

We describe the temporal development of alterations in wall mechanics and vascular reactivity of middle cerebral arteries of obese Zucker rats through evolution of the metabolic syndrome. While novel, this study is particularly noteworthy, as we also use clinically relevant agents against the metabolic syndrome given from an early age to determine vascular outcomes.
Keywords: metabolic syndrome, cerebrovascular remodeling, arterial stiffness, reactive oxygen species
Abstract
The metabolic syndrome (MetS) is highly prevalent in the North American population and is associated with increased risk for development of cerebrovascular disease. This study determined the structural and functional changes in the middle cerebral arteries (MCA) during the progression of MetS and the effects of chronic pharmacological interventions on mitigating vascular alterations in obese Zucker rats (OZR), a translationally relevant model of MetS. The reactivity and wall mechanics of ex vivo pressurized MCA from lean Zucker rats (LZR) and OZR were determined at 7–8, 12–13, and 16–17 wk of age under control conditions and following chronic treatment with pharmacological agents targeting specific systemic pathologies. With increasing age, control OZR demonstrated reduced nitric oxide bioavailability, impaired dilator (acetylcholine) reactivity, elevated myogenic properties, structural narrowing, and wall stiffening compared with LZR. Antihypertensive therapy (e.g., captopril or hydralazine) starting at 7–8 wk of age blunted the progression of arterial stiffening compared with OZR controls, while treatments that reduced inflammation and oxidative stress (e.g., atorvastatin, rosiglitazone, and captopril) improved NO bioavailability and vascular reactivity compared with OZR controls and had mixed effects on structural remodeling. These data identify specific functional and structural cerebral adaptations that limit cerebrovascular blood flow in MetS patients, contributing to increased risk of cognitive decline, cerebral hypoperfusion, and ischemic stroke; however, these pathological adaptations could potentially be blunted if treated early in the progression of MetS.
NEW & NOTEWORTHY
We describe the temporal development of alterations in wall mechanics and vascular reactivity of middle cerebral arteries of obese Zucker rats through evolution of the metabolic syndrome. While novel, this study is particularly noteworthy, as we also use clinically relevant agents against the metabolic syndrome given from an early age to determine vascular outcomes.
the metabolic syndrome (MetS), a clustering of metabolic abnormalities such as obesity, impaired glycemic control, atherogenic dyslipidemia, and hypertension, with the additional contributing conditions of a prooxidant, prothrombotic, and proinflammatory state, is prevalent in ∼56 million adults in the United States (21). Correspondingly, MetS is associated with a threefold increased risk of cardiovascular mortality (21) and a 50% increased risk of stroke (5) and is a known risk factor for cognitive decline with aging (41). It is therefore imperative that translationally relevant models of MetS be effectively interrogated to guide our understanding of the cerebrovascular adaptations that are associated with the development of MetS, its physiological, molecular, and genomic mechanistic underpinnings, and the effectiveness of established and novel therapeutic agents in either blunting or ameliorating MetS-associated cerebrovascular dysfunction.
The obese (fa/fa) Zucker rat (OZR) represents a translationally relevant animal model for MetS in humans, with its high utility for studying negative cerebrovascular outcomes as a result of the constellation of systemic pathologies (1). We previously reported that cerebral resistance arteries from OZR develop significant alterations in vascular reactivity (6) and demonstrated a progressive rarefaction of cerebral cortical microvessels with the progression of MetS. Of relevance, recent data suggest that cerebral infarct size is greater in OZR than in age-matched control lean Zucker rats (LZR) (39) following ischemic stroke and that this disparity in infarct severity with MetS development could be blunted with improved control of the chronic hypertension that develops (40). While these are compelling initial observations regarding the impact of MetS on stroke outcomes and the potential importance of risk factor control as a means to lessen stroke severity, further information is necessary to discern the role of altered cerebrovascular function in the compromised outcomes.
The MetS in OZR is embodied by a few consistent overarching characteristics. Among these are a global reduction in the bioavailability of vascular-derived nitric oxide (NO) (and the associated alterations in vascular reactivity), the genesis of a systemic proinflammatory condition, and a progressive remodeling of the resistance and exchange microvasculature at multiple levels of resolution, including a change in the mechanics of the vascular wall and a reduction in tissue capillarity that have the potential to elevate perfusion resistance and impair processes of mass transport and exchange. However, the temporal development of these adaptations in the cerebral circulation throughout the progression of MetS is not well understood, and our ability to blunt them with relevant interventions remains virtually unknown. As such, the purpose of the present study was 1) to study OZR at key age ranges throughout the development of the MetS to gain a fuller understanding of the temporal development of cerebrovascular adaptation/dysfunction with the evolution of the systemic pathologies and 2) to determine the extent to which interventions against the major constituent pathologies of MetS are effective in blunting the development of alterations in vascular reactivity and vascular wall mechanics in the middle cerebral arteries (MCA) of OZR.
MATERIALS AND METHODS
Animals.
Male LZR and OZR (Harlan) arrived at the West Virginia University Health Sciences Center (WVUHSC) at 6–7 wk of age and, after 1 wk of acclimation to the local environment, were placed into a specific protocol cohort for the subsequent 9–10 wk. Unless otherwise stated, animals were fed standard chow and tap water ad libitum for all experiments. All rats were housed in the animal care facility at the WVUHSC, and all protocols received prior approval from the WVUHSC Animal Care and Use Committee. At 7–8 wk of age, LZR (n = 6, time-control groups only) and OZR (n = 6 for each group, at each age) were randomly placed into one of the following treatments for the subsequent 9–10 wk: 1) time control (normal food and water ad libitum), 2) antihypertensive treatment with captopril (angiotensin-converting enzyme inhibitor, 60 mg·kg−1·day−1, in drinking water) (3), 3) antihypertensive treatment with hydralazine (systemic vasodilator, 50 mg·kg−1·day−1, in drinking water) (3), 4) antidiabetic treatment with metformin (hepatic gluconeogenesis inhibitor, 300 mg·kg−1·day−1, in drinking water) (3, 53), 5) antidiabetic treatment with rosiglitazone (insulin-sensitizing agent, 10 mg·kg−1·day−1, mixed with food) (3, 47), 6) antidyslipidemia treatment with atorvastatin (3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitor, 25 mg·kg−1·day−1, mixed with food) (19), 7) antidyslipidemia treatment with gemfibrozil (peroxisome proliferator-activated receptor-α activator, 100 mg·kg−1·day−1, mixed with food) (19), 8) antioxidant treatment with tempol (10−3 M, in drinking water) (16), and 9) NO synthase (NOS) inhibition with NG-nitro-l-arginine methyl ester (l-NAME, 10−4 M, in drinking water) (16).
Doses of therapeutic agents were selected on the basis of their previously demonstrated effectiveness in reducing the severity of the specific elevations in cardiovascular disease risk in rats. The results presented in Table 1 support the use of the selected doses.
Table 1.
