

Abstract
Preeclampsia (PE) is a pregnancy-specific disorder typically presenting as new-onset hypertension and proteinuria. While numerous epidemiological studies have demonstrated that obesity increases the risk of PE, the mechanisms have yet to be fully elucidated. Growing evidence from animal and human studies implicate placental ischemia in the etiology of this maternal syndrome. It is thought that placental ischemia is brought about by dysfunctional cytotrophoblast migration and invasion into the uterus and subsequent lack of spiral arteriole widening and placental perfusion. Placental ischemia/hypoxia stimulates the release of soluble placental factors into the maternal circulation where they cause endothelial dysfunction, particularly in the kidney, to elicit the clinical manifestations of PE. The most recognized of these factors are the anti-angiogenic sFlt-1 and pro-inflammatory TNF-α and AT1-AA, which promote endothelial dysfunction by reducing levels of the provasodilator nitric oxide and stimulating production of the potent vasoconstrictor endothelin-1 and reactive oxygen species. We hypothesize that obesity-related metabolic factors increase the risk for developing PE by impacting various stages in the pathogenesis of PE, namely, 1) cytotrophoblast migration and placental ischemia; 2) release of soluble placental factors into the maternal circulation; and 3) maternal endothelial and vascular dysfunction. This review will summarize the current experimental evidence supporting the concept that obesity and metabolic factors like lipids, insulin, glucose, and leptin affect placental function and increase the risk for developing hypertension in pregnancy by reducing placental perfusion; enhancing placental release of soluble factors; and by increasing the sensitivity of the maternal vasculature to placental ischemia-induced soluble factors.
Keywords: body mass index, inflammation, placental ischemia, pregnancy, RUPP, sFlt-1
pe is a pregnancy-specific disorder typically identified by new-onset hypertension in the second half of pregnancy. Although often accompanied by new-onset proteinuria, PE can be associated with many other signs and symptoms including headaches, visual disturbances, epigastric pain, and the development of edema (4, 192). Globally, this disease affects over 8 million pregnancies per year and is estimated to account for 40–60% of maternal deaths in developing countries (125). In the United States, there was a 25% increase in the rate of PE between the years 1987–2004 (189). This significant increase highlights the importance of understanding how risk factors like obesity play a role in the pathogenesis of this maternal syndrome. Obesity is defined as a body mass index (BMI) of greater than or equal to 30 kg/m2. Greater than one-half of pregnant women are overweight or obese (48), and more than two-thirds of reproductive-aged women in the United States are overweight or obese (49). Although mounting epidemiological evidence supports obesity as a major risk factor for PE, the mechanisms linking these two morbidities are unclear. In this review, we use epidemiological and experimental studies to propose potential mechanisms linking obesity and the development of PE. We also highlight the importance of utilizing animal models to investigate the relative importance of metabolic factors in the pathophysiology of PE.
We will discuss numerous studies that reinforce that obesity increases the risk for the development of PE. However, less is known regarding the specific mechanisms whereby obesity poses this risk. Mounting evidence supports that the pathogenesis of PE encompasses a two-stage process. The first stage is thought to involve improper placental development as a result of dysfunctional proliferation, migration, and invasion of fetus-derived cytotrophoblast cells into the uterus (170). Ultimately, this leads to inappropriate uterine spiral arteriole widening, decreased placental blood flow, and placental hypoxia. A number of experiments using the RUPP (reduced uterine perfusion pressure) model of placental ischemia in pregnant rats, mice, and baboons have clearly demonstrated that placental ischemia is a strong stimulus for the release of soluble placental factors into the maternal circulation, which facilitate the second stage in the pathogenesis of PE: maternal endothelial and vascular dysfunction. Table 1 lists the similarities between the RUPP rat model and the clinical pathologies in preeclamptic women. These placental ischemia-associated factors include the anti-angiogenic soluble fms-like tyrosine kinase 1 (sFlt-1) and pro-inflammatory factors like tumor necrosis factor (TNF)-α and agonistic autoantibodies to the angiotensin II type 1 receptor (AT1-AA). Similarly, each one of these factors is increased in preeclamptic women. A direct role for each of these factors in increasing blood pressure during pregnancy was demonstrated by studies showing that infusion of each of these factors into normal pregnant rats increases blood pressure. This was accompanied by endothelial dysfunction driven by reductions in the pro-vasodilatory molecule nitric oxide (NO) and production of the pro-vasoconstrictor peptide endothelin (ET)-1 in endothelial cells. In the kidney, vascular dysfunction leads to altered renal hemodynamics and reduced renal excretory function. The kidney is important in the long-term control of blood pressure and endothelial dysfunction in the kidney manifests as hypertension and proteinuria (31). Because this cascade of events is thought to play an important role in the pathogenesis of PE, it is likely that obesity-related metabolic factors may increase the risk of developing PE by impinging on these pathophysiological processes. Therefore, in this review, we propose that obesity impacts the various stages thought to encompass the pathogenesis of PE by: 1) acting on the placenta causing cytotrophoblast dysfunction and placental ischemia; 2) enhancing ischemia/hypoxia-induced release of soluble placental factors; and 3) by increasing the sensitivity by which placental ischemia-factors are able to promote endothelial dysfunction and hypertension.
Table 1.
Similarities in cardiovascular outcomes; circulating placental ischemia factors; and fetal consequences compared between human preeclampsia and the RUPP rat model of preeclampsia
| Parameter | Human | RUPP |
|---|---|---|
| Hypertension | + | + |
| Proteinuria | ± | ± |
| Renal vasoconstriction | + | + |
| ↑TPR/↓CO | + | + |
| Endothelial dysfunction | + | + |
| ↑sFlt-1/↓VEGF | + | + |
| ↑Proinflammatory factors (TNF-α, AT1-AA) | + | + |
| Placental insufficiency (Fetal/placental weight) | + | + |
| Intrauterine growth restriction | ± | + |
RUPP, reduced uterine perfusion pressure; TPR, total peripheral resistance; CO, cardiac output; sFlt-1, soluble fms-like tyrosine kinase 1; VEGF, vascular endothelial growth factor; TNF-α, tumor necrosis factor-α; AT1-AA, agonistic autoantibodies to the angiotensin II type 1 receptor. Plus sign (+) denotes the presence, whereas ± indicates that the symptom is not always present in preeclamptic women and RUPP rats.
Epidemiological Evidence Supporting That Obesity Increases the Risk for the Development of PE
One of the early observations indicating that obese pregnancies are at greater risk for PE was reported in 1969 by Tracy and Miller (181). This study was published before the widespread use of BMI to classify body weight status. In their cohort of 48 women weighing greater than 250 pounds at any point during their pregnancies, there was found a staggering 31% prevalence of PE. In fact, PE was the number one obstetric complication found in their sample population. Similar statistics have been reported in more recent studies designed to compare obese and normal weight pregnant women. Indeed, Roberts and colleagues (15) showed that prepregnancy obesity attributes a 30% risk for PE in pregnant women with a BMI > 35 compared with lean pregnant counterparts. Furthermore, another study reported that prepregnancy obesity confers a three- to fourfold elevated risk for PE in triple gestations (152). The significance of BMI status as a predictor for PE is emphasized by an almost linear-response effect of BMI on the incidence of PE: incidence has been shown to be 7% in those gravidas with class I obesity (BMI = 30–34.9), 9% with class II (BMI = 35–39.9), 11% with class III obesity (BMI = 40–49.9), 13% in super-obese women (BMI = 50), whereas normal weight gravidas (BMI = 18.5–24.9) had a 3% chance of developing PE (110) (Fig. 1). The differences in the percent risk for PE within the same BMI class in these reports may be due to environmental exposure differences; the type of study (prospective vs. retrospective); and/or inclusion criteria for study volunteers. The vast majority of studies have demonstrated a detrimental impact of obesity on the rates of PE where this association between prepregnancy obesity and PE has been found in several countries across the world. In the United Kingdom, for example, nulliparous, morbidly obese women with a BMI > 40 determined at 10 wk of gestation were found to have a 30% chance of developing PE (13). This strong positive correlation between obesity and the prevalence of PE has also been observed in Americans (110), New Zealanders (103), and Canadians (43).
Fig. 1.

Rates of preeclampsia (PE) increase with increasing body mass index (BMI). From Mbah AK et al. (110).
