

Cardiomyocyte-specific Bmal1 gene deletion in heart progresses to 1) diastolic dysfunction with significant age-dependent hypertrophy; 2) dilative hypertrophy marked with endocardial fibrosis and interstitial fibrosis in an age-dependent manner; and 3) age-dependent ventricular fibrosis displaying aggravated extracellular matrix deposition and defective resolution of the inflammation response.
Keywords: aging, Bmal1, circadian clock, extracellular matrix, inflammation, diastolic dysfunction
Abstract
The mammalian circadian clock consists of multiple transcriptional regulators that coordinate biological processes in a time-of-day-dependent manner. Cardiomyocyte-specific deletion of the circadian clock component, Bmal1 (aryl hydrocarbon receptor nuclear translocator-like protein 1), leads to age-dependent dilated cardiomyopathy and decreased lifespan in mice. We investigated whether cardiomyocyte-specific Bmal1 knockout (CBK) mice display early alterations in cardiac diastolic function, extracellular matrix (ECM) remodeling, and inflammation modulators by investigating CBK mice and littermate controls at 8 and 28 wk of age (i.e., prior to overt systolic dysfunction). Left ventricles of CBK mice exhibited (P < 0.05): 1) progressive abnormal diastolic septal annular wall motion and reduced pulmonary venous flow only at 28 wk of age; 2) progressive worsening of fibrosis in the interstitial and endocardial regions from 8 to 28 wk of age; 3) increased (>1.5 fold) expression of collagen I and III, as well as the matrix metalloproteinases MMP-9, MMP-13, and MMP-14 at 28 wk of age; 4) increased transcript levels of neutrophil chemotaxis and leukocyte migration genes (Ccl2, Ccl8, Cxcl2, Cxcl1, Cxcr2, Il1β) with no change in Il-10 and Il-13 genes expression; and 5) decreased levels of 5-LOX, HO-1 and COX-2, enzymes indicating impaired resolution of inflammation. In conclusion, genetic disruption of the cardiomyocyte circadian clock results in diastolic dysfunction, adverse ECM remodeling, and proinflammatory gene expression profiles in the mouse heart, indicating signs of early cardiac aging in CBK mice.
NEW & NOTEWORTHY
Cardiomyocyte-specific Bmal1 gene deletion in heart progresses to 1) diastolic dysfunction with significant age-dependent hypertrophy; 2) dilative hypertrophy marked with endocardial fibrosis and interstitial fibrosis in an age-dependent manner; and 3) age-dependent ventricular fibrosis displaying aggravated extracellular matrix deposition and defective resolution of the inflammation response.
the circadian clock is a timekeeping system that regulates physiological performance and behavior relative to day-night cycles. Oscillations in cardiovascular functions are firmly established, including time-of-day-dependent fluctuations in blood pressure, heart rate, and cardiac output (9, 10, 31). Night shift work and frequent time zone changes result in a dissociation between this intrinsic timekeeping mechanism and the environment, which is associated with increased risk of adverse cardiovascular effects (such as myocardial infarction and sudden cardiac death) (5, 15, 33). In mammals, the timekeeping system can be divided into two subsystems, the central (located in the superchiasmatic nucleus) and the peripheral (present in essentially all cells of the body) clocks. Central and peripheral clocks coordinate biological processes in a time-of-day-dependent manner, thus facilitating homoeostasis (34). In the past two decades, it has become clear that the circadian clock modulates numerous physiological processes, in addition to influences on pathological events (e.g., myocardial infarction) (43). The circadian clock mechanism is composed of several core components (PER1, PER2, PER3, CRY1, CRY2, CK1e, and TIM), including CLOCK (circadian locomotor output cycles protein kaput), Bmal1 (synonym: aryl hydrocarbon receptor nuclear translocator-like protein 1), and REV-ERBα. Genetic disruption of clock components in mouse models initiates metabolic abnormalities, including an increased incidence of adiposity and insulin resistance (38). In humans, interrupted sleep duration, altered sleep-wake cycle, or insomnia directly associates with disturbed circadian rhythm and sudden sensorineural hearing loss marked with reduced levels of PER1 and CRY2 in peripheral leukocytes (41). In rodent hearts, the impact of disruption of two main transcription factors, CLOCK and BMAL1, has been investigated (42). These studies reveal important roles for these clock components in the heart at the levels of contractility, electrophysiology, metabolism, signaling, translation, and transcription (7). Of these models, genetic ablation of BMAL1 in the heart elicits the most striking phenotype from 28 wk onwards: age-onset development of dilated cardiomyopathy associated with reduced life span. The mean survival for cardiomyocyte-specific Bmal1 deletion (CBK) mice was 33 wk, while no deaths were observed in littermate controls (i.e., Bmal1flox/flox) or wild-type (C57BL/6J) within one year of the study (42).
The mechanism(s) by which genetic ablation of Bmal1 in the heart results in cardiomyopathy and an accelerated aging phenotype is (are) currently unknown. We determined that Bmal1 deletion initiates an extracellular matrix (ECM) response which proceeds to an inflammatory response by 28 wk of age. Compared with littermate controls, germline Bmal1 deletion results in the development of chronic inflammation (6). Here, we report that ECM genes are increased in CBK mice at the age of 28 wk. As expected, with increased ECM deposition, the transforming growth factor (TGF-β) signaling cascade, including SMAD2 (Mothers against decapentaplegic homolog 2), was activated in CBK hearts by 28 wk of age. Thus Bmal1 deletion in mice initiated dilative hypertrophy, diastolic dysfunction, and ECM responses and an impaired resolution of inflammation axis with marked signs of accelerated early cardiac aging by 28 wk of age.
MATERIALS AND METHODS
Mice.
All animal procedures were conducted according to the “Guide for the Care and Use of Laboratory Animals” (8th ed., 2011) and were approved by the Institutional Animal Care and Use Committees at the University of Alabama at Birmingham. CBK (BMAL1flox/flox/α-MHC-CRE+/−) mice on the C57Bl/6J background were developed as previously described (12, 42). Male CBK and C57BL/6J background littermate control mice (control) were maintained on a standard diet. Both littermate control and CBK mice were subjected to pulsed-wave and Doppler echocardiography at 8 and 28 wk of age. A total of 33 mice was used in the experiment, 6 for control at 8 wk of age, 12 for control at 28 wk of age, 5 for 8-wk-old CBK, and 10 for 28-wk-old CBK.
Diastolic function measurements using echocardiography.
Diastolic function was assessed in mice using echocardiography; assessments were made in mice under 1.5–2.0% isoflurane in an oxygen mix using Vevo 770 (VisualSonics, Canada) high-resolution imaging system. This system was equipped with a RMV707B transducer containing a 30-MHz high-frequency probe, and data were analyzed using VisualSonics software. Temperature and heart rates of mice were monitored and kept in ranges of 37.0 ± 0.5°C and >450 ± 100 beats/min, respectively. The mouse was placed in supine position on adjustable rail to allow coordination of the ultrasound transducer. The placement of the transducer was manipulated to obtain multiple views, such as parasternal LV long- and short-axis, apical four-chamber, and suprasternal views. Parasternal long-axis views were obtained by aligning the scan head at 30–45° counterclockwise to the head of the mouse. The images obtained included LV long-axis B mode, left atrium M-mode, tissue Doppler imaging of LV, and pulsed-wave Doppler of pulmonary venous flow. Parasternal short-axis B mode images were acquired by rotating ∼90° clockwise from the parasternal long-axis view. Pulsed-wave Doppler images of the mitral valve, showing an apical four-chamber view, were obtained by moving the scan head transverse at the lower left side of the thorax. Aortic valve velocity was obtained via a pulsed-wave Doppler image in suprasternal view.
Necropsy of control and CBK mice.
Mice were anesthetized with 2% isoflurane in an oxygen mix and injected with 4 IU/g heparin. After 5 min, the carotid artery was cut and plasma from the blood was collected. The left ventricle (LV) was injected with 2 ml cardioplegic solution to arrest the heart in a diastolic state, and the whole heart and lungs were then removed. The right ventricle and LV were separated. Lungs were removed and all masses were recorded. The tibia was removed and placed in 1.5 M potassium hydroxide to digest for 24 h and the length was measured with a vernier caliper. The LV was cut into 3 sections, with the middle being stored in 10% formalin for 24 h, then preserved in 70% ethanol for histological analysis and the remaining LV sections were snap-frozen and stored at −80 Co for further cellular and molecular analysis.
LV wheat germ agglutinin staining.
To analyze myocyte area, wheat germ agglutinin (WGA) staining was performed. Formalin-fixed, paraffin-embedded LV blocks were sectioned at 5-μm thickness, deparaffinized, and rehydrated, as previously described (26). Alexa Fluor 488-conjugated WGA (Invitrogen) solution was added to the tissue in 1:1,000 dilution for 1 h. Samples were then washed with PBS and mounted with prolonged gold antifade reagent (Invitrogen). Myocyte area and cardiac fibrosis were quantified from 5–6 high-power fields per section using ImageJ software (NIH). Data for each group were calculated from 30 cardiomyocyte sections and 4–5 mice/group.
LV picrosirius red staining.
For picosirius red (PSR) staining, paraffin-embedded, unstained sections of LV tissue for control and CBK mice were deparaffinized in Citrisolv and rehydrated through subsequent washes of ethanol. After a wash with water, phosphomolybdic acid 0.2% aqueous was added to the slides. Another subsequent wash with water was followed by addition of sirius red, 0.1% in saturated picric acid (26357-02) and then application of 0.01 N hydrochloric acid. Following this step, the slides were dehydrated and then mounted using permount. The slides were allowed to dry for image analysis. The collagen staining in polarized light was determined using Image Pro Premier 64-bit software.
RT2 profiler inflammatory and ECM PCR array.
Frozen samples of LV tissue from CBK and control mice were used for RNA extraction. Four to eight milligrams of LV tissue was homogenized using a sonic dismembrator (Fisher Scientific, Amplitude: 10–100) and RNA was isolated with TRIzol as per manufacturer's instruction. RNA concentration was determined using the ND1000 nanodrop, and cDNA synthesis was performed with a RT2 first strand kit using 0.5 μg RNA per sample. Each sample was prepared on a RT2-PCR plate for inflammatory genes (Inflammatory Cytokine and Receptor by Qiagen PAMM-011E) and extracellular matrix genes (Mouse ECM and Adhesion molecules by Qiagen PAMM-011E), then run on an ABI 7900HT. Gene levels were normalized to hypoxanthine phosphoribosyltransferase 1 (HPRT-1) as the housekeeping gene control. The results were reported as 2−ΔCt values.
RT-PCR for measurements of resolving gene transcripts.
For qPCR, reverse transcription was performed with 2.5 μg of total RNA using SuperScript VILO cDNA Synthesis Kit (Invitrogen). Quantitative PCR for proresolving (hmox-1, alox-5, ptgs-1) and proinflammatory (alox-12 and alox-15) genes was performed using Taqman probes (Applied Biosystems) on master cycler ABI, 7900HT. The mRNA expression was normalized with the reference genes (β-Actin). The results were reported as 2−ΔCt (ΔΔCt) values. All the experiments were performed in duplicates with n = 3–4 mice/group.
LV protein extraction and immunoblotting.
The LV tissue from CBK and control mice was homogenized in reagent A [1× PBS from Invitrogen, without calcium and 1× proteinase inhibitor (PI)]. Homogenates were centrifuged at 14,000 rpm for 5 min at 4°C, and supernatant was snap-frozen in liquid nitrogen and used as the fraction A soluble protein. The pellet was resuspended reagent B (Reagent 4 from Sigma and 1× PI). The new homogenous solution was snap-frozen and used as fraction B insoluble protein. Total protein content was determined using the Bradford assay (26). A total of 10 μg protein was loaded, electrophoresed on Criterion XT Bis-Tris 4–12% 18-gel, MOPS buffer and transferred to a nitrocellulose membrane using material from Bio-Rad. The blots were probed with primary antibody [COX-2 (abcam) 1:1,000, 5-LOX (abcam) 1:1,000, HO-1 (Enzo lifescience) 1:2,000. Collagen I (abcam) 1:1,000, Collagen III (abcam) 1:1,000, TIMP-1(abcam) 1:1,000, MMP-9 (abcam) 1:1,000, phospho-Smad2/3 (Ser465/467) 1:1,000, and total smad2 (millipore) 1:1,000] overnight at 4°C followed by secondary antibody (Bio-Rad). The blots were stripped using stripping buffer (ThermoFisher, cat no. 46428) and reprobed for β-actin and total-smad2 as loading control. The proteins were detected using the femto chemiluminescence detection system (Pierce Chemical, Rockford, IL). Densitometry was performed using ImageJ software.
Statistical analysis.
Data are expressed as means and SE. Statistical analyses were performed using Graphpad Prism 5. Analysis of variance (ANOVA) followed by Newman-Keuls post hoc test was used for multiple comparisons. All immunoblotting densitometry data were normalized to β-actin/total protein lane. For 2-group comparison, the Student's t-test (unpaired) was applied. P < 0.05 was considered as statistically significant.
RESULTS
CBK mice develop age-related cardiac hypertrophy.
To determine if CBK mice develop age-dependent changes in LV mass, we measured the weights of the right and left ventricles at 8 and 28 wk. At 28 wk of age, CBK mice had higher RV mass, RV/body weight and larger LV mass/body weight and LV mass/tibia ratios than littermate 28-wk-old controls (Table 1); in contrast, no genotype-dependent effects were observed at 8 wk of age. LV cardiac hypertrophy was next assessed by measuring myocyte area cross-sections using WGA staining. Myocyte area was not different between control and CBK mice at 8 wk of age (Fig. 1, A and B). However, at 28 wk of age, CBK mice showed a threefold increase in the surface area of myocyte, indicative of cardiomyocyte hypertrophy (Fig. 1, A and B).
Table 1.
Necropsy parameters at 8 wk and 28 wk in control and CBK mice
| Groups: | Control |
CBK |
||
|---|---|---|---|---|
| Age/Sample Size: | 8 wk (n = 6) | 28 wk (n = 12) | 8 wk (n = 5) | 28 wk (n = 10) |
| Body weight, g | 26 ± 1 | 32 ± 1 | 25.8 ± 0.8 | 28.9 ± 0.9* |
| Tibia, mm | 17 ± 0.1 | 18 ± 0.1 | 17 ± 0.1 | 18 ± 0.2 |
| Spleen, mg | 73 ± 7 | 90 ± 13 | 75 ± 8.6 | 67 ± 3 |
| RV, mg | 18 ± 1 | 24 ± 1 | 22 ± 1 | 26 ± 2 |
| LV, mg | 83 ± 2 | 97 ± 3 | 96 ± 3* | 109 ± 8 |
| RV/BW | 0.70 ± 0.03 | 0.77 ± 0.04 | 0.86 ± 0.04 | 0.91 ± 0.07 |
| LV/BW | 3.1 ± 0.07 | 3.0 ± 0.09 | 3.7 ± 0.05 | 3.82 ± 0.31* |
| Lung wet weight, mg | 155 ± 13 | 167 ± 12 | 144 ± 7 | 180 ± 9 |
| Lung dry weight, mg | 29 ± 2 | 35 ± 2 | 31 ± 2 | 37 ± 3 |
| LV/tibia | 4.8 ± 0.1 | 5.6 ± 0.2 | 5.4 ± 0.2 | 6.1 ± 0.4 |
Values are means ± SE.
RV, right ventricle; LV, left ventricle; BW, body weight; Control, littermate; CBK, cardiomyocyte-specific Bmal-1 deleted knockout.
P < 0.05 vs. wild type.
Fig. 1.

Cardiomyocyte-specific Bmal1 knockout (CBK) mice promoted age-onset dilative hypertrophy and diastolic dysfunction. A: images of wheat germ agglutinin (WGA) at 8 and 28 wk representing increase in cardiomyocyte size indicating age-associated hypertrophy at 28 wk in CBK mice (scale bars: 10 μm). B: quantitation of cardiomyocyte area/section per mouse indicated hypertrophic response at 28 wk of age in CBK mice. *P < 0.05 vs. age-matched control (CON); n = 5 mice/group. C: representative echocardiographic images with abnormal diastolic septal annular wall motion indicating diastolic dysfunction in CBK mice at 28 wk compared with age-matched controls.
CBK mice develop age-dependent diastolic dysfunction.
CBK mice have been reported to develop an age-dependent dilated cardiomyopathy (initially observed after 28 wk of age), with a shorter lifespan compared with control mice (42). To determine whether development of diastolic dysfunction occurred at a younger age in CBK mice, we measured diastolic function in CBK and control mice using Doppler echocardiography at 8 and 28 wk. At 8 wk of age, no significant differences were observed for markers of diastolic function between control and CBK mice (Table 2). However, at 28 wk of age, CBK mice showed changes in several key functional parameters, including increased mitral valve E/A ratio, abnormal diastolic septal annular wall motion, and reduced pulmonary venous flow (Table 2; Fig. 1C). Mitral valve E/A ratio was ∼1.5 in control mice at both 8 and 28 wk; in comparison, the E/A ratio was progressively increased >2 from 8 to 28 wk in CBK mice. The 28-wk-old CBK mice showed increased left atrium diameter and increase in left atrium filling pressure compared with both age-matched controls, indicating multiple signs of diastolic dysfunction. End-diastolic volume (EDV) and end-systolic volume (ESV) were increased, and ejection fraction (EF; P < 05) was significantly lower for CBK mice at 28 wk of age compared with all other groups, implying a diastolic dysfunction and clear indication of systolic dysfunction in aging-prone CBK mice.
Table 2.
Summary of diastolic function measurements using pulsed-wave and tissue Doppler echocardiography in control and CBK mice at 8 and 28 wk of age
| Groups: | Control |
CBK |
||
|---|---|---|---|---|
| Age/Sample Size: | 8 wk (n = 6) | 28 wk (n = 7) | 8 wk (n = 5) | 28 wk (n = 5) |
| E/A | 1.69 ± .07 | 1.55 ± 0.05 | 2.06 ± 0.17 | 2.74 ± 0.79* |
| Isovolumic relaxation time, ms | 14.6 ± 1.6 | 15.0 ± 1.5 | 20.4 ± .9 | 18.1 ± 2.1 |
| Isovolumic contraction time, ms | 16.0 ± 1.0 | 18.0 ± 1.6 | 16.0 ± 1.0 | 17.0 ± 2.3 |
| Left atrium diameter, mm | 2.4 ± 0.2 | 2.5 ± 0.07 | 2.4 ± 0.13 | 3.0 ± 0.33* |
| E′/A′ | 1.44 ± 0.03 | 1.40 ± 0.04 | 1.46 ± 0.02 | 1.5 ± 0.11 |
| E/E′ | 29.2 ± 2.1 | 31.3 ± 3.1 | 40.1 ± 3.6 | 38.0 ± 5.6 |
| Pulmonary venous flow-systole, ms | 237 ± 33 | 257 ± 22 | 257 ± 28 | 153 ± 55* |
| Pulmonary venous flow-diastole, ms | 475 ± 14 | 489 ± 41 | 510 ± 39 | 264 ± 88* |
| Pulmonary venous flow-AR, ms | 143 ± 21 | 122 ± 12 | 117 ± 14 | 100 ± 21 |
| Pulmonary venous flow-AR duration | 19 ± 1 | 21 ± 1 | 17 ± 1 | 22.7 ± 2 |
| End-diastolic volume, μl | 71 ± 4 | 74 ± 4 | 77 ± 7 | 112 ± 32* |
| End-systolic volume, μl | 32 ± 4 | 32 ± 6 | 37 ± 4 | 85 ± 31* |
| Ejection fraction, % | 56 ± 3 | 58 ± 5 | 52 ± 3 | 28 ± 7* |
Data are means ±SE.
Control, littermate control; E, velocity of early mitral flow; A, velocity of late mitral flow; E′, early peak velocity of septal annulus; A′, late peak velocity of septal annulus; E/E′ index of left atrial filling pressure; AR, arterial reverse.
P < 0.05 vs. wild type.
CBK mice exhibit interstitial and endocardial fibrosis at 28 wk of age.
Since diastolic dysfunction and cardiac hypertrophy primarily contribute to stiffening of the left ventricle, we next examined the intensity of fibrosis in control and CBK hearts at 8 and 28 wk of age. Myocardial collagen content was slightly higher at 8 wk of age in CBK, compared with control, examined using picrosirius red staining (Fig. 2A, PSR images, and Fig. 2B, %stained area). However, at 28 wk of age, CBK mice showed even greater interstitial and endocardial ECM deposition than age-matched controls, as shown in PSR-stained LV images and quantification of stained areas (Fig. 2, A–C). At 28 wk of age, CBK mice also displayed a clear hypertrophic response with larger dilation compared with control mice, as shown by representative 1.25× LV middle pieces (Fig. 2D). Along with 1.25× magnification, 40× magnification hematoxylin and eosin images are shown for 28-wk-old mice to indicate the hypertrophy of the cardiomyocytes in CBK mice (Fig. 2D) at 28 wk but not at 8 wk. Further, LV sections at 28 wk of age were also imaged using polarized light, where large collagen fibers appear yellow/orange, and thinner fibers appear green (20). Plane polarized light images showed that CBK and control mice collagen deposition varied in thickness, with CBK displaying elevated levels of the thicker, orange-tinted type I collagen fiber and controls showing more of the thinner, green/yellow type III collagen fibers (Fig. 2C, bottom panel). The orange collagen fibers in CBK mice at 28 wk of age appeared to be denser compared with age-matched controls, indicating an accelerated fibrotic response.
Fig. 2.

CBK mice triggered collagen deposition and dilation in left ventricle (LV) compared with controls. A: LV middle region images (40× magnification) stained with picrosirius red (PSR) at 8 and 28 wk for wild-type (WT) and CBK mice indicated higher interstitial collagen deposition in CBK mice at 28 wk. Intense red color indicates collagen deposition. B: quantification of collagen density in CBK mice compared with control mice at 28 wk of age; 4–5 images/mouse, n = 5 mice/group. C: 10× magnification images of PSR-stained 28 wk WT and CBK mice show interstitial and endocardial fibrosis (black arrows, top panel). Under plane-polarized light endocardial fibrosis is marked by an orange color (white arrows, bottom panel). D: hematoxylin- and eosin (H&E)-stained LV middle cavity (1.25× magnification) indicates hypertrophy and dilation at 28 wk compared with age-matched control. H&E LV staining indicates dilative hypertrophy at 28 wk of age (40× images). *P < 0.05 vs. age-matched controls (CON).
CBK mice displayed pathological remodeling of ECM transcripts.
Data presented thus far suggest that ventricular hypertrophy in CBK mice, indicated by an increase in size, shape, mass and LV dysfunction, is associated with the changes in collagen density. Thus, to elucidate the mechanism which initiated the reactive or reparative fibrosis in CBK hearts, we performed an 84-gene ECM array at 8 and 28 wk for control and CBK mice. At 8 wk, none of the 84 genes were statistically different indicating reparative or adaptive fibrosis (Fig. 3, A and B). However, at 28 wk, CBK mice showed a robust upregulation in 20 genes, indicating reactive fibrosis (Fig. 3A). The upregulation of MMP-9, MMP-13, MMP-14, and MMP-1a is consistent with a rapid alteration of the collagen weave, known to lead to increased stiffness, muscle fiber slippage, and an increase in chamber size. Further activation of genes encoding for collagen types I-V (Col1a1, Col2a1, Col3a1 Col4a1, Col4a2, Col4a3, Col5a1) in CBK mice suggested the presence of active interstitial fibrosis. Furthermore, increased Timp-1 and Timp-3, along with MMPs, in CBK mice suggests a potential stimulation of feedback loop. The upregulation of Icam-1, Thbs3, Vcan, Itgax, Lamc1, Tnc, and Emilin-1 indicates an activated reparative fibrosis (Fig. 3B). Thus our data suggest a hypertrophic and fibrotic response in CBK mice at 28 wk, which overlaps with myocyte hypertrophy and accelerated ECM deposition indicating signs of early cardiac aging.
Fig. 3.

Aging-prone CBK mice stimulated early ventricular fibrotic response and activated phosphorylation of Smad2 at 28 wk of age. A: Venn diagram presenting changes in ECM and cell adhesion genes expression in LV of CBK mice compared with age-matched control littermate control. Black color genes indicate no change in gene expression. Red color genes indicate an increased expression and blue color genes indicate decrease in gene expression compared with control littermate control. P < 0.05 for all the increased and decreased genes compared with age-matched control. B: change in extracellular matrix (ECM)-mediated pathways at 28 wk of age in CBK mice. C: immunoblot representing phosphorylation of smad2/3 (ser465/467) in control and CBK mice. D: densitometry analysis of phospho-Smad2/3 control and CBK mice at 8 and 28 wk. *P < 0.05 vs. age-matched control. $P < 0.01 vs. 8-wk-old CBK mice.
CBK mice evidenced the activation TGF-β pathway.
Upregulation of TGF-β isoforms, and activation of the Smad2/3 and Smad1/5 pathways, is typically observed during the cardiac fibrotic response (40). Thus we investigated the activation status of Smad2/3 in CBK hearts. Phosphorylation of Smad2/3 at serine 465/467 was increased in LV of CBK mice at 28 wk of age (Fig. 3, C and D), suggesting TGF-β activation. In addition, we also observed phosphorylation of Smad2/3 at 8 wk in CBK mice indicating activation of Smad2/3 occurs as an earlier event prior to reparative fibrosis. Thus an early event in the progression of LV remodeling and development of dilated cardiomyopathy in CBK hearts appears to be Smad2/3 activation and initiation of fibrosis.
Aging-prone CBK mice showed accelerated ECM remodeling.
CBK mice displayed higher collagen content, accelerated fibrosis, and ECM gene response in mRNA levels, which was initiated as reparative fibrosis by 8 wk of age, and progressed to reactive pathological fibrosis by 28 wk of age. To confirm these changes at the protein level at 28 wk, we performed immunoblotting. Protein analysis revealed an increase in the expression levels of collagen I and III in CBK hearts (Fig. 4, A–E). Similarly, matrix metalloproteinase-9 (MMP-9) with TIMP-1 were increased in CBK hearts at 28 wk of age.
Fig. 4.

Aging-prone CBK mice showed accelerated extracellular matrix remodeling. A: immunoblot representing increase in expression of MMP-9, TIMP-1, and collagen I and III in LV of CBK mice compared with littermate controls at 28 wk of age. Densitometric analyses of MMP-9 (B), TIMP-1 (C), and collagen I (D) and III (E); β-actin was used as loading control. *P < 0.05 vs. age-matched control; n = 5 mice/group.
CBK hearts showed impaired resolution of inflammation with aging.
Given that cardiac dysfunction is often associated with delicate imbalances in proinflammatory and resolution of inflammation mechanisms, an array of inflammatory transcripts was determined in control and CBK hearts at 8 and 28 wk of age. Inflammatory cytokine and receptor array showed that at 8 wk there were only 3 genes (i.e., Il2rb, Il6rb, spp1; all P < 0.05) increased in CBK mice and 1 gene decreased (i.e., Mif; P < 0.05) compared with age-matched controls (Fig. 5, A and B). These changes revealed no signs of inflammation noticed at 8 wk. In contrast, by 28 wk of age, several inflammatory pathways were upregulated in CBK mice compared with controls (Fig. 5A). The CBK mice showed several genes commonly involved in chemokine binding, neutrophil chemotaxis, T cell migration, and leukocyte chemotaxis (Cxcl1, Cxcr2, Ccr2, Ccl6, Cxcl5) which were significantly upregulated. Simultaneously, the gene markers for lymphocyte proliferation (Ccr7, Ccr5, and CXcl13) were downregulated in CBK mice. Thus dysregulation of several cytokines/chemokines and adhesion molecules genes indicates that Bmal1 deletion developed unresolved and chronic inflammation in CBK mice at 28 wk, and aggravated ECM gene response. At 28 wk of age, CBK mice showed rapid increase in proinflammatory cytokines (i.e., ccl8 and il-1β increased, without change in il-10 and il-13), which are differentiated cytokines for proresolving genes (Fig. 5C), indicating a defective resolution axis. Bmal1 deletion in CBK mice triggers a defective inflammation resolution axis and signs of early cardiac aging, and adverse ECM remodeling, and thereby impaired LV function. Thus, to understand how Bmal1 deletion affects the inflammation-resolution axis at 28 wk, we determined mRNA levels of resolving genes. The CBK mice showed a significant decrease in proresolving transcripts of Hmox-1, Alox5, and Ptgs-2 (3, 17, 19) with no change in levels of inflammation promoting transcripts Alox12 and Alox15 (21) compared with age-matched controls (Fig. 6, A and B). Further, our immunoblotting analyses validated that not only the mRNA levels were affected in CBK mice, but also the protein levels of 5-LOX, COX-2, and HO-1 were significantly lower in CBK mice at 28 wk of age compared with age-matched controls (Fig. 6, C and D), indicating that altered circadian clock from Bmal1 deletion leads to an impaired inflammation resolution axis compared with control mice.
Fig. 5.

CBK mice displayed higher inflammation than age-matched control. A: Venn diagram presenting change in inflammatory cytokines and receptor genes expression in LV of CBK mice at 28 wk of age compared with age-matched control littermate control. Black color genes indicate no change in expression. Red color genes indicate an increase in expression and blue color genes indicate decrease in expression. *P < 0.05 for all the increased or decreased genes compared with age-matched control. B: change in inflammation-mediated pathways at 28 wk of age in CBK mice. C: bar graph depicting change in proinflammatory and proresolving gene at 28 wk of age in CBK mice compared with control. *P < 0.05 vs. age-matched control; n = 4 mice/group.
Fig. 6.

CBK mice develop a defective inflammation-resolution axis. mRNA expression of Alox-5, Hmox-1, ptgs-2 (A) and Alox-12, Alox-15 (B). C: immunoblot representing 5-LOX, COX-2, and HO-1 in control and CBK mice at 28 wk of age. D: densitometry analyses of 5-LOX, COX-2, and HO-1. β-Actin was used as loading control. *P < 0.05 vs. age-matched control; n = 5 mice/group.
DISCUSSION
Central and peripheral circadian clocks influence multiple aspects of cellular physiology, and as a consequence, disruption of clock function invariably results in cardiovascular pathology (11, 32). In this study, we defined the role of Bmal1 in age-related LV remodeling and diastolic dysfunction, through characterization of control and CBK mice at 8 and 28 wk of age. Importantly, the present study was aimed towards addressing the role of Bmal1 in age-onset cardiac remodeling rather than time-of-day events. Our results highlighted that cardiac-specific deletion of Bmal1 in mice 1) initiated dilative ventricular hypertrophy and diastolic dysfunction; 2) activated phospho-Smad2 (Ser465/467), triggered ECM responsive genes, and increased collagen deposition; and 3) impaired the inflammation-resolution axis. Thus our study validates that deletion of Bmal1 showed early signs of cardiac aging by 28 wk of age, indicated by LV diastolic dysfunction, with increased fibrotic response and impaired resolution of inflammation (see Fig. 7).
Fig. 7.
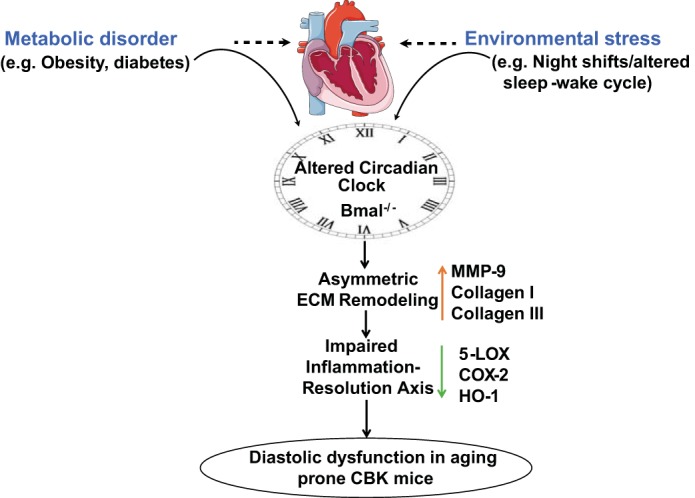
Schematic design showing altered circadian rhythm in CBK mice leads to an early age onset hypertrophy and fibrosis accompanied by diastolic dysfunction leading to defective resolution of inflammation. Adapted with Servier Medical Art with permission for use of clip art under CC-BY license (http://www.servier.com/content/servier-medical-art-now-licensed-under-creative-commons).
At the molecular level, the clock consists of rhythmic transcription, such as the core clock genes Bmal1 and CLOCK that are essential and are expressed in all mammalian cells, including cardiomyocytes (30). Germline ablation of Bmal1 results in a premature aging phenotype, as described previously, suggesting a central role of Bmal1 in lifespan and survival. A recently published study shows that cardiomyocyte-specific Bmal1 deletion leads to development of dilated cardiomyopathy associated with reduced lifespan (42). The present study indicates that cardiac-specific deletion of Bmal1 leads to ventricular hypertrophy by the age of 28 wk and was initiated by a reparative form of fibrosis at 8 wk of age (triggered ECM response but not inflammatory response). In fact, CBK mice displayed lower Mif (macrophage migration inhibitory factor) expression during puberty at 8 wk of age, but not 28 wk of age. Mif possesses oxidoreductase activity that may regulate macrophage migration and antagonizes myocardial hypertrophy, as the Mif deficient mice are prone to fibrosis and ventricular hypertrophy (22).
In CBK mice, increased LV mass-to-body weight ratio is an indication of cardiac hypertrophy which occurs due to continuous pressure load that may transit to heart failure due to diastolic dysfunction, systolic dysfunction, or a combination of both. CBK mice displayed indifferent inflammatory gene expression at 8 wk of age but marked induction of reactive fibrosis and signs of chronic inflammatory responses at 28 wk of age. These proinflammatory cytokines activate inhibitory mediators, such as TGF-β, that counteract inflammation, but promotes interstitial and perivascular fibrosis. Since ventricular hypertrophy is not associated with significant cardiomyocyte loss, future perspective studies are warranted to identify the mechanism related to TGF-β-mediated matrix deposition. Thus fibrotic response of ventricle is associated with early hypertrophy leading to the chamber dilation, ECM responses, and systolic dysfunction in CBK mice. These subsequent changes lead to the age-related LV dilative hypertrophy, ultimately leading to dilative cardiomyopathy. Present findings are consistent with an earlier study reported by Young et al. (42) that CBK mice begin to develop systolic dysfunction at 20–24 wk of age. Bmal1 plays a direct role in maintaining cardiomyocyte physiological function and a lack of Bmal1 in the heart results in development of premature and early cardiac aging. This was clearly indicated by development of the dilative cardiomyopathy at 28 wk of age as indicated by diastolic dysfunction.
The doubling of E/A ratio in CBK mice suggested a restrictive filling pattern and LV stiffness, indicating a severe form of diastolic dysfunction (44). The CBK mice also showed an increase in left atrium size, and decrease in S wave velocity confirmed an increased left atrial filling pressure with increase in E/E′, ratio which indicated early development of dilative cardiomyopathy. Lefta et al. (25) have shown that the genetic deletion of Bmal1 leads to dilative cardiomyopathy, indicating the importance of Bmal1 in cardiac pathology. Our study demonstrated a direct and definitive role of Bmal1 in development of dilative cardiomyopathy. Cardiac-specific Bmal1 deletion implicates the mechanical input that transduces into a biochemical event and modifies fetal gene transcription triggering to ECM response. As Bmal1 is a transcriptional regulator and regulates many posttranslational modifications (14), we observed that the deletion of Bmal1 led to minimal phosphorylation of SMAD2 at serine (465/467) at 8 wk which is then amplified at 28 wk of age. The multiple tyrosine-phosphorylated kinases and serine-threonine kinases are implicated in the signaling of hypertrophy found in the ECM (24, 37). Several pieces of evidence suggest that the disruptions of cell-cell and cell-ECM contact are sufficient in themselves to modulate changes which lead to ventricular hypertrophy. Bmal1 deletion leads to early cardiac aging and development of cardiovascular disorders. The role of TGF-β/SMAD signaling has been well demonstrated in the ECM deposition and fibrotic response (40). Ventricular hypertrophy lead to an increase in collagen production and pathological collagen deposition, which is characterized by both perivascular and interstitial fibrosis (4, 39). These studies linked a direct role of Bmal1 in controlling ECM genes, thereby leading to the deposition of interstitial and endocardial fibrosis. Increase in expression of the transcript levels of Col1a1, Col2a1, Col3a1 Col4a1, Col4a2, Col4a3, and Col5a1 along with Icam-1, Thbs3, Vcan, Itgax, Lamc1, Tnc, and Emilin-1 indicated an activated fibrotic response in 28-wk-old aging-prone CBK mice. Thus our study has defined the role of disturbed Bmal1 signaling in ventricular remodeling in the heart and presents the notion that Bmal1 has a control on multiple factors that regulate ECM remodeling and age-related inflammation.
A persistent, ungoverned inflammatory microenvironment drives myocardial infarction, and results in diurnal clustering in humans (28). Further, continuous, chronic slow-grade inflammation leads to accelerated aging (8). Previous studies have demonstrated chronic inflammation leads to progressive development of reparative to reactive fibrosis (13). A recent study by Nguyen et al. (29) demonstrated Bmal1 regulates the oscillation of inflammatory Ly6chi monocyte, which potentiates the chronic inflammatory response both locally and systemically. This potentially leads to chronic inflammatory diseases, such as myocardial infarction leading to heart failure. Predominantly, CBK mice show one of the most prevailing phenotypes of early cardiac aging with chronic inflammation (6, 23). Adrenal-specific Bmal1 knockout mice exhibit a pulsating plasma corticosterone secretion, while mice with a Bmal1 deficiency exclusively within pancreatic β cells display hypoinsulinemia and diabetes (27, 36). Not only cardiomyocytes but also immune cells i.e., particularly leukocytes, are influenced by sleep duration and sleep-wake up cycle, which peaks in the circulation during the resting phase and decreases during the active period. The proinflammatory cytokines tumor necrosis factor (TNF) and interleukin-1β (IL-1β) peak during the onset of the active phase, implicating association of circadian rhythms and the onset of diseases (16). Recent studies have shown that mice with disrupted circadian clock expressed substantially lower levels of Ccl2 mRNA compared with wild-type mice (35). The studies also showed oscillations in the serum levels of IL-6, IL-12, CCL2, CCL5, and CXCL1 when the mice are stimulated with lipopolysaccharide (LPS); however, these oscillations are absent in Bmal1−ma macrophages and Nr1d1−r1 mice (35). As leukocytes are known to display higher phagocytic ability and cytotoxicity during the active phase, the disruption of the circadian clocks leads to acute inflammatory insults (2, 18). Moreover, after myocardial injury diurnal rhythm disruptions alter neutrophil and macrophage kinetics and delays the resolving phase (1). In consistent with previous study, the current report also displayed that the disruption of cardiomyocyte-specific Bmal1 gene displayed chronic inflammation at the age of 28 wk. Significant increase in the proinflammatory transcripts viz ccl2, Il-1β, ccl8, and ccr2 (neutrophil chemotaxis and leukocyte migration genes), with no change in the proresolving/alternative transcript Il-10 and Il-13, in CBK mice indicates an impaired defensive mechanism to repair inflammation at 28 wk of age. This was further supported by decreased levels of 5-LOX, COX-2, and HO-1 (3, 17, 19), impairing the resolution of the inflammation axis. Thus an age-related defective resolution of inflammation and dilative ventricular dysfunction leads to reduced survival (42) with marked cardiomyopathy.
In summary, the current study reveals diastolic dysfunction, ventricular hypertrophy, and cardiac pathology in CBK mice. Thus genetic ablation of Bmal1 in the cardiomyocyte leads to dilative cardiomyopathy through Smad2/3 that triggered fibrosis, stimulated inflammation, and impaired the resolution of inflammation.
GRANTS
We acknowledge support from NIH-NCCAM K99/R00 AT006704 and UAB start-up funds, awarded to G. V. Halade.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: K.A.I., V.K., M.G., and G.V.H. performed experiments; K.A.I., V.K., and G.V.H. analyzed data; K.A.I., V.K., and G.V.H. prepared figures; K.A.I., V.K., M.E.Y., and G.V.H. drafted manuscript; S.D.P., M.E.Y., and G.V.H. conception and design of research; M.E.Y. and G.V.H. interpreted results of experiments; M.E.Y. and G.V.H. edited and revised manuscript; G.V.H. approved final version of manuscript.
REFERENCES
- 1.Alibhai FJ, Tsimakouridze EV, Chinnappareddy N, Wright DC, Billia F, O'Sullivan ML, Pyle WG, Sole MJ, Martino TA. Short-term disruption of diurnal rhythms after murine myocardial infarction adversely affects long-term myocardial structure and function. Circ Res 114: 1713–1722, 2014. [DOI] [PubMed] [Google Scholar]
- 2.Arjona A, Sarkar DK. Circadian oscillations of clock genes, cytolytic factors, and cytokines in rat NK cells. J Immunol 174: 7618–7624, 2005. [DOI] [PubMed] [Google Scholar]
- 3.Blomer N, Pachel C, Hofmann U, Nordbeck P, Bauer W, Mathes D, Frey A, Bayer B, Vogel B, Ertl G, Bauersachs J, Frantz S. 5-Lipoxygenase facilitates healing after myocardial infarction. Basic Res Cardiol 108: 367, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boluyt MO, O'Neill L, Meredith AL, Bing OH, Brooks WW, Conrad CH, Crow MT, Lakatta EG. Alterations in cardiac gene expression during the transition from stable hypertrophy to heart failure. Marked upregulation of genes encoding extracellular matrix components. Circ Res 75: 23–32, 1994. [DOI] [PubMed] [Google Scholar]
- 5.Carson PA, O'Connor CM, Miller AB, Anderson S, Belkin R, Neuberg GW, Wertheimer JH, Frid D, Cropp A, Packer M. Circadian rhythm and sudden death in heart failure: results from Prospective Randomized Amlodipine Survival Trial. J Am Coll Cardiol 36: 541–546, 2000. [DOI] [PubMed] [Google Scholar]
- 6.Castanon-Cervantes O, Wu M, Ehlen JC, Paul K, Gamble KL, Johnson RL, Besing RC, Menaker M, Gewirtz AT, Davidson AJ. Dysregulation of inflammatory responses by chronic circadian disruption. J Immunol 185: 5796–5805, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chatham JC, Young ME. Regulation of myocardial metabolism by the cardiomyocyte circadian clock. J Mol Cell Cardiol 55: 139–146, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chung HY, Cesari M, Anton S, Marzetti E, Giovannini S, Seo AY, Carter C, Yu BP, Leeuwenburgh C. Molecular inflammation: underpinnings of aging and age-related diseases. Ageing Res Rev 8: 18–30, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Degaute JP, Van Cauter E, van de Borne P, Linkowski P. Twenty-four-hour blood pressure and heart rate profiles in humans. A twin study. Hypertension 23: 244–253, 1994. [DOI] [PubMed] [Google Scholar]
- 10.Delp MD, Manning RO, Bruckner JV, Armstrong RB. Distribution of cardiac output during diurnal changes of activity in rats. Am J Physiol Heart Circ Physiol 261: H1487–H1493, 1991. [DOI] [PubMed] [Google Scholar]
- 11.Durgan DJ, Trexler NA, Egbejimi O, McElfresh TA, Suk HY, Petterson LE, Shaw CA, Hardin PE, Bray MS, Chandler MP, Chow CW, Young ME. The circadian clock within the cardiomyocyte is essential for responsiveness of the heart to fatty acids. J Biol Chem 281: 24254–24269, 2006. [DOI] [PubMed] [Google Scholar]
- 12.Durgan DJ, Tsai JY, Grenett MH, Pat BM, Ratcliffe WF, Villegas-Montoya C, Garvey ME, Nagendran J, Dyck JR, Bray MS, Gamble KL, Gimble JM, Young ME. Evidence suggesting that the cardiomyocyte circadian clock modulates responsiveness of the heart to hypertrophic stimuli in mice. Chronobiol Int 28: 187–203, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fielding CA, Jones GW, McLoughlin RM, McLeod L, Hammond VJ, Uceda J, Williams AS, Lambie M, Foster TL, Liao CT, Rice CM, Greenhill CJ, Colmont CS, Hams E, Coles B, Kift-Morgan A, Newton Z, Craig KJ, Williams JD, Williams GT, Davies SJ, Humphreys IR, O'Donnell VB, Taylor PR, Jenkins BJ, Topley N, Jones SA. Interleukin-6 signaling drives fibrosis in unresolved inflammation. Immunity 40: 40–50, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harfmann BD, Schroder EA, Esser KA. Circadian rhythms, the molecular clock, and skeletal muscle. J Biol Rhythms 30: 84–94, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harma MI, Ilmarinen JE. Towards the 24-hour society—new approaches for aging shift workers? Scand J Work Environ Health 25: 610–615, 1999. [DOI] [PubMed] [Google Scholar]
- 16.Haus E, Smolensky MH. Biologic rhythms in the immune system. Chronobiol Int 16: 581–622, 1999. [DOI] [PubMed] [Google Scholar]
- 17.Howes LG. Selective COX-2 inhibitors, NSAIDs and cardiovascular events— is celecoxib the safest choice? Ther Clin Risk Manag 3: 831–845, 2007. [PMC free article] [PubMed] [Google Scholar]
- 18.Hriscu ML. Modulatory factors of circadian phagocytic activity. Ann NY Acad Sci 1057: 403–430, 2005. [DOI] [PubMed] [Google Scholar]
- 19.Issan Y, Kornowski R, Aravot D, Shainberg A, Laniado-Schwartzman M, Sodhi K, Abraham NG, Hochhauser E. Heme oxygenase-1 induction improves cardiac function following myocardial ischemia by reducing oxidative stress. PLoS One 9: e92246, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Junqueira LC, Bignolas G, Brentani RR. Picrosirius staining plus polarization microscopy, a specific method for collagen detection in tissue sections. Histochem J 11: 447–455, 1979. [DOI] [PubMed] [Google Scholar]
