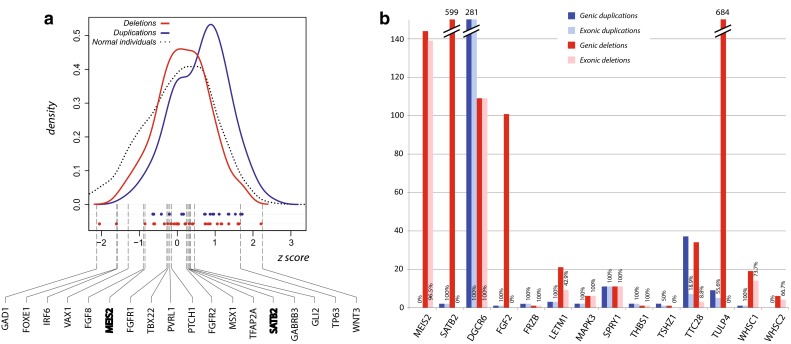
Genomic variability of overlapping deletions and duplications shared by OFC patients and identified candidate genes. a Kernel density plot shows variability z scores of genomic regions with 1 MB windows in healthy individuals reported in DGV (−5.80 ≤ z ≤ + 3.65, dotted curve) and of the windows encompassing overlapping deletions (−2.92 ≤ z ≤ + 3.02, red solid curve) or duplications (−2.30 ≤ z ≤ + 3.05, blue solid curve) shared by OFC patients. The dots on the X-axis represent the variability z scores of genomic windows encompassing deleted (red dots) and duplicated (blue dots) candidate genes in CNVs (Supplementary Table 5). The variability z scores of windows encompassing 18 OFC-associated genes (OFC-AGs) from the literature are indicated by dashed lines. Among these, two genes (MEIS2, SATB2) highlighted in bold are identified from the overlapping deletions in this study (Table 3; Supplementary Table 2). Y-axis: density; X-axis: z score (variability). b The histogram of duplications and deletions at the genomic regions of the 14 known and potential OFC genes identified in healthy individuals, based on DGV dataset (Database of Genomic Variants, http://dgv.tcag.ca/). Duplications in the genic regions of the genes are indicated in dark blue, and those affecting exonic regions are indicated in light blue. Deletions in genic regions of the genes are indicated in dark red, and those affecting exonic regions are indicated in light red. Y-axis number of CNVs, X-axis genes
Finally, we assessed the total number of genomic and exonic CNVs in healthy individuals affecting the two known cleft genes in our analysis, SATB2 and MEIS2, and the 12 proposed OFC genes (Fig. 3b). SATB2 and MEIS2 appear to be partially deleted in a large number of DGV individuals, 149 and 599, respectively (Fig. 3b; Supplementary Figure 3). Interestingly, all the deletions within SATB2 in healthy individuals are not located in the exonic regions but map to intronic regions (Fig. 3b; Supplementary Figure 3b), whereas deletions within MEIS2 disrupt the promoter regions (Fig. 3b; Supplementary Figure 3a). Except for DGCR6 that is affected by a large number of exonic deletions and duplications, the other eleven genes are not frequently affected by CNVs in healthy population (Fig. 3b). These data suggest that most of the identified candidate genes in OFC CNV patients are not often disrupted in healthy individuals, and therefore the deletion or duplication of these genes likely contributes to the etiology.
Taken together, our data suggest that our systematic large-scale CNV analysis of OFC patients and prioritization of genes in these CNV regions can identify potential candidate OFC genes.
Discussion
Craniofacial abnormalities including OFCs are among the most significant phenotypes where large CNVs are involved in the etiology (Cooper et al. 2011). However, due to the limited number of OFC patients with CNVs in individual centers, analyses of CNVs from a large number of OFC patients are difficult to perform. In the present study, we analyzed a set of CNVs from a large cohort of OFC patients collected from publicly available CNV databases to identify common genomic deleted or duplicated regions and potential causative genes. Specifically, 312 OFC patients were collected from DECIPHER and ECARUCA databases with a total of 249 genomic deletions and 226 duplications. Overlapping genomic regions shared by patients were analyzed, and RefSeq genes in these regions were prioritized to identify candidate causative genes for OFCs, resulting in 45 potential candidate genes for deletions and 27 for duplications. Furthermore, statistical analyses showed that the overlapping CNV regions in OFC patients are not randomly located, and that the identified candidate genes do not lie within hypervariable genomic regions of the human genome.
Several considerations need to be taken into account with regards to our analysis pipeline for the identification of candidate OFC genes. Firstly, we collected unique OFC patients from DECIPHER and ECARUCA databases, to ensure a single registration of patients and to avoid overrepresentation due to repeated data. Unique registration is required for the submission of CNV patients to the ECARUCA database (personal communication) (de Leeuw et al. 2012). The uniqueness of the collected patients is further confirmed by their different genomic locations (Supplementary Table 1). We note that not all CNVs in these databases may be validated. Secondly, different types of OFC patients including CP, CL and CLP were collected to have a large cohort of patients for statistical analyses, and the analysis pipeline is likely to identify the common mechanisms involved in different OFCs. It should be noted that distinct mechanisms have been proposed for CP, CL and CLP, which likely cannot be distinguished using this approach. Thirdly, we used a cutoff of fewer than five genes in CNV regions to prioritize candidate genes, because it is difficult to evaluate the contribution of each gene when a large number of genes are deleted or duplicated. Given that there are not many CNVs containing more than 5 genes, as compared to smaller CNVs (with ≤5 genes) (Supplementary Figure 1), it is not likely that we miss many OFC relevant genes using this criteria. However, this approach cannot identify genes that have a minor contribution individually but act synergistically when located in the same CNV regions. It is known that one of the intrinsic problems of CNV studies is to identify the dosage-sensitive genes that underlie the phenotypes because CNVs usually encompass many genes (Cooper et al. 2011). Fourthly, one of the strengths in our analysis pipeline is that our study focuses on genes located in overlapping CNVs shared by several patients whose common phenotype is OFC. This strategy allows to identify common genetic mechanisms of OFCs, rather than those of individual patients. Finally, we used gene expression levels in mouse embryonic palatal shelves (Supplementary Figure 2) to prioritize OFC candidate genes. This is based on the hypothesis that genes expressed in embryonic palate are likely involved in the pathogenesis of OFCs. However, some known OFC genes such as TBX1 that do not have high expression levels in mouse embryonic palates were not selected as the candidate genes using our systematic approach (Supplementary Table 4). TBX1 is located in the 22q11.2 region that is highly susceptible to rearrangements including microdeletions and microduplications. Patients with 22q11.2 microdeletion syndrome, also known as DiGeorge syndrome [OMIM #188400] or Velocardiofacial syndrome [OMIM #192430], and with 22q11.2 microduplication syndrome often have orofacial abnormalities together with many other phenotypes (Yagi et al. 2003; Torres-Juan et al. 2007; Wentzel et al. 2008). TBX1 plays a role in early progenitor cells relevant for palate development and is generally considered to be the causative gene in this region (Torres-Juan et al. 2007; Funato et al., 2012; Herman et al. 2012). In our analysis, TBX1 is present in our duplicated gene list before the prioritization using RNA-seq data (Supplementary Table 4) but is filtered out by this step. This shows that our selection criteria are rather stringent, increasing the probability that identified candidate genes are involved in OFCs. However, OFC genes that are not expressed in embryonic palates might be missed.
Our analysis pipeline identified two known OFC genes, MEIS2 that is deleted in five CP patients in our analysis and SATB2 that is deleted in eight CP patients (Table 3; Supplementary Tables 3, 5, 6). Many studies have confirmed the contribution of SATB2 to CP both in non-syndromic and syndromic forms, causing Glass syndrome (OMIM #612313), Pierre-Robin sequence with or without ankyloglossia and cleft-associated intellectual disability (Supplementary Table 2) (FitzPatrick et al. 2003; Beaty et al. 2006; Britanova et al. 2006; Dobreva et al. 2006; Leoyklang et al. 2007; Rainger et al. 2014). MEIS2 is one of the recently identified OFC genes, which has been proposed as the main contributor to the pathogenesis of chromosome 15q14 deletion syndrome (Supplementary Table 2) (Erdogan et al. 2007; Crowley et al. 2010; Johansson et al. 2014; Louw et al. 2015). Intriguingly, although these two genes have been classified as OFC causative genes, they appear to be frequently affected by small deletions in healthy individuals (144 deletions in MEIS2 and 599 deletions in SATB2) (Fig. 3b; Supplementary Figure 3). The deletions affecting SATB2 are all located in intronic regions, suggesting they are not pathogenic in those individuals (Supplementary Figure 3b). In contrast, the majority of the deletions at MEIS2 (139 out of 144) affect not only introns but also exons and promoters (Supplementary Figure 3a). This raises the question about how ‘healthy’ the normal individuals in the DVG database are. It is known that some of the OFC phenotypes are not directly evident (e.g., submucous CP) and hence difficult to diagnose (Souza et al. 2013). In addition, genetic defects of OFCs are not 100 % penetrant. Therefore, even if small CNVs are found in the gene body regions of potential candidate genes (Table 3; Fig. 3b; Supplementary Figure 3) in ‘healthy’ individual, these genes may still be relevant to OFCs.