Data illustrated in Fig. 1 suggest that obesity adds risk to the development of both early-onset and late-onset PE [odds ratios (OR) = 2.94; 95% confidence intervals (CI) = 2.87–3.01]. This risk was stronger for late PE (OR = 2.97; 95% CI = 2.90–3.04) than early PE (OR = 2.22; 95% CI = 2.06–2.40) (110). It has been proposed that the etiology of early and late PE is different, being abnormal placentation implicated in the development of early-onset PE, whereas an abnormal maternal metabolic milieu at conception has been linked to late-onset PE (144). It is likely that an abnormal metabolic environment is tied to increased secretion of factors by adipose tissue, which is not only a triglyceride-storing depot, but an endocrine organ that synthesizes and secretes a variety of hormones and inflammatory factors (182). Because increased body fat is associated with elevated circulating cytokine levels, obese women are more likely to begin pregnancy in a subclinical inflammatory state than normal weight women. Studies have shown that BMI is positively correlated with neutrophil infiltration into blood vessels, vascular inflammation, and blood pressure in women (159). On top of this, clinical studies have pointed to a positive relationship between BMI and activation of inflammatory pathways within the placenta (154), suggesting that metabolic factors exaggerate placental ischemia-induced release of inflammatory factors. Thus a harmful prepregnant pro-inflammatory status is most likely accompanied by baseline vascular dysfunction resulting in a lowered threshold and increased sensitivity to the combined ability of obesity-related metabolic factors and soluble placental ischemic inflammatory factors to elicit maternal endothelial dysfunction and hypertension. This represents a potential mechanism whereby obesity increases the risk for the development of late-onset PE.
These above reports highlight that it is important to understand how prepregnancy and increased early pregnancy BMI predisposes obese women to the development of PE. Some studies have also assessed whether gestational weight gain poses additional risk for this maternal disorder. The Avon Longitudinal Study of Parents and Children conducted at the University of Bristol confirmed that increased prepregnancy weight is associated with increased PE risk. These investigators went on to also show that, after adjustment for prepregnancy weight, weight gain of 200 g/wk in early pregnancy before 18 wk of gestation was independently associated with increased blood pressure during mid (18–29 wk) and very late (>36 wk) but not late (29–36 wk) pregnancy. However, it was found that if this amount of weight gain was reached during each of these gestational periods, there was an increase in blood pressure within that respective period (101). Studies such as this lead to recommendations by the Institute of Medicine (IOM) that women gain between 6.8 and 11.3 kg during pregnancy. In a study examining cohorts of women in Missouri having gestational weight gain of either <6.8, 6.8–11.3, or >11.3 kg, it was found that, compared with women with normal gestational weight gain, those with lower weight gain had reduced risk, whereas those with the highest weight gain were at greater risk for PE (94). Future studies, especially in experimental animal models, should tease out the relative importance of prepregnancy body weight versus gestational weight gain in promoting and exaggerating the mechanisms that promote PE. This could be accomplished in experimental animals having high-fat diet or genetic obesity in combination with adipose tissue transplant studies to increase fat content at different time points through pregnancy.
With concern to fetal growth, a discrepant feature between PE and obesity is that while PE is regularly accompanied by intrauterine growth restriction, obesity is often associated with macrosomia (201). However, different reports have found that maternal obesity may also increase the risk for low birth weight and intrauterine growth-restricted babies (135, 139, 141, 150). Additionally, epidemiological studies have shown that gestational weight gain is independently associated with birth weight and can contribute to the impact of obesity on fetal growth (39, 40, 46, 51, 105) in a way that limiting gestational weight gain in obese women reduces the odds of having a baby with high birth weight. Furthermore, using a high-fat diet-induced obesity model, Hayes et al. (64) observed that fetuses of obese pregnant animals are lighter than fetuses of lean counterparts. Interestingly, we have reported that individual metabolic factors lead to distinct adverse fetal outcomes: whereas euglycemic hyperinsulinemic pregnant rats presented fetuses with increased body weight (128), hyperleptinemic pregnant rats exhibited fetuses with decreased body weight compared with normal pregnant controls (126). In vivo and in vitro studies have demonstrated that obesity-related metabolic factors can alter the placental transport of nutrients and then lead to abnormal fetal growth (185). Therefore, it is possible that the development of adverse fetal outcomes in obese pregnancies might depend on the specific metabolic profile and the amount of weight gained during gestation.
Effects of Obesity on the Placental Function and Perfusion
The placenta is a pregnancy-specific organ of which its existence foremost depends on its own ability to remodel blood vessels in the uterine wall to meet its metabolic needs. For this purpose, fetus-derived cytotrophoblasts are tasked with invading the uterine spiral arteries where they differentiate into an endothelial cell-like phenotype while incorporating and repopulating both the tunica intima and tunica media. This results in transformation of the spiral arteries from high-resistance, low-flow blood vessels into high capacitance conduits. In normal pregnancy, sufficient remodeling occurs to meet the metabolic demands of the growing uteroplacental-fetal unit. In contrast, PE is associated with abnormal migration and invasion and reductions in spiral artery remodeling and placental perfusion (17, 184).
Reduced cytotrophoblast-mediated remodeling of spiral arteries represents an attractive mechanism whereby obesity could potentially increase the risk for the development of PE. It has been shown that reduced serum levels of the angiogenic factor placental growth factor (PlGF), which is an important factor in the proliferative nature of trophoblast cells, is better at predicting the development of PE in obese pregnant women compared with underweight/lean counterparts when assessed early in the second trimester (5, 56). This corroborates observations that obese women have reduced fetoplacental vascularity characterized by villi having large diameters and low numbers of capillaries (42). In addition, there is evidence showing that the degree of muscularity in the uteroplacental vasculature of obese women is greater than nonobese controls (147).
Therefore, it is reasonable to hypothesize in some cases that obesity may promote placental ischemia. This hypothesis is also supported by indirect evidence. It has been shown that there is a reduction in the fetus/placenta weight ratio (a marker of placental efficiency) with increased maternal BMI, which is in stark contrast to the 40-fold increase in this value seen from 6 wk of gestation to term in normal pregnancy (9, 187). Indeed, reductions in placental efficiency are linked to compensatory increases in placental growth and hypertrophy (7, 112). Higgins et al. (67) found that obesity is associated with placental hypertrophy along with reduced cell turnover, which are both characteristics of a fetal hypoxic insult (37, 85). These findings suggest that obesity-related metabolic factors may attenuate cytotrophoblast-mediated spiral artery remodeling thereby contributing to the development of placental ischemia-induced hypertension.
Experimental evidence supports that high-fat diet (HFD)-induced obesity adversely affects placental perfusion. Strong findings by Frias et al. (52) showed that 4 years of HFD (32% calories from fat) in pregnant monkeys increased maternal body weight and reduced placental perfusion measured in the third trimester. They calculated placental volume blood flow (cQUV) to estimate the blood flow on the fetal side of the placenta. The authors indicated that cQUV is reduced in pregnancies accompanied by intrauterine grow restriction. Interestingly, their HFD-fed monkeys segregated into those that had statistically significant increases in body weight (HFD sensitive) and those that did not (HFD resistant) compared with their control diet (14% calories from fat) counterparts. The HFD-resistant group had reduced uterine artery volume blood flow that was even greater in the HFD-sensitive group with placental volume blood flow being reduced only in the latter (Fig. 2). These data indicate that obesity per se is deleterious to placental perfusion. To begin probing the mechanisms responsible for the reduced placental perfusion, these authors found that circulating levels of the obesity-related metabolic factors leptin and insulin were significantly increased only in the HFD-sensitive group, and this was accompanied by higher placental protein levels of the pro-inflammatory factors toll-like receptor (TLR)-4, monocyte chemoattractant protein (MCP-1), and interleukin-1β. Blood pressure was not examined in this study in monkeys (52). However, it was found in rats maintained on a HFD from 3 to 19 wk of age that there were not only increases in body weight, serum leptin levels, and a immunohistochemical marker of placental hypoxia (carbonic anhydrase) but also elevated systolic blood pressure as measured by tail-cuff plethysmography on gestational day 15 (52, 64). These animal studies provide credence to the hypothesis that obesity-related metabolic factors and inflammation can induce placental ischemia or decrease the threshold whereby placental ischemia leads to hypertension in pregnancy.
Fig. 2.

High-fat diet (HFD) increases maternal body weight (A) and reduces placental volume blood flow (B), which is accompanied by increased plasma leptin (C) and insulin (D) levels in the third trimester of pregnancy in baboons. Control, control diet; R, HFD resistant; S, HFD sensitive; cQutv, placental volume blood flow. *P < 0.05 vs. control and HFD R. From Frias AE et al. (52).
While human and experimental studies in animals suggest that diet-induced obesity may affect placental function and perfusion, the pathophysiological mechanisms whereby metabolic factors alter the proliferation, migration, and invasion of cytrotrophoblast cells and uteroplacental vascular remodeling and these influences on placental ischemia have yet to be fully elucidated. Although the pathophysiological mechanisms responsible for the abnormal placental trophoblast invasion and vascular remodeling in PE are unclear, a number of factors have been recently implicated in placentation including the Notch signaling pathway. This pathway consists of the Notch receptors numbered 1–4 and the Notch ligands Jagged 1–2 and Delta 1 and 3–4. The formation of Notch receptor-ligand complexes on adjacent cells or even by acting in an autocrine fashion is important in orchestrating cell movements (138). Indeed, Dr. Susan Fisher's group demonstrated that Notch 2 is crucial for proper migration and integration of cytotrophoblasts into the spiral arteries of mice. They showed that mice lacking Notch 2 in trophoblast cells had significant reductions in uteroplacental vascular diameters (76). Blood pressure was not examined in her study. However, it has been shown in human placental tissue collected from hypertensive pregnancies and intrauterine growth restricted pregnancies that there is reduced Notch 1 (target p21), Notch 2, Notch 4, and reduced Jagged 1 and Jagged 2 (29, 155). These data support a connection between reduced Notch signaling and the pathogenesis of PE.
At this time there is only indirect evidence supporting that obesity disrupts Notch signaling in cytotrophoblast cells. It has been shown that adipose tissue multipotent stem cells isolated from morbidly obese individuals express reduced levels of Hey proteins, which are downstream targets of Notch signaling (151). Furthermore, circulating levels of Delta(DLK)1, which binds and stimulates apoptosis of Notch 2 expressing cells, are increased in obese humans (23, 197). While these data provide some evidence that obesity may attenuate Notch signaling, cytotrophoblast migration, and uteroplacental vascular morphogenesis, more research needs to evaluate how metabolic factors impact the Notch signaling pathway.
The mechanisms whereby obesity may cause disruption of cytotrophoblast migration and placental uterovascular morphogenesis are unclear but may be associated with the metabolic disturbances that occur in obesity such as hyperlipidemia, hyperinsulinemia, or hyperleptinemia. These factors are known to be elevated in plasma of obese pregnant women and in many cases even higher in women with PE. Indeed, total serum cholesterol levels in the first and second trimesters predict the onset of PE (38). Cekmen et al. (21) showed increases in low-density lipoprotein (LDL) cholesterol and its transport protein apolipoprotein B along with reductions in high-density lipoprotein (HDL) cholesterol in PE over normal pregnancy. Mechanistically, LDL, in particular its oxidized form, can reduce extravillous cytotrophoblast migration and promotes trophoblast apoptosis (95, 131). Oxidized cholesterols, termed oxysterols, inhibit first trimester trophoblast cell function and decrease glial cell missing-1 (GCM-1) (6). GCM-1 is important for fusion of syncytiotrophoblasts and the formation of the maternal-fetal interface (97). In addition, hypertriglyceridemia increases the risk for PE (70, 186). Genetic manipulations that increase triglycerides by knocking out apolipoprotein E in mice promote hypertension and proteinuria by the end of pregnancy (104). Placentas from these animals have increased levels of apoptotic markers including BAX, bcl-1, and caspase-3, and electron microscopy illustrated increased trophoblast apoptosis. Free fatty acid levels are increased in obesity and are elevated in PE (44, 45). Furthermore, free fatty acids can activate the nuclear receptor peroxisome proliferator-activated receptor-γ (PPAR-γ), whose expression is increased in placentas from preeclamptic pregnancies and signal to inhibit the invasiveness of trophoblast cells (69, 175). However, normal pregnant rats treated with a PPAR-γ antagonist from gestational days 11–15 developed increased blood pressure, proteinuria, endothelial dysfunction, reduced fetal weight, and placental abnormalities. In addition, reduced plasma VEGF and increased plasma and placental sFlt-1 were observed following administration of the PPAR-γ antagonist (111). Conversely, treatment of RUPP rats with a PPAR-γ agonist ameliorated hypertension, improved vascular function, and reduced microalbumin-to-creatinine ratios. Therefore, the role of PPAR-γ in mediating trophoblastic invasion and other features of PE requires further investigation.
A hallmark of many forms of obesity is insulin resistance and hyperinsulinemia. A higher rate of hyperinsulinemia occurs in preeclamptic versus normal pregnancy in humans (107). Experimental evidence directly linking hyperinsulinemia to the development of hypertension in pregnancy was demonstrated in pregnant rats where chronic infusion of insulin beginning 1 week before pregnancy and then continued throughout gestation raised insulin levels and increased blood pressure when measured at the end of pregnancy (136). Subsequent studies in this model showed that insulin reduced PlGF and arrested trophoblast invasion in the uterus (168). Intriguingly, it was noted that the trophoblasts in placentas from the hyperinsulinemic rats expressed greater endothelin converting enzyme (82). The major cleavage product of this enzyme is ET-1. Trophoblast cells express ET precursors and receptors (163). Fiore et al. (47) showed that exaggerated levels of ET-1 trigger placental villi to produce reactive oxygen species, which promote trophoblast apoptosis (47, 116). Placentas from preeclamptic pregnancies have increased placental oxidative stress and ET-1 binding (8, 22). These data are interesting in light of the findings that mice with HFD-induced obesity and insulin resistance have placental oxidative stress and reductions in trophoblast numbers (96).
As a caveat, the studies showing that increased insulin levels over the course of pregnancy increases blood pressure in rats were confounded by the effect of insulin-induced hypoglycemia. However, we have recently shown that increasing insulin levels toward the end of pregnancy (from gestational days 14–19) while maintaining euglycemia also raises blood pressure in rats (128) (Fig. 3). These data highlight that hyperinsulinemia could be a major player in the mechanisms whereby obesity increases the risk for PE.
Fig. 3.
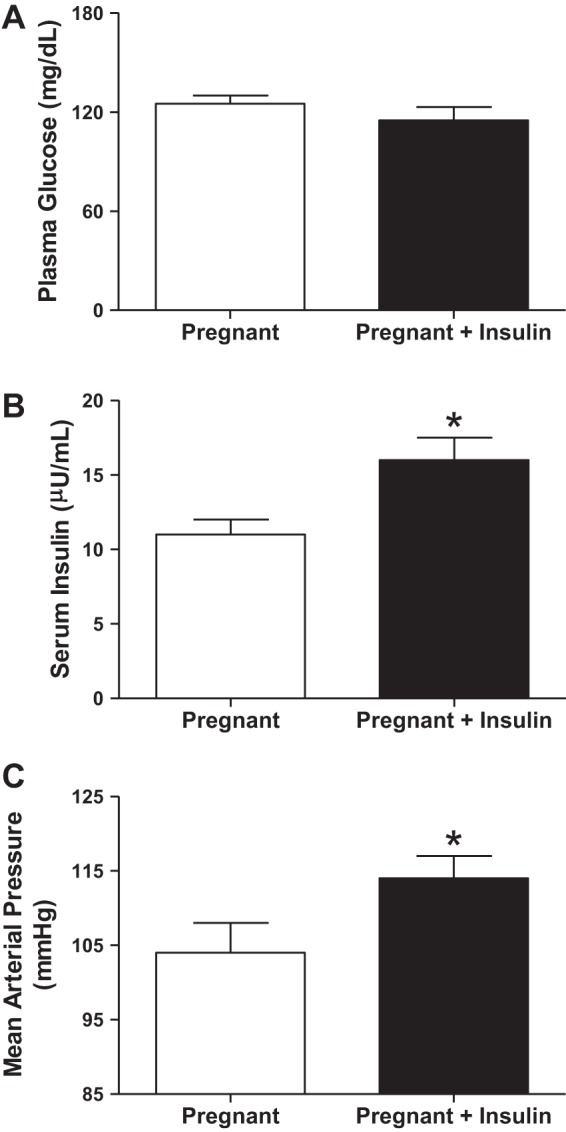
Euglycemic (A) hyperinsulinemia (B) elicits hypertension (C) in Sprague-Dawley pregnant rats. Insulin was infused at 1.5 mU·kg−1·day−1 from gestational day 14–19 while rats were provided with 20% glucose in drinking water. *P < 0.05 vs. pregnant. From Palei AC et al. (128).
Leptin is an adipokine whose levels rise in parallel with increasing adiposity and has been shown to mediate obesity-induced hypertension (160). Obese preeclamptic women have the greatest circulating leptin levels compared with lean preeclamptic women and lean and obese normotensive pregnant women (65, 115). Furthermore, leptin levels are greater in severely preeclamptic women compared with women whose pregnancies are solely complicated by chronic hypertension, which suggests that leptin is a link between obesity and PE (180). Indeed, studies have shown that leptin reduces cytotrophoblast proliferation (98) and reduces PlGF production from BeWo cells (202). Trophoblastic cells do express leptin receptors (199). Furthermore, leptin has been shown to activate the transcription factor hypoxia inducible factor (HIF)-1α in breast cancer cells (61). Because cytotrophoblast proliferation occurs early in pregnancy, before migration and invasion of these cells into the uterus (54), these data suggest that hyperleptinemia may play a role in obesity-induced placental ischemia and PE.
Although it has been consistently found that placental ischemia/hypoxia induces hypertension in pregnant experimental models, the mechanisms whereby placental dysfunction elicits maternal hypertension in humans seem to be much more complicated. It has been shown that early development of placental dysfunction may not always be the consequence of chronic ischemia/hypoxia. There are studies with findings ranging from data showing that placentas from women with PE have elevated expression of the hypoxia marker HIF-1α to data that there are increased oxygen tensions in PE placentas. Although these data may conflict one another at first glance, there are events that may occur within the placenta during the development of PE that may unify such opposing data. This includes ischemia-reperfusion (IR) injury. For example, it seems that low oxygen tension is considered to be in the physiological range for placental tissue and allows for proper trophoblast proliferation in early pregnancy. In a review by Huppertz et al. (77), it was detailed that during the first trimester, the embryo develops at low oxygen tension below 20 mmHg. This is before maternal blood flow is established through the uterine spiral arteries and into the intervillous spaces bathing the fetal villous structures where nutrient and oxygen exchange take place (77). If there were to be an early escape of maternal blood from spiral arteries, this could lead to premature increases oxygen levels in the vicinity of the developing trophoblast cells. It has been demonstrated that abnormal spiral artery remodeling is associated with increased, not reduced, oxygen levels in placentas from pregnancies complicated by hypertension (with and without proteinuria) or intrauterine growth restriction (79–81, 164). Thus this sequence of events seems similar to what is seen with IR insults (72–75). First, it is known that IR injury induces increased expression of the transcription factor HIF-1α in tissues such as liver (32). HIF-1α levels are greater in placental tissue from preeclamptic versus normal pregnant women (140). Second, it is established in tissues such as the heart, liver, and brain that bouts of ischemia reduce oxygen consumption (133, 179, 196), which would seemingly result in heightened levels of oxygen and oxidative stress during reperfusion. In vitro IR injury increases oxidative stress in the term human placenta in villous endothelial, trophoblast, and stromal cells (71). Regarding trophoblast function, oxidative stress produced by xanthine oxidase elicits apoptosis in isolated extravillous trophoblast cells, which are those that are derived from proliferating trophoblast cells (118). There is increased apoptosis of first trimester extravillous trophoblasts from pregnancies at higher risk for developing PE with elevated uterine artery resistance (198). It is important to direct our understanding of how obesity and its accompanying metabolic factors affect early placental development and that high oxygen tensions may in fact be deleterious to placental development and function. Indeed, it has been shown in vitro that high glucose levels reduce proliferation of a first trimester trophoblast cell line only at high oxygen tensions (53). Immunohistochemical data from term villous tissue from obese patients have decreased villous proliferation (67). Therefore, the development of a model of placental IR injury may mimic more closely the development of PE in humans, and we predict this injury would be exacerbated in the face of obesity.
Effects of Obesity on the Release of Soluble Placental Factors
Angiogenic imbalance.
The cascade of events linking placental ischemia to maternal hypertension has been a subject of intense focus in our laboratory and others. While both placental and circulating levels of VEGF and PlGF are reduced in preeclamptic women, the levels of the VEGF/PlGF receptor sFlt-1 are elevated (27, 102, 143, 161, 193). By using the RUPP model, we have shown that placental ischemia/hypoxia elicits the release of sFlt-1 (58). The latter is thought to occur largely via activation of the transcription factor HIF-1α (176). Sequestration of circulating VEGF and PlGF by sFlt-1 inhibits their proangiogenic and provasodilatory actions. Supplementation of VEGF significantly attenuates placental ischemia-induced hypertension (59). Confirmation that sFlt-1 is capable of elevating blood pressure during PE is evidenced from infusion studies whereby increasing sFlt-1 levels to mimic those found in preeclamptic women promotes the development of hypertension in pregnant rats (120).
There are fewer investigations examining sFlt-1 levels in obese pregnancies. For instance, Masuyama et al. (108) reported that sFlt-1 levels in an obese hypertensive pregnancy were greater than those in obese normotensive pregnancy in early and late pregnancy. Supportive of this increase in sFlt-1 is the finding that obese preeclamptic women have lower levels of PlGF, which typically rise during healthy pregnancy, than nonobese preeclamptic women (108). There are several lines of evidence suggesting that this may be due to the actions of obesity-related metabolic factors. Indeed, leptin and insulin, which are elevated in obesity and PE, both reduce PlGF release from BeWo human trophoblastic cells (202). Furthermore, we have collected data using an obese pregnant rat model having increased body weight and fat mass at gestational day 19, namely melanocortin-4 receptor heterozygous (MC4R+/−) pregnant rats, suggesting that they have baseline levels (without surgically induced placenta ischemia) of sFlt-1 that are slightly elevated in the circulation; significantly increased in adipose tissue; but not altered in placental tissue levels (Fig. 4) (171). These animals also had slight elevations in leptin and blood pressure. Intriguingly, we have data using ex vivo placental explant experiments suggesting that sFlt-1 release is greater under hypoxic conditions in obese pregnant MC4R+/− versus control MC4R+/+ pregnant rats (Fig. 5), which suggests that placental ischemia raises sFlt-1 to a greater extent in obese pregnancies. These findings implicate metabolic factors as a potential cause of angiogenic imbalance and further reduced vasorelaxation in obese pregnancies. There are novel adipokine factors including the serine protease adipsin, also known as factor D in the complement system, that are shown to be released to a greater extent from placentas from obese pregnant women (167) and whose levels are found to be greater in the urine from preeclamptic women (167, 190).
Fig. 4.
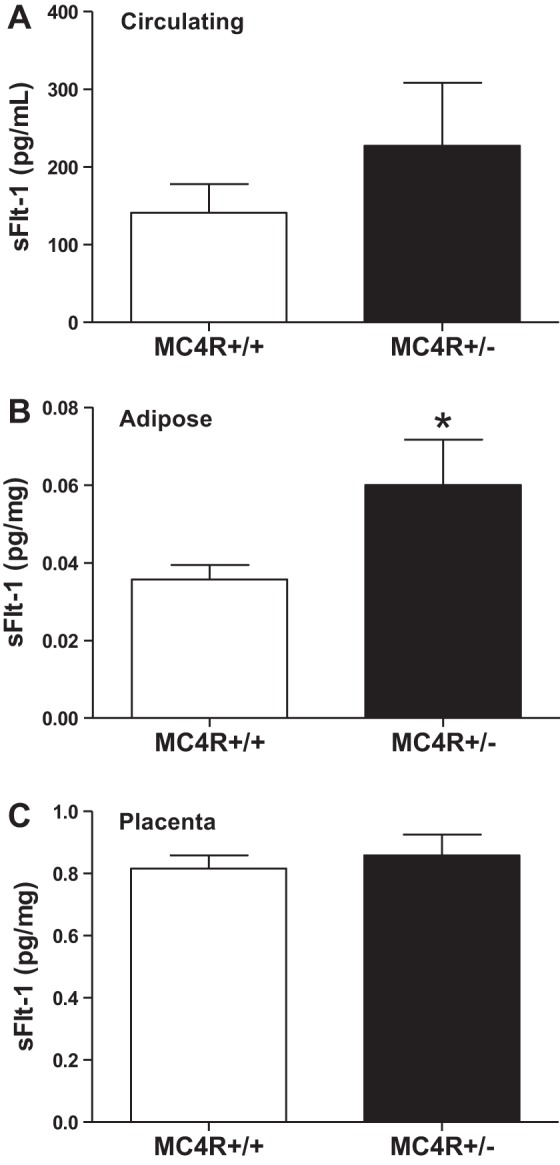
The soluble fms-like tyrosine kinase 1 (sFlt-1) levels in obese MC4R+/− versus MC4R+/+ pregnant rats are slightly but not significantly elevated the circulation (A), elevated in adipose tissue (B); but similar in placental tissue (C). The sFlt-1 was quantified using a mouse Flt-1 ELISA from R&D Systems. *P < 0.05 vs. MC4R+/+ pregnant rats. From Spradley FT et al. (171).
Fig. 5.
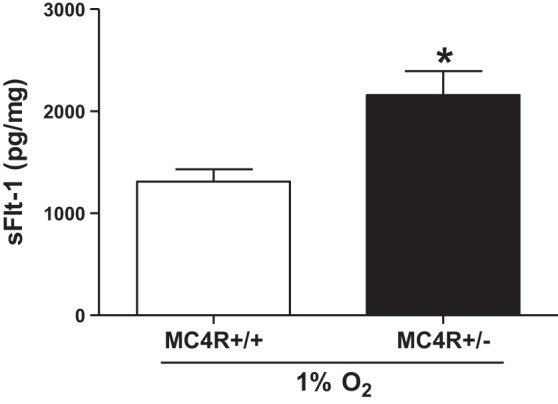
The release of sFlt-1 is greater under hypoxic conditions in placental villous explants isolated from obese MC4R+/− (n = 10) versus lean MC4R+/+ (n = 6) pregnant rats. Placental explant studies were conducted as previously described (55). The sFlt-1 was quantified using a mouse Flt-1 ELISA from R&D Systems. Data are means ± SE. *P < 0.05 vs. MC4R+/+ pregnant rats. These data are unpublished.
Human studies have also demonstrated reduced levels of placental factors such as sFlt-1 in obese preeclamptic women. Straughen et al. (174) demonstrated that there are reduced sFlt-1 levels in obese compared with normal weight preeclamptic women. However, one confounding factor could be increased plasma volume during obese pregnancies, which has been demonstrated in pregnant rats fed a cafeteria-style diet (2). This may result in dilution of these soluble placental factors in the maternal circulation. Also, the extracellular matrix of adipose tissue expresses heparin sulfate proteoglycans (106), which may lead to tissue retention of the sFlt-1 produced and sequestration of sFlt-1 from the circulation, in addition to increased local action of sFlt-1 in pregnant women with greater adipose mass. Therefore, it may not be that sFlt-1 production is reduced, but that there are confounding factors (e.g., increased plasma volume and tissue retention) clouding these measurements in obese pregnancies if their levels have not reached the steady state between placental metabolic production and renal clearance rates.
Inflammatory factors.
Proper maternal immune cell function is critical for establishment of the uteroplacental circulation. Evidence implicates a strong role of the innate immune system in the regulation of trophoblast function. Indeed, resident uterine natural killer (uNK) cells, which are the most abundant leukocytes in early human and mouse decidua, regulate the invasiveness of trophoblast cells and uterine vascular remodeling and angiogenesis. Knockout mouse studies have emphasized this whereby adoptive transfer of NK cells from Rag2−/− mice (having NK cells but not T or B lymphocytes) into BALB/c-Rag2−/−Il2−/− mice (having no NK, T, or B cells) fully reversed the uterine vascular defects found in the latter mouse (68). Moreover, coculture experiments of human uNK and extravillous trophoblast cells with isolated spiral arteries demonstrated that uNK cells certainly mediate spiral artery remodeling (149). In contrast, these cells isolated from women destined to develop PE secrete less trophoblast invasion-promoting factors (145).
There are also findings that excessive activation of the innate immune system promotes the etiopathology of PE (134). Immunohistochemical studies revealed that there is increased expression of the CD14 macrophage marker in the decidua basalis from preterm preeclamptic versus preterm control pregnancies (157). Interestingly, placental tissue from Type 1 diabetic women and from streptozotocin (STZ) diabetic rats had markers of increased pro-inflammatory M1 and decreased anti-inflammatory M2 macrophages (166). In vitro experiments from these investigators found that high glucose levels activated pro-inflammatory genes belonging to TLR-dependent pathways isolated rat placental macrophages. These data implicate obesity and metabolic factors in promoting a pro-inflammatory environment in the placenta.
The adaptive immune system has caught the attention of researchers in the field of PE. Whereas normal pregnancy is associated with a T helper (Th) 2 anti-inflammatory state, a subpopulation of pro-inflammatory CD4+ T lymphocytes termed Th17 cells characterized by production of the cytokine IL-17 are increased in the peripheral blood of preeclamptic patients in the third trimester compared with a control group (35). A direct role for this immune cell population was defined by LaMarca's group (30) whereby an IL-17 inhibitor significantly reduced RUPP-induced hypertension and AT1-AA levels and that adoptive transfer of Th17 cells from RUPP rats elicits increases in blood pressure and AT1-AA in normal pregnant rats. Additionally, administration of a CD20 blocker that acts through B lymphocyte depletion into RUPP rats decreased circulating levels of AT1-AA and resulted in a blunted blood pressure response to placental ischemia (90). This group had also previously shown that CD4+ cells contribute to circulating placental ischemia pro-inflammatory factors like TNF-α and can also activate B cells to produce AT1-AA (124, 188). A direct role for obesity in activation of the immune system during pregnancy and PE should be addressed as studies in other pro-inflammatory diseases such as colitis have found that leptin receptors on CD4+ cells quicken disease progression (165).
Inflammatory mechanisms have been shown to mediate placental ischemia-induced production of sFlt-1 is via action of AT1-AA on AT1 receptors in the placenta. We have shown that RUPP increases circulating levels of AT1-AA (91). Moreover, AT1-AA infusion into normal pregnant rats increases blood pressure and plasma sFlt-1 levels along with release of sFlt-1 from placental explants from the same animals. These effects were blocked by the AT1 receptor antagonist losartan (130). Interestingly, we have recent evidence suggesting that the placenta may not be the only source of circulating sFlt-1 levels in response to elevated circulating AT1-AA. With respect to obesity, we showed in normal pregnant obese versus lean rats that adipose tissue levels of sFlt-1 were significantly greater accompanying a slight but nonstatistical increase in circulating sFlt-1 with no difference in placental levels (171). These data suggest that adipose tissue may contribute to the circulating pool of sFlt-1. Data from Herse and colleagues (66) showed that mature adipocytes isolated from humans do express and release sFlt-1 (Fig. 6). To determine whether in fact AT1-AA can stimulate the release of sFlt-1 from adipose tissue, we conducted adipose tissue explants studies. AT1-AA was capable of increasing the release of sFlt-1 into culture media verging on statistical significance (Fig. 6). However, it is important to note that these acute studies were conducted using adipose tissue from normal pregnant Sprague-Dawley rats. Therefore, in the face of placental ischemia, obese pregnant animals may have exaggerated circulating levels of sFlt-1, as a consequence of a synergistic action of AT1-AA in the placenta and adipose tissue, and a more profound pro-hypertensive phenotype.
Fig. 6.

Top, left: mRNA expression of sFlt-1 in isolated adipose-derived stem cells (ADSCs) and isolated mature adipocytes from human adipose tissue; *P < 0.01. Middle: release of sFlt-1 from ADSCs incubated for 5 days and mature adipocytes incubated for 3 or 5 days; *P < 0.05 for 3 vs. 5 days; n.d., not detectable. Right: release of sFlt-1 from adipose tissue nonfat cells, stromal vascular cells (SV cells) and mature adipocytes; *P < 0.01 vs. adipose tissue nonfat cells. From Herse et al. (66). Bottom: stimulation with AT1-AA promotes sFlt-1 release from adipose tissue explants isolated from normal pregnant (NP) Sprague-Dawley rats. Adipose explant (∼100 mg) were incubated in F12K supplemented with media (DMEM containing 4 mM l-glutamin, 4,500 mg/l glucose, 1 mM sodium pyruvate, 1,500 mg/l sodium bicarbonate, 4 g/l BSA, and 1% PenStrep) in the presence or absence of AT1-AA (1:100 dilution) for 48 h, at which time media were collected and assayed for sFlt-1 as previously described (171). n = 3 rats/group. Data are means ± SE. Data in bottom panel are unpublished.
Release of the pro-inflammatory cytokine TNF-α is increased in response to placental ischemia/hypoxia (132). We have evidence showing that placental ischemia-induced hypertension increases placental mRNA and protein levels of TNF-α in pregnant rats (89). Supplementation with the TNF-α inhibitor etanercept significantly attenuates the development of placental ischemia-induced hypertension in rats (89). In addition, we have recently demonstrated in vitro that acute hypoxia increases TNF-α secretion from normal pregnant rat placental explants (122). Similarly, hypoxia stimulated the release of TNF-α from placental explants isolated from normal pregnant women (11, 12, 132). Although hypoxia stimulated the same degree of release of TNF-α from placental explants from preeclamptic women, the levels under room oxygen and hypoxia were actually less in this group (12). These data are supported by in vivo studies from Conrad's group (12) suggesting that, at delivery, the placenta does not secrete greater amounts of TNF-α during PE; this was because the peripheral and uterine venous TNF-α levels were 1.0, which was similar to the values found in normal pregnant women. Even so, these preeclamptic women did have greater peripheral and uterine venous levels of TNF-α than normal pregnant women. This study concluded that other sources than the placenta must contribute to the greater circulating levels of TNF-α during PE. In support of this notion, in already-preeclamptic women, TNF-α mRNA levels were greater in peripheral blood leukocytes from healthy pregnant women versus those with PE (26). Future investigations should be carried out to determine whether initial insults to the placenta, such as in placental ischemia, lead first to increases in placental TNF-α levels that may subsequently feedforward to activation of placental and circulating immune cells levels that then autonomously maintain elevated cytokine levels instead of the diseased placenta during the clinical manifestations of PE.
With regard to obesity, TNF-α mRNA levels have been demonstrated to be greater in peripheral blood mononuclear cells and placental macrophages isolated from obese compared with lean pregnant women (24). We found that circulating levels of TNF-α are increased in obese MC4R+/− pregnant rats along with elevations in circulating leptin (171). While human data have shown that hyperleptinemia is positively associated with PE (114), the long-term effect of hyperleptinemia on blood pressure during pregnancy is unknown. We recently tested the hypothesis that chronic circulating leptin elevations in pregnant rats increases blood pressure and placental factors known to play a role in PE (126). On gestational day 14, normal pregnant rats were infused with leptin at a rate to mimic plasma levels in obesity. We found that hyperleptinemia in pregnancy resulted in significant increases in blood pressure and placental TNF-α expression despite reductions in food intake and body weight. The blood pressure response to leptin observed in our pregnant rats [change in mean arterial pressure (ΔMAP) ∼ 20 mmHg] was higher than that documented in previous studies using male rats (ΔMAP ∼ 6–8 mmHg) (160). Interestingly, a recent report found that acute intracerebroventricular infusion of leptin enhances renal sympathetic activity, but not MAP, in nonpregnant female rats (162). This effect of leptin on stimulating renal nerves was observed only during the estrous phase, suggesting that high estrogen levels may exaggerate the blood pressure response to hyperleptinemia. Whether the exaggerated blood pressure response to leptin during pregnancy is due to the hyperestrogenemic state of pregnancy or to an effect of leptin on placental function is unknown and remains to be investigated.
There are limitations in currently available animal models of PE. Although the RUPP model serves as an excellent system to study the impact of placental ischemia on maternal cardiovascular-renal function, which will be discussed in the succeeding section, this system does not provide mechanistic insight into earlier causes of placental dysfunction such as trophoblast-mediated spiral artery remodeling. This is because the symptoms in the RUPP model are induced after midpregnancy. However, a spontaneous model of superimposed PE in the Dahl salt-sensitive rat has been evaluated (60). These animals have hypertension before pregnancy and develop dramatic pregnancy-induced increases in blood pressure all the while being maintained on normal-salt rodent chow compared with spontaneously hypertensive rats (SHR) that are hypertensive before pregnancy and normotensive Sprague-Dawley controls. This model has great promise for studying the early pathogenesis of PE and the potential for deleterious actions of obesity and related metabolic factors on this process.
Effects of Obesity on Placental Ischemia-Induced Endothelial and Arterial Smooth Muscle Cell Dysfunction and Hypertension
The endothelium is very important in the control of vascular tone homeostasis and blood pressure. Studies using the RUPP rat model of placental ischemia have shown that placental ischemia/hypoxia stimulates production of soluble inflammatory and anti-angiogenic placental factors that are capable of entering into the maternal circulation where they target the endothelium. These factors cause vascular dysfunction by reducing nitric oxide (NO) bioavailability and increasing ET-1 production (36, 83). Endothelial dysfunction has been detected in obese pregnancies. A study comparing obese versus lean pregnant women demonstrated that obesity was associated with elevated blood pressure and reduced endothelium-dependent vasorelaxation in skin arteries (142, 173). It was not shown whether this was dependent on reductions in NO levels or activity of the endothelial enzyme that produces NO (endothelial NOS; NOS3). However, these investigators did find that there was reduced responsiveness to the exogenous NO donor sodium nitroprusside (SNP) in these arteries suggesting that this dysfunction lies in part to reduced smooth muscle responsiveness to NO. These data are reminiscent to findings in male rabbits fed a high-cholesterol diet, which has been shown in other studies to elicit increases in serum total and LDL cholesterol and reduced serum HDL cholesterol, where there was reduced proximal descending aortic response to acetylcholine as well as NO (195). With regard to the latter, it was found that isolated smooth muscle cells from these hypercholesterolemic rats had reduced responsiveness to NO-mediated inhibition of ANG II-induced increases in intracellular calcium suggestive of attenuation in the capacity of NO to buffer vasoconstriction. These data collectively show an association between obesity and its accompanying metabolic dysfunction, such as hypercholesterolemia, and disruption of NO signaling in control of blood vessel tone. Furthermore, they suggest that obesity increases the sensitivity for developing endothelial and smooth muscle dysfunction, and thus hypertension, in response to placental ischemia factors due to reduced bioavailability and the vasoconstrictive buffering power of NO.
The studies mentioned above also showed that obese women enter pregnancy with preexisting endothelial dysfunction. However, it seems that the degree that pregnancy increased endothelium-dependent relaxation was similar between the obese versus lean groups even though the absolute values were still greater in the lean pregnant women (173). These data indicate that pregnancy-induced vasorelaxation is not altered by obesity. It should be examined whether these mechanisms are similar between obese and lean pregnancies. A complete understanding of these mechanisms could lead to discoveries about novel targets of placental ischemia during obesity. Furthermore, studies should examine the association of maternal obesity with PE in the context of obesity-induced preexisting endothelial dysfunction versus pregnancy-induced vascular dysfunction that may occur during excessive gestational weight gain. It was found in women having supra-physiological gestational weight gain, that is, rates of weight gain of >0.42 kg/wk, presented with markers of reduced NOS3 activity and vasorelaxation in isolated umbilical vein rings (129).
Studies in humans and animals support that elevated maternal sFlt-1 promotes the development of hypertension accompanied by reduced NO production in systemic and renal circulations (109, 120). The infusion of sFlt-1 into pregnant Sprague-Dawley rats elicits hypertension and reduces both endothelium-dependent vasorelaxation responses to acetylcholine and smooth muscle responsiveness to SNP in carotid artery rings (16, 178). This endothelial dysfunction was significantly curtailed by treating the rings with the oxidative stress scavenger tiron, whereas the reduced response to SNP was not as influenced by tiron. Thus sFlt-1-induced hypertension is accompanied by oxidative stress-mediated endothelial dysfunction. The reduced NO responsiveness of the underlying vascular smooth muscle cells seems to be dependent on alterations in calcium flux across the plasma membrane. Indeed, smooth muscle cells isolated from renal interlobular arteries from RUPP rats, which have increased circulating levels of sFlt-1, have enhanced and maintained ANG II-, KCl-, and calcium channel antagonist-induced intracellular calcium levels and cell contraction compared with normal pregnant rats (119). These data support that placental ischemia elicits endothelial and vascular smooth muscle cell dysfunction.
Indirect evidence to support that obesity exaggerates the deleterious effects of sFlt-1 on pregnancy was demonstrated by the finding that diet-induced obese pregnant mice infused with sFlt-1 have offspring that go on to develop the highest blood pressures when compared nonobese and noninfused controls (20). More direct data about the combined actions of sFlt-1 and obesity on maternal vascular function comes from studies showing that pro-angiogenic factors are important for vasorelaxation responses to hormones during healthy pregnancy. Indeed, treatment of rat and mouse small renal arteries with recombinant human relaxin significantly attenuated myogenic constriction, which was blocked by VEGF receptor antagonism, VEGF neutralizing antibodies, or NOS inhibition. Intriguingly, it has been shown in high-fat diet-induced overweight female Wistar Hannover rats that there is reduced effectiveness of relaxin to promote flow-mediated vasodilation and blunt phenylephrine-induced vasoconstriction and the myogenic response, which was in part mediated by reduced signaling to NOS (113, 183). Thus aberrations in the action of VEGF to promote the ability of relaxin to stimulate NOS in the blood vessel wall could consequently allow for exaggerated dysfunction in response to placental factors like sFlt-1 in obesity. This is perhaps especially true if the blood vessel is strongly dependent on NO signaling to maintain vascular tone. Foremost, we have shown that pregnancy dramatically increases the control of blood pressure on NOS whereby NOS inhibition in Sprague-Dawley rats increases blood pressure more in pregnant rats versus age-matched nonpregnant controls. We have shown that sFlt-1 infusion blunts glomerular NO production much to the extent as seen with maximal NOS inhibition implicating the NO system as the target of sFlt-1-mediated hypertension during placental ischemia. With regard to obesity, it seems that obese females have increased NO bioavailability whereby nitrite/nitrate levels, as a surrogate marker of NO, are positively correlated with body fat in obese women (28). This may be an explanation to why high-fat diet-induced obesity promotes greater hypertension in male than female mice (63). Along these lines, we have shown greater endothelium-dependent vasorelaxation to acetylcholine (ACh) and to the NO donor SNP in small mesenteric arteries from genetically obese MC4R+/− pregnant rats (Fig. 7) (171). The latter suggests that vascular tone control in these obese rats is more dependent on NO signaling. However, it is not yet understood what is the cause of this increased NO signaling. Perhaps there is a change in the components of ACh-induced relaxation in these mesenteric arteries from the genetically obese pregnant rats, such as a shift from endothelial hyperpolarizing factor (EDHF)-mediated to NO-mediated relaxation. It was shown in male Wistar rats with diet-induced obesity that ACh-mediated vasorelaxation in coronary arterioles is maintained similar to that of their lean counterparts because of increased smooth muscle response to NO, which was mediated by increased soluble guanylate cyclase (sGC) function (78). As a result, the SNP response was increased in these obese rats. This is in contrast to conduit arteries like the aorta, which typically have reduced ACh response with no change in SNP responsiveness all accompanied by reduced endothelial NOS expression. For instance, this occurs in the face of high-fat diet-induced obesity in female mice (84). These data suggest that the potentially compensatory increase in NO signaling in small and resistance arteries during obesity in pregnancy may predispose these types of blood vessels to greater vascular dysfunction in response to soluble placental ischemic factors, which are known to reduce NO bioavailability. Future studies will probe whether this holds true in the uteroplacental and renal circulations. Further studies should address whether there is increased sGC function in our obese model and whether there are regional differences NO-sGC signaling during obesity in pregnancy, such as in mesenteric versus uteroplacental arteries. Whether this increased NO vasorelaxation is also dependent on deficiency of the MC4R receptor itself or if it is mediated by increased body weight or altered circulating metabolic factors such as their elevated leptin levels is yet to be determined. In this regard, it is has been shown that exogenous leptin has the capability to increase NO dependency of blood pressure regulation (87). Moreover, leptin infusion into male mice resulting in levels similar to those in our obese pregnant rats attenuated ANG II-induced endothelial dysfunction (10). Thus the contribution of NO and NO-dependent mechanisms to vascular dysfunction during obesity in pregnancy and preeclampsia is still unclear. In summary, it is not clear what role NO sensitivity of blood vessel tone has on placental ischemia-induced hypertension in obesity. From the above discussions, we propose that obesity exaggerates placental ischemia-induced release of soluble placental factors such as sFlt-1 subsequently leading to greater reduction in NO bioavailability culminating in accentuated placental ischemia-induced hypertension during obesity.
Fig. 7.

Endothelial-dependent responses to acetylcholine (ACh) (A; left: concentration-response curve, right: logEC50 sensitivity) and endothelial-independent responses to sodium nitroprusside (SNP) (B; left: concentration-response curve, right: logEC50 sensitivity) are enhanced in third-order mesenteric arteries from obese MC4R+/− pregnant versus lean MC4R+/+ pregnant rats. *P < 0.05 vs. MC4R+/+ pregnant rats. From Spradley et al. (171).
Inflammatory cytokines such TNF-α have also been implicated in PE and many other cardiovascular diseases including patients with essential hypertension (191, 200). Our previous studies showed that placental ischemia stimulates production of placental TNF-α and increases circulating levels of this cytokine in pregnant rats (92). Furthermore, we have shown that infusion of TNF-α into normal pregnant rats elicits hypertension and reductions in renal and systemic vascular NO systems (3, 57). Circulating, adipose tissue, and placental levels of TNF-α levels are increased in obese women (18, 19, 24, 86). Founds et al. (50) performed a study to examine whether TNF-α may be a potential mechanistic link between obesity and PE. They used four groups of pregnant women, namely, lean, lean preeclamptic, obese, and obese preeclamptic (50). There are several important observations to be gleaned from this study: 1) obese preeclamptic women had the greatest blood pressures, 2) blood pressure and TNF-α levels were increased in obese versus lean control subjects, and 3) TNF levels were the greatest in the preeclamptic patients but were not different from the obese and lean preeclamptic groups. On the surface, this third finding may seem disappointing. However, this might indicate that more TNF-α is bound to its receptor thereby exerting more biological activity and vascular dysfunction in obese preeclamptic women or that, in obesity, less TNF-α is needed to promote vascular dysfunction. In support of this theory, although only performed in male mice, it was demonstrated that HFD-induced obesity is associated with greater TNF-α-induced impairment of endothelium/NO-mediated vasorelaxation in resistance arteries isolated from white adipose tissue even though plasma levels of this cytokine were similar between normal diet and HFD groups (41). It should be considered that this phenomenon may result from local production of TNF-α in adipose or the vasculature per se that can impact arterial function without reaching the circulation. Thus future studies should assess both circulating TNF-α levels and local vascular levels of not only TNF-α but also the TNF receptor in obese preeclamptic women. While existing data suggest that placental ischemia-induced endothelial dysfunction and hypertension may be exaggerated in obese versus lean pregnancies via a TNF-α-dependent mechanism, the importance of inflammatory cytokines in the pathophysiological mechanisms whereby obesity increases the risk for developing PE remains unclear. Moreover, whether maternal immune tolerance mechanisms that include interactions between regulatory CD4+ T cells and uNK cells play a role in obese pregnancies also remains to be determined.
Recent studies have also suggested an important role for ET-1 in the pathophysiology of PE. ET-1 is the most potent vasoconstrictor known. ET-1 is derived from a longer 203 amino acid precursor known as preproendothelin, and the active peptide is proteolytically cleaved into its final 21 amino acid form. ET-1 exerts its powerful vasoconstrictor effects via the endothelin type A (ETA) receptor located on vascular smooth muscle cells. Multiple studies have examined circulating levels of ET-1 in normal and preeclamptic pregnant women. They have found elevated levels of plasma ET-1 in the preeclamptic group (153), with some studies suggesting that the level of circulating ET-1 correlates with the severity of the disease symptoms (123), although this is not a universal finding (158). ET-1, however, is produced locally in the vasculature and plasma levels typically do not reflect tissue levels of the peptide. Local renal and endothelial production of ET-1 is exaggerated in placental ischemic conditions (121, 148). We have shown that the hypertensive responses to infusion of the soluble placental ischemia factors TNF-α, AT1-AA, or sFlt-1 into normal pregnant rats and in response to RUPP are prevented by antagonism of the ETA receptor (88, 93, 121, 177).
It is thought that the increased production of ET-1 and its hypertensive actions during placental ischemia are a result of reduced NO bioavailability. Support for this comes from in vivo studies in male rats showing that falls in single nephron GFR (SNGFR) and glomerular plasma flow following systemic NOS inhibition with NG-nitro-l-arginine methyl ester (l-NAME) were prevented with the ETA/ETB antagonist bosentan (137). More recent ex vivo functional data have gone on to demonstrate that the concomitant ET-1-mediated reductions in SNGFR and plasma flow seem to be dependent on signaling through the ETA receptor to produce stronger vasoconstriction in mouse microperfused afferent over efferent arterioles. l-NAME was able to exaggerate ET-1-induced constriction only in the latter (156). With regard to pregnancy, l-NAME reduces renal blood flow by 35% by the end of gestation in Sprague-Dawley rats (1), and we have shown that sFlt-1 and l-NAME induce similar degrees of hypertension and reductions in glomerular production of NO (120). In sFlt-1-infused pregnant rats, the increase in renal cortical mRNA levels of preproET-1, which is the precursor to ET-1, was reduced by supplementation with the NOS cofactor l-arginine. Together, support that NOS functionally tempers the production and hemodynamic effects of ET-1 in the kidney, and that when NOS is targeted and inhibited by placental ischemic factors like sFlt-1, this allows ET-1 to elicit the development of maternal hypertension.
Obesity is associated with over activation of the ET system. Overweight and obese humans have enhanced ET-1-mediated endothelial dysfunction (194). An ETA receptor antagonist attenuated HFD-induced hypertension in rats having increased visceral obesity and leptin levels (34). Indirect evident suggests that this pathway is augmented in obese preeclamptic situations. For example, studies in nonpregnant animals and humans have demonstrated that obesity is associated with increased ET-1-induced vasoconstriction and ET-1-mediated reductions in endothelium-dependent vasorelaxation (117, 194). Intriguingly, there are data showing that the adipokine chemerin, whose levels are increased in PE (172), augments ET-1-induced vasoconstriction (99). In summary, we believe that one potential mechanism whereby obesity and obesity-related metabolic factors may increase the risk of developing PE is by enhancing placental factor (sFlt-1, TNF-α, AT-1AA)-induced production of ET-1. In fact, we have novel data showing that TNF-α-induced ET-1 production from human umbilical vein endothelial cells (HUVECs) is exaggerated in the presence of high obese levels of insulin (Fig. 8).
Fig. 8.
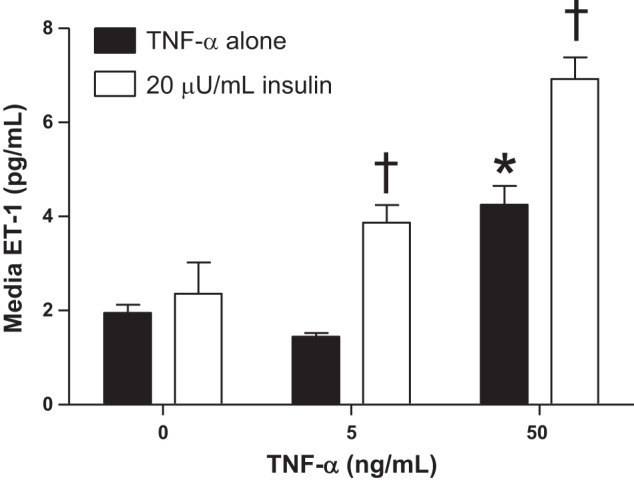
Insulin exaggerates tumor nerosis factor-α (TNF-α)-induced release of endothelin-1 (ET-1) from human umbilical vein endothelial cells (HUVECs). One million cells were seeded into 6-well plates and allowed to grow until 70% confluent at which time they were serum starved for 36 h then treated with TNF-α or in combination with insulin at levels observed in obese preeclamptic women (20 μU/ml) for 8 h. ET-1 levels were measured in HUVEC-conditioned media as previously described (89). n = 3–6 wells/group. Data are means ± SE. *P < 0.05 vs. 0 TNF-α; †P < 0.05 vs. corresponding TNF-α group. These data are unpublished.
It is recognized that both obesity and PE are complex health disorders associated with multiple genetic and environmental traits. Although they share many pathophysiological mechanisms as we have discussed in the review, not all obese pregnant women develop PE. Indeed, a 30% increase in the risk, as most epidemiological studies examining the impact of obesity on the rates of PE have found, predicts that only 10% of obese women will develop the disease (146). Human studies suggest that preeclamptic patients express exaggerated metabolic profiles, including hyperleptinemia, hyperinsulinemia, insulin resistance, and hyperlipidemia, which precedes the appearance of the clinical symptoms (25, 38, 100, 169). Intriguingly, in vitro and in vivo studies have shown that depending on the concentration of obesity-related metabolic factors, there are beneficial or detrimental effects on placental-fetal development and vascular function (33, 62). We and others (146) believe that obese pregnant women having an extremely abnormal metabolic profile are at increased risk to develop PE. Furthermore, as obese pregnant women have endothelial dysfunction before the development of PE, it is likely that obesity may decrease the threshold at which the combination of obesity-related metabolic factors and soluble placental ischemic factors elicit cardiovascular dysfunction and hypertension. Thus, the greater the BMI is during pregnancy, the greater the likelihood of decreasing this threshold for developing PE.
Perspectives and Significance
Growing evidence implicates obesity as a major risk factor for the development of PE, a hypertensive disease of pregnancy (14). This is bolstered by demographic data indicating that there is a high prevalence of PE in the southern United States, a region with significantly high obesity rates. Importantly, the concurrence of obesity and PE is a worldwide phenomenon. Confirmation of a relationship between obesity and PE has been shown in Brazil, Colombia, United Kingdom, Finland, Japan, Kazakhstan, Portugal, Trinidad, Tobego, and Australia. This highlights the importance of understanding the mechanisms linking these two pathologies. Foremost, the pathophysiology of PE is a two-stage process with placental and maternal origins. Reductions in cytotrophoblast migration and uterine spiral artery remodeling are strongly tied to placental ischemia. This insult stimulates the placenta to release soluble antiangiogenic and inflammatory factors into the maternal circulation favoring endothelial and vascular dysfunction and hypertension. Because this cascade of events is thought to play an important role in the pathogenesis of PE, it is likely that obesity-related metabolic factors may increase the risk of developing PE by impinging on these pathophysiological processes. Therefore, in this review, we propose that obesity impacts the various stages thought to encompass the pathogenesis of PE by 1) acting on the placenta causing cytotrophoblast dysfunction and placental ischemia and dysfunction, 2) enhancing ischemia/hypoxia-induced release of soluble placental factors, and 3) increasing the sensitivity by which placental ischemia-factors are able to promote endothelial dysfunction and hypertension (Fig. 9). Because of the alarming increase in the prevalence of obesity, the strong correlation between BMI and PE rates, and the fact that PE is a major cause of maternal and perinatal morbidity and mortality, a better understanding of how obesity impacts the pathogenesis of PE remains to be a critical area of investigation.
Fig. 9.

Hypothetical scheme whereby obesity and obesity-related metabolic factors act in the cascade of events leading to enhancement of placental ischemia-induced hypertension. We propose that these metabolic factors may act individually or in combination to potentiate dysfunctional uteroplacental vascular remodeling, exaggerate the release of soluble placental factors into the maternal circulation, and to exaggerate the maternal cardiovascular-renal responses to these soluble placental-ischemia factors. This may occur in a feed-forward fashion subsequently exaggerating the downstream actions of these metabolic factors.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants 5P01HL051971-22 and 1R01HL108618-01 (to J. P. Granger), 2T32HL105324-06 (to F. T. Spradley), and 14POST18970005 (to A. C. Palei).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: F.T.S., A.C.P., and J.P.G. conception and design of research; F.T.S. and A.C.P. interpreted results of experiments; F.T.S. prepared figures; F.T.S. drafted manuscript; F.T.S., A.C.P., and J.P.G. edited and revised manuscript; F.T.S., A.C.P., and J.P.G. approved final version of manuscript.
REFERENCES
- 1.Ajne G, Nisell H, Jansson T. Effects of blockade of the endothelin receptor A and inhibition of nitric oxide synthesis on uteroplacental and renal blood flow in awake pregnant rats. Am J Obstet Gynecol 192: 295–301, 2005. [DOI] [PubMed] [Google Scholar]
- 2.Akyol A, Langley-Evans SC, McMullen S. Obesity induced by cafeteria feeding and pregnancy outcome in the rat. Br J Nutr 102: 1601–1610, 2009. [DOI] [PubMed] [Google Scholar]
- 3.Alexander BT, Cockrell KL, Massey MB, Bennett WA, Granger JP. Tumor necrosis factor-alpha-induced hypertension in pregnant rats results in decreased renal neuronal nitric oxide synthase expression. Am J Hypertens 15: 170–175, 2002. [DOI] [PubMed] [Google Scholar]
- 4.American College of Obstetricians and Gynecologists; Task Force on Hypertension in Pregnancy. Hypertension in pregnancy. Report of the American College of Obstetricians and Gynecologists' Task Force on Hypertension in Pregnancy. Obstet Gynecol 122: 1122–1131, 2013. [DOI] [PubMed] [Google Scholar]
- 5.Athanassiades A, Hamilton GS, Lala PK. Vascular endothelial growth factor stimulates proliferation but not migration or invasiveness in human extravillous trophoblast. Biol Reprod 59: 643–654, 1998. [DOI] [PubMed] [Google Scholar]
- 6.Aye IL, Waddell BJ, Mark PJ, Keelan JA. Oxysterols inhibit differentiation and fusion of term primary trophoