

Abstract
The polyphenolic flavone chrysin has been evaluated as a natural chemopreventive agent due to its anti-cancer effects in a variety of cancer cell lines. However, the mechanism of the chemopreventive effect has been not well established, especially in human colorectal cancer cells. We evaluated the chemopreventive effect of chrysin in three different human colorectal cancer cell lines. We found that chrysin treatment consequently reduced cell viability via induction of apoptosis. We identified that the involvement of up-regulation of pro-apoptotic cytokines tumor necrosis factor (Tnf) α and β genes and consequent activation of the TNF-mediated transcriptional pathway in chrysin-induced apoptosis. Using our generated AHR siRNA expressing colorectal cancer cells, we demonstrated that the chrysin-induced up-regulation of Tnfα and β gene expression was dependent on the aryl hydrocarbon receptor (AHR), which is a ligand-receptor for chrysin. Subsequently, we found that the AHR siRNA expressing colorectal cancer cells were resistant to chrysin-induced apoptosis. Therefore, we concluded that AHR is required for the chrysin-induced apoptosis and the up-regulation of Tnfα and β gene expression in human colorectal cancer cells.
Keywords: Aryl hydrocarbon receptor, Chrysin, Apoptosis, Colorectal cancer, Tumor necrosis factor
1. INTRODUCTION
Despite advances in screening for and treatment of colorectal cancer, the impact of this malignancy on men and women both globally and in the United States remains substantial [1]. In 2014, there were an estimated 137,000 new cases of colorectal cancer in the United States with approximately 50,000 deaths, which ranks as the third leading cause of cancer death in both men and women [2]. The improved screening techniques for colorectal cancer have resulted in reduced incidence and mortality, with the advantage of detecting tumors at an earlier stage [1]. Additionally, advances in surgical intervention (i.e. total mesorectal excision in rectal cancer) with radiation therapy have reduced local recurrence rates and newer chemotherapeutic regimens have improved overall survival in patients with later stage disease [3–6]. Currently, strategies aimed at inhibiting malignant transformation have gained interest, because carcinogenesis within the colon and rectum is a multistep process that generally assumes a pre-cancerous state requiring sequential genetic defects to progress to invasive disease [7]. One putative avenue for disease prevention is characterized by the use of natural or synthetic agents to either suppress initiation of tumorigenesis or inhibit progression of premalignant cells to fully invasive complements, also known as chemoprevention [8].
Chemopreventive agents modulate tumorigenic pathways as opposed to chemotherapeutics that function to eliminate vast populations of premalignant or malignant cells [9, 10]. Several studies have revealed an association between diet (dietary compounds) and cancer prevention [11–13]. The natural polyphenolic substances known as “flavonoids” which are prevalent in vegetables, fruits and nuts and constitute supplements with excellent safety profile and low toxicity, have gained significant interest by many in the field of chemoprevention [14, 15]. Flavonoids encompass more than 4000 biologically active compounds with diverse functions such as enzyme inhibition, ligand-activation of signaling pathways and signal transduction [14, 15]. Multiple subclasses of flavonoids possess anti-inflammatory, anti-oxidant and anti-proliferative capabilities [14, 15]. Anti-carcinogenic properties have been attributed to free radical scavenging, modification of enzymes involved in carcinogen metabolism, inhibition of transcription factors and induction of apoptosis [14, 15]. Epidemiologic investigations have highlighted the association of reduced risk of development of various malignancies with flavonoid consumption [14, 15].
One such flavonoid that has received considerable attention is chrysin, a compound present in honey, propolis and various plant extracts [16]. Among the molecular pathways investigated, chrysin has been shown to induce cell cycle arrest, increase degradation of hypoxia inducible factor α, inhibit tumor cell-induced angiogenesis, inhibit cell adhesion, and induce apoptosis in a variety of cancer cells [16]. However, despite numerous investigations for the anti-cancer effects of chrysin in several different cancer cells, the mechanism is not well established, especially in colorectal cancer cells. Therefore, we evaluated the anti-cancer effect of chrysin in human colorectal cancer cells. Additionally, we investigated the molecular mechanism of the chemopreventive effect by chrysin in colorectal cancer cells. We elucidated that chrysin induces apoptotic cell death in human colorectal cancer cells and that induction of pro-apoptotic cytokines tumor necrosis factor (TNF) α and β are involved in chrysin-induced apoptosis. Previous study demonstrated that chrysin is a natural agonist of the aryl hydrocarbon receptor (AHR) known as a xenobiotic receptor [17], therefore we hypothesized that activation of AHR would be required for chrysin’s ability to kill colorectal cancer cells. As predicted, chrysin-induced apoptosis and induction of Tnfα/β gene expression are mediated via the AHR.
2. MATERIALS AND METHODS
2.1. Cell culture
Colon (HCT116, DLD1) and rectal (SW837) cancer cell lines were obtained from ATCC (Manassas, VA). Cells were maintained in DMEM medium (Life Technologies, Carlsbad, CA) supplemented with 10% fetal bovine serum (Life Technologies), 1% non-essential amino acids (Life Technologies), 1% penicillin-streptomycin (Life Technologies), and 1% glutamine (Life Technologies) at 37°C and 5% CO2.
2.2. Cell viability
Cells were seeded in 96-well plates with approximately 1.0 × 104 cells / well and incubated in DMEM supplemented medium for 24 hours. Cells were then treated with chrysin (Sigma-Aldrich, St. Louis, MO) (10 µM, 50 µM, 100 µM) or vehicle (DMSO) for 24 hours and the number of viable cells determined using an XTT proliferation assay (Roche Life Science, Indianapolis, IN). The absorbance (460nm) and reference (750 nm) were measured using a spectrophotometer (Spectramax, Molecular Devices, Sunnyvale, CA). For the fluorescence cell viability assay, cells were seeded to 96-well plates with approximately 1.0 × 104 cells / well and incubated in DMEM medium for 24 hours. Cells were treated with chrysin or vehicle for 6, 12, 24 and 48 hours. Cell viability was measured using CellTiter-Fluor™ cell viability assay kit (Promega, Madison WI). The fluorescence (excitation 390nm, emission 460nm) was detected using spectramax plus 384 microplate reader (Molecular Devices).
2.3. Cytotoxicity and apoptosis assay
To investigate the mechanism of decreased cell viability induced by chrysin, we used the ApoTox-Glo™ Triplex Assay (Promega). Approximately 1.0 × 104 cells / well were seeded to 96-well plate and treated with 100 µM chrysin or 0.1% DMSO for 6, 12, 24 and 48h. Live-cells (cell viability) and dead-cells (cytotoxicity) were detected with treatment of fluorogenic peptide substrates glycylphenylalanyl-aminofluorocoumarin (GF-AFC) and bis-alanylalanylphenylalanyl-rhodamine 110 (bis-AAF-R110), respectively. Fluorescence (GF-AFC (excitation 390nm/emission 460nm) and bis-AAF-R110 (excitation 485 nm/ emission 520 nm)) were measured using spectramax plus 384 microplate reader (Molecular Devices). Apoptosis activity was detected using Caspase-Glo® 3/7 Reagent (Promega). After addition of the reagent to cell culture medium, luminescence was measured by MicroLumat plus (Berthold).
2.4. TUNEL assay
DeadEnd™ Fluorometric TUNEL System (Promega) was utilized to evaluate cell apoptosis (DNA fragmentation) via incorporation of fluorescein-12-dUTP at 3’-OH DNA ends by recombinant terminal deoxynucleotidyl transferase (rTdT). Cells were treated with 100 µM chrysin or 0.1% DMSO for 48 hours and transferred to slides, which were then fixed, permeabilized, and treated with equilibration buffer followed by rTDT and nucleotide mix. The cells were then stained with propidium iodide (PI) and analyzed using fluorescence microscopy in which PI (apoptotic and nonapoptotic cells) and fluorescein-12-dUTP (apoptotic cells) were visualized. The number of terminal deoxynucleotidyl transferase-dUTP nick end labeling (TUNEL) positive cells and total cell number were counted.
2.5. Gene expression analysis
Cells were treated with chrysin, 6-formylindolo (3,2-b) carbazole (FICZ) or vehicle (DMSO) as described. Total RNA was isolated from cells using the Qiagen RNeasy kit (Qiagen, Valencia CA). The isolated RNAs were reverse-transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). The mRNA levels were measured with TaqMan Universal PCR Master Mix (Applied Biosystems) and custom-designed probes (Assay ID: CYP1A1; Hs01054797_g1, Tnf-α; Hs01113624_g1, Ltα (Tnf-β); Hs04188773_g1, Ahr: Hs00169233_m1, c-fos; Mm00487425_m1, β-actin; Hs01060665_gl). β-actin mRNA levels were measured as internal controls.