- 21.Kayama Y, Minamino T, Toko H, Sakamoto M, Shimizu I, Takahashi H, Okada S, Tateno K, Moriya J, Yokoyama M, Nojima A, Yoshimura M, Egashira K, Aburatani H, Komuro I. Cardiac 12/15 lipoxygenase-induced inflammation is involved in heart failure. J Exp Med 206: 1565–1574, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koga K, Kenessey A, Ojamaa K. Macrophage migration inhibitory factor antagonizes pressure overload-induced cardiac hypertrophy. Am J Physiol Heart Circ Physiol 304: H282–H293, 2013. [DOI] [PubMed] [Google Scholar]
- 23.Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV, Antoch MP. Early aging and age-related pathologies in mice deficient in BMAL1, the core component of the circadian clock. Genes Dev 20: 1868–1873, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuppuswamy D, Kerr C, Narishige T, Kasi VS, Menick DR, Cooper Gt. Association of tyrosine-phosphorylated c-Src with the cytoskeleton of hypertrophying myocardium. J Biol Chem 272: 4500–4508, 1997. [DOI] [PubMed] [Google Scholar]
- 25.Lefta M, Campbell KS, Feng HZ, Jin JP, Esser KA. Development of dilated cardiomyopathy in Bmal1-deficient mice. Am J Physiol Heart Circ Physiol 303: H475–H485, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lopez EF, Kabarowski JH, Ingle KA, Kain V, Barnes S, Crossman DK, Lindsey ML, Halade GV. Obesity superimposed on aging magnifies inflammation and delays the resolving response after myocardial infarction. Am J Physiol Heart Circ Physiol 308: H269–H280, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marcheva B, Ramsey KM, Buhr ED, Kobayashi Y, Su H, Ko CH, Ivanova G, Omura C, Mo S, Vitaterna MH, Lopez JP, Philipson LH, Bradfield CA, Crosby SD, JeBailey L, Wang X, Takahashi JS, Bass J. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature 466: 627–631, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Muller JE, Stone PH, Turi ZG, Rutherford JD, Czeisler CA, Parker C, Poole WK, Passamani E, Roberts R, Robertson T, et al. Circadian variation in the frequency of onset of acute myocardial infarction. N Engl J Med 313: 1315–1322, 1985. [DOI] [PubMed] [Google Scholar]
- 29.Nguyen KD, Fentress SJ, Qiu Y, Yun K, Cox JS, Chawla A. Circadian gene Bmal1 regulates diurnal oscillations of Ly6C(hi) inflammatory monocytes. Science 341: 1483–1488, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Panda S, Antoch MP, Miller BH, Su AI, Schook AB, Straume M, Schultz PG, Kay SA, Takahashi JS, Hogenesch JB. Coordinated transcription of key pathways in the mouse by the circadian clock. Cell 109: 307–320, 2002. [DOI] [PubMed] [Google Scholar]
- 31.Richards AM, Nicholls MG, Espiner EA, Ikram H, Cullens M, Hinton D. Diurnal patterns of blood pressure, heart rate and vasoactive hormones in normal man. Clin Exp Hypertens A 8: 153–166, 1986. [DOI] [PubMed] [Google Scholar]
- 32.Ridker PM, Manson JE, Buring JE, Muller JE, Hennekens CH. Circadian variation of acute myocardial infarction and the effect of low-dose aspirin in a randomized trial of physicians. Circulation 82: 897–902, 1990. [DOI] [PubMed] [Google Scholar]
- 33.Rutters F, Lemmens SG, Adam TC, Bremmer MA, Elders PJ, Nijpels G, Dekker JM. Is social jetlag associated with an adverse endocrine, behavioral, and cardiovascular risk profile? J Biol Rhythms 29: 377–383, 2014. [DOI] [PubMed] [Google Scholar]
- 34.Saini C, Suter DM, Liani A, Gos P, Schibler U. The mammalian circadian timing system: synchronization of peripheral clocks. Cold Spring Harb Symp Quant Biol 76: 39–47, 2011. [DOI] [PubMed] [Google Scholar]
- 35.Silver AC, Arjona A, Walker WE, Fikrig E. The circadian clock controls toll-like receptor 9-mediated innate and adaptive immunity. Immunity 36: 251–261, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Son GH, Chung S, Choe HK, Kim HD, Baik SM, Lee H, Lee HW, Choi S, Sun W, Kim H, Cho S, Lee KH, Kim K. Adrenal peripheral clock controls the autonomous circadian rhythm of glucocorticoid by causing rhythmic steroid production. Proc Natl Acad Sci USA 105: 20970–20975, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Terracio L, Rubin K, Gullberg D, Balog E, Carver W, Jyring R, Borg TK. Expression of collagen binding integrins during cardiac development and hypertrophy. Circ Res 68: 734–744, 1991. [DOI] [PubMed] [Google Scholar]
- 38.Turek FW, Joshu C, Kohsaka A, Lin E, Ivanova G, McDearmon E, Laposky A, Losee-Olson S, Easton A, Jensen DR, Eckel RH, Takahashi JS, Bass J. Obesity and metabolic syndrome in circadian Clock mutant mice. Science 308: 1043–1045, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weber KT, Brilla CG. Pathological hypertrophy and cardiac interstitium. Fibrosis and renin-angiotensin-aldosterone system. Circulation 83: 1849–1865, 1991. [DOI] [PubMed] [Google Scholar]
- 40.Xia Y, Lee K, Li N, Corbett D, Mendoza L, Frangogiannis NG. Characterization of the inflammatory and fibrotic response in a mouse model of cardiac pressure overload. Histochem Cell Biol 131: 471–481, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang CH, Hwang CF, Lin PM, Chuang JH, Hsu CM, Lin SF, Yang MY. Sleep disturbance and altered expression of circadian clock genes in patients with sudden sensorineural hearing loss. Medicine 94: e978, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Young ME, Brewer RA, Peliciari-Garcia RA, Collins HE, He L, Birky TL, Peden BW, Thompson EG, Ammons BJ, Bray MS, Chatham JC, Wende AR, Yang Q, Chow CW, Martino TA, Gamble KL. Cardiomyocyte-specific BMAL1 plays critical roles in metabolism, signaling, and maintenance of contractile function of the heart. J Biol Rhythms 29: 257–276, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao R, Li D, Zuo P, Bai R, Zhou Q, Fan J, Li C, Wang L, Yang X. Influences of age, gender, and circadian rhythm on deceleration capacity in subjects without evident heart diseases. Ann Noninvasive Electrocardiol 20: 158–166, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zile MR, Brutsaert DL. New concepts in diastolic dysfunction and diastolic heart failure: Part I: diagnosis, prognosis, and measurements of diastolic function. Circulation 105: 1387–1393, 2002. [DOI] [PubMed] [Google Scholar]