Baseline characteristics of animal groups at 7–8, 12–13, and 16–17 wk of age
| OZR |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| LZR | Control | CAP | HDZ | MET | RGZ | ATOR | GEM | TEM | l-NAME | |
| Body mass, g | ||||||||||
| 7–8 wk | 145 ± 6 | 339 ± 11* | 335 ± 7* | 344 ± 9* | 338 ± 11* | 342 ± 9* | 331 ± 11* | 345 ± 9* | 350 ± 10* | 338 ± 11* |
| 12–13 wk | 224 ± 6 | 452 ± 10* | 454 ± 9* | 450 ± 10* | 448 ± 8* | 456 ± 9* | 453 ± 11* | 454 ± 9* | 450 ± 10* | 458 ± 9* |
| 16–17 wk | 365 ± 9 | 691 ± 13* | 685 ± 10* | 683 ± 11* | 692 ± 13* | 685 ± 9* | 681 ± 12* | 685 ± 12* | 690 ± 11* | 678 ± 12* |
| MAP, mmHg | ||||||||||
| 7–8 wk | 104 ± 4 | 103 ± 7 | 102 ± 6 | 99 ± 5 | 101 ± 8 | 103 ± 5 | 101 ± 4 | 105 ± 6 | 104 ± 6 | 106 ± 4 |
| 12–13 wk | 105 ± 5 | 114 ± 5 | 98 ± 5† | 97 ± 5† | 113 ± 4 | 112 ± 4 | 106 ± 5 | 109 ± 6 | 100 ± 4† | 131 ± 6*† |
| 16–17 wk | 103 ± 6 | 135 ± 5* | 107 ± 6† | 101 ± 6† | 129 ± 6* | 130 ± 6* | 114 ± 6† | 119 ± 5*† | 109 ± 6† | 142 ± 6* |
| Glucose, mg/dl | ||||||||||
| 7–8 wk | 84 ± 5 | 99 ± 6 | 94 ± 4 | 94 ± 5 | 88 ± 7 | 84 ± 6 | 90 ± 5 | 88 ± 5 | 90 ± 5 | 94 ± 6 |
| 12–13 wk | 92 ± 5 | 121 ± 5* | 116 ± 6* | 120 ± 7* | 101 ± 4† | 94 ± 6† | 119 ± 5* | 121 ± 6* | 112 ± 5* | 123 ± 5* |
| 16–17 wk | 94 ± 4 | 148 ± 8* | 141 ± 9* | 147 ± 7* | 116 ± 5*† | 114 ± 5*† | 145 ± 6* | 139 ± 6* | 134 ± 6* | 151 ± 6* |
| Insulin, ng/ml | ||||||||||
| 7–8 wk | 0.9 ± 0.3 | 5.4 ± 0.5* | 5.2 ± 0.4* | 5.5 ± 0.5* | 5.3 ± 0.6* | 5.1 ± 0.7* | 5.5 ± 0.6* | 5.4 ± 0.5* | 5.5 ± 0.6* | 5.4 ± 0.4* |
| 12–13 wk | 1.2 ± 0.2 | 6.9 ± 0.5* | 5.5 ± 0.5* | 6.8 ± 0.6* | 5.0 ± 0.6*† | 4.8 ± 0.5*† | 6.3 ± 0.5* | 6.5 ± 0.6* | 5.2 ± 0.6*† | 7.1 ± 0.4* |
| 16–17 wk | 1.4 ± 0.4 | 8.8 ± 0.6* | 6.8 ± 0.5*† | 8.4 ± 0.6* | 5.5 ± 0.6*† | 5.2 ± 0.4*† | 7.3 ± 0.6*† | 7.7 ± 0.6* | 6.5 ± 0.5*† | 9.4 ± 0.5* |
| Cholesterol, mg/dl | ||||||||||
| 7–8 wk | 76 ± 6 | 84 ± 8 | 88 ± 7 | 94 ± 11 | 83 ± 8 | 90 ± 9 | 79 ± 5 | 82 ± 7 | 79 ± 8 | 84 ± 8 |
| 12–13 wk | 81 ± 5 | 104 ± 6* | 106 ± 5* | 105 ± 7* | 110 ± 9* | 109 ± 9* | 89 ± 5† | 86 ± 5† | 98 ± 6* | 106 ± 6* |
| 16–17 wk | 86 ± 6 | 134 ± 9* | 130 ± 6* | 134 ± 11* | 133 ± 10* | 129 ± 11* | 96 ± 5† | 102 ± 7† | 129 ± 9* | 138 ± 11* |
| N-tyrosine, ng/ml | ||||||||||
| 7–8 wk | 8 ± 3 | 15 ± 4 | 13 ± 4 | 16 ± 5 | 15 ± 4 | 17 ± 5 | 14 ± 4 | 15 ± 5 | 13 ± 4 | 16 ± 5 |
| 12–13 wk | 11 ± 3 | 34 ± 5* | 29 ± 4* | 33 ± 5* | 24 ± 4* | 22 ± 5*† | 15 ± 4† | 23 ± 4*† | 13 ± 4† | 36 ± 5* |
| 16–17 wk | 15 ± 4 | 56 ± 6* | 42 ± 5*† | 49 ± 7* | 46 ± 7* | 39 ± 5*† | 38 ± 4*† | 46 ± 7* | 27 ± 4*† | 66 ± 8* |
| TNF-α, pg/ml | ||||||||||
| 7–8 wk | 1.1 ± 0.4 | 3.4 ± 0.5* | 3.2 ± 0.6* | 3.5 ± 0.5* | 2.9 ± 0.5* | 3.1±.4* | 3.2 ± 0.4* | 3.5 ± 0.5* | 2.8 ± 0.4* | 3.3 ± 0.6* |
| 12–13 wk | 1.8 ± 0.2 | 10.4 ± 1.0* | 9.4 ± 0.8* | 9.5 ± 0.6* | 7.2 ± 0.5*† | 8.0 ± 0.4*† | 6.2 ± 0.5*† | 7.5 ± 0.5*† | 5.8 ± 0.5*† | 10.8 ± 0.6* |
| 16–17 wk | 2.6 ± 0.4 | 13.1 ± 1.0* | 10.2 ± 0.6*† | 12.5 ± 0.7* | 9.2 ± 0.6*† | 8.6 ± 0.7*† | 7.2 ± 0.6*† | 9.5 ± 0.5*† | 5.0 ± 0.6*† | 14.3 ± 1.1* |
| MCP-1, pg/ml | ||||||||||
| 7–8 wk | 29 ± 4 | 35 ± 6 | 34 ± 5 | 37 ± 4 | 30 ± 5 | 31 ± 6 | 34 ± 6 | 33 ± 5 | 30 ± 4 | 36 ± 5 |
| 12–13 wk | 30 ± 5 | 90 ± 10* | 68 ± 6*† | 87 ± 5* | 74 ± 6*† | 71 ± 5*† | 64 ± 6*† | 73 ± 5*† | 54 ± 6*† | 96 ± 6*† |
| 16–17 wk | 38 ± 6 | 104 ± 8* | 80 ± 5*† | 90 ± 9* | 84 ± 8* | 71 ± 7*† | 64 ± 6*† | 93 ± 9* | 58 ± 6*† | 116 ± 10* |
Values are means ± SE.
LZR, lean Zucker rat; OZR, obese Zucker rat; CAP, captopril, HDZ, hydralazine; MET, metformin; RGZ, rosiglitazone; ATOR, atorvastatin; GEM, gemfibrozil; TEM, tempol; l-NAME, NG-nitro-l-arginine methyl ester; MAP, mean arterial pressure; TNF-α, tumor necrosis factor-α; MCP-1, monocyte chemotactic protein-1.
P < 0.05 vs. LZR;
P < 0.05 vs. OZR.
After completion of the treatment period, each rat was anesthetized with pentobarbital sodium (50 mg/kg ip) and tracheally intubated to facilitate maintenance of a patent airway. In all rats, a carotid artery and an external jugular vein were cannulated for determination of arterial pressure and for infusion of additional substances, respectively, as necessary (e.g., anesthetic and heparin). Under anesthesia, an aliquot of blood was drawn from the jugular vein to be used for the subsequent determination of plasma insulin, tumor necrosis factor-α (TNF-α), and nitrotyrosine concentrations from each animal with use of commercially available kits (Millipore, Billerica, MA).
Under deep anesthesia, each rat was decapitated, and the brain was removed from the skull case and placed in cold (4°C) physiological salt solution (PSS). Subsequently, a MCA was dissected from its origin at the circle of Willis. Each MCA was doubly cannulated in a heated (37°C) chamber that allowed perfusion and superfusion of the lumen and exterior of the vessel, respectively, with PSS from separate reservoirs. The PSS (in mM: 119 NaCl, 4.7 KCl, 1.17 MgSO4, 1.6 CaCl2, 1.18 NaH2PO4, 24 NaHCO3, 0.026 EDTA, and 5.5 glucose) was equilibrated with a 21% O2-5% CO2-74% N2 gas mixture. Any side branches were ligated using a single strand teased from 6-0 suture. Vessel diameter was measured using television microscopy and an on-screen video micrometer.
Measurements of vascular reactivity in isolated MCA.
After cannulation, MCA were extended to their in situ length and equilibrated at 80% of the animal's mean arterial pressure (MAP, 88 ± 4 mmHg for LZR and 109 ± 5 mmHg for OZR) to approximate in vivo perfusion pressure (33). Any vessel that did not demonstrate significant active tone at the equilibration pressure was discarded. Active tone at the equilibration pressure was calculated as (ΔD/Dmax)·100, where ΔD is the diameter increase from rest in response to Ca2+-free PSS and Dmax is the maximum diameter measured at the equilibration pressure in Ca2+-free PSS. Active tone for vessels in the present study averaged 35 ± 2% in LZR and 32 ± 3% in OZR.