2.6. PCR array
Gene expression associated with apoptosis was evaluated using the RT2 Profiler PCR array (PAHS-012Z, Qiagen). HCT116 cells were treated with 100 µM chrysin or 0.1% DMSO for 24 hours. Total RNA for RT2 Profiler PCR array was extracted using RNeasy mini QIAcube kit. The data analysis was performed by web-based RT2 Profiler™ PCR Array Data Analysis program. Genes that demonstrated a two-fold change or greater (chrysin (n=4) vs. DMSO (n=4), p < 0.05) were selected for further correlation analyses.
2.7. Stable si-RNA expression cell lines
For generation of small interfering RNA (siRNA) stable expression cell lines, HCT116 cells were transfected in 6-cm diameter dishes with 5 µg of pRNAT-U6-siAHR (5’-GGATCCCAAGATGGATCAATACTTCCACTTGATATCCGGTGGAAGTATTGATCCATCTTTTTT TTCCAAAAGCTT-3’) or pRNAT-U6-siScramble (5’-GGATCCCACATGATCGACTATAACACGTTTGATATCCGGTGGAAGTATTGATCCATCTTTTTTTTCCAAAAGCTT-3’) (Genscript) using Lipofectamine 2000 (Life technologies). Six hours after transfection, cells were transferred to 15-cm diameter dishes and allowed to recover for a further 24 hours. The transfected cells were selected by culturing cells in DMEM medium (with 10% fetal bovine serum) containing 900 µg/ml G418. After 5–7 days of selection, resistant colonies were isolated and expanded in DMEM medium containing 250 µg/ml G418. The resistant clones were screened by Ahr and CYP1A1 mRNA levels using RT-PCR.
2.8. Luciferase reporter gene assay
To determine the alteration of TNF-mediated transcriptional regulation from treatment with chrysin, cells were transiently transfected with TNF signaling pathway analysis luciferase reporter vector (pNF-κB-RE LUC (pGL4.32), pSRE-LUC (pGL4.33) or pAP1-RE LUC (pGL4.44) (Promega)) and pTK-renilla luciferase plasmid (Promega) using Lipofectamine 2000 (Life technologies). Six hours after transfection, cells were cultured with DMEM media containing 100 µM chrysin or 0.1% DMSO for 6, 12 and 24 hours. The luciferase activity was measured using the dual luciferase reporter assay system (Promega) according to manufacturer's instructions, and MicroLumat Plus luminometer (Berthold Technologies, Hartfordshire, UK).
2.9. Statistical analysis
All statistical analyses were performed using GraphPad Prism software (version 5, LaJolla, CA). Statistically significant differences between treatment and control groups were determined using unpaired t-test with Welch’s correction. Data was presented as mean ± standard error of mean and was considered statistically significant when p value was < 0.05.
3. RESULTS
3.1. Chrysin induces cell apoptosis in colorectal cancer cell lines
The anti-cancer effects of chrysin have been observed in numerous cancer cell lines [16]. However, its effects in colon and rectal cancer cell lines have not been well studied. To evaluate chrysin effects on colorectal cancer cells, we first examined changes in cell viability with treatment of chrysin using HCT116, DLD-1 and SW837 colorectal tumor cell lines (Fig. 1A). With treatment of 10–100 µM chrysin for 24 hours, each cell line demonstrated a dose-dependent decrease in cell viability (Fig 1A). Cell viability in all three cell lines was significantly decreased at 50 µM and 100 µM chrysin. HCT116 cells demonstrated the highest susceptibility to chrysin-induced cell death among the three cell lines with cell viability (61.4% (50 µM) and 37.9% (100 µM) of control, respectively). Therefore, we primarily used HCT116 cells as an experimental model for investigating the mechanism of chrysin’s effects.
Fig.1. Chrysin induced cell apoptosis in colorectal cancer cells.
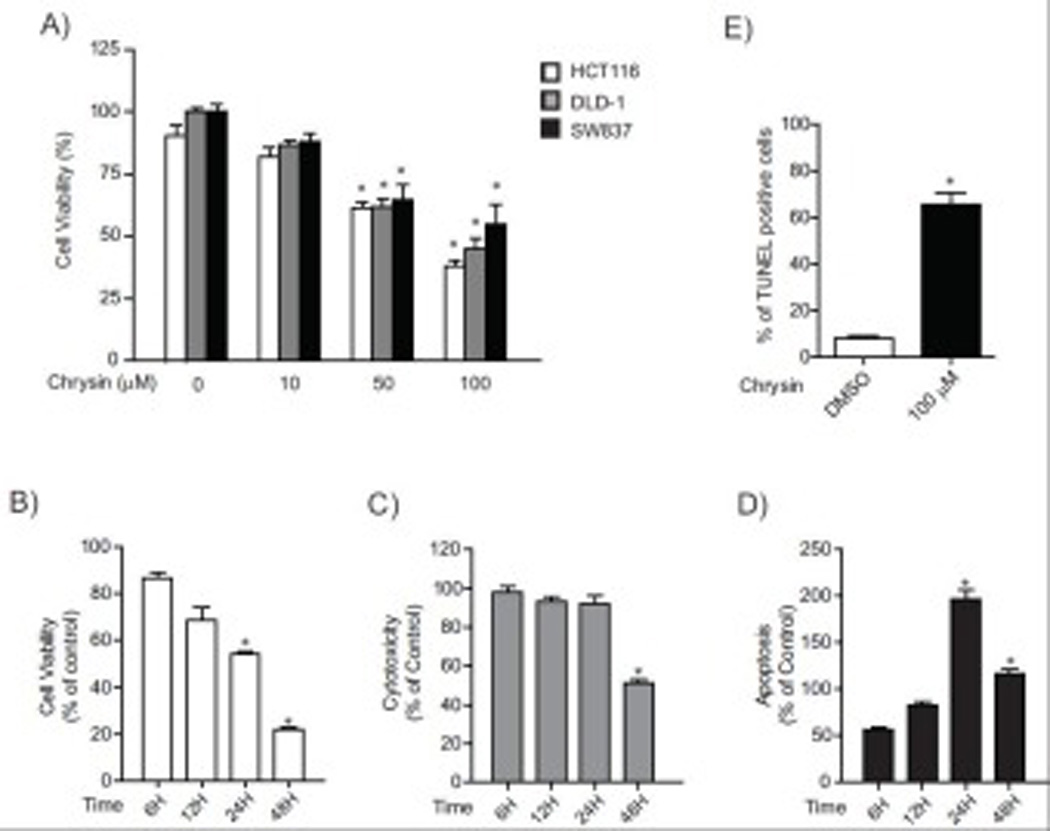
(A) Cell viability of HCT116, DLD-1 and SW837 cells after treatment with DMSO or chrysin (10, 50 and 100 µM) for 24 hours. Each group contained 8–18 samples. Error bars represent SEM. *significantly different relative to DMSO-treated cells (p<0.05), (B) Cell viability, cytotoxicity and apoptosis of HCT116 cells after treatment with 100 µM chrysin or DMSO for a period of 48 hours. Each group contained 8 samples. Error bars represent SEM. *significantly increased relative to DMSO-treated cells (p<0.05), (C) Percent of TUNEL positive HCT116 cells after treatment with DMSO or 100 µM chrysin (number of TUNEL positive cells divided by total number of cells). Each group contained 3 samples. Error bars represent SEM. *significantly different relative to DMSO-treated cells (p<0.05)
To identify the basis of decreased cell viability after treatment with chrysin, we assessed cell viability, cytotoxicity and apoptosis (caspase3/7 activity) using ApoTox-Glo™ Triplex assay system (Promega) (Fig 1B–D). The number of viable HCT116 cells after treatment with 100 µM chrysin decreased in a time-dependent manner from 86.7 % of control at 6 hours to 21.8% at 48 hours (6h; 86.7% (% of control), 12h; 68.7% 24h: 54.1% 48h; 21.8%) (Fig. 1B). Concomitant with decrease in cell viability, treatment with 100 µM chrysin resulted increases in indicating increased apoptosis (6h; 57.1% (% of control), 12h; 85.4% 24h: 200.0% 48h; 118.7%) (Fig. 1D). Cytotoxicity was not similarly elevated compared to control but actually decreased to 51.1 % of control at 48 hours (6h; 98.2% (% of control), 12h; 92.1% 24h: 91.7% 48h; 51.1%) (Fig 1C). These results suggest that the decrease in viable cells was consequent to apoptosis, which surpassed levels in control cells between 24–48 hours.
Further evidence for apoptosis was provided by evaluation of nuclear DNA fragmentation, a biomarker of the advanced stage of apoptosis, with a terminal deoxynucleotidyl transferase-dUTP nick end labeling (TUNEL) assay in chrysin (100 µM) and vehicle (DMSO) treated cells. A marked increase in nuclear DNA fragmentation was observed at 48 hours, approximately 65% of chrysin treated HCT116 cells detected as TUNEL positive cells (apoptotic cells), while only 8% apoptotic cells were present in the control group (p < 0.05) (Fig 1E). Together, these results indicate that chrysin induces apoptotic cell death in colon and rectal cancer cells.
3.2. Tumor necrosis factor (Tnf) α and β genes were up-regulated by chrysin
To identify the signaling pathways involved in chrysin-induced apoptotic cell death, we performed a gene profiling analysis using the apoptosis RT2 Profiler PCR Array System (Qiagen). Among the 84 apoptosis-related genes profile, gene expressions of lymphotoxin α (LTα) (known as tumor necrosis factor β (TNFβ)), TNFα and PYD and CARD domain containing (PYCARD) were significantly increased (>2 fold change, p < 0.05) in HCT116 cells treated with 100 µM chrysin for 24 hours compared to DMSO treated HCT116 cells (Table 1). TNFα and TNFβ are major ligands for the TNF-receptor (TNFR)-mediated apoptosis pathway [18, 19] and PYCARD is controlled by TNF in KG1a cells [20]. Therefore, we suspected that increased Tnfα and Tnfβ gene expression were essential components of chrysin-induced apoptotic cell death.
Table 1.
Induction of apoptosis-related gene expression in chrysin-treated HCT116 cells
| Gene Symbol | Description | Fold differencea |
|---|---|---|
| LTA (TNFβ) | Lymphotoxin alpha (TNF superfamily, member 1) | 11.2 |
| TNF (TNFα) | Tumor necrosis factor | 7.2 |
| PYCARD | PYD and CARD domain containing | 3.1 |
Genes that demonstrated a two-fold change or greater (chrysin-treated cells (n=4) vs.DMSO-treated cells (n=4), p<0.05)
To further examine how the up-regulation of Tnfα and Tnfβ gene expression participated in chrysin-induced cell apoptosis, we monitored the time-course of Tnfα and Tnfβ induction in chrysintreated HCT116 cells using quantitative RT-PCR (Taqman). Tnfα and Tnfβ mRNA expression increased at 3 hours and expression levels were more than 10-fold induced between 6–12 hours (Fig. 2A–B). Peak levels occurred at 6–12 hours, but significant induction remained as long as 48 hours. Elevated expression of Tnfα and Tnfβ was also observed in DLD-1 and SW837 cells (Figure 2C–F) demonstrating that chrysin treatment increased Tnfα and Tnfβ expression in all colorectal cancer cell lines. Notably, elevated Tnfα and Tnfβ expression preceded the increase in caspase 3/7 activity (24–48 hours). Since Tnfα and Tnfβ are principal ligands for the TNFR-1 mediated apoptosis signaling pathway, this is consisted with a critical role for the TNFR-1 pathway in chrysin-induced apoptosis.
Fig.2. Tnfα and Tnfβ genes expression were induced by chrysin.

Tnfα and Tnfβ mRNA levels in HCT116 cells treated with DMSO or 100 µM chrysin for time periods up to 48 hours (1, 3, 6, 12, 18, 24, 48 hours). Results are expressed as relative mRNA level compared to DMSO-treated controls (relative fold induction). Tnfα and Tnfβ mRNA levels (A-F). (A) Tnfα (HCT116), (B) Tnfβ (HCT116), (C) Tnfα (DLD-1), (D) Tnfβ (DLD-1), (E) Tnfα (SW837), (F) Tnfβ (SW837). Each group contained more than 4 samples. Error bars represent SEM. *significantly different relative to DMSO-treated controls (p<0.05)
3.3. Chrysin activated TNF signaling pathways
To test this hypothesis that TNF-regulated signaling pathway(s) should also be activated by chrysin, we measured the transcriptional activity of TNF-controlled nuclear factor κ-light-chain enhancer of activated B cells (NF-κB), mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) and activator protein (AP-1) pathways using luciferase reporter vectors containing NF-κB response element (NF-κB RE), serum response element (SRE) or AP-1 response element (AP-1 RE), respectively (Fig. 3).
Fig.3. Chrysin activated TNF signaling pathways.

Luciferase activity for (A) HCT116 or (B) DLD-1 cells transiently transfected with TNF signaling pathway analysis luciferase reporter vector pNF-κB-RE LUC, pSRE-LUC or pAP1-RE LUC (Promega) and pTK-renilla luciferase plasmid (Promega). The relative luciferase activity was calculated by normalizing the firefly luciferase activity to the transfection control TK-renilla luciferase activity. Each group contained 4 samples. Error bars represent SEM. *significantly different relative to DMSO-treated controls (p<0.05).
In HCT116 cells, all three transcriptional pathways were activated by chrysin between 6–24 hours and these transcriptional activities were increased in time-dependent manner (Fig. 3A). Notably, there was a robust increase in the transcriptional activity of the MAPK/ERK pathway mediated through SRE, which was increased approximately 66 fold by chrysin at 24 hours. The transcriptional activity of NF-κB and AP-1 pathways was increased approximately 4 and 3 fold (24 hours) in HCT116 cells, respectively (Fig. 3A). In DLD-1 cells, the time-dependent increases of NF-κB RE and SRE-mediated transcriptional activity were also observed (Fig. 3B). The induction level of SRE-MAPK/ERK transcriptional activity by treatment of chrysin at 24 hours was higher than those of NF-κB and AP-1 mediated transcriptional activity (NF-κB RE, 3.2 fold; SRE, 8.7 fold; AP-1 RE, 1.6 fold) (Fig. 3B).
3.4. Chrysin activates AHR signaling
Chrysin has been reported as an agonist of the transcriptional receptor AHR in several cell lines [17] and certain AHR agonists (e.g. 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)) induce TNF gene expression in vitro and in vivo [21, 22]. Therefore, we hypothesized that the chrysin-induced elevation of Tnfα and Tnfβ gene expression was regulated by AHR. We first examined whether or not chrysin activates AHR signaling in the colorectal cancer cell lines (Fig. 4). In HCT116 cells, the expression of CYP1A1, a highly sensitive and specific marker of AHR transactivation, was increased by treatment with chrysin in a dose-dependent manner (approximate 5-fold at 50 µM chrysin and 8-fold at 100 µM chrysin) (Figure 4A). Similar to the time-course of Tnfα and Tnfβ induction (Fig. 2A–B), CYP1A1 mRNA levels were increased between 3–6 hours after chrysin treatment (Fig. 4B). CYP1A1 induction by chrysin was also observed in DLD-1 and SW837 cells (Fig 4C–D). These results indicated that chrysin activated AHR signaling in the colorectal cancer cell lines and the induction was occurred within 3–6 hours post-treatment..
Fig.4. Chrysin activated AHR signaling pathway.
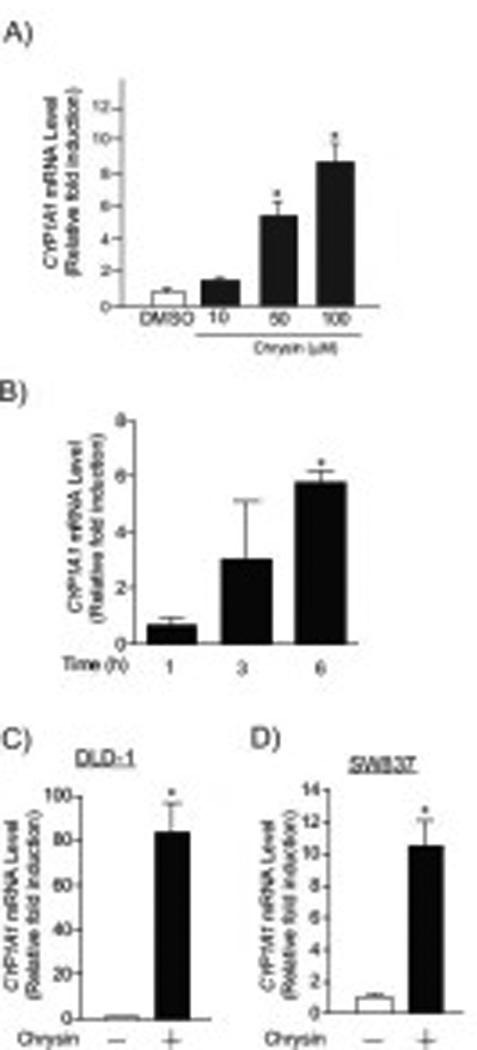
(A) CYP1A1 mRNA expression in HCT116 cells treated with DMSO or chrysin (10, 50 and 100 µM) for 6 hours. (B) Time-dependent increases in CYP1A1 mRNA levels in HCT116 cells treated with 100 µM chrysin. (C-D) CYP1A1 mRNA expression in (C) DLD-1 and (D) SW837 cells treated with DMSO or 100 µM chrysin for 6 hours. Each group contained more than 4 samples. Error bars represent SEM. *significantly different relative to DMSO-treated controls (p<0.05).
3.5. Inhibition of AHR expression blocked induction of AHR activity by chrysin
Although chrysin activates AHR signaling and promotes colon cancer cell apoptosis, the relationship between the AHR pathway and apoptosis remains unclear. The dependence of chrysin-induced transcriptional and apoptotic events on AHR was investigated by generating HCT116 cell lines stably transfected with si-AHR or si-scramble (control) expression vectors. The si-AHR stable-expression HCT116 cells demonstrated greater than 60% knock-down in Ahr mRNA expression compared to si-scramble stable-expression control cells (Fig. 5A). More importantly, the si-AHR stable expression cells demonstrated a substantial inhibition of AHR activity with an approximately 80% decrease in CYP1A1 induction compared to si-scramble cells after treatment with 6-formylindolo (3,2-b) carbazole (FICZ, a potent AHR ligand) (si-scramble, 60 fold; P<0.05, si-AHR, 13 fold, p < 0.05) (Fig. 5B). Additionally, we compared the induction levels of CYP1A1 gene expression after chrysin treatment between AHR and scramble si-RNA expression cells (Fig. 5C). The induction level of CYP1A1 gene expression by chrysin in si-AHR expression cells (2.1 fold compared to vehicle, P=0.07) was significantly lower than that in si-scramble expression control cells (4.5 fold compared to vehicle, P<0.05) (Fig. 5C). From these studies, it appeared that chrysin-AHR-mediated signaling was significantly reduced in the si-AHR expression cells.
Fig.5. Decrease of AHR and AHR-driven gene expression in HCT116 cells expressing si-AHR.

(A) Ahr mRNA levels in si-scramble and si-AHR expressing cell lines. Each group contained 3 samples. Error bars represent SEM. *significantly different relative to si-scramble cells (p<0.05). (B–C) CYP1A1 mRNA expression in si-scramble and si-AHR expressing cells treated with (B) 200 nM FICZ or (C) 100 µM chrysin for 6 hours. Each group contained 4 samples. Error bars represent SEM. *significantly different relative to DMSO-treated si-scramble cells (p<0.05), ** significantly different relative to chrysin-treated si-scramble cells (p<0.05).
3.6. Inhibition of AHR signaling suppressed chrysin-induced cell apoptosis and increases in Tnfα and Tnfβ expression
We compared cell viability and apoptotic caspase activity after treatment with chrysin in si-AHR and si-scramble cell lines (Fig. 6A–B). At each treatment dose of chrysin (25 µM, 50 µM, 100 µM), si-AHR cells demonstrated resistance to chrysin induced cell death compared to si-scramble cells (si-scramble: 61.8% (% of DMSO-treated control) (25 µM), 48.6% (50 µM), 36.9% (100 µM); si-AHR: 80.0% (25 µM), 76.6% (50 µM), 47.6% (100 µM)) (Figure 6A). Moreover, in the si-AHR expression HCT116 cells, the increases in caspase-3/7 activity by chrysin (50 µM and 100 µM) were suppressed compared to si-scramble expression cells (si-scramble: 168.7% (50 µM), 171.1% (100 µM); si-AHR: 101.8% (50 µM), 133.3% (100 µM)) (Fig. 6B). These results demonstrated that AHR is involved in chrysin-induced apoptotic cell death.
Fig.6. The si-AHR expressing HCT116 cells resisted to the decrease of cell viability and the increase of cell apoptosis induced by chrysin.
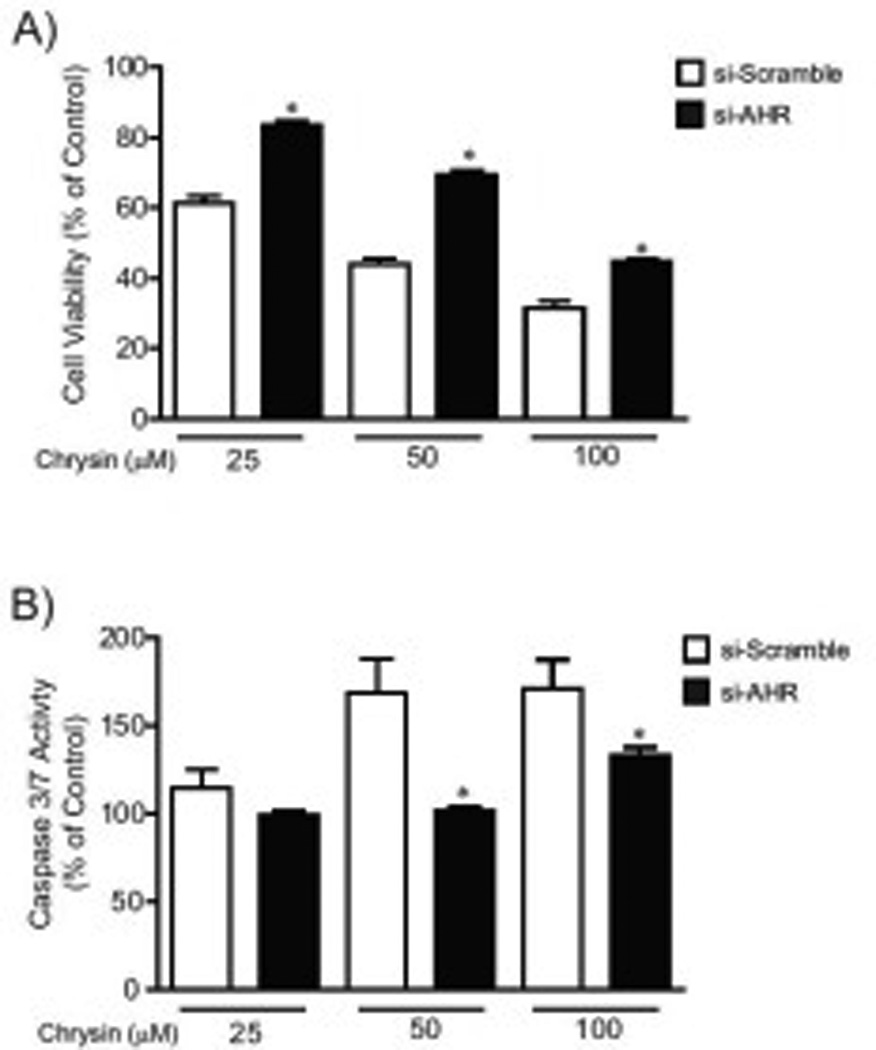
(A) Cell viability of si-scramble and si-AHR expressing cells treated with DMSO or chrysin (25, 50 and 100 µM) for 48 hours. (B) Caspase 3/7 activity in si-scramble and si-AHR expressing cells treated with DMSO or chrysin for 24 hours. Each group contained 8 samples. Error bars represent SEM. *significantly different relative to chrysin-treated si-scramble cells (p<0.05).
Furthermore, we evaluated induction levels of Tnfα and Tnfβ mRNA after chrysin treatment in si-AHR and si-scramble cell lines (Fig. 7A–B). Tnfα and Tnfβ expression was substantially reduced (approximately 81% and 77%, respectively) in si-AHR cells compared to si-scramble control cells following treatment with chrysin. Interestingly, the induction of SRE-mediated transcriptional activity by chrysin was significantly suppressed in si-AHR expression cells (Fig. 8A), although NF-κB and AP-1 mediated transcriptional activity was more induced in si-AHR cells compared to control cells (Fig. 8A). The expression of c-fos gene, a well-studied SRE-driven gene [23, 24], was increased by treatment of chrysin in control cells, while the increase was not observed in si-AHR expressing cells (Fig. 8B). These results suggested that AHR controls Tnfα and β gene expression and SRE-mediated transcriptional pathway.
Fig.7. The increases of Tnfα and Tnfβ genes expression by chrysin were suppressed in HCT116 cells expressing si-AHR.
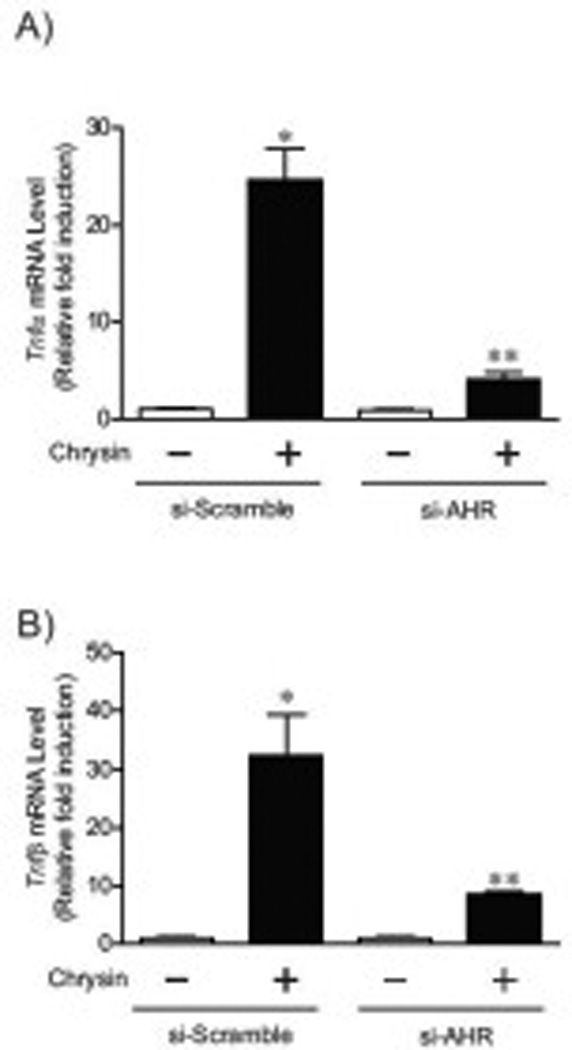
Tnfα (A) and Tnfβ (B) expression in si-scramble and si-AHR expressing cells treated with DMSO or 100 µM chrysin for 6 hours. Each group contained more than 4 samples. Error bars represent SEM. *significantly different relative to DMSO-treated si-scramble cells (p<0.05), ** significantly different relative to chrysin-treated si-scramble cells (p<0.05).
Fig.8. AHR-dependent upregulation of SRE-mediated transcriptional pathway by chrysin.
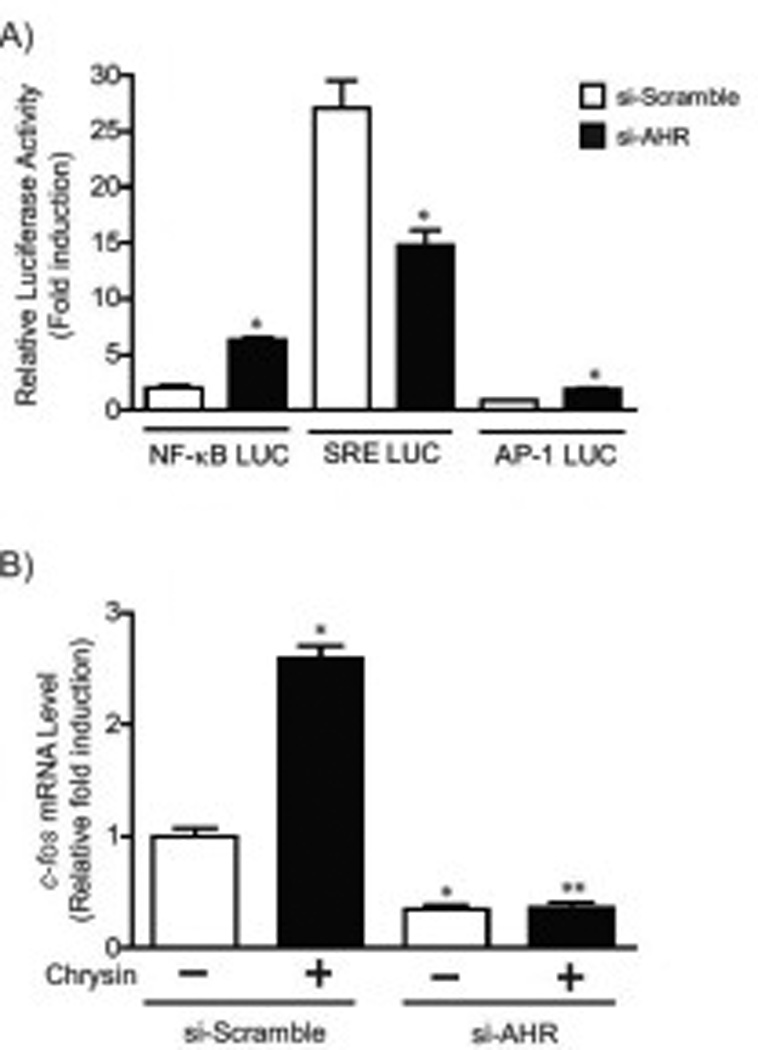
(A) Siscramble or si-AHR cells transiently transfected with TNF signaling pathway analysis luciferase reporter vector pNF-κB-RE LUC, pSRE-LUC or pAP1-RE LUC (Promega) and pTK-renilla luciferase plasmid were treated with DMSO or chrysin (100 µM) for 24 hours. The relative luciferase activity was calculated by normalizing the firefly luciferase activity to the transfection control TK-renilla luciferase activity. (B) c-fos mRNA expression in si-scramble or si-AHR cells treated with DMSO or chrysin (100 µM) for 24 hours. Each group contained more than 4 samples. Error bars represent SEM. *significantly different relative to DMSO-treated si-scramble cells (p<0.05), ** significantly different relative to chrysin-treated si-scramble cells (p<0.05).
4. DISCUSSION
In this study, we proposed that polyphenolic flavone chrysin would be a prospective chemopreventive flavonoid for human colorectal cancer cells, because chrysin has demonstrated an anti-cancer effect though cell cycle arrest, reduction of cell viability, and induction of apoptosis in a variety of human cancer cell lines [16]. Therefore, to investigate our hypothesis, we examined the effect on cell viability after chrysin treatment in three different human colorectal cancer cell lines. Our results indicated that chrysin induced apoptotic cell death in colorectal cancer cells after exposure to 50–100 µM chrysin for 24 hours (Fig. 1). Moreover, we identified that apoptosis-related cytokines Tnfα and Tnfβ genes were up-regulated in chrysin-treated colorectal cancer cells (Table 1).
TNFα and TNFβ are members of the TNF superfamily and are ligands of TNF receptors (TNFR) 1 and 2. Binding of TNF to TNFR induces pleiotropic cellular responses including cell proliferation, inflammation and cell apoptosis. TNF/TNFR binding leads to formation and activation of “deathincluding signaling complex” composed of TNFR-associated death domain protein (TRADD), Fas associated protein with death domain (FADD), TNFR-associated factor 1 (TRAF1) and caspase-8 [18, 19]. Alternatively, the complex also stimulates oxidative stress responses with activation of pro-apoptotic Bcl-2 members (i.e., Bax, Bid), mitochondrial permeabilization and resultant release of cytochrome c, and activation of caspase-9 [18, 19]. These two pathways ultimately promote to activate executioner caspases-3, -6 and -7, which mediate the cascade of apoptotic cell death [19]. We hypothesized that the TNF-TNFR mediated apoptosis pathway constitutes the apoptotic mechanism in chrysin treated human colorectal cells, due to the up-regulation of Tnfα and Tnfβ gene expression with chrysin treatment (Table 1). Because the up-regulation in Tnfα and Tnfβ expression were preceded the elevation of apoptotic caspase- 3/7 activity by 12 to 18 hours, and the Tnfα/β genes induction was sustained up to 48 hours (Fig. 2A–B) and the up-regulation of Tnfα/β genes expression was identified in the other two colorectal cell lines (Fig. 2C–F).
In response to TNF binding TNFR, regulation of several signaling pathways occurs, with activation of transcriptional factors and kinases such as nuclear factor κ-light-chain enhancer of activated B cells (NF-κB), extracellular signal-regulated kinase (ERK), mitogen-activated protein kinase (MAPK), c-jun N-terminal kinase (JNK), activator protein (AP-1), as well as the apoptosis cascades [19]. Therefore, the transcriptional activity of three distinct TNF-response pathways (NF-κB-RE SRE (MAPK/ERK), AP-1-RE (AP-1)) were measured and found to be induced by chrysin. Consistent with the temporal relationship of Tnf genes expression and caspase activity, the induction of TNF-response pathways was observed 12–18 hours prior to the increase in caspase-3/7 activity (Fig. 3A–B). Taken together, these results suggest that the increase of Tnfα and Tnfβ expression by chrysin would trigger activation of TNF/TNFR-mediated signaling which in turn regulates the chrysin-induced apoptosis through the TNF-caspase cascade in colorectal cancer cells.
Li and colleagues found that pretreatment of 40 µM chrysin for 2 hours sensitized TNFα-induced apoptosis in TNFα-treated HCT116 cells through suppression of NF-κB signaling thereby inhibiting an anti-apoptosis pathway [25]. In contrast, our results revealed NF-κB signaling was up-regulated from 6 to 24 hours after treatment of chrysin. Discrepant results may be due to the different treatment conditions (chrysin followed by treatment with TNF-α vs chrysin alone) and experimental duration.
SRE-mediated transcriptional activity was substantially enhanced by chrysin compared to NF-κB-RE and AP-1-RE-driven transcription (Fig. 3A–B). Most interestingly, the up-regulation of SRE-mediated transcriptional activity by chrysin was suppressed in AHR-dependent manner (Fig. 8A), while NF-κB-RE and AP-1-RE-mediated transcription was upregulated in si-AHR cells (Fig. 8A). These results suggest that the SRE-mediated transcriptional pathway would be involved in the AHR-dependent cell apoptosis by chrysin. The SRE-driven transcription is regulated by serum response factor (SRF)-ternary and complex factors (TCFs) complexes which are activated through ERK/MAPK and Rho/actin signaling pathways. The SRF/TCFs-SRE signal controls the transcriptions of several immediate early genes (IEGs) such as c-fos, JunB, Fos-B and Egr-1[24]. The most studied SRE-driven gene c-fos is known to play a key role in cell proliferation, transformation and differentiation [26]. Several reports have demonstrated that c-fos expression influences the induction of cell apoptosis in nerve cells, immune cells and colorectal cancer cell lines [27–31]. The c-fos gene expression was also increased by treatment of chrysin in AHRdependent way (Fig. 8B). It has been reported that TNFα enhances SRF-SRE binding activity and SREdrive IEGs expression [32–33]. Therefore, we concluded that the AHR-dependent induction of Tnfα and β genes expression by chrysin would lead to upregulate the SRE-mediated transcriptional pathway and the expression of SRE-driven IEGs (Fig. 9). The increased SRE-driven IEGs may contribute to the mechanism of chrysin-induced apoptosis (Fig. 9).
Fig.9. Schematic representation of the mechanism in chrysin-induced cell apoptosis through AHR.
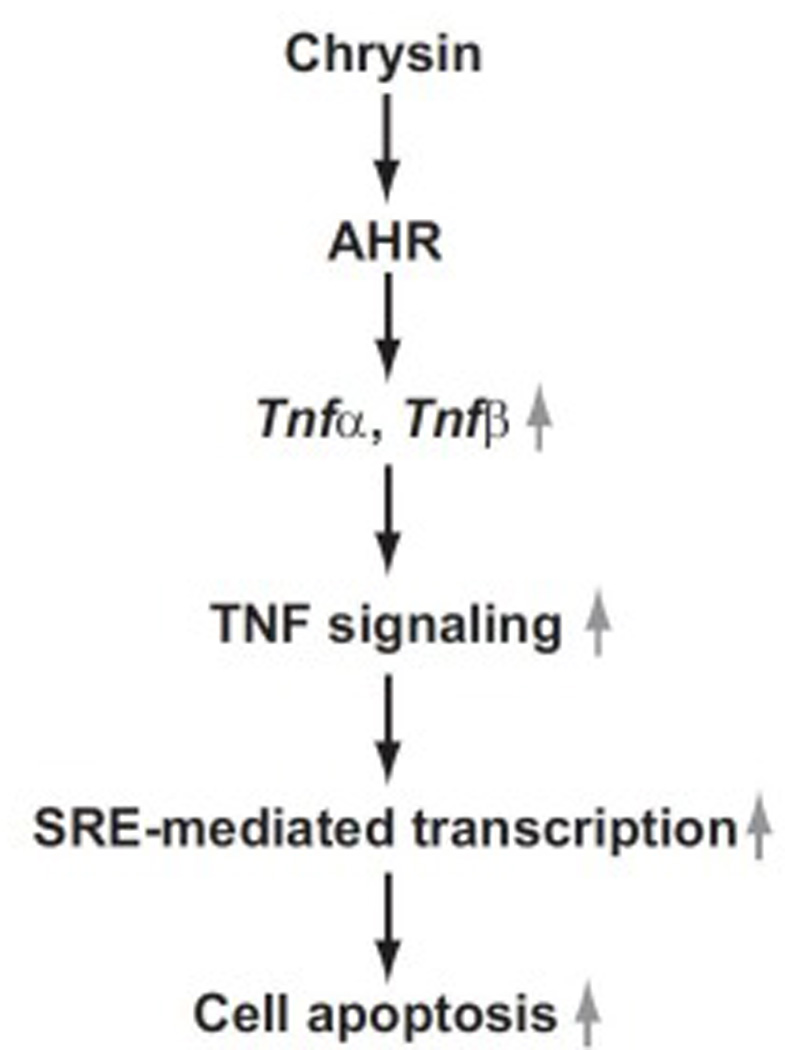
The Aryl hydrocarbon receptor (AHR) is a cytoplasmic receptor, transcriptional factor and member of the PAS protein family which responds to environmental and nutritional compounds such as halogenated aromatic hydrocarbons and 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) [34]. Once the AHR ligand (agonist) binds to AHR, AHR dimerizes with AHR nuclear translocator (ARNT) in the nucleus [35, 36]. The AHR/ARNT dimer recognizes dioxin-responsive elements (DREs) located in the enhancer region of AHR-driven genes such as the xenobiotic metabolizing enzyme cytochrome P450 1A1 (CYP1A1) [35, 36]. The AHR-mediated transcriptional regulation facilitates diverse biological responses such as adaptive and toxic responses to xenobiotics in the body [35, 36]. We hypothesized that AHR was involved in chrysin-induced apoptosis by controlling Tnfα and Tnfβ gene expression. This suspicion arose from previous studies demonstrating interactions between AHR and chrysin as well as AHR and TNF. Shu et al. screened the agonist/antagonist activity of 15 chemical compounds including chrysin and revealed chrysin as a potent agonist of AHR in hepatoma and breast cancer cell lines [17]. Additionally, we previously demonstrated that TNFR1, TNFR2 and interleukin-1 receptor were required for AHR-mediated liver toxicity induced by TCDD in mice [37, 38]. Finally, Vogel and Abel have reported that Tnfα gene expression was induced by exposure of TCDD in human breast cancer cells [21]. Therefore, to investigate our hypothesis of AHR regulating chrysin-induced apoptosis, we generated AHR si-RNA expressing HCT116 cells characterized by reduced AHR mRNA expression (Fig. 5A). In the si-AHR expression cells, the elevation of AHR activity by FICZ and chrysin was also suppressed. (Fig. 5B–C). In addition to inhibition of AHR activity, chrysin induction of Tnfα and Tnfβ expression was significantly suppressed (Fig. 7A–B) and resistance to chrysin-induced apoptotic cell death arose in the si-AHR expressing cells compared to control cells (Fig. 6A–B). These results indicate that chrysin-induced upregulation of Tnfα and Tnfβ genes and cell apoptosis is controlled in AHR-dependent fashion. Presumably the AHR-dependent suppression of Tnf genes expression may yield the resistance to chrysin-induced apoptosis.
The mechanism of AHR-dependent regulation of Tnfα and Tnfβ genes expression has not been elucidated. The proximal −1kb promoter region of human Tnfα gene does not reveal an AHR-binding DRE sequence [39]. In contrast to the Tnfα gene, human Tnfβ gene has one copy of core sequence of DRE (5’-GCGTG-3’ or 5’-CACTG-3’) on distal, proximal and downstream promoter regions (distal: −1,079 to −1,074, proximal: −59 to −54, downstream: +399 to +404) [40]. We speculate that both Tnfα and Tnfβ genes may be regulated through the DRE sequences in promoter regions of Tnfβ gene similar to the regulation mechanism of Cyp1a1 and Cyp1a2 genes. Cyp1a1 and Cyp1a2 genes are characteristic AHRdriven batteries and located in a “head to head” configuration on chromosomal Cyp1a locus [41]. In mice, Cyp1a1 and Cyp1a2 genes are regulated by the same DREs located upstream of the Cyp1a1 gene, with the DREs enhancer region 13.9 kb removed from the transcriptional start site of Cyp1a2 gene [41, 42]. Interestingly, human Tnfα and Tnfβ genes are adjacent on the TNF/LT gene locus of human chromosome 6 [43, 44]. Although Tnfα and Tnfβ genes are sequentially and transcriptionally oriented, not “head to head” [44], Tsytsykova et al. illustrated that the Tnfα gene was regulated by an enhancer region that lies 5 kb upstream of the Tnfβ gene and 9kb downstream of Tnfα due to intrachromosomal looping [43]. Therefore, given the concomitant TNFα and β gene induction by chrysin (Fig. 2A–B), Tnfα and β genes may be regulated through the same DREs located in the promoter regions of Tnfβ gene due to the mechanism of intrachromosomal interaction.
In our current study, we have demonstrated that chrysin showed significant chemopreventive effect in human colorectal cancer cell lines, mainly as a consequence of apoptotic cell death. Because chrysin treatment induced Tnfα and Tnfβ gene expression and activated multiple TNF-mediated signaling pathways, our findings suggest that TNF-mediated apoptosis underlies chrysin’s effects. Additionally, we have identified a novel pathway in which the transcriptional factor AHR appears to orchestrate TNF-mediated apoptosis following ligand binding of chrysin. Chrysin-AHR binding presumably regulates the Tnfα and β genes expression and the TNF-mediated SRE pathway, which in turn drive apoptosis (Fig. 9), since it is known that several SRE-driven genes are associated with induction of apoptosis [26, 30, 45] and are identified as up-regulated genes by exposure of AHR agonist [46, 47]. Therefore, future studies can focus on further clarifying the pathway by which AHR regulates TNF-SRE mediated apoptosis after chrysin treatment. Characterizing the mechanism of chrysin-induced cell death in colon cancer cells will allow better understanding of its putative role as a cancer chemopreventive agent.
Research Highlights.
-
►
The treatment of flavonoid chrysin showed chemopreventive effect in human colorectal cancer cells via apoptosis pathway.
-
►
Up-regulation of pro-apoptotic cytokines tumor necrosis factor (Tnf) α and β genes and consequent activation of the TNF-mediated transcriptional pathway were involved in the chrysin-induced apoptosis.
-
►
The aryl hydrocarbon receptor, a ligand-receptor for chrysin, is required for the chrysin-induced apoptosis and the up-regulation of Tnfα and β gene expression in human colorectal cancer cells.
ACKNOWLEDGEMENTS
This work was supported by National Institutes of Health Grants K08ES017283 and R01ES020900 (GDK), and T32CA090217 (SMRK). We thank Anna L. Shen and Melanie M. Ivancic for reviewing the manuscript.
Abbreviations used are as follows
- AHR
aryl hydrocarbon receptor
- AP-1
activator protein 1
- ARNT
AHR nuclear translocator
- BAD
Bcl-2-associated death promoter
- CYP
cytochrome P450
- DRE
dioxin-responsive element
- ERK
extracellular signal-regulated kinase
- FICZ
6-formylindolo (3,2-b) carbazole
- IEGs
immediate early genes
- JNK
c-jun N-terminal kinase
- LTα
lymphotoxin α
- MAPK
mitogen-activated protein kinase
- NF-κB
nuclear factor κ-light-chain enhancer of activated B cells
- PYCARD
PYD and CARD domain containing
- rTdT
recombinant terminal deoxynucleotidyl transferase
- siRNA
small interfering RNA
- SRE
serum response element
- SRF
serum response factor
- TCDD
2,3,7,8-tetrachlorodibenzo-p-dioxin
- TCFs
ternary and complex factors
- TNF
tumor necrosis factor
- TNFR
TNF receptor
- TUNEL
terminal deoxynucleotidyl transferase-dUTP nick end labeling
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
None
REFERENCES
- 1.Edwards BK, Ward E, Kohler BA, Eheman C, Zauber AG, Anderson RN, Jemal A, Schymura MJ, Lansdorp-Vogelaar I, Seeff LC, van Ballegooijen M, Goede SL, Ries LA. Annual report to the nation on the status of cancer, 1975–2006, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer. 2010;116:544–573. doi: 10.1002/cncr.24760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel R, Desantis C, Jemal A. Colorectal cancer statistics, 2014. CA Cancer J Clin. 2014;64:104–117. doi: 10.3322/caac.21220. [DOI] [PubMed] [Google Scholar]
- 3.Andre T, Boni C, Navarro M, Tabernero J, Hickish T, Topham C, Bonetti A, Clingan P, Bridgewater J, Rivera F, de Gramont A. Improved overall survival with oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment in stage II or III colon cancer in the MOSAIC trial. J Clin Oncol. 2009;27:3109–3116. doi: 10.1200/JCO.2008.20.6771. [DOI] [PubMed] [Google Scholar]
- 4.van Gijn W, Marijnen CA, Nagtegaal ID, Kranenbarg EM, Putter H, Wiggers T, Rutten HJ, Pahlman L, Glimelius B, van de Velde CJ G. Dutch Colorectal Cancer. Preoperative radiotherapy combined with total mesorectal excision for resectable rectal cancer: 12-year follow-up of the multicentre, randomised controlled TME trial. Lancet Oncol. 2011;12:575–582. doi: 10.1016/S1470-2045(11)70097-3. [DOI] [PubMed] [Google Scholar]
- 5.Sauer R, Liersch T, Merkel S, Fietkau R, Hohenberger W, Hess C, Becker H, Raab HR, Villanueva MT, Witzigmann H, Wittekind C, Beissbarth T, Rodel C. Preoperative versus postoperative chemoradiotherapy for locally advanced rectal cancer: results of the German CAO/ARO/AIO-94 randomized phase III trial after a median follow-up of 11 years. J Clin Oncol. 2012;30:1926–1933. doi: 10.1200/JCO.2011.40.1836. [DOI] [PubMed] [Google Scholar]
- 6.Benson AB, 3rd, Venook AP, Bekaii-Saab T, Chan E, Chen YJ, Cooper HS, Engstrom PF, Enzinger PC, Fenton MJ, Fuchs CS, Grem JL, Hunt S, Kamel A, Leong LA, Lin E, Messersmith W, Mulcahy MF, Murphy JD, Nurkin S, Rohren E, Ryan DP, Saltz L, Sharma S, Shibata D, Skibber JM, Sofocleous CT, Stoffel EM, Stotsky-Himelfarb E, Willett CG, Gregory KM, Freedman-Cass DA N. National Comprehensive Cancer. Colon cancer, version 3.2014. J Natl Compr Canc Netw. 2014;12:1028–1059. doi: 10.6004/jnccn.2014.0099. [DOI] [PubMed] [Google Scholar]
- 7.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 8.Steward WP, Brown K. Cancer chemoprevention: a rapidly evolving field. Br J Cancer. 2013;109:1–7. doi: 10.1038/bjc.2013.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dorai T, Aggarwal BB. Role of chemopreventive agents in cancer therapy. Cancer Lett. 2004;215:129–140. doi: 10.1016/j.canlet.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 10.Wu X, Lippman SM. An intermittent approach for cancer chemoprevention. Nat Rev Cancer. 2011;11:879–885. doi: 10.1038/nrc3167. [DOI] [PubMed] [Google Scholar]
- 11.Key TJ, Schatzkin A, Willett WC, Allen NE, Spencer EA, Travis RC. Diet, nutrition and the prevention of cancer. Public Health Nutr. 2004;7:187–200. doi: 10.1079/phn2003588. [DOI] [PubMed] [Google Scholar]
- 12.Pan MH, Ho CT. Chemopreventive effects of natural dietary compounds on cancer development. Chem Soc Rev. 2008;37:2558–2574. doi: 10.1039/b801558a. [DOI] [PubMed] [Google Scholar]
- 13.Gonzalez CA, Riboli E. Diet and cancer prevention: Contributions from the European Prospective Investigation into Cancer and Nutrition (EPIC) study. Eur J Cancer. 2010;46:2555–2562. doi: 10.1016/j.ejca.2010.07.025. [DOI] [PubMed] [Google Scholar]
- 14.Batra P, Sharma AK. Anti-cancer potential of flavonoids: recent trends and future perspectives, 3. Biotech. 2013;3:439–459. doi: 10.1007/s13205-013-0117-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ren W, Qiao Z, Wang H, Zhu L, Zhang L. Flavonoids: promising anticancer agents. Med Res Rev. 2003;23:519–534. doi: 10.1002/med.10033. [DOI] [PubMed] [Google Scholar]
- 16.Khoo BY, Chua SL, Balaram P. Apoptotic effects of chrysin in human cancer cell lines. Int J Mol Sci. 2010;11:2188–2199. doi: 10.3390/ijms11052188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang S, Qin CH, Safe SH. Flavonoids as aryl hydrocarbon receptor agonists/antagonists: Effects of structure and cell context. Environ Health perspect. 2003;111:1877–1882. doi: 10.1289/ehp.6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rath PC, Aggarwal BB. TNF-induced signaling in apoptosis. J Clin Immunol. 1999;19:350–364. doi: 10.1023/a:1020546615229. [DOI] [PubMed] [Google Scholar]