As OFCs are often not fully penetrant, we briefly assessed the OFC penetrance in CNVs containing the OFC candidate genes. Indeed, in DECIPHER and ECARUCA databases, many individuals with deleted or duplicated OFC candidate genes show other disease phenotypes but lack an OFC (non-OFC individuals) (Supplementary Table 6). For several deleted genes including ACBD3, FAM98B, H3F3A, RNPS1, SPRED1, and THBS1, the number of non-OFC individuals with deletions of these genes is lower than that of the OFC patients, suggesting that the deletion of these genes might be more penetrant. Furthermore, patients with deletions and duplications of the OFC candidate genes exhibit heterogeneous phenotypes with many other disease features such as defects in the cardiovascular, nervous and skeletal systems (Supplementary Tables 3, 6). A systematic analysis of these additional features may provide insights into novel OFC syndromes.
In addition to the two known OFC causative genes SATB2 and MEIS2, we identified other 12 genes, nine in deletions (FGF2, FRZB,LETM1, SPRY1, THBS1, TSHZ1, TTC28, WHSC1, WHSC2) and three in duplications (DGCR6, TULP4 and MAPK3), that have been previously proposed as orofacial development regulators or as potential causative genes for OFCs (Table 3; Supplementary Table 5). WHSC1, WHSC2 (aka NELFA) and LETM1 have been proposed to be primarily involved in Wolf–Hirschhorn syndrome (OMIM #194190), whose features include OFC in almost half of the cases (Wright et al. 1997, 1999; Stec et al. 1998; Zollino et al. 2000, 2003; Schlickum et al. 2004; Maas et al. 2008; Shimizu et al. 2014; Liu et al. 2015). Among the OFC patients we collected, all three genes were deleted simultaneously in the same patients, two affected by CP and one by CL (Supplementary Tables 3, 6), consistent with the hypothesis that the deletion of all these three genes is required to cause more severe craniofacial features including OFCs. In a case report describing a patient with a 22q12.2 microdeletion, TTC28 has been suggested as the likely gene responsible for Pierre-Robin sequence including CP (Davidson et al. 2012). Interestingly, our data show the deletion of TTC28 in three CP patients (Supplementary Tables 3, 6), which is in agreement with this hypothesis. In human, TSHZ1 is considered as one of the candidate causative genes for chromosome 18q deletion syndrome (OMIM#601808), which present OFC in 25 % of the cases (Dostal et al. 2009). In addition, knock-out of this gene in mice gives rise to abnormal skeletal morphogenesis and craniofacial defects (Coré et al. 2007). In our cohort, this gene is deleted in two CLP and four CP patients (Supplementary Tables 3, 6). THBS1 which has been shown to play a role in the etiology of Peters-plus syndrome (OMIM #261540) with OFC as one of the clinical features is deleted in two CP patients (Supplementary Tables 3, 6) (Nishiwaki et al. 2006; Heinonen et al. 2009). Other two genes, FRZB and SPRY1, found deleted in two and three patients (Supplementary Tables 3, 6), respectively, have been studied in animal models. Specifically, FRZB is locally expressed in primary mouth tissues and involved in primary ossification of craniofacial regions by interacting with the WNT pathway (Hoang et al. 1996, 1998; Lin et al. 1997; Dickinson et al. 2009; Kamel et al. 2013). SPRY1, characterized by structural and functional similarity with the proposed OFC gene SPRY2, has been shown to cause cardiac defects as well as facial cleft and CP in transgenic mice (Supplementary Table 2) (Yang et al. 2010). The Sprouty family proteins inhibit the FGF pathway where several causative OFC genes such as FGFR1 and FGFR2 are involved (Supplementary Table 2). Interestingly, one of the main ligands of FGFR1/2, FGF2, was detected as one of the top deleted candidate genes in our analysis (Supplementary Tables 2, 5). Indeed, the role of FGF signaling and FGF2 in craniofacial development, specifically in osteogenesis and cranial suture homeostasis, has been demonstrated by a number of studies (Mansukhani et al. 2000; Britto et al. 2002; Moore et al. 2002; Ignelzi et al. 2003; Sasaki et al., 2006; Szabo-Rogers et al. 2008; Li et al. 2010; Porntaveetus et al. 2010). In addition, a statistically significant association between FGF2 markers and OFCs has also been reported (Wang et al. 2013; Nikopensius et al. 2010).
Three genes involved in duplications, DGRC6, TULP4 and MAPK3, have also been previously proposed to associate with OFC or orofacial development (Supplementary Table 2). DGCR6, duplicated in five patients affected by CL, CLP and CP (Supplementary Tables 3, 6), has been proposed in literature to be involved in the developmental defects associated with 22q11.2 deletions syndrome (aka DiGeorge syndrome) (OMIM*601279) (Demczuk et al. 1996; Das Chakraborty et al. 2012). In mouse, it is highly expressed during embryogenesis, probably contributing to neural crest cell migration (Lindsay et al. 1997). A statistically significant association of TULP4 with OFC has been reported in a recent study based on 6q23.1 fine mapping in a cohort of five hundred OFC patients (Vieira et al. 2015). In our analysis, this gene has been found duplicated in two patients (Supplementary Tables 3, 6). The MAPK/ERK pathway has been shown to be involved in craniofacial development and related diseases (Yamamoto et al. 2003; Singh et al. 2007; Newbern et al. 2008; Nakamura et al. 2009; Parada et al. 2015). Although orofacial defects have been observed in the Mapk1 (aka Erk2) knockout but not in the Mapk3 (aka Erk1) knockout mice (Newbern et al. 2008; Parada et al. 2015), the cooperation of these two genes has been shown in many cell types and tissues (Srivastava et al. 2011; O’Brien et al. 2015), suggesting a modulating role of MAPK3 in craniofacial development. In our analysis, MAPK3 duplication is present in two patients, one affected by CP and the other by CL (Supplementary Tables 3, 6).
In addition to above potential OFC genes, we identified 34 deleted and 24 duplicated genes that have not yet been associated with OFC. These novel candidates cover a broad range of protein types with various functions, such as transcription factors, metabolic enzymes, kinases and phosphatases, structural proteins, signaling mediators, membrane proteins including receptors, and several uncharacterized proteins (Supplementary Table 5). Although each of them has not yet been linked to orofacial development directly, we found that many of these genes are involved in several signaling pathways important for the process, such as RAC1, BMP and MAPK. Some of these genes are involved in multiple pathways, suggesting the combinatory role of these pathways in orofacial development. Specifically, several identified genes are related to the RAC1 pathway. Recent mouse studies suggest the causative role of RAC1 signaling in CP etiology through fibronectin regulation and cytoskeletal reorganization during neural crest cell development and palatal shelf elevation (Thomas et al. 2010; Tang et al. 2015). RAC1 is a ubiquitously expressed Rho small GTPase, inducible by a number of cell-surface receptors and transmembrane adhesion molecules to stimulate different cellular responses, mainly based on actin cytoskeleton remodeling, ROS production and gene expression regulation (Didsbury et al. 1989; Polakis et al. 1989; Ando et al. 1992; Ménard et al. 1992; Ridley et al. 1992; Heyworth et al. 1993; Minden et al. 1995; Westwick et al. 1997; Ridley 2000; Schmitz et al. 2001). Within this pathway, some of our candidate genes are involved in regulating RAC1 activity, such as IGF1R, FGF2, FARP2, THBS1, YES1 and SMURF1 (Adams 1995; Adams and Schwartz 2000; Kubo et al. 2002; Pennisi et al. 2002; Jackson et al. 2003; Madura et al. 2003; Wang et al. 2003, 2006; Fera et al. 2004; Shin et al. 2004; Asanuma et al. 2006; Giehl et al. 2008; Kanazawa et al. 2010; Takahashi et al. 2010; Takahashi and Suzuki 2010; Takegahara et al. 2010; Ding et al. 2011; Lee and Kay 2011; He et al. 2013; Deng and Huang 2014; Chatterji et al. 2015). Some of the candidate genes are regulated by RAC1, such as NCKAP1 and CYFIP1 (Miki et al. 1998; Schenck et al. 2001, 2003; Eden et al. 2002; Billuart and Chelly 2003; Kurisu et al. 2005; Anitei et al. 2010; De Rubeis et al. 2013) and some others are paralogs of RAC1 interactors such as PARD3B and STK38, primary paralogs of PARD3 and CDC42, respectively. In addition, other candidates belong to protein families whose members are known to interact with RAC1 pathway, such as SEPT2 and SPRY1 (Gross et al. 2001; Yigzaw et al. 2001; Lim et al. 2002; Lee et al. 2004; Poppleton et al. 2004; Nagata and Inagaki 2005; Lito et al. 2009; Ballou et al. 2013; Ireton et al. 2014; Assinder et al. 2015). Related to this pathway, FAT4 encodes for a cell–cell interaction molecule, a member of the protocadherin family, which has been though to regulate planar cell polarity (Fukata et al. 1999; Kuroda et al. 1999; Evers et al. 2000; Frebourg et al. 2006; Suo et al. 2012; Keeler et al. 2015). FAT4 is described in OMIM as causative of two non-OFC syndromes, Van Maldergerm syndrome type 2 (OMIM #615546) and Hennekam lymphangiectasia-lymphedema syndrome type 2 (OMIM # 616006). Nevertheless, a 4q deletion syndrome has been characterized in 20 patients, among them four affected by CP and two by CL or CLP (Strehle et al. 2012). The deletions of these six OFC patients encompass nine of our deleted candidate genes including FAT4 but also ANKRD50, DCTD, FGF2, FRG1, NEIL3, SPATA5, SPRY1, WWC2, and one duplicated candidate gene, FAM149A (Strehle et al. 2012).