After equilibration, the dilator reactivity of MCA was assessed in response to increasing concentrations (10−10–10−6 M) of acetylcholine (ACh). Vascular responses to ACh were also measured following acute (45–60 min) incubation with l-NAME (10−4 M) and tempol (10−4 M) to assess the contributions of NO and oxidative stress, respectively, to modulation of vascular reactivity.
Myogenic properties were assessed in MCA over the range 40–140 mmHg, in 20-mmHg increments. Pressure was changed nonsequentially, and vessels were allowed 10 min to equilibrate at each pressure before arterial inner and outer diameters were recorded.
After the experimental procedures for measuring ex vivo vascular reactivity, the perfusate and superfusate PSS were replaced with Ca2+-free PSS containing the metal ion chelators EDTA (0.03 mM) and EGTA (2.0 mM). Vessels were challenged with 10−7 M serotonin until all active tone was lost. Subsequently, intraluminal pressure within the isolated vessel was altered, in 20-mmHg increments, between 0 and 160 mmHg. To ensure that a negative intraluminal pressure was not exerted on the vessel, 5 mmHg was used as the “0-mmHg” intraluminal pressure point; all other intraluminal pressure values were multiples of 20 mmHg up to 160 mmHg. After 7 min at each intraluminal pressure, the inner and outer diameters of the passive MCA were determined.
Measurement of vascular NO bioavailability.
The abdominal aorta was removed from each rat, and vascular NO production was assessed using amperometric sensors (World Precision Instruments, Sarasota, FL). Briefly, aortae were isolated, sectioned longitudinally, pinned in a Silastic-coated dish, and superfused with warmed (37°C) PSS equilibrated with 95% O2-5% CO2. A NO sensor (ISO-NOPF 100) was placed in close apposition to the endothelial surface, and a baseline level of current was obtained. Subsequently, increasing concentrations (10−10–10−6 M) of methacholine were added to the bath, and the changes in current were determined. To verify that responses represented NO release, these procedures were repeated following pretreatment of the aortic strip with l-NAME (10−4 M).
Data and statistical analyses.
Mechanical responses following challenge with logarithmically increasing doses of ACh were fit with the three-parameter logistic equation
where y represents the vessel diameter, “min” and “max” represent the lower (minimum) and upper (maximum) bounds, respectively, of the change in diameter with agonist concentration, x is the logarithm of the agonist concentration, and logED50 represents the logarithm of the agonist concentration (x) where the response (y) is halfway between the bounds. For the presentation of results, we have focused on the changes in the upper bounds as a representation of vessel reactivity, as the lower bound (defined as the prechallenge diameter) remained consistent between all groups and we did not determine a consistent or significant change in the logED50 values between treatment groups. As a result of this approach, the upper bound represents that statistically determined asymptote for the concentration-response relationship and does not assume that the vascular response at the highest utilized concentration of the agonist represents the maximum possible response. Rather, the sigmoidal relationship of best fit to the data will predict the statistical upper bound of the response given the data points entered into the model. As such, the upper bound is frequently slightly larger than the dilator response of the vessel at the highest concentration of the agonist.
The myogenic properties of MCA from each experimental group were plotted as mean diameter at each intraluminal pressure and fitted with a linear regression (y = α0 + βx), where the slope coefficient β represents the degree of myogenic activation (δdiameter/δpressure). Increasingly negative values of β, therefore, represent a greater degree of myogenic activation in response to changes in intraluminal pressure. A similar analysis was used to determine NO bioavailability in response to increasing concentrations of methacholine, where β represents the rate of change in NO released by the vessels in response to agonist challenge.
All calculations of passive arteriolar wall mechanics (used as indicators of structural alterations in the individual microvessel) are based on those used previously (4), with minor modification. Vessel wall thickness was calculated as
where WT represents wall thickness (μm) and OD and ID represent arteriolar outer and inner diameter (μm), respectively.
Incremental arteriolar distensibility (DISTINC, %change in arteriolar diameter/mmHg) was calculated as
where ΔID represents the change in internal arteriolar diameter for each incremental change in intraluminal pressure (ΔPIL).
For the calculation of circumferential stress, intraluminal pressure was converted from mmHg to N/m2, where 1 mmHg = 1.334 × 102 N/m2. Circumferential stress (σ) was then calculated as
Circumferential strain (ε) was calculated as
where ID5 represents the internal arteriolar diameter at the lowest intraluminal pressure (i.e., 5 mmHg). The stress-strain relationship from each vessel was fit (ordinary least-squares analysis, r2 > 0.85) with the following exponential equation
where σ5 represents circumferential stress at ID5 and β is the slope coefficient describing arterial stiffness. Higher levels of β are indicative of increasing arterial stiffness (i.e., requiring a greater degree of distending pressure to achieve a given level of wall deformation).
Values are means ± SE. Differences in passive mechanical characteristics, slope coefficients describing vascular reactivity or NO bioavailability, or descriptive characteristics between LZR and OZR groups were assessed using analysis of variance, with Student-Newman-Keuls test post hoc as appropriate. In all cases, P < 0.05 was taken to reflect statistical significance.
RESULTS
Table 1 presents body mass, MAP, and plasma biomarker data for each of the animal groups in the present study at three time points: 7–8, 12–13, and 16–17 wk of age. With increasing age, control OZR developed characteristic symptoms of MetS compared with LZR control, including elevated MAP, hyperglycemia, and elevated plasma insulin. Chronic treatment of OZR with specific pharmacological agents improved the expected outcomes for each drug's targeted risk factors. Specifically, treatment with captopril or hydralazine abrogated the development of hypertension, treatment with metformin or rosiglitazone improved glycemic control, treatment with atorvastatin or gemfibrozil blunted the severity of the developing dyslipidemia in OZR, and chronic treatment with tempol blunted the oxidant stress levels in OZR compared with LZR. Conversely, chronic NOS inhibition by treatment with l-NAME accelerated the development of hypertension and increased the level of oxidant stress.
Figure 1 presents mechanical properties, dilator reactivity, and myogenic properties of ex vivo MCA from OZR and LZR under untreated conditions for all three age ranges, as well as arterial NO bioavailability under untreated, l-NAME-treated, and tempol-treated conditions. By 16–17 wk, LZR and OZR demonstrated a significant decrease in inner diameter at elevated intraluminal pressure compared with their values at 7–8 wk, with OZR also exhibiting a decreased inner diameter by 12–13 wk (Fig. 1A), with a significant leftward shift of the circumferential stress-strain relationship in OZR at 12–13 and 16–17 wk compared with 7–8 wk (Fig. 1B). LZR exhibited a robust vasodilation in response to ACh in each age group, while reactivity to ACh was steadily reduced with increasing age in OZR (Fig. 1C). The slope of myogenic activation did not change between age groups in LZR, while the β-coefficients became increasingly negative with increasing age in OZR, indicating a greater pressure-induced constrictor response (Fig. 1D). NO bioavailability did not change with age in LZR, while it decreased steadily in OZR at 12–13 wk and again at 16–17 wk (Fig. 1E). Treatment with tempol did not significantly alter NO bioavailability in LZR but did improve NO bioavailability in OZR at 12–13 and 16–17 wk. At all ages, in both LZR and OZR, treatment with l-NAME nearly eliminated NO bioavailability.
Fig. 1.

Ex vivo middle cerebral artery (MCA) wall mechanics and reactivity in lean Zucker rats (LZR) and obese Zucker rats (OZR) at 7–8, 12–13, and 16–17 wk of age (n = 6 in each age group for each strain). A: MCA inner diameter with passive expansion. B: circumferential stress-strain relationships. C: dilator responses to increasing concentrations of acetylcholine (ACh). D: myogenic properties. E: vascular nitric oxide (NO) bioavailability with increasing concentrations of methacholine (Met). Values are means ± SE. In A–D, *P < 0.05 vs. LZR at 7–8 wk; †P < 0.05 vs. LZR at 12–13 wk; ‡P < 0.05 vs. OZR at 7–8 wk; #P < 0.05 vs. OZR at 12–13 wk. In E, *P < 0.05 vs. LZR at that age; †P < 0.05 vs. OZR at that age.
The impact of chronic treatment of OZR with the antihypertensive agents captopril and hydralazine on arterial wall mechanics, reactivity, and NO bioavailability at 12–13 and 16–17 wk is presented in Fig. 2. Generally, chronic treatment with either agent blunted remodeling of the MCA wall in OZR, blunting changes to the MCA inner diameter (Fig. 2A) and the stress-strain relationship (Fig. 2B). The effect on vascular reactivity was heterogeneous, as dilator reactivity to ACh was better maintained in captopril-treated OZR than OZR control, while the hydralazine-treated cohort demonstrated no improvement (Fig. 2C). Both antihypertensive treatments reduced the β-coefficient of myogenic activation to levels comparable to LZR (Fig. 2D). Captopril significantly improved NO bioavailability at 12–13 and 16–17 wk, while hydralazine had no effect at 12–13 wk and only effected a modest (not statistically significant) improvement in NO bioavailability at 16–17 wk.
Fig. 2.

Changes in ex vivo MCA wall mechanics and reactivity in LZR and OZR at 12–13 and 16–17 wk of age vs. 7–8 wk of age (n = 6 at each age group for each strain) following chronic treatment with the antihypertensive agents captopril (CAP) and hydralazine (HDZ) starting at 8 wk of age. A: MCA maximum inner diameter with passive expansion. B: β-coefficient for circumferential stress-strain relationships. C: upper bound of ACh concentration-response relationship. D: β-coefficient for myogenic activation. E: β-coefficient for vascular NO bioavailability with increasing concentrations of methacholine. Results following acute administration of tempol to the vessels are designated +TEM (a). Values are means ± SE. *P < 0.05 vs. LZR at that age; †P < 0.05 vs. OZR at that age; ‡P < 0.05 vs. that condition at 12–13 wk.
Data describing the impact of chronic treatment of OZR with the antidiabetic treatments rosiglitazone and metformin on vessel wall mechanics, vascular reactivity, and NO bioavailability at 12–13 and 16–17 wk are presented in Fig. 3. While neither rosiglitazone nor metformin significantly altered MCA inner diameter at 12–13 wk, both showed small, but significant, improvements at 16–17 wk (Fig. 3A). Treatment with rosiglitazone reduced the changes in the stress-strain β-coefficient primarily at 16–17 wk; metformin had only a slight effect at 16–17 wk (Fig. 3B). Both metformin and rosiglitazone moderately improved reactivity to ACh at 16–17 wk (Fig. 3C), and both drugs were also able to improve the β-coefficient of the myogenic response at 16–17 wk (Fig. 3D). While both metformin and rosiglitazone significantly improved NO bioavailability at 12–13 wk, only rosiglitazone remained effective at 16–17 wk (Fig. 3E).
Fig. 3.

Changes in ex vivo MCA wall mechanics and reactivity in LZR and OZR at 12–13 and 16–17 wk of age vs. 7–8 wk of age (n = 6 at each age group for each strain) following chronic treatment with the antidiabetes agents rosiglitazone (ROSI) and metformin (MET) starting at 8 wk of age. A: MCA maximum inner diameter with passive expansion. B: β-coefficient for circumferential stress-strain relationships. C: upper bound of ACh concentration-response relationship. D: β-coefficient for myogenic activation. E: β-coefficient for vascular NO bioavailability with increasing concentrations of methacholine. Results following acute administration of tempol to the vessels are designated +TEM (a). Values are means ± SE. *P < 0.05 vs. LZR at that age; †P < 0.05 vs. OZR at that age; ‡P < 0.05 vs. that condition at 12–13 wk.
The impact of chronic treatment of OZR with the antidyslipidemia agents atorvastatin and gemfibrozil on arterial wall mechanics, reactivity, and NO bioavailability at 12–13 and 16–17 wk is presented in Fig. 4. In contrast to the effects of the glycemic control agents described above, chronic treatment with the dyslipidemia agents significantly improved (i.e., increased) the inner diameter of MCA from OZR in response to elevated intraluminal pressure compared with LZR control (Fig. 4A). For atorvastatin, this effect was present at 12–13 and 16–17 wk; for gemfibrozil, this effect was present at 16–17 wk only. As a result, atorvastatin treatment significantly reduced the stress-strain β-coefficient at 12–13 and 16–17 wk, while gemfibrozil had no effect at 12–13 wk and a modest impact at 16–17 wk (Fig. 4B). Similarly, atorvastatin was highly effective at improving reactivity to ACh (Fig. 4C) and NO bioavailability (Fig. 4E) at both ages, while gemfibrozil had no effect on either metric at any age. Both agents were able to significantly improve the β-coefficient of the myogenic response at 12–13 and 16–17 wk (Fig. 4D).
Fig. 4.

Changes in ex vivo MCA wall mechanics and reactivity in LZR and OZR at 12–13 and 16–17 wk of age vs. 7–8 wk of age (n = 6 at each age group for each strain) following chronic treatment with the antidyslipidemia agents atorvastatin (ATOR) and gemfibrozil (GEM) starting at 8 wk of age. A: MCA maximum inner diameter with passive expansion. B: β-coefficient for circumferential stress-strain relationships. C: upper bound of ACh concentration-response relationship. D: β-coefficient for myogenic activation. E: β-coefficient for vascular NO bioavailability with increasing concentrations of methacholine. Results following acute administration of tempol to the vessels are designated +TEM (a). Values are means ± SE. *P < 0.05 vs. LZR at that age; †P < 0.05 vs. OZR at that age; ‡P < 0.05 vs. under that condition at 12–13 wk.
Figure 5 shows the impact of chronic treatment with the antioxidant agent tempol and chronic treatment with the competitive NOS inhibitor l-NAME on arterial wall mechanics, reactivity, and NO bioavailability in OZR at 12–13 and 16–17 wk. Tempol, which targets the increases in oxidant stress that accompany the onset of MetS in OZR, reduced the narrowing of the MCA inner diameter to levels matching LZR control (Fig. 5A) and significantly reduced the stress-strain β-coefficients (Fig. 5B). In contrast, chronic treatment with l-NAME had minimal impact on structural narrowing or the stress-strain β-coefficient at either age in OZR. Chronic treatment with tempol resulted in a maintained robust dilation to ACh in OZR, while l-NAME nearly abolished all responses to ACh across both strains and all ages (Fig. 5C). While tempol improved the β-coefficient of the myogenic response of MCA from OZR at 12–13 and 16–17 wk, l-NAME had a minimal impact on the slope coefficient (Fig. 5D). In agreement with the reactivity data, tempol significantly improved NO bioavailability in OZR at 12–13 and 16–17 wk, while l-NAME severely reduced NO bioavailability to levels even lower than OZR control (Fig. 5E).
Fig. 5.

Changes in ex vivo MCA wall mechanics and reactivity in LZR and OZR at 12–13 and 16–17 wk of age vs. 7–8 wk of age (n = 6 at each age group for each strain) following chronic treatment with the antioxidant tempol (TEM) or the NO synthase (NOS) inhibitor NG-nitro-l-arginine methyl ester (LNM) starting at 8 wk of age. A: MCA maximum inner diameter with passive expansion. B: β-coefficient for circumferential stress-strain relationships. C: upper bound of ACh concentration-response relationship. D: β-coefficient for myogenic activation. E: β-coefficient for vascular NO bioavailability with increasing concentrations of methacholine. Results following acute administration of tempol to the vessels are designated +TEM (a). Values are means ± SE. *P < 0.05 vs. LZR at that age; †P < 0.05 vs. OZR at that age; ‡P < 0.05 vs. that condition at 12–13 wk.
The relationship between NO bioavailability, β-coefficients from the stress-strain curves, and the upper bound of ACh-induced vasodilation for each animal in each of the three age ranges is displayed in Fig. 6. At 7–8 wk, OZR have not yet fully developed the MetS pathologies, which is reflected by the clustering of data points into one small area (Fig. 6A). By 12–13 wk, OZR have developed MetS and demonstrate increased stress-strain β-coefficients along with reduced NO bioavailability and decreased ACh upper bounds (Fig. 6B). By 12–13 wk, treatment of OZR with atorvastatin or tempol best preserved NO bioavailability and ACh upper bound, while also maintaining stress-strain β-coefficients with increasing severity of MetS. In contrast, gemfibrozil and metformin, which were, overall, two of the least-effective treatments, already demonstrated a significant displacement in these relationships from their values at 7–8 wk by only 12–13 wk of age. This trend continues at 16–17 wk, when the OZR controls are extremely divergent from LZR, representing significantly increased stress-strain β-coefficients and poor NO bioavailability/vascular reactivity. The effectiveness of gemfibrozil and metformin, as well as hydralazine, has similarly declined, suggesting that while hydralazine is able to delay the deleterious effects of MetS at 12–13 wk, it is no longer able to do so by 16–17 wk. Atorvastatin and tempol remain toward the upper-right quadrant, while captopril and rosiglitazone populate the middle quadrants (Fig. 6C).
Fig. 6.
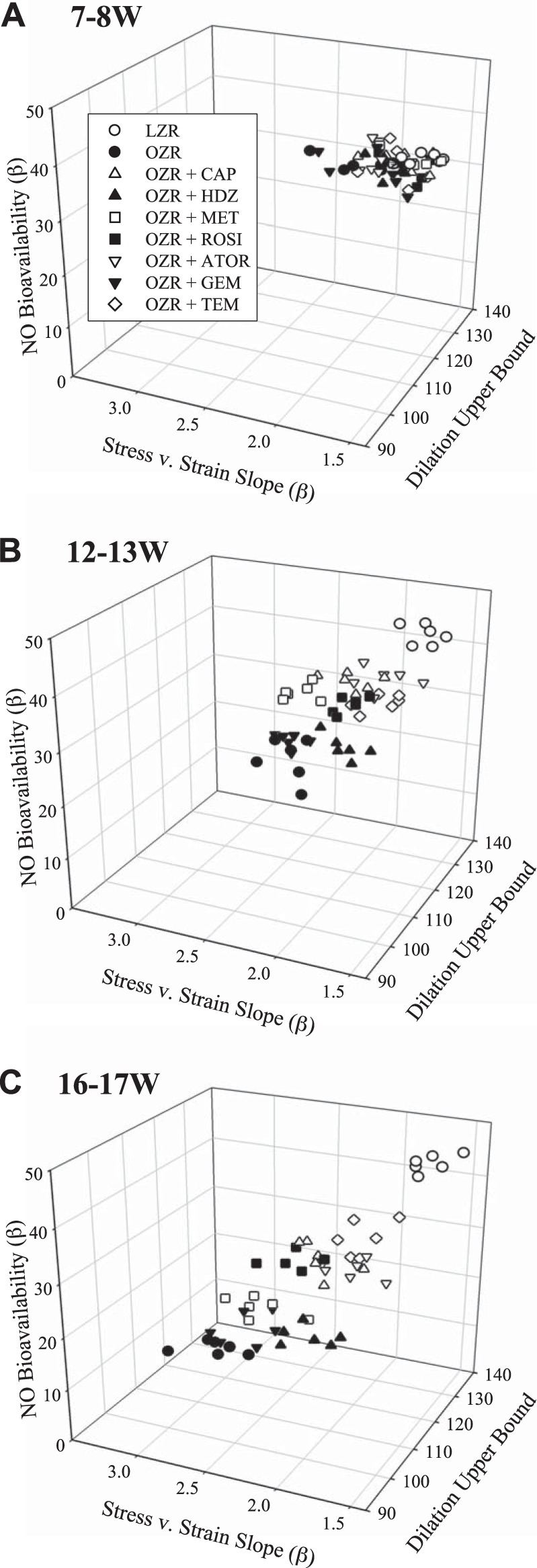
Relationships between vascular NO bioavailability, dilator reactivity to ACh, and wall mechanics in LZR under control conditions and OZR under control conditions and following treatment with captopril (CAP), hydralazine (HDZ), rosiglitazone (ROSI), metformin (MET), atorvastatin (ATOR), gemfibrozil (GEM), and tempol (TEM) at 7–8 wk (A), 12–13 wk (B), and 16–17 wk (C) of age (n = 6 at each age group for each strain). Vascular NO bioavailability is presented as β-coefficient for vascular NO concentration with increasing concentrations of methacholine, dilator reactivity is presented as the upper bound of the ACh concentration-response relationship for ex vivo MCA, and wall mechanics are presented as β-coefficient for the stress-strain relationship.
Figure 7 plots the stress-strain β-coefficients vs. MAP at 16–17 wk. The four treatments that most effectively controlled hypertension in OZR, tempol, atorvastatin, hydralazine, and captopril, also exhibited β-coefficients in the stress-strain curve that were most similar to those for LZR control MCA. These treatment groups with the lowest β-coefficients had a final MAP at 17 wk that was not significantly different from LZR control. For the other four treatments, β-coefficients were >2.2 and MAP was significantly higher than for LZR control.
Fig. 7.
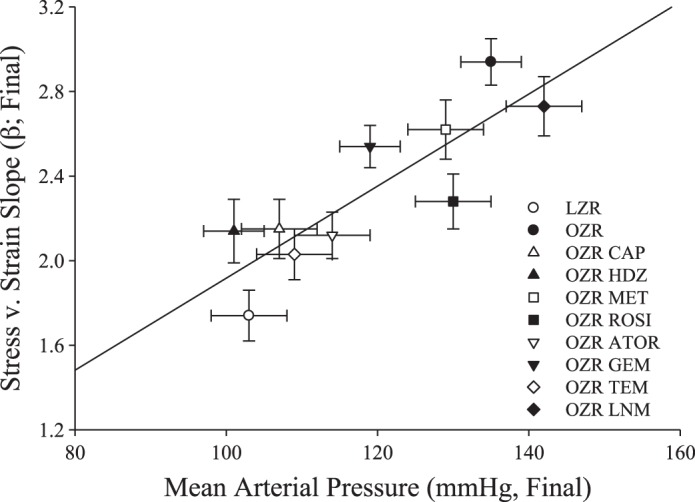
Relationships between β-coefficient for circumferential stress-strain relationships and mean arterial pressure for LZR and OZR at 16–17 wk of age (n = 6 for each strain in each group). Data are presented as the aggregate for the results from LZR under control conditions and OZR under control conditions and following treatment with captopril (CAP), hydralazine (HDZ), rosiglitazone (ROSI), metformin (MET), atorvastatin (ATOR), gemfibrozil (GEM), tempol (TEM), and NG-nitro-l-arginine methyl ester (LNM). Values are means ± SE. Linear regression equation of best fit is as follows: y = −0.259 + 0.021x (r2 = 0.724).
DISCUSSION
In the absence of intervention, OZR experience a progressive decline in vascular structure, mechanics, and reactivity in the MCA, associated with the progressing MetS state. However, changes in mechanical properties (i.e., distensibility) precede changes in lumen diameter and wall thickness in MetS. At 7–8 wk of age, OZR are significantly heavier and have elevated levels of TNF-α and insulin but show minimal changes in markers of oxidant stress and blood pressure and minor changes in vascular reactivity. The MCA of OZR and LZR remain dimensionally similar at 7–8 wk, but substantial arterial stiffening is already present, as reflected by the leftward-shifted stress-strain curve and increased β-coefficient. At 12–13 wk, inflammation and insulin levels have risen further, while substantial oxidative damage has accumulated, along with an increase in blood pressure. This is reflected in further mechanical stiffening of the MCA, likely through increased deposition and cross-linking of collagen plus fragmentation of elastin shift load to collagen at lower pressures, thereby making the vessel less distensible (30). Oxidative and inflammatory damage has negatively impacted the endothelium and reduced NO bioavailability, resulting in impaired dilator reactivity, which is alleviated by acute treatment with the antioxidant tempol. Additionally, eutrophic structural remodeling (lumen and outer diameters decrease with little change in wall thickness) of the MCA is present in OZR, and stiffness of the MCA continues to worsen as measured by increased β-coefficients of the stress-strain relationship. By 16–17 wk, as inflammation, oxidative stress, and blood pressure have increased with age and severity of the MetS, the degree of arterial dysfunction, remodeling, and stiffening continues to worsen. It is possible that the progressive loss of vascular wall distensibility may have contributed to the reduction of dilator reactivity between LZR and OZR and across the different interventions. However, the extent to which wall distensibility contributed to impaired reactivity is difficult to assess owing to the concurrent impairment of the mechanisms of dilator reactivity (i.e., loss of NO bioavailability).
The increase in arterial stiffness was closely associated with the onset of hypertension, indicative of the well-established relationship between elevated arterial pressure and vascular remodeling (42). In the present study, treatment with atorvastatin, tempol, captopril, or hydralazine blunted the onset of hypertension and was most effective at mitigating the increased arterial stiffness. While atorvastatin, tempol, and captopril reduced inflammation and oxidative stress, as well as blood pressure, hydralazine had minimal effects on inflammation and oxidative stress, suggesting that their efficacy in control of arterial stiffness may be primarily related to their antihypertensive actions, rather than any pleiotropic effect. Of particular relevance, clinical observations in hypertensive human subjects have shown that chronic treatment with angiotensin-converting enzyme inhibitors can reduce the severity of large artery stiffness with prolonged hypertension (35). However, it should be noted that a limitation of the present study is that we do not have chronic measurements of arterial pressure under the different conditions of the study and only present data from the three age ranges.
While the relationship between the onset of MetS in OZR and the presence of arterial dysfunction has been well established, the present study found that four of the interventions (atorvastatin, tempol, captopril, and rosiglitazone) improved NO bioavailability and endothelium-dependent dilation in 16- to 17-wk-old OZR. As shown in Table 1, these pharmacological interventions reduce levels of TNF-α, monocyte chemotactic protein-1, and nitrotyrosine, suggesting that treatments conferring both anti-inflammatory and antioxidant benefits are most effective at improving NO bioavailability and arterial function in OZR. Atorvastatin, for example, has well-defined roles in improving endothelial function, increasing NO bioavailability, and reducing MAP and has anti-inflammatory/antioxidant effects in addition to its cholesterol-lowering mechanism (11, 29).
To assess the effects of impaired glycemic control on arterial remodeling and dysfunction, OZR were treated with metformin or rosiglitazone. Both drugs improved insulin resistance, with minimal effects on arterial pressure. Although metformin had little effect on the remodeling or stiffness of the MCA in OZR, rosiglitazone did blunt both structural narrowing and the loss of MCA elasticity. Additionally, rosiglitazone was more effective at improving dilator responses to ACh and arterial NO bioavailability than metformin. The divergence in the effectiveness of these antidiabetic agents for MCA structure and function may lie within the additional effects of rosiglitazone on improving endothelial function, inflammation, remodeling, and blood pressure that do not appear to be as robust with metformin (27, 28). It is possible that metformin may be more effective at preventing remodeling at later stages of insulin resistance/type 2 diabetes after prolonged treatment, when it shows greatest cardiovascular risk reduction clinically (31), although this speculation is beyond the scope of the present study.
Dyslipidemia is common in MetS, and statins are the most popular front-line pharmacological agents for treatment of this condition. As stated above, atorvastatin has numerous pleiotropic effects in the context of cardiovascular disease, such as improving endothelial function via increasing NO bioavailability and boosting the innate antioxidant system (10). In this study, atorvastatin resulted in significant improvements in arterial reactivity and coupled a decrease in oxidative load with a robust anti-inflammatory effect. These effects corresponded with limited remodeling of the MCA and reduced arterial stiffness compared with OZR control. These observations support those from previous studies demonstrating that atorvastatin improved endothelial function in diabetes and cardiomyopathy (12, 32). In contrast, gemfibrozil, a peroxisome proliferator-activated receptor agonist with potent triglyceride-lowering actions (52), had little effect on arterial stiffness, distensibility, or reactivity, especially at 16–17 wk in OZR. This disparity provides additional support for the concept that the beneficial effects of atorvastatin stem not necessarily from management of cholesterol but, rather, from its pleiotropic cardiovascular benefits (9, 19). Similarly, gemfibrozil, alone or in combination with niacin (as an additional treatment to improve HDL, LDL, and triglyceride profiles), did not significantly improve endothelial function in patients with normal or elevated cholesterol levels (2).
Tempol was included in this study as a positive treatment standard for chronic low-level inflammation and oxidative stress associated with MetS (15). Oxidative stress is known to play an important role in the pathogenesis of vascular dysfunction (50). Not only does a prooxidative stress [reactive oxygen species (ROS)] environment impact the NO pathway and its regulation of arterial remodeling, but ROS can independently interact with the components of the perivascular matrix and drive collagen cross-linking, collagen deposition, and the fracturing of elastin (8). Previous evidence suggests that eutrophic inward remodeling was dependent on ROS activation of specific matrix metalloproteinases (that degrade and reorganize the extracellular matrix of the vessel wall) (36). Importantly, tempol was highly effective at lowering nitrotyrosine, TNF-α, and monocyte chemotactic protein-1 concentrations in the OZR (Table 1), in addition to improving arterial reactivity, reducing stiffness, and restoring NO bioavailability. It is important to note that all measurements of NO bioavailability were made using conduit artery segments (aorta), and not harvested MCA. As such, results should be interpreted appropriately.
The observation of increased myogenic properties of ex vivo MCA in the present study is consistent with previous observations and across multiple tissues and organs from OZR and other similar models of MetS (17, 22, 24, 43, 49). While mechanistic contributors to increased pressure-induced constriction remain to be fully elucidated, it does appear that the progressive dysfunction of the vascular endothelium may act to remove a “buffer” on the robustness of the constrictor response and contribute to the enhanced myogenic properties. It is unclear if this reflects the loss of vascular NO bioavailability or a broader condition involving other mechanisms of endothelial dysfunction and alterations in vascular smooth muscle responses per se. However, on the basis of the results from the interventional procedures in the present study, this appears to be more complicated than a simple “recovery of endothelial function,” as correlations between improvements in vascular NO bioavailability (Figs. 2E, 3E, 4E, and 5E) were not perfectly associated with normalization of myogenic constriction in treated OZR (Figs. 2D, 3D, 4D, and 5D). This suggests that there may be alterations in vascular smooth muscle itself and responses to intraluminal pressure with the development of the MetS (23, 37, 44) that are not as readily correctable by improved endothelial function alone.
Translational relevance.
Recent work from Stepp's group clearly demonstrated an increase in stroke infarct severity in OZR (39, 40). Given that cognitive impairments and vascular dementia have also been linked to impairments of cerebral blood flow regulation, the cerebrovascular adaptations in MetS may represent key contributors to multiple cerebral pathologies (45, 46, 48). We previously reported a decrease in cerebral cortical microvascular density associated with MetS and suggested that this rarefaction may contribute to either accumulated stroke risk or poor health outcomes in afflicted individuals (7). In the present study we have further added to the potential mechanisms by which MetS may contribute to impaired cerebral perfusion and increased risk of ischemic stroke by describing cerebrovascular dysfunction along with reduced cerebral arterial wall distensibility. In combination with microvessel rarefaction, these additional alterations may serve as physiological contributors to significant impairments in cerebral blood flow regulation and perfusion, which have been linked to cognitive decline with aging. Indeed, provocative recent work by Tucsek et al. (51), who found that aging in combination with high-fat diet-induced obesity in mice led to an exacerbation of cerebrovascular rarefaction and neurovascular uncoupling previously observed in obese mice and associated cognitive impairments, may provide additional insight. MetS-associated cerebrovascular changes can also lead to elevated risk for ischemic stroke in MetS, as well as worsened stroke outcomes in patients in whom MetS is a preexisting condition, through compromised processes of mass transport and exchange, leading to an increased risk for areas of ischemic damage.
Previous research has demonstrated that MetS in OZR is associated with cognitive and memory impairments through hippocampal-dependent pathways (54), and similar impairments in cognitive function and memory have been observed in other rat strains fed high-fat Western diets (20, 25, 26). Furthermore, rats experiencing transient cerebral ischemia developed both cognitive and memory impairments (55), while rats exposed to elevated oxidative stress developed cognitive deficits as a result of direct impairments in the hippocampus and frontal cortex (18). Taken together, these findings suggest that cerebral vasculopathies resulting from the MetS can contribute to impaired cerebral blood flow regulation and to an ensuing cognitive decline. With a growing body of clinical and epidemiological literature implicating MetS and other chronic cardiovascular disease risk factors for the development of cognitive impairment (13, 38, 41), the structural and functional impairments in the cerebral vasculature highlighted in this study may provide novel insight for prevention or treatment of cognitive impairments in at-risk patients.
Thus, early recognition of MetS and therapeutic intervention targeted at protecting the cerebral vasculature are critical. Although blood pressure control alone was effective in dampening arterial stiffening, in the present study we found atorvastatin to be the most effective treatment for ameliorating cerebrovascular dysfunction. It is noteworthy that there is substantial evidence to support the beneficial effect of statins on limiting infarct size as a result of stroke and improving recovery (34). It has also been demonstrated that the use of statins during hospitalization is associated with improved survival and a better discharge disposition among patients following ischemic stroke (14). This cumulative evidence would suggest that early treatment with 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors to utilize their pleiotropic effects on blood pressure, inflammation, and oxidative stress might be a useful clinical tool for combating MetS-related cerebrovascular disorders.
GRANTS
This study was supported by American Heart Association Grants IRG 14330015, PRE 16850005, and EIA 0740129N and National Institutes of Health Grants 1P20 GM-109098, RR-2865AR, and P20 RR-016477. This research was supported by the National Institute of General Medical Sciences Grant U54 GM-104942.
DISCLAIMER
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
S.D.B., E.D., J.C.F., and P.D.C. performed the experiments; S.D.B., E.D., S.J.F., J.C.F., and P.D.C. analyzed the data; S.D.B., E.D., A.C.d'A., S.J.F., L.E.T., C.D.S., J.C.F., and P.D.C. interpreted the results of the experiments; S.D.B., E.D., J.C.F., and P.D.C. prepared the figures; S.D.B., E.D., J.C.F., and P.D.C. drafted the manuscript; S.D.B., E.D., A.C.d'A., S.J.F., L.E.T., C.D.S., J.C.F., and P.D.C. edited and revised the manuscript; S.D.B., E.D., A.C.d'A., S.J.F., L.E.T., C.D.S., J.C.F., and P.D.C. approved the final version of the manuscript; A.C.d'A., L.E.T., C.D.S., J.C.F., and P.D.C. developed the concept and designed the research.
ACKNOWLEDGMENTS
The authors thank Milinda James for expert technical assistance. The authors acknowledge the support provided through Center for Cardiovascular and Respiratory Sciences and the Clinical and Translational Sciences Institute at the West Virginia University Health Sciences Center.
REFERENCES
- 1.Aleixandre de Artinano A, Miguel Castro M. Experimental rat models to study the metabolic syndrome. Br J Nutr 102: 1246–1253, 2009. [DOI] [PubMed] [Google Scholar]
- 2.Andrews TC, Whitney EJ, Green G, Kalenian R, Personius BE. Effect of gemfibrozil ± niacin ± cholestyramine on endothelial function in patients with serum low-density lipoprotein cholesterol levels <160 mg/dl and high-density lipoprotein cholesterol levels <40 mg/dl. Am J Cardiol 80: 831–835, 1997. [DOI] [PubMed] [Google Scholar]
- 3.Anonymous. Goodman and Gilman's The Pharmacological Basis of Therapeutics. New York: McGraw-Hill Professional, 2005. [Google Scholar]
- 4.Baumbach GL, Hajdu MA. Mechanics and composition of cerebral arterioles in renal and spontaneously hypertensive rats. Hypertension 21: 816–826, 1993. [DOI] [PubMed] [Google Scholar]
- 5.Boden-Albala B, Sacco RL, Lee HS, Grahame-Clarke C, Rundek T, Elkind MV, Wright C, Giardina EG, DiTullio MR, Homma S, Paik MC. Metabolic syndrome and ischemic stroke risk: Northern Manhattan Study. Stroke 39: 30–35, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Butcher JT, Goodwill AG, Stanley SC, Frisbee JC. Differential impact of dilator stimuli on increased myogenic activation of cerebral and skeletal muscle resistance arterioles in obese Zucker rats. Microcirculation 20: 579–589, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chantler P, Shrader CD, Tabone LE, d'Audiffret AC, Huseynova K, Brooks SD, Branyan KW, Grogg KA, Frisbee JC. Cerebral cortical microvascular rarefaction in metabolic syndrome is dependent on insulin resistance and loss of nitric oxide bioavailability. Microcirulation 22: 435–445, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chantler PD, Frisbee JC. Arterial function in cardio-metabolic diseases: from the microcirculation to the large conduits. Prog Cardiovasc Dis 57: 489–496, 2015. [DOI] [PubMed] [Google Scholar]
- 9.Chironi G, Simon A, Gariepy J, Balice M, Del-Pino M, Levenson J. Differential associations of statin and fibrate treatment with carotid arterial remodeling. Am J Hypertens 18: 1476–1481, 2005. [DOI] [PubMed] [Google Scholar]
- 10.Crisby M, Nordin-Fredriksson G, Shah PK, Yano J, Zhu J, Nilsson J. Pravastatin treatment increases collagen content and decreases lipid content, inflammation, metalloproteinases, and cell death in human carotid plaques: implications for plaque stabilization. Circulation 103: 926–933, 2001. [DOI] [PubMed] [Google Scholar]
- 11.Davignon J. Beneficial cardiovascular pleiotropic effects of statins. Circulation 109: III39–III43, 2004. [DOI] [PubMed] [Google Scholar]
- 12.Economides PA, Caselli A, Tiani E, Khaodhiar L, Horton ES, Veves A. The effects of atorvastatin on endothelial function in diabetic patients and subjects at risk for type 2 diabetes. J Clin Endocrinol Metab 89: 740–747, 2004. [DOI] [PubMed] [Google Scholar]
- 13.Eskelinen MH, Ngandu T, Helkala EL, Tuomilehto J, Nissinen A, Soininen H, Kivipelto M. Fat intake at midlife and cognitive impairment later in life: a population-based CAIDE study. Int J Geriatr Psychiatry 23: 741–747, 2008. [DOI] [PubMed] [Google Scholar]
- 14.Flint AC, Kamel H, Navi BB, Rao VA, Faigeles BS, Conell C, Klingman JG, Sidney S, Hills NK, Sorel M, Cullen SP, Johnston SC. Statin use during ischemic stroke hospitalization is strongly associated with improved poststroke survival. Stroke 43: 147–154, 2012. [DOI] [PubMed] [Google Scholar]
- 15.Ford ES. The metabolic syndrome and C-reactive protein, fibrinogen, and leukocyte count: findings from the Third National Health and Nutrition Examination Survey. Atherosclerosis 168: 351–358, 2003. [DOI] [PubMed] [Google Scholar]
- 16.Frisbee JC. Reduced nitric oxide bioavailability contributes to skeletal muscle microvessel rarefaction in the metabolic syndrome. Am J Physiol Regul Integr Comp Physiol 289: R307–R316, 2005. [DOI] [PubMed] [Google Scholar]
- 17.Frisbee JC, Maier KG, Stepp DW. Oxidant stress-induced increase in myogenic activation of skeletal muscle resistance arteries in obese Zucker rats. Am J Physiol Heart Circ Physiol 283: H2160–H2168, 2002. [DOI] [PubMed] [Google Scholar]
- 18.Fukui K, Omoi NO, Hayasaka T, Shinnkai T, Suzuki S, Abe K, Urano S. Cognitive impairment of rats caused by oxidative stress and aging, and its prevention by vitamin E. Ann NY Acad Sci 959: 275–284, 2002. [DOI] [PubMed] [Google Scholar]
- 19.Goodwill AG, Frisbee SJ, Stapleton PA, James ME, Frisbee JC. Impact of chronic anticholesterol therapy on development of microvascular rarefaction in the metabolic syndrome. Microcirculation 16: 667–684, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greenwood CE, Winocur G. Cognitive impairment in rats fed high-fat diets: a specific effect of saturated fatty-acid intake. Behav Neurosci 110: 451–459, 1996. [DOI] [PubMed] [Google Scholar]
- 21.Grundy SM, Cleeman JI, Daniels SR, Donato KA, Eckel RH, Franklin BA, Gordon DJ, Krauss RM, Savage PJ, Smith SC, Spertus JA, Costa F. Diagnosis and management of the metabolic syndrome: an American Heart Association/National Heart, Lung, and Blood Institute scientific statement. Curr Opin Cardiol 21: 1–6, 2006. [DOI] [PubMed] [Google Scholar]
- 22.Hayashi K, Kanda T, Homma K, Tokuyama H, Okubo K, Takamatsu I, Tatematsu S, Kumagai H, Saruta T. Altered renal microvascular response in Zucker obese rats. Metab Clin Exp 51: 1553–1561, 2002. [DOI] [PubMed] [Google Scholar]
- 23.Hodnett BL, Hester RL. Regulation of muscle blood flow in obesity. Microcirculation 14: 273–288, 2007. [DOI] [PubMed] [Google Scholar]
- 24.Jarajapu YP, Guberski DL, Grant MB, Knot HJ. Myogenic tone and reactivity of cerebral arteries in type II diabetic BBZDR/Wor rat. Eur J Pharmacol 579: 298–307, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jurdak N, Lichtenstein AH, Kanarek RB. Diet-induced obesity and spatial cognition in young male rats. Nutr Neurosci 11: 48–54, 2008. [DOI] [PubMed] [Google Scholar]
- 26.Kanoski SE, Meisel RL, Mullins AJ, Davidson TL. The effects of energy-rich diets on discrimination reversal learning and on BDNF in the hippocampus and prefrontal cortex of the rat. Behav Brain Res 182: 57–66, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kelly AS, Thelen AM, Kaiser DR, Gonzalez-Campoy JM, Bank AJ. Rosiglitazone improves endothelial function and inflammation but not asymmetric dimethylarginine or oxidative stress in patients with type 2 diabetes mellitus. Vasc Med 12: 311–318, 2007. [DOI] [PubMed] [Google Scholar]
- 28.Kiyici S, Ersoy C, Kaderli A, Fazlioglu M, Budak F, Duran C, Gul OO, Sigirli D, Baran I, Tuncel E, Erturk E, Imamoglu S. Effect of rosiglitazone, metformin and medical nutrition treatment on arterial stiffness, serum MMP-9 and MCP-1 levels in drug naive type 2 diabetic patients. Diabetes Res Clin Pract 86: 44–50, 2009. [DOI] [PubMed] [Google Scholar]
- 29.Kuklinska AM, Mroczko B, Musial WJ, Sawicki R, Kozieradzka A, Usowicz-Szarynska M, Kaminski K, Knapp M, Szmitkowski M. Influence of atorvastatin on blood pressure control in treated hypertensive, normolipemic patients: an open, pilot study. Blood Pressure 19: 260–266, 2010. [DOI] [PubMed] [Google Scholar]
- 30.Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises. I. Aging arteries: a “set up” for vascular disease. Circulation 107: 139–146, 2003. [DOI] [PubMed] [Google Scholar]
- 31.Lamanna C, Monami M, Marchionni N, Mannucci E. Effect of metformin on cardiovascular events and mortality: a meta-analysis of randomized clinical trials. Diabetes Obes Metab 13: 221–228, 2011. [DOI] [PubMed] [Google Scholar]
- 32.Liu M, Wang F, Wang Y, Jin R. Atorvastatin improves endothelial function and cardiac performance in patients with dilated cardiomyopathy: the role of inflammation. Cardiovasc Drugs Ther 23: 369–376, 2009. [DOI] [PubMed] [Google Scholar]
- 33.Lombard JH, Liu Y, Fredricks KT, Bizub DM, Roman RJ, Rusch NJ. Electrical and mechanical responses of rat middle cerebral arteries to reduced Po2 and prostacyclin. Am J Physiol Heart Circ Physiol 276: H509–H516, 1999. [DOI] [PubMed] [Google Scholar]
- 34.Lu D, Mahmood A, Goussev A, Schallert T, Qu C, Zhang ZG, Li Y, Lu M, Chopp M. Atorvastatin reduction of intravascular thrombosis, increase in cerebral microvascular patency and integrity, and enhancement of spatial learning in rats subjected to traumatic brain injury. J Neurosurg 101: 813–821, 2004. [DOI] [PubMed] [Google Scholar]
- 35.Mahmud A, Feely J. Reduction in arterial stiffness with angiotensin II antagonist is comparable with and additive to ACE inhibition. Am J Hypertens 15: 321–325, 2002. [DOI] [PubMed] [Google Scholar]
- 36.Martinez-Lemus LA, Zhao G, Galinanes EL, Boone M. Inward remodeling of resistance arteries requires reactive oxygen species-dependent activation of matrix metalloproteinases. Am J Physiol Heart Circ Physiol 300: H2005–H2015, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matrougui K. Diabetes and microvascular pathophysiology: role of epidermal growth factor receptor tyrosine kinase. Diabetes Metab Res Rev 26: 13–16, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morris MC, Evans DA, Bienias JL, Tangney CC, Wilson RS. Dietary fat intake and 6-year cognitive change in an older biracial community population. Neurology 62: 1573–1579, 2004. [DOI] [PubMed] [Google Scholar]
- 39.Osmond JM, Mintz JD, Dalton B, Stepp DW. Obesity increases blood pressure, cerebral vascular remodeling, and severity of stroke in the Zucker rat. Hypertension 53: 381–386, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Osmond JM, Mintz JD, Stepp DW. Preventing increased blood pressure in the obese Zucker rat improves severity of stroke. Am J Physiol Heart Circ Physiol 299: H55–H61, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Panza F, Frisardi V, Capurso C, Imbimbo BP, Vendemiale G, Santamato A, D'Onofrio G, Seripa D, Sancarlo D, Pilotto A, Solfrizzi V. Metabolic syndrome and cognitive impairment: current epidemiology and possible underlying mechanisms. J Alzheimers Dis 21: 691–724, 2010. [DOI] [PubMed] [Google Scholar]
- 42.Payne RA, Wilkinson IB, Webb DJ. Arterial stiffness and hypertension: emerging concepts. Hypertension 55: 9–14, 2010. [DOI] [PubMed] [Google Scholar]
- 43.Phillips SA, Sylvester FA, Frisbee JC. Oxidant stress and constrictor reactivity impair cerebral artery dilation in obese Zucker rats. Am J Physiol Regul Integr Comp Physiol 288: R522–R530, 2005. [DOI] [PubMed] [Google Scholar]
- 44.Pires PW, Dams Ramos CM, Matin N, Dorrance AM. The effects of hypertension on the cerebral circulation. Am J Physiol Heart Circ Physiol 304: H1598–H1614, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Raffaitin C, Gin H, Empana JP, Helmer C, Berr C, Tzourio C, Portet F, Dartigues JF, Alperovitch A, Barberger-Gateau P. Metabolic syndrome and risk for incident Alzheimer's disease or vascular dementia: the Three-City Study. Diabetes Care 32: 169–174, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Roberts RO, Geda YE, Knopman DS, Cha RH, Boeve BF, Ivnik RJ, Pankratz VS, Tangalos EG, Petersen RC. Metabolic syndrome, inflammation, and nonamnestic mild cognitive impairment in older persons: a population-based study. Alzheimer Dis Assoc Disord 24: 11–18, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sardone LD, Renlund R, Willett TL, Fantus IG, Grynpas MD. Effect of rosiglitazone on bone quality in a rat model of insulin resistance and osteoporosis. Diabetes 60: 3271–3278, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Solfrizzi V, Scafato E, Capurso C, D'Introno A, Colacicco AM, Frisardi V, Vendemiale G, Baldereschi M, Crepaldi G, Di Carlo A, Galluzzo L, Gandin C, Inzitari D, Maggi S, Capurso A, Panza F, Italian Longitudinal Study on Ageing Working Group . Metabolic syndrome and the risk of vascular dementia: the Italian Longitudinal Study on Ageing. J Neurol Neurosurg Psychiatry 81: 433–440, 2010. [DOI] [PubMed] [Google Scholar]
- 49.Stabley JN, Prisby RD, Behnke BJ, Delp MD. Type 2 diabetes alters bone and marrow blood flow and vascular control mechanisms in the ZDF rat. J Endocrinol 225: 47–58, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stocker R, Keaney JF Jr. Role of oxidative modifications in atherosclerosis. Physiol Rev 84: 1381–1478, 2004. [DOI] [PubMed] [Google Scholar]
- 51.Tucsek Z, Toth P, Tarantini S, Sosnowska D, Gautam T, Warrington JP, Giles CB, Wren JD, Koller A, Ballabh P, Sonntag WE, Ungvari Z, Csiszar A. Aging exacerbates obesity-induced cerebromicrovascular rarefaction, neurovascular uncoupling, and cognitive decline in mice. J Gerontol A Biol Sci Med Sci 69: 1339–1352, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wagner AM, Jorba O, Bonet R, Ordonez-Llanos J, Perez A. Efficacy of atorvastatin and gemfibrozil, alone and in low dose combination, in the treatment of diabetic dyslipidemia. J Clin Endocrinol Metab 88: 3212–3217, 2003. [DOI] [PubMed] [Google Scholar]
- 53.Wessels B, Ciapaite J, van den Broek NM, Nicolay K, Prompers JJ. Metformin impairs mitochondrial function in skeletal muscle of both lean and diabetic rats in a dose-dependent manner. PLos One 9: e100525, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Winocur G, Greenwood CE, Piroli GG, Grillo CA, Reznikov LR, Reagan LP, McEwen BS. Memory impairment in obese Zucker rats: an investigation of cognitive function in an animal model of insulin resistance and obesity. Behav Neurosci 119: 1389–1395, 2005. [DOI] [PubMed] [Google Scholar]
- 55.Zhang L, Wei WS, Li YJ, Wang Y. A rat model of mild cognitive impairment associated with vascular factor. Neuropathology 31: 112–121, 2011. [DOI] [PubMed] [Google Scholar]