- 19.Aggarwal BB, Gupta SC, Kim JH. Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood. 2012;119:651–665. doi: 10.1182/blood-2011-04-325225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mhyre AJ, Marcondes AM, Spaulding EY, Deeg HJ. Stroma-dependent apoptosis in clonal hematopoietic precursors correlates with expression of PYCARD. Blood. 2009;113:649–658. doi: 10.1182/blood-2008-04-152686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vogel C, Abel J. Effect of 2,3,7,8-Tetrachlorodibenzo-P-Dioxin on Growth-Factor Expression in the Human Breast-Cancer Cell-Line Mcf-7. Arch Toxicol. 1995;69:259–265. doi: 10.1007/s002040050168. [DOI] [PubMed] [Google Scholar]
- 22.Fan F, Yan B, Wood G, Viluksela M, Rozman KK. Cytokines (IL-1beta and TNFalpha) in relation to biochemical and immunological effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in rats. Toxicology. 1997;116:9–16. doi: 10.1016/s0300-483x(96)03514-7. [DOI] [PubMed] [Google Scholar]
- 23.Kovács KJ. c-Fos as a transcription factor: a stressful (re)view from a functional map. Neurochem Int. 1998;33:287–297. doi: 10.1016/s0197-0186(98)00023-0. [DOI] [PubMed] [Google Scholar]
- 24.Knoll B, Nordheim A. Functional versatility of transcription factors in the nervous system: the SRF paradigm. Trends Neurosci. 2009;32:432–442. doi: 10.1016/j.tins.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 25.Li X, Huang Q, Ong CN, Yang XF, Shen HM. Chrysin sensitizes tumor necrosis factor-alphainduced apoptosis in human tumor cells via suppression of nuclear factor-kappaB. Cancer Lette. 2010;293:109–116. doi: 10.1016/j.canlet.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 26.Angel P, Karin M. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta. 1991;1072:129–157. doi: 10.1016/0304-419x(91)90011-9. [DOI] [PubMed] [Google Scholar]
- 27.Oshitari T, Dezawa M, Okada S, Takano M, Negishi H, Horie H, Sawada H, Tokuhisa T, Adachi-Usami E. The role of c-fos in cell death and regeneration of retinal ganglion cells. Invest Ophth Vis Sci. 2002;43:2442–2449. [PubMed] [Google Scholar]
- 28.Colotta F, Polentarutti N, Sironi M, Mantovani A. Expression and involvement of c-fos and c-jun protooncogenes in programmed cell death induced by growth factor deprivation in lymphoid cell lines. J Biol Chem. 1992;267:18278–18283. [PubMed] [Google Scholar]
- 29.Smeyne RJ, Vendrell M, Hayward M, Baker SJ, Miao GG, Schilling K, Robertson LM, Curran T, Morgan JI. Continuous c-fos expression precedes programmed cell death in vivo. Nature. 1993;363:166–169. doi: 10.1038/363166a0. [DOI] [PubMed] [Google Scholar]
- 30.Preston GA, Lyon TT, Yin Y, Lang JE, Solomon G, Annab L, Srinivasan DG, Alcorta DA, Barrett JC. Induction of apoptosis by c-Fos protein. Mol Cell Biol. 1996;16:211–218. doi: 10.1128/mcb.16.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Inada K, Okada S, Phuchareon J, Hatano M, Sugimoto T, Moriya H, Tokuhisa T. c-Fos induces apoptosis in germinal center B cells. J immunol. 1998;161:3853–3861. [PubMed] [Google Scholar]
- 32.Li YP. TNF-alpha is a mitogen in skeletal muscle. Am J Physiol Cell Physiol. 2003;285:C370–C376. doi: 10.1152/ajpcell.00453.2002. [DOI] [PubMed] [Google Scholar]
- 33.Li YP, Schwartz RJ. TNF-alpha regulates early differentiation of C2C12 myoblasts in an autocrine fashion. FASEB J. 2001;15:1413–1415. doi: 10.1096/fj.00-0632fje. [DOI] [PubMed] [Google Scholar]
- 34.Nguyen LP, Bradfield CA. The search for endogenous activators of the aryl hydrocarbon receptor. Chem Res Toxicol. 2008;21:102–116. doi: 10.1021/tx7001965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hankinson O. The aryl hydrocarbon receptor complex. Annu Rev Pharmacol. 1995;35:307–340. doi: 10.1146/annurev.pa.35.040195.001515. [DOI] [PubMed] [Google Scholar]
- 36.Schmidt JV, Bradfield CA. Ah receptor signaling pathways. Annu Rev Cell Dev Bi. 1996;12:55–89. doi: 10.1146/annurev.cellbio.12.1.55. [DOI] [PubMed] [Google Scholar]
- 37.Pande K, Moran SM, Bradfield CA. Aspects of dioxin toxicity are mediated by interleukin 1-like cytokines. Mol Pharmacol. 2005;67:1393–1398. doi: 10.1124/mol.105.010983. [DOI] [PubMed] [Google Scholar]
- 38.Kennedy GD, Nukaya M, Moran SM, Glover E, Weinberg S, Balbo S, Hecht SS, Pitot HC, Drinkwater NR, Bradfield CA. Liver tumor promotion by 2,3,7,8-tetrachlorodibenzo-p-dioxin is dependent on the aryl hydrocarbon receptor and TNF/IL-1 receptors. Toxicol Sci. 2014;140:135–143. doi: 10.1093/toxsci/kfu065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lai ZW, Pineau T, Esser C. Identification of dioxin-responsive elements (DREs) in the 5' regions of putative dioxin-inducible genes. Chem Biol Interact. 1996;100:97–112. doi: 10.1016/0009-2797(96)03691-5. [DOI] [PubMed] [Google Scholar]
- 40.Yokley BH, Selby ST, Posch PE. A Stimulation-Dependent Alternate Core Promoter Links Lymphotoxin alpha Expression with TGF-beta 1 and Fibroblast Growth Factor-7 Signaling in Primary Human T Cells. J immunol. 2013;190:4573–4584. doi: 10.4049/jimmunol.1201068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nukaya M, Bradfield CA. Conserved genomic structure of the Cyp1a1 and Cyp1a2 loci and their dioxin responsive elements cluster. Biochem pharmacol. 2009;77:654–659. doi: 10.1016/j.bcp.2008.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nukaya M, Moran S, Bradfield CA. The role of the dioxin-responsive element cluster between the Cyp1a1 and Cyp1a2 loci in aryl hydrocarbon receptor biology. Proc Natl Acad Sci USA. 2009;106:4923–4928. doi: 10.1073/pnas.0809613106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsytsykova AV, Rajsbaum R, Falvo JV, Ligeiro F, Neely SR, Goldfeld AE. Activation-dependent intrachromosomal interactions formed by the TNF gene promoter and two distal enhancers. Proc Natl Acad Sci USA. 2007;104:16850–16855. doi: 10.1073/pnas.0708210104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Falvo JV, Jasenosky LD, Kruidenier L, Goldfeld AE. Epigenetic control of cytokine gene expression: regulation of the TNF/LT locus and T helper cell differentiation. Adv Immunol. 2013;118:37–128. doi: 10.1016/B978-0-12-407708-9.00002-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shaulian E, Karin M. AP-1 as a regulator of cell life and death. Nat Cell Biol. 2002;4:E131–E136. doi: 10.1038/ncb0502-e131. [DOI] [PubMed] [Google Scholar]
- 46.Puga A, Nebert DW, Carrier F. Dioxin induces expression of c-fos and c-jun proto-oncogenes and a large increase in transcription factor AP-1. DNA and Cell Biol. 1992;11:269–281. doi: 10.1089/dna.1992.11.269. [DOI] [PubMed] [Google Scholar]
- 47.Hoffer A, Chang CY, Puga A. Dioxin induces transcription of fos and jun genes by Ah receptor-dependent and -independent pathways. Toxicol Appl Pharmacol. 1996;141:238–247. doi: 10.1006/taap.1996.0280. [DOI] [PubMed] [Google Scholar]