Other candidates are involved in BMP signaling include SMURF1 and SPRED1. SMURF1 is a ubiquitin-protein ligase specific for SMAD proteins in the BMP pathway. It interacts as a negative regulator of BMP signaling pathway and regulates cell motility, signaling and polarity. This interaction with the BMP signaling may be the key point to explain a possible association between SMURF1 and OFC etiology, as two members of this pathway, BMP2 and BMP4, are already known to be associated with OFCs (Supplementary Table 2) (Zhang et al. 2002; Liu et al. 2005; Marazita 2007; Lin et al. 2008; Suzuki et al. 2009; Suazo et al. 2010; Sahoo et al. 2011; Williams et al. 2012). SPRED1, recognized as the causative gene for Legius syndrome (OMIM #611431), interacts with SPRY2 that has been described as a causative OFC gene (Supplementary Table 2) (Vieira et al. 2005; Goodnough et al. 2007; Welsh et al. 2007; Spurlock et al. 2009; Matsumura et al. 2011; Song et al. 2015). In addition, SPRED1 and SPRY2 act as negative regulators of the FGF and MAPK pathways (Katoh and Katoh 2006; Di Bari et al. 2009; Sylvestersen et al. 2011; Zhao et al. 2015), both shown to affect orofacial development in human or in mouse models (Reardorn et al. 1994; Wilkie et al. 1995; Sasaki et al. 2001; Dodé et al. 2003; Yamamoto et al. 2003; Riley and Murray 2007; Singh et al. 2007; Newbern et al. 2008; Nakamura et al. 2009; García-Domínguez et al. 2011).
Same as for WHSC1, WHSC2 and LETM1, some of our novel candidates lie in the same duplicated or deleted regions (Supplementary Table 5). The co-localization of these genes in the same CNVs together with their high expression levels in mouse embryonic palate support the hypothesis of a combinatory function. For instance, NIPA1, a duplicated novel candidate, whose deletion has been recently confirmed to be pathogenic (Cooper et al. 2011), maps to a duplicated region that contains two other functionally related candidates, CYFIP1 and TUBGCP5.
In conclusion, this study developed a systematic analysis pipeline for the CNV analysis of a large cohort of OFC patients, and for the identification of potential OFC-related genes. As the result, we identified 45 genes in large genomic deletions and 27 in duplications, including several known causative genes for OFC, such as SATB2 and MEIS2. Our study enriches the reservoir of potential causative OFC genes for genetic studies and provides a disease link to many of these genes that are known to be involved in several signaling pathways. Future human mutation analyses and animal model studies are necessary to confirm the role of the identified potential causative OFC genes in OFC-related diseases and in orofacial development.
Electronic supplementary material
Acknowledgments
The author acknowledges the support received from the European Science Foundation within the EUROCleftNet Programme (FC, ESF exchange visit grants, n. 4712, 4918). This work was supported by NWO/ALW/MEERVOUD/836.12.010 (HZ), NGI/BioRange program of the NBIC (MO), Radboud University fellowship (HZ), the Healing Foundation (JD) and Wellcome Trust grant 097820 (JD). This study makes use of data generated by the DECIPHER Consortium. A full list of centers who contributed to the generation of the data is available from http://decipher.sanger.ac.uk and via email from decipher@sanger.ac.uk. Funding for the DECIPHER project was provided by the Wellcome Trust. DECIPHER Consortium declares that those who carried out the original analysis and collection of the data bear no responsibility for the further analysis or interpretation of it. Furthermore, we would like to thank the ECARUCA Scientific Advisory Board members, as well as N. de Leeuw and R. Dirks from the Department of Human Genetics, Radboud University Medical Centre, Nijmegen, for their support and suggestions.
Compliance with ethical standards
Conflict of interest
The authors have declared that no conflict of interest exists.
Contributor Information
Michele Rubini, Phone: +39 0532 974473, Email: michele.rubini@unife.it.
Huiqing Zhou, Phone: +31 24 3616850, Email: j.zhou@science.ru.nl, Email: jo.zhou@radboudumc.nl.
References
- Abel EL. Fetal alcohol syndrome: the ‘American Paradox’ alcohol. Alcohol. 1998;33:195–201. doi: 10.1093/oxfordjournals.alcalc.a008382. [DOI] [PubMed] [Google Scholar]
- Abrishamchian AR, Khoury MJ, Calle EE. The contribution of maternal epilepsy and its treatment to the etiology of oral clefts: a population based case-control study. Genet Epidemiol. 1994;11:343–351. doi: 10.1002/gepi.1370110404. [DOI] [PubMed] [Google Scholar]
- Adams JC. Formation of stable microspikes containing actin and the 55 kDa actin bundling protein, fascin, is a consequence of cell adhesion to thrombospondin-1: implications for the anti-adhesive activities of thrombospondin-1. J Cell Sci. 1995;108(Pt 5):1977–1990. doi: 10.1242/jcs.108.5.1977. [DOI] [PubMed] [Google Scholar]
- Adams JC, Schwartz MA. Stimulation of fascin spikes by thrombospondin-1 is mediated by the GTPases Rac and Cdc42. J Cell Biol. 2000;150:807–822. doi: 10.1083/jcb.150.4.807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando S, et al. Post-translational processing of rac p21 s is important both for their interaction with the GDP/GTP exchange proteins and for their activation of NADPH oxidase. J Biol Chem. 1992;267:25709–25713. [PubMed] [Google Scholar]
- Anitei M, et al. Protein complexes containing CYFIP/Sra/PIR121 coordinate Arf1 and Rac1 signalling during clathrin-AP-1-coated carrier biogenesis at the TGN. Nat Cell Biol. 2010;12:330–340. doi: 10.1038/ncb2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asanuma K, Yanagida-Asanuma E, Faul C, Tomino Y, Kim K, Mundel P. Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat Cell Biol. 2006;8:485–491. doi: 10.1038/ncb1400. [DOI] [PubMed] [Google Scholar]
- Assinder SJ, Beniamen D, Lovicu FJ. Cosuppression of sprouty and sprouty-related negative regulators of FGF signalling in prostate cancer: a working hypothesis. Biomed Res Int. 2015;2015:827462. doi: 10.1155/2015/827462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballou ER, Kozubowski L, Nichols CB, Alspaugh JA. Ras1 acts through duplicated Cdc42 and Rac proteins to regulate morphogenesis and pathogenesis in the human fungal pathogen Cryptococcus neoformans. PLoS Genet. 2013;9:e1003687. doi: 10.1371/journal.pgen.1003687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber JC, et al. 8p23.1 duplication syndrome; common, confirmed, and novel features in six further patients. Am J Med Genet A. 2013;161A(3):487–500. doi: 10.1002/ajmg.a.35767. [DOI] [PubMed] [Google Scholar]
- Beaty TH, et al. Analysis of candidate genes on chromosome 2 in oral cleft case-parent trios from three populations. Hum Genet. 2006;120:501–518. doi: 10.1007/s00439-006-0235-9. [DOI] [PubMed] [Google Scholar]
- Beaty TH, et al. A genome-wide association study of cleft lip with and without cleft palate identifies risk variants near MAFB and ABCA4. Nat Genet. 2010;42:525–529. doi: 10.1038/ng.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billuart P, Chelly J. From fragile X mental retardation protein to Rac1 GTPase: new insights from Fly CYFIP. Neuron. 2003;38:843–845. doi: 10.1016/S0896-6273(03)00362-3. [DOI] [PubMed] [Google Scholar]
- Birnbaum S, et al. Key susceptibility locus for nonsyndromic cleft lip with or without cleft palate on chromosome 8q24. Nat Genet. 2009;41:473–477. doi: 10.1038/ng.333. [DOI] [PubMed] [Google Scholar]
- Bragin E, Chatzimichali EA, Wright CF, Hurles ME, Firth HV, Bevan AP, Swaminathan GJ. DECIPHER: database for the interpretation of phenotype-linked plausibly pathogenic sequence and copy-number variation. Nucleic Acids Res. 2014;42:D993–D1000. doi: 10.1093/nar/gkt937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britanova O, Depew MJ, Schwark M, Thomas BL, Miletich I, Sharpe P, Tarabykin V. Satb2 haploinsufficiency phenocopies 2q32-q33 deletions, whereas loss suggests a fundamental role in the coordination of jaw development. Am J Hum Genet. 2006;79:668–678. doi: 10.1086/508214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britto JA, Evans RD, Hayward RD, Jones BM. Toward pathogenesis of Apert cleft palate: FGF, FGFR, and TGF beta genes are differentially expressed in sequential stages of human palatal shelf fusion . Cleft palate-craniofac J Off Publ Am Cleft Palate-Craniofac Assoc. 2002;39:332–340. doi: 10.1597/1545-1569(2002)039<0332:TPOACP>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Canfield MA, et al. Changes in the birth prevalence of selected birth defects after grain fortification with folic acid in the United States: findings from a multi-state population-based study. Birth Defects Res A Clin Mol Teratol. 2005;73:679–689. doi: 10.1002/bdra.20210. [DOI] [PubMed] [Google Scholar]
- Celli J, et al. Heterozygous germline mutations in the p53 homolog p63 are the cause of EEC syndrome. Cell. 1999;99:143–153. doi: 10.1016/S0092-8674(00)81646-3. [DOI] [PubMed] [Google Scholar]
- Chatterji T, et al. Yes-mediated phosphorylation of focal adhesion kinase at tyrosine 861 increases metastatic potential of prostate cancer cells. Oncotarget. 2015;6:10175–10194. doi: 10.18632/oncotarget.3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobourne MT. The complex genetics of cleft lip and palate. Eur J Orthod. 2004;26:7–16. doi: 10.1093/ejo/26.1.7. [DOI] [PubMed] [Google Scholar]
- Cooper GM, et al. A copy number variation morbidity map of developmental delay. Nat Genet. 2011;43:838–846. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core N, Caubit X, Metchat A, Boned A, Djabali M, Fasano L. Tshz1 is required for axial skeleton, soft palate and middle ear development in mice. Dev Biol. 2007;308:407–420. doi: 10.1016/j.ydbio.2007.05.038. [DOI] [PubMed] [Google Scholar]
- Crowley MA, Conlin LK, Zackai EH, Deardorff MA, Thiel BD, Spinner NB. Further evidence for the possible role of MEIS2 in the development of cleft palate and cardiac septum. Am J Med Genet A. 2010;152A(5):1326–1327. doi: 10.1002/ajmg.a.33375. [DOI] [PubMed] [Google Scholar]
- Das Chakraborty R, et al. Dysregulation of DGCR6 and DGCR6L: psychopathological outcomes in chromosome 22q11.2 deletion syndrome. Transl Psychiatry. 2012;2:e105. doi: 10.1038/tp.2012.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson TB, et al. Microdeletion del(22)(q12.2) encompassing the facial development-associated gene, MN1 (meningioma 1) in a child with Pierre-Robin sequence (including cleft palate) and neurofibromatosis 2 (NF2): a case report and review of the literature. BMC Med Genet. 2012;13:19. doi: 10.1186/1471-2350-13-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leeuw N, et al. Diagnostic interpretation of array data using public databases and internet sources. Hum Mutat. 2012;33:930–940. doi: 10.1002/humu.22049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rubeis S, et al. CYFIP1 coordinates mRNA translation and cytoskeleton remodeling to ensure proper dendritic spine formation. Neuron. 2013;79:1169–1182. doi: 10.1016/j.neuron.2013.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demczuk S, Thomas G, Aurias A. Isolation of a novel gene from the DiGeorge syndrome critical region with homology to Drosophila gdl and to human LAMC1 genes. Hum Mol Genet. 1996;5:633–638. doi: 10.1093/hmg/5.5.633. [DOI] [PubMed] [Google Scholar]
- Deng S, Huang C. E3 ubiquitin ligases in regulating stress fiber, lamellipodium, and focal adhesion dynamics. Cell Adh Migr. 2014;8:49–54. doi: 10.4161/cam.27480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derijcke A, Eerens A, Carels C. The incidence of oral clefts: a review. Br J Oral Maxillofac Surg. 1996;34:488–494. doi: 10.1016/S0266-4356(96)90242-9. [DOI] [PubMed] [Google Scholar]
- di Bari MG, et al. TGF-beta modulates the functionality of tumor-infiltrating CD8+ T cells through effects on TCR signaling and Spred1 expression. Cancer Immunol Immunother. 2009;58:1809–1818. doi: 10.1007/s00262-009-0692-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson AJ, Sive HL. The Wnt antagonists Frzb-1 and Crescent locally regulate basement membrane dissolution in the developing primary mouth. Development. 2009;136:1071–1081. doi: 10.1242/dev.032912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Didsbury J, Weber RF, Bokoch GM, Evans T, Snyderman R. rac, a novel ras-related family of proteins that are botulinum toxin substrates. J Biol Chem. 1989;264:16378–16382. [PubMed] [Google Scholar]
- Ding F, Yin Z, Wang HR. Ubiquitination in Rho signaling. Curr Top Med Chem. 2011;11:2879–2887. doi: 10.2174/156802611798281357. [DOI] [PubMed] [Google Scholar]
- Dixon MJ, Marazita ML, Beaty TH, Murray JC. Cleft lip and palate: understanding genetic and environmental influences. Nat Rev Genet. 2011;12:167–178. doi: 10.1038/nrg2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobreva G, et al. SATB2 is a multifunctional determinant of craniofacial patterning and osteoblast differentiation. Cell. 2006;125:971–986. doi: 10.1016/j.cell.2006.05.012. [DOI] [PubMed] [Google Scholar]
- Dode C, et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat Genet. 2003;33:463–465. doi: 10.1038/ng1122. [DOI] [PubMed] [Google Scholar]
- Dostal A, Nemeckova J, Gaillyova R. The 18q deletion syndrome and analysis of the critical region for orofacial cleft at 18q22.3. J Craniomaxillofac Surg. 2009;37:272–275. doi: 10.1016/j.jcms.2008.12.002. [DOI] [PubMed] [Google Scholar]
- Dravet C, et al. Epilepsy, antiepileptic drugs, and malformations in children of women with epilepsy: a French prospective cohort study. Neurology. 1992;42:75–82. [PubMed] [Google Scholar]
- Eden S, Rohatgi R, Podtelejnikov AV, Mann M, Kirschner MW. Mechanism of regulation of WAVE1-induced actin nucleation by Rac1 and Nck. Nature. 2002;418:790–793. doi: 10.1038/nature00859. [DOI] [PubMed] [Google Scholar]
- Erdogan F, et al. Characterization of a 5.3 Mb deletion in 15q14 by comparative genomic hybridization using a whole genome “tiling path” BAC array in a girl with heart defect, cleft palate, and developmental delay. Am J Med Genet A. 2007;143A(2):172–178. doi: 10.1002/ajmg.a.31541. [DOI] [PubMed] [Google Scholar]
- Evers EE, Zondag GC, Malliri A, Price LS, ten Klooster JP, van der Kammen RA, Collard JG. Rho family proteins in cell adhesion and cell migration. Eur J Cancer. 2000;36:1269–1274. doi: 10.1016/S0959-8049(00)00091-5. [DOI] [PubMed] [Google Scholar]
- Fera E, O’Neil C, Lee W, Li S, Pickering JG. Fibroblast growth factor-2 and remodeled type I collagen control membrane protrusion in human vascular smooth muscle cells: biphasic activation of Rac1. J Biol Chem. 2004;279:35573–35582. doi: 10.1074/jbc.M400711200. [DOI] [PubMed] [Google Scholar]
- FitzPatrick DR, et al. Identification of SATB2 as the cleft palate gene on 2q32-q33. Hum Mol Genet. 2003;12:2491–2501. doi: 10.1093/hmg/ddg248. [DOI] [PubMed] [Google Scholar]
- Frebourg T, et al. Cleft lip/palate and CDH1/E-cadherin mutations in families with hereditary diffuse gastric cancer. J Med Genet. 2006;43:138–142. doi: 10.1136/jmg.2005.031385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata M, Nakagawa M, Kuroda S, Kaibuchi K. Cell adhesion and Rho small GTPases. J Cell Sci. 1999;112(Pt 24):4491–4500. doi: 10.1242/jcs.112.24.4491. [DOI] [PubMed] [Google Scholar]
- Funato N, Nakamura M, Richardson JA, Srivastava D, Yanagisawa H. Tbx1 regulates oral epithelial adhesion and palatal development. Hum Mol Genet. 2012;21:2524–2537. doi: 10.1093/hmg/dds071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Dominguez CA, et al. Sprouty2 and Spred1-2 proteins inhibit the activation of the ERK pathway elicited by cyclopentenone prostanoids. PLoS One. 2011;6:e16787. doi: 10.1371/journal.pone.0016787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giehl K, Graness A, Goppelt-Struebe M. The small GTPase Rac-1 is a regulator of mesangial cell morphology and thrombospondin-1 expression. Am J Physiol Renal Physiol. 2008;294:F407–F413. doi: 10.1152/ajprenal.00093.2007. [DOI] [PubMed] [Google Scholar]
- Goodnough LH, Brugmann SA, Hu D, Helms JA. Stage-dependent craniofacial defects resulting from Sprouty2 overexpression. Dev Dyn. 2007;236:1918–1928. doi: 10.1002/dvdy.21195. [DOI] [PubMed] [Google Scholar]
- Gross I, Bassit B, Benezra M, Licht JD. Mammalian sprouty proteins inhibit cell growth and differentiation by preventing ras activation. J Biol Chem. 2001;276:46460–46468. doi: 10.1074/jbc.M108234200. [DOI] [PubMed] [Google Scholar]
- He X, Kuo YC, Rosche TJ, Zhang X. Structural basis for autoinhibition of the guanine nucleotide exchange factor FARP2. Structure. 2013;21:355–364. doi: 10.1016/j.str.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinonen TY, Maki M. Peters′-plus syndrome is a congenital disorder of glycosylation caused by a defect in the beta1, 3-glucosyltransferase that modifies thrombospondin type 1 repeats. Ann Med. 2009;41:2–10. doi: 10.1080/07853890802301975. [DOI] [PubMed] [Google Scholar]
- Herman SB, et al. Overt cleft palate phenotype and TBX1 genotype correlations in velo-cardio-facial/DiGeorge/22q11.2 deletion syndrome patients. Am J Med Genet Part A. 2012;158:2781–2787. doi: 10.1002/ajmg.a.35512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Diaz S, Werler MM, Walker AM, Mitchell AA. Folic acid antagonists during pregnancy and the risk of birth defects. N Engl J Med. 2000;343:1608–1614. doi: 10.1056/NEJM200011303432204. [DOI] [PubMed] [Google Scholar]
- Heyworth PG, Knaus UG, Settleman J, Curnutte JT, Bokoch GM. Regulation of NADPH oxidase activity by Rac GTPase activating protein(s) Mol Biol Cell. 1993;4:1217–1223. doi: 10.1091/mbc.4.11.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang B, Moos M, Jr, Vukicevic S, Luyten FP. Primary structure and tissue distribution of FRZB, a novel protein related to Drosophila frizzled, suggest a role in skeletal morphogenesis. J Biol Chem. 1996;271:26131–26137. doi: 10.1074/jbc.271.42.26131. [DOI] [PubMed] [Google Scholar]
- Hoang BH, Thomas JT, Abdul-Karim FW, Correia KM, Conlon RA, Luyten FP, Ballock RT. Expression pattern of two Frizzled-related genes, Frzb-1 and Sfrp-1, during mouse embryogenesis suggests a role for modulating action of Wnt family members. Dev Dyn. 1998;212:364–372. doi: 10.1002/(SICI)1097-0177(199807)212:3<364::AID-AJA4>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Ignelzi MA, Jr, Wang W, Young AT. Fibroblast growth factors lead to increased Msx2 expression and fusion in calvarial sutures. J bone Mineral Res Off J Am Soc Bone Mineral Res. 2003;18:751–759. doi: 10.1359/jbmr.2003.18.4.751. [DOI] [PubMed] [Google Scholar]
- Ireton K, Rigano LA, Dowd GC. Role of host GTPases in infection by Listeria monocytogenes. Cell Microbiol. 2014;16:1311–1320. doi: 10.1111/cmi.12324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo G, et al. A microduplication of 5p15.33 reveals CLPTM1L as a candidate gene for cleft lip and palate. Eur J Med Genet. 2013;56:222–225. doi: 10.1016/j.ejmg.2013.01.002. [DOI] [PubMed] [Google Scholar]
- Jackson TA, Koterwas DM, Morgan MA, Bradford AP. Fibroblast growth factors regulate prolactin transcription via an atypical Rac-dependent signaling pathway. Mol Endocrinol (Baltimore, Md) 2003;17:1921–1930. doi: 10.1210/me.2003-0167. [DOI] [PubMed] [Google Scholar]
- Jezewski PA, et al. Complete sequencing shows a role for MSX1 in non-syndromic cleft lip and palate. J Med Genet. 2003;40:399–407. doi: 10.1136/jmg.40.6.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson S, et al. Haploinsufficiency of MEIS2 is associated with orofacial clefting and learning disability. Am J Med Genet A. 2014;164:1622–1626. doi: 10.1002/ajmg.a.36498. [DOI] [PubMed] [Google Scholar]
- Jugessur A, Murray JC. Orofacial clefting: recent insights into a complex trait. Curr Opin Genet Dev. 2005;15:270–278. doi: 10.1016/j.gde.2005.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalay E, et al. Mutations in RIPK4 cause the autosomal-recessive form of popliteal pterygium syndrome. Am J Hum Genet. 2012;90:76–85. doi: 10.1016/j.ajhg.2011.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamel G, et al. Requirement for frzb and fzd7a in cranial neural crest convergence and extension mechanisms during zebrafish palate and jaw morphogenesis. Dev Biol. 2013;381:423–433. doi: 10.1016/j.ydbio.2013.06.012. [DOI] [PubMed] [Google Scholar]
- Kanazawa S, et al. bFGF regulates PI3-kinase-Rac1-JNK pathway and promotes fibroblast migration in wound healing. PLoS One. 2010;5:e12228. doi: 10.1371/journal.pone.0012228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh Y, Katoh M. FGF signaling inhibitor, SPRY4, is evolutionarily conserved target of WNT signaling pathway in progenitor cells. Int J Mol Med. 2006;17:529–532. [PubMed] [Google Scholar]
- Keeler AB, Molumby MJ, Weiner JA. Protocadherins branch out: multiple roles in dendrite development. Cell Adh Migr. 2015;9:214–226. doi: 10.1080/19336918.2014.1000069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo S, et al. Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat Genet. 2002;32:285–289. doi: 10.1038/ng985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo T, Yamashita T, Yamaguchi A, Sumimoto H, Hosokawa K, Tohyama M. A novel FERM domain including guanine nucleotide exchange factor is involved in Rac signaling and regulates neurite remodeling. J Neurosci. 2002;22:8504–8513. doi: 10.1523/JNEUROSCI.22-19-08504.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurisu S, Suetsugu S, Yamazaki D, Yamaguchi H, Takenawa T. Rac-WAVE2 signaling is involved in the invasive and metastatic phenotypes of murine melanoma cells. Oncogene. 2005;24:1309–1319. doi: 10.1038/sj.onc.1208177. [DOI] [PubMed] [Google Scholar]
- Kuroda S, Fukata M, Nakagawa M, Kaibuchi K. Cdc42, Rac1, and their effector IQGAP1 as molecular switches for cadherin-mediated cell-cell adhesion. Biochem Biophys Res Commun. 1999;262:1–6. doi: 10.1006/bbrc.1999.1122. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JG, Kay EP. PI 3-kinase/Rac1 and ERK1/2 regulate FGF-2-mediated cell proliferation through phosphorylation of p27 at Ser10 by KIS and at Thr187 by Cdc25A/Cdk2. Invest Ophthalmol Vis Sci. 2011;52:417–426. doi: 10.1167/iovs.10-6140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CC, Putnam AJ, Miranti CK, Gustafson M, Wang LM, Vande Woude GF, Gao CF. Overexpression of sprouty 2 inhibits HGF/SF-mediated cell growth, invasion, migration, and cytokinesis. Oncogene. 2004;23:5193–5202. doi: 10.1038/sj.onc.1207646. [DOI] [PubMed] [Google Scholar]
- Leoyklang P, Suphapeetiporn K, Siriwan P, Desudchit T, Chaowanapanja P, Gahl WA, Shotelersuk V. Heterozygous nonsense mutation SATB2 associated with cleft palate, osteoporosis, and cognitive defects. Hum Mutat. 2007;28:732–738. doi: 10.1002/humu.20515. [DOI] [PubMed] [Google Scholar]
- Leslie EJ, Marazita ML. Genetics of cleft lip and cleft palate. Am J Med Genet C Semin Med Genet. 2013;163C(4):246–258. doi: 10.1002/ajmg.c.31381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Liu J, Hudson M, Kim S, Hatch NE. FGF2 promotes Msx2 stimulated PC-1 expression via Frs2/MAPK signaling. J Cell Biochem. 2010;111:1346–1358. doi: 10.1002/jcb.22861. [DOI] [PubMed] [Google Scholar]
- Lidral AC, et al. Association of MSX1 and TGFB3 with nonsyndromic clefting in humans. Am J Hum Genet. 1998;63:557–568. doi: 10.1086/301956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Yusoff P, Wong ES, Chandramouli S, Lao DH, Fong CW, Guy GR. The cysteine-rich sprouty translocation domain targets mitogen-activated protein kinase inhibitory proteins to phosphatidylinositol 4,5-bisphosphate in plasma membranes. Mol Cell Biol. 2002;22:7953–7966. doi: 10.1128/MCB.22.22.7953-7966.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K, Wang S, Julius MA, Kitajewski J, Moos M, Jr, Luyten FP. The cysteine-rich frizzled domain of Frzb-1 is required and sufficient for modulation of Wnt signaling. Proc Natl Acad Sci USA. 1997;94:11196–11200. doi: 10.1073/pnas.94.21.11196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JY, et al. Association of bone morphogenetic protein 4 gene polymorphisms with nonsyndromic cleft lip with or without cleft palate in Chinese children DNA. Cell Biol. 2008;27:601–605. doi: 10.1089/dna.2008.0777. [DOI] [PubMed] [Google Scholar]
- Lindsay EA, Baldini A. A mouse gene (Dgcr6) related to the Drosophila gonadal gene is expressed in early embryogenesis and is the homolog of a human gene deleted in DiGeorge syndrome. Cytogenet Cell Genet. 1997;79:243–247. doi: 10.1159/000134736. [DOI] [PubMed] [Google Scholar]
- Lito P, Mets BD, Appledorn DM, Maher VM, McCormick JJ. Sprouty 2 regulates DNA damage-induced apoptosis in Ras-transformed human fibroblasts. J Biol Chem. 2009;284:848–854. doi: 10.1074/jbc.M808045200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little J, Cardy A, Munger RG. Tobacco smoking and oral clefts: a meta-analysis. Bull World Health Organ. 2004;82:213–218. [PMC free article] [PubMed] [Google Scholar]
- Liu W, Sun X, Braut A, Mishina Y, Behringer RR, Mina M, Martin JF. Distinct functions for Bmp signaling in lip and palate fusion in mice. Development. 2005;132:1453–1461. doi: 10.1242/dev.01676. [DOI] [PubMed] [Google Scholar]
- Liu S, Higashihori N, Yahiro K, Moriyama K. Retinoic acid inhibits histone methyltransferase Whsc1 during palatogenesis. Biochem Biophys Res Commun. 2015;458:525–530. doi: 10.1016/j.bbrc.2015.01.148. [DOI] [PubMed] [Google Scholar]
- Louw JJ, Corveleyn A, Jia Y, Hens G, Gewillig M, Devriendt K. MEIS2 involvement in cardiac development, cleft palate, and intellectual disability. Am J Med Genet A. 2015;167A(5):1142–1146. doi: 10.1002/ajmg.a.36989. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maas NM, et al. Genotype-phenotype correlation in 21 patients with Wolf–Hirschhorn syndrome using high resolution array comparative genome hybridisation (CGH) J Med Genet. 2008;45:71–80. doi: 10.1136/jmg.2007.052910. [DOI] [PubMed] [Google Scholar]
- MacDonald JR, Ziman R, Yuen RK, Feuk L, Scherer SW. The database of genomic variants: a curated collection of structural variation in the human genome. Nucleic Acids Res. 2014;42:D986–D992. doi: 10.1093/nar/gkt958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madura T, Yamashita T, Kubo T, Taniguchi M, Kawakita A, Hosokawa K, Tohyama M. Expression of FERM domain including guanine nucleotide exchange factor mRNA in adult rat brain. Brain Res Mol Brain Res. 2003;114:163–167. doi: 10.1016/S0169-328X(03)00166-9. [DOI] [PubMed] [Google Scholar]
- Mansukhani A, Bellosta P, Sahni M, Basilico C. Signaling by fibroblast growth factors (FGF) and fibroblast growth factor receptor 2 (FGFR2)-activating mutations blocks mineralization and induces apoptosis in osteoblasts. J Cell Biol. 2000;149:1297–1308. doi: 10.1083/jcb.149.6.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marazita ML. Subclinical features in non-syndromic cleft lip with or without cleft palate (CL/P): review of the evidence that subepithelial orbicularis oris muscle defects are part of an expanded phenotype for CL/P. Orthod Craniofac Res. 2007;10:82–87. doi: 10.1111/j.1601-6343.2007.00386.x. [DOI] [PubMed] [Google Scholar]
- Matsumura K, et al. Sprouty2 controls proliferation of palate mesenchymal cells via fibroblast growth factor signaling. Biochem Biophys Res Commun. 2011;404:1076–1082. doi: 10.1016/j.bbrc.2010.12.116. [DOI] [PubMed] [Google Scholar]
- McGrath JA, et al. Hay-Wells syndrome is caused by heterozygous missense mutations in the SAM domain of p63. Hum Mol Genet. 2001;10:221–229. doi: 10.1093/hmg/10.3.221. [DOI] [PubMed] [Google Scholar]
- Menard L, Tomhave E, Casey PJ, Uhing RJ, Snyderman R, Didsbury JR. Rac1, a low-molecular-mass GTP-binding-protein with high intrinsic GTPase activity and distinct biochemical properties. Eur J Biochem. 1992;206:537–546. doi: 10.1111/j.1432-1033.1992.tb16957.x. [DOI] [PubMed] [Google Scholar]
- Miki H, Suetsugu S, Takenawa T. WAVE, a novel WASP-family protein involved in actin reorganization induced by Rac. EMBO J. 1998;17:6932–6941. doi: 10.1093/emboj/17.23.6932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minden A, Lin A, Claret FX, Abo A, Karin M. Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell. 1995;81:1147–1157. doi: 10.1016/S0092-8674(05)80019-4. [DOI] [PubMed] [Google Scholar]
- Mitchell K, et al. Exome sequence identifies RIPK4 as the Bartsocas-Papas syndrome locus. Am J Hum Genet. 2012;90:69–75. doi: 10.1016/j.ajhg.2011.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore R, Ferretti P, Copp A, Thorogood P. Blocking endogenous FGF-2 activity prevents cranial osteogenesis. Develop Biol. 2002;243:99–114. doi: 10.1006/dbio.2001.0533. [DOI] [PubMed] [Google Scholar]
- Mossey PA, Catilla EE (2003) Global registry and database on craniofacial anomalies: report of a WHO Registry Meeting on Craniofacial Anomalies. WHO Meeting on International Collaborative Research on Craniofacial Anomalies (3rd, 2001; Bauru, Brazil). http://apps.who.int/iris/handle/10665/42840#sthash.yPHt56gA.dpuf
- Mossey PA, Little J, Munger RG, Dixon MJ, Shaw WC. Cleft lip and palate. Lancet. 2009;374:1773–1785. doi: 10.1016/S0140-6736(09)60695-4. [DOI] [PubMed] [Google Scholar]
- Mulatinho M, Llerena J, Leren TP, Rao PN, Quintero-Rivera F. Deletion (1)(p32.2-p32.3) detected by array-CGH in a patient with developmental delay/mental retardation, dysmorphic features and low cholesterol: A new microdeletion syndrome? Am J Med Genet A. 2008;146A(17):2284–2290. doi: 10.1002/ajmg.a.32454. [DOI] [PubMed] [Google Scholar]
- Munger RG, Romitti PA, Daack-Hirsch S, Burns TL, Murray JC, Hanson J. Maternal alcohol use and risk of orofacial cleft birth defects. Teratology. 1996;54:27–33. doi: 10.1002/(SICI)1096-9926(199607)54:1<27::AID-TERA4>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Murthy J, Bhaskar L. Current concepts in genetics of nonsyndromic clefts. Indian J Plast Surg. 2009;42:68–81. doi: 10.4103/0970-0358.53004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata K, Inagaki M. Cytoskeletal modification of Rho guanine nucleotide exchange factor activity: identification of a Rho guanine nucleotide exchange factor as a binding partner for Sept9b, a mammalian septin. Oncogene. 2005;24:65–76. doi: 10.1038/sj.onc.1208101. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Gulick J, Pratt R, Robbins J. Noonan syndrome is associated with enhanced pERK activity, the repression of which can prevent craniofacial malformations. Proc Natl Acad Sci USA. 2009;106:15436–15441. doi: 10.1073/pnas.0903302106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newbern J, et al. Mouse and human phenotypes indicate a critical conserved role for ERK2 signaling in neural crest development. Proc Natl Acad Sci USA. 2008;105:17115–17120. doi: 10.1073/pnas.0805239105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SB, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010;42:790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SB, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikopensius T, et al. Genetic variants in COL2A1, COL11A2, and IRF6 contribute risk to nonsyndromic cleft palate Birth defects research Part A. Clin Mol Teratol. 2010;88:748–756. doi: 10.1002/bdra.20700. [DOI] [PubMed] [Google Scholar]
- Nishiwaki T, et al. Reduced expression of thrombospondins and craniofacial dysmorphism in mice overexpressing Fra1. J Bone Miner Res. 2006;21:596–604. doi: 10.1359/jbmr.051216. [DOI] [PubMed] [Google Scholar]
- O’Brien DE, et al. ERK2 alone drives inflammatory pain but cooperates with ERK1 in sensory neuron survival. J Neurosci Off J Soc Neurosci. 2015;35:9491–9507. doi: 10.1523/JNEUROSCI.4404-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parada C, et al. Disruption of the ERK/MAPK pathway in neural crest cells as a potential cause of Pierre Robin sequence. Dev (Cambridge, England) 2015 doi: 10.1242/dev.125328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennisi PA, Barr V, Nunez NP, Stannard B, Le Roith D. Reduced expression of insulin-like growth factor I receptors in MCF-7 breast cancer cells leads to a more metastatic phenotype. Cancer Res. 2002;62:6529–6537. [PubMed] [Google Scholar]
- Polakis PG, Weber RF, Nevins B, Didsbury JR, Evans T, Snyderman R. Identification of the ral and rac1 gene products, low molecular mass GTP-binding proteins from human platelets. J Biol Chem. 1989;264:16383–16389. [PubMed] [Google Scholar]
- Poppleton HM, Edwin F, Jaggar L, Ray R, Johnson LR, Patel TB. Sprouty regulates cell migration by inhibiting the activation of Rac1 GTPase. Biochem Biophys Res Commun. 2004;323:98–103. doi: 10.1016/j.bbrc.2004.08.070. [DOI] [PubMed] [Google Scholar]
- Porntaveetus T, Oommen S, Sharpe PT, Ohazama A. Expression of Fgf signalling pathway related genes during palatal rugae development in the mouse. Gene Expr Patterns GEP. 2010;10:193–198. doi: 10.1016/j.gep.2010.03.004. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahimov F, et al. Disruption of an AP-2alpha binding site in an IRF6 enhancer is associated with cleft lip. Nat Genet. 2008;40:1341–1347. doi: 10.1038/ng.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainger JK, et al. Disruption of SATB2 or its long-range cis-regulation by SOX9 causes a syndromic form of Pierre Robin sequence. Hum Mol Genet. 2014;23:2569–2579. doi: 10.1093/hmg/ddt647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon W, Winter RM, Rutland P, Pulleyn LJ, Jones BM, Malcolm S. Mutations in the fibroblast growth factor receptor 2 gene cause Crouzon syndrome. Nat Genet. 1994;8:98–103. doi: 10.1038/ng0994-98. [DOI] [PubMed] [Google Scholar]
- Ridley A. Rho GTPases. Integrating integrin signaling. J Cell Biol. 2000;150:F107–F109. doi: 10.1083/jcb.150.4.F107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- Riley BM, Murray JC. Sequence evaluation of FGF and FGFR gene conserved non-coding elements in non-syndromic cleft lip and palate cases. Am J Med Genet A. 2007;143A:3228–3234. doi: 10.1002/ajmg.a.31965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson PN, Mundlos S. The human phenotype ontology. Clin Genet. 2010;77:525–534. doi: 10.1111/j.1399-0004.2010.01436.x. [DOI] [PubMed] [Google Scholar]
- Sahoo T, et al. Microdeletion 20p12.3 involving BMP2 contributes to syndromic forms of cleft palate. Am J Med Genet A. 2011;155A(7):1646–1653. doi: 10.1002/ajmg.a.34063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki A, Taketomi T, Wakioka T, Kato R, Yoshimura A. Identification of a dominant negative mutant of Sprouty that potentiates fibroblast growth factor- but not epidermal growth factor-induced ERK activation. J Biol Chem. 2001;276:36804–36808. doi: 10.1074/jbc.C100386200. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Ito Y, Bringas P, Jr, Chou S, Urata MM, Slavkin H, Chai Y. TGFbeta-mediated FGF signaling is crucial for regulating cranial neural crest cell proliferation during frontal bone development. Dev (Cambridge, England) 2006;133:371–381. doi: 10.1242/dev.02200. [DOI] [PubMed] [Google Scholar]
- Schenck A, Bardoni B, Moro A, Bagni C, Mandel JL. A highly conserved protein family interacting with the fragile X mental retardation protein (FMRP) and displaying selective interactions with FMRP-related proteins FXR1P and FXR2P. Proc Natl Acad Sci USA. 2001;98:8844–8849. doi: 10.1073/pnas.151231598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenck A, Bardoni B, Langmann C, Harden N, Mandel JL, Giangrande A. CYFIP/Sra-1 controls neuronal connectivity in Drosophila and links the Rac1 GTPase pathway to the fragile X protein. Neuron. 2003;38:887–898. doi: 10.1016/S0896-6273(03)00354-4. [DOI] [PubMed] [Google Scholar]
- Schlickum S, Moghekar A, Simpson JC, Steglich C, O’Brien RJ, Winterpacht A, Endele SU. LETM1, a gene deleted in Wolf-Hirschhorn syndrome, encodes an evolutionarily conserved mitochondrial protein. Genomics. 2004;83:254–261. doi: 10.1016/j.ygeno.2003.08.013. [DOI] [PubMed] [Google Scholar]
- Schmitz U, Thommes K, Beier I, Wagner W, Sachinidis A, Dusing R, Vetter H. Angiotensin II-induced stimulation of p21-activated kinase and c-Jun NH2-terminal kinase is mediated by Rac1 and Nck. J Biol Chem. 2001;276:22003–22010. doi: 10.1074/jbc.M102450200. [DOI] [PubMed] [Google Scholar]
- Seto-Salvia N, Stanier P. Genetics of cleft lip and/or cleft palate: association with other common anomalies. Eur J Med Genet. 2014;57:381–393. doi: 10.1016/j.ejmg.2014.04.003. [DOI] [PubMed] [Google Scholar]
- Shi M, et al. Identification of microdeletions in candidate genes for cleft lip and/or palate. Birth Defects Res A Clin Mol Teratol. 2009;85:42–51. doi: 10.1002/bdra.20571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu K, et al. Microarray and FISH-based genotype-phenotype analysis of 22 Japanese patients with Wolf-Hirschhorn syndrome. Am J Med Genet A. 2014;164A(3):597–609. doi: 10.1002/ajmg.a.36308. [DOI] [PubMed] [Google Scholar]
- Shin EY, et al. Basic fibroblast growth factor stimulates activation of Rac1 through a p85 betaPIX phosphorylation-dependent pathway. J Biol Chem. 2004;279:1994–2004. doi: 10.1074/jbc.M307330200. [DOI] [PubMed] [Google Scholar]
- Shkoukani MA, Chen M, Vong A. Cleft lip—a comprehensive review. Front Pediatr. 2013;1:53. doi: 10.3389/fped.2013.00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S, Yin X, Pisano MM, Greene RM. Molecular profiles of mitogen activated protein kinase signaling pathways in orofacial development. Birth Defects Res A Clin Mol Teratol. 2007;79:35–44. doi: 10.1002/bdra.20320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song T, et al. Association between NOGGIN and SPRY2 polymorphisms and nonsyndromic cleft lip with or without cleft palate. Am J Med Genet A. 2015;167A:137–141. doi: 10.1002/ajmg.a.36802. [DOI] [PubMed] [Google Scholar]
- Souza J, Raskin S. Clinical and epidemiological study of orofacial clefts. J Pediatr (Rio J) 2013;89:137–144. doi: 10.1016/j.jped.2013.03.010. [DOI] [PubMed] [Google Scholar]
- Spurlock G, et al. SPRED1 mutations (Legius syndrome): another clinically useful genotype for dissecting the neurofibromatosis type 1 phenotype. J Med Genet. 2009;46:431–437. doi: 10.1136/jmg.2008.065474. [DOI] [PubMed] [Google Scholar]
- Srivastava N, Sudan R, Saha B. CD40-modulated dual-specificity phosphatases MAPK phosphatase (MKP)-1 and MKP-3 reciprocally regulate Leishmania major infection. J Immunol. 2011;186(10):5863–5872. doi: 10.4049/jimmunol.1003957. [DOI] [PubMed] [Google Scholar]
- Stec I, et al. WHSC1, a 90 kb SET domain-containing gene, expressed in early development and homologous to a Drosophila dysmorphy gene maps in the Wolf-Hirschhorn syndrome critical region and is fused to IgH in t(4;14) multiple myeloma. Hum Mol Genet. 1998;7:1071–1082. doi: 10.1093/hmg/7.7.1071. [DOI] [PubMed] [Google Scholar]
- Strauss RP, Cassell CH. Critical issues in craniofacial care: quality of life, costs of care, and implications of prenatal diagnosis. Acad Pediatr. 2009;9:427–432. doi: 10.1016/j.acap.2009.09.019. [DOI] [PubMed] [Google Scholar]
- Strehle EM, et al. Genotype-phenotype analysis of 4q deletion syndrome: proposal of a critical region. Am J Med Genet Part A. 2012;158:2139–2151. doi: 10.1002/ajmg.a.35502. [DOI] [PubMed] [Google Scholar]
- Suazo J, Santos JL, Jara L, Blanco R. Association between bone morphogenetic protein 4 gene polymorphisms with nonsyndromic cleft lip with or without cleft palate in a chilean population. DNA Cell Biol. 2010;29:59–64. doi: 10.1089/dna.2009.0944. [DOI] [PubMed] [Google Scholar]
- Suo L, Lu H, Ying G, Capecchi MR, Wu Q. Protocadherin clusters and cell adhesion kinase regulate dendrite complexity through Rho GTPase. J Mol Cell Biol. 2012;4:362–376. doi: 10.1093/jmcb/mjs034. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, et al. In a Vietnamese population, MSX1 variants contribute to cleft lip and palate. Genet Med. 2004;6:117–125. doi: 10.1097/01.GIM.0000127275.52925.05. [DOI] [PubMed] [Google Scholar]
- Suzuki S, et al. Mutations in BMP4 are associated with subepithelial, microform, and overt cleft lip. Am J Hum Genet. 2009;84:406–411. doi: 10.1016/j.ajhg.2009.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sylvestersen KB, Herrera PL, Serup P, Rescan C. Fgf9 signalling stimulates Spred and Sprouty expression in embryonic mouse pancreas mesenchyme. Gene Expr Patterns. 2011;11:105–111. doi: 10.1016/j.gep.2010.10.001. [DOI] [PubMed] [Google Scholar]
- Szabo-Rogers HL, Geetha-Loganathan P, Nimmagadda S, Fu KK, Richman JM. FGF signals from the nasal pit are necessary for normal facial morphogenesis. Dev Biol. 2008;318:289–302. doi: 10.1016/j.ydbio.2008.03.027. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Suzuki K. WAVE2 targeting to phosphatidylinositol 3,4,5-triphosphate mediated by insulin receptor substrate p53 through a complex with WAVE2. Cell Signal. 2010;22:1708–1716. doi: 10.1016/j.cellsig.2010.06.011. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanaka T, Suzuki K. Directional control of WAVE2 membrane targeting by EB1 and phosphatidylinositol 3,4,5-triphosphate. Cell Signal. 2010;22:510–518. doi: 10.1016/j.cellsig.2009.11.005. [DOI] [PubMed] [Google Scholar]
- Takegahara N, et al. Integral roles of a guanine nucleotide exchange factor, FARP2, in osteoclast podosome rearrangements. FASEB J. 2010;24:4782–4792. doi: 10.1096/fj.10-158212. [DOI] [PubMed] [Google Scholar]
- Tamura T, Munger RG, Corcoran C, Bacayao JY, Nepomuceno B, Solon F. Plasma zinc concentrations of mothers and the risk of nonsyndromic oral clefts in their children: a case-control study in the Philippines. Birth Defects Res A Clin Mol Teratol. 2005;73:612–616. doi: 10.1002/bdra.20179. [DOI] [PubMed] [Google Scholar]
- Tang Q, Li L, Jin C, Lee JM, Jung HS. Role of region-distinctive expression of Rac1 in regulating fibronectin arrangement during palatal shelf elevation. Cell Tissue Res. 2015 doi: 10.1007/s00441-015-2169-9. [DOI] [PubMed] [Google Scholar]
- Thomas PS, Kim J, Nunez S, Glogauer M, Kaartinen V. Neural crest cell-specific deletion of Rac1 results in defective cell-matrix interactions and severe craniofacial and cardiovascular malformations. Dev Biol. 2010;340:613–625. doi: 10.1016/j.ydbio.2010.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Juan L, et al. Mutations in TBX1 genocopy the 22q11.2 deletion and duplication syndromes: a new susceptibility factor for mental retardation. European J Hum Genet EJHG. 2007;15:658–663. doi: 10.1038/sj.ejhg.5201819. [DOI] [PubMed] [Google Scholar]
- van den Boogaard MJ, Dorland M, Beemer FA, van Amstel HK. MSX1 mutation is associated with orofacial clefting and tooth agenesis in humans. Nat Genet. 2000;24:342–343. doi: 10.1038/74155. [DOI] [PubMed] [Google Scholar]
- Vieira AR, Meira R, Modesto A, Murray JC. MSX1, PAX9, and TGFA contribute to tooth agenesis in humans. J Dent Res. 2004;83:723–727. doi: 10.1177/154405910408300913. [DOI] [PubMed] [Google Scholar]
- Vieira AR, et al. Medical sequencing of candidate genes for nonsyndromic cleft lip and palate. PLoS Genet. 2005;1:e64. doi: 10.1371/journal.pgen.0010064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira AR, de Carvalho FM, Johnson L, DeVos L, Swailes AL, Weber ML, Deeley K. Fine Mapping of 6q23.1 Identifies TULP4 as Contributing to Clefts. Cleft Palate Craniofac J. 2015;52:128–134. doi: 10.1597/13-023. [DOI] [PubMed] [Google Scholar]
- Villamor E, Sparen P, Cnattingius S. Risk of oral clefts in relation to prepregnancy weight change and interpregnancy interval. Am J Epidemiol. 2008;167:1305–1311. doi: 10.1093/aje/kwn065. [DOI] [PubMed] [Google Scholar]
- Vulto-van Silfhout AT, et al. An update on ECARUCA, the European cytogeneticists association register of unbalanced chromosome aberrations. Eur J Med Genet. 2013;56:471–474. doi: 10.1016/j.ejmg.2013.06.010. [DOI] [PubMed] [Google Scholar]
- Wang HR, Zhang Y, Ozdamar B, Ogunjimi AA, Alexandrova E, Thomsen GH, Wrana JL. Regulation of cell polarity and protrusion formation by targeting RhoA for degradation. Science. 2003;302:1775–1779. doi: 10.1126/science.1090772. [DOI] [PubMed] [Google Scholar]
- Wang Y, et al. Tyrosine phosphorylated Par3 regulates epithelial tight junction assembly promoted by EGFR signaling. EMBO J. 2006;25:5058–5070. doi: 10.1038/sj.emboj.7601384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, et al. The FGF and FGFR gene family and risk of cleft lip with or without cleft palate. Cleft Palate-Craniofac Jo Off Publ Am Cleft Palate-Craniofac Assoc. 2013;50:96–103. doi: 10.1597/11-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehby GL, Cassell CH. The impact of orofacial clefts on quality of life and healthcare use and costs. Oral Dis. 2010;16:3–10. doi: 10.1111/j.1601-0825.2009.01588.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh IC, Hagge-Greenberg A, O’Brien TP. A dosage-dependent role for Spry2 in growth and patterning during palate development. Mech Dev. 2007;124:746–761. doi: 10.1016/j.mod.2007.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentzel C, Fernstrom M, Ohrner Y, Anneren G, Thuresson AC. Clinical variability of the 22q11.2 duplication syndrome. European J Med Genet. 2008;51:501–510. doi: 10.1016/j.ejmg.2008.07.005. [DOI] [PubMed] [Google Scholar]
- Westwick JK, Lambert QT, Clark GJ, Symons M, Van Aelst L, Pestell RG, Der CJ. Rac regulation of transformation, gene expression, and actin organization by multiple, PAK-independent pathways. Mol Cell Biol. 1997;17:1324–1335. doi: 10.1128/MCB.17.3.1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkie AO, et al. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet. 1995;9:165–172. doi: 10.1038/ng0295-165. [DOI] [PubMed] [Google Scholar]
- Williams ES, Uhas KA, Bunke BP, Garber KB, Martin CL. Cleft palate in a multigenerational family with a microdeletion of 20p12.3 involving BMP2. Am J Med Genet A. 2012;158A(10):2616–2620. doi: 10.1002/ajmg.a.35594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright TJ, et al. A transcript map of the newly defined 165 kb Wolf-Hirschhorn syndrome critical region. Hum Mol Genet. 1997;6:317–324. doi: 10.1093/hmg/6.2.317. [DOI] [PubMed] [Google Scholar]
- Wright TJ, Costa JL, Naranjo C, Francis-West P, Altherr MR. Comparative analysis of a novel gene from the Wolf-Hirschhorn/Pitt-Rogers-Danks syndrome critical region. Genomics. 1999;59:203–212. doi: 10.1006/geno.1999.5871. [DOI] [PubMed] [Google Scholar]
- Wyszynski DF, Beaty TH, Maestri NE. Genetics of nonsyndromic oral clefts revisited. Cleft Palate Craniofac J. 1996;33:406–417. doi: 10.1597/1545-1569(1996)033<0406:GONOCR>2.3.CO;2. [DOI] [PubMed] [Google Scholar]
- Yagi H, et al. Role of TBX1 in human del22q11.2 syndrome. Lancet (London, England) 2003;362:1366–1373. doi: 10.1016/S0140-6736(03)14632-6. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Cui XM, Shuler CF. Role of ERK1/2 signaling during EGF-induced inhibition of palatal fusion. Dev Biol. 2003;260:512–521. doi: 10.1016/S0012-1606(03)00275-6. [DOI] [PubMed] [Google Scholar]
- Yang X, Kilgallen S, Andreeva V, Spicer DB, Pinz I, Friesel R. Conditional expression of Spry1 in neural crest causes craniofacial and cardiac defects. BMC Dev Biol. 2010;10:48. doi: 10.1186/1471-213X-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yigzaw Y, Cartin L, Pierre S, Scholich K, Patel TB. The C terminus of sprouty is important for modulation of cellular migration and proliferation. J Biol Chem. 2001;276:22742–22747. doi: 10.1074/jbc.M100123200. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Song Y, Zhao X, Zhang X, Fermin C, Chen Y. Rescue of cleft palate in Msx1-deficient mice by transgenic Bmp4 reveals a network of BMP and Shh signaling in the regulation of mammalian palatogenesis. Development. 2002;129:4135–4146. doi: 10.1242/dev.129.17.4135. [DOI] [PubMed] [Google Scholar]
- Zhao G, Wojciechowski MC, Jee S, Boros J, McAvoy JW, Lovicu FJ. Negative regulation of TGFbeta-induced lens epithelial to mesenchymal transition (EMT) by RTK antagonists. Exp Eye Res. 2015;132:9–16. doi: 10.1016/j.exer.2015.01.001. [DOI] [PubMed] [Google Scholar]
- Zollino M, et al. Genotype-phenotype correlations and clinical diagnostic criteria in Wolf-Hirschhorn syndrome. Am J Med Genet. 2000;94:254–261. doi: 10.1002/1096-8628(20000918)94:3<254::AID-AJMG13>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Zollino M, et al. Mapping the Wolf-Hirschhorn syndrome phenotype outside the currently accepted WHS critical region and defining a new critical region, WHSCR-2. Am J Hum Genet. 2003;72:590–597. doi: 10.1086/367925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucchero TM, et al. Interferon regulatory factor 6 (IRF6) gene variants and the risk of isolated cleft lip or palate. N Engl J Med. 2004;351:769–780. doi: 10.1056/NEJMoa032909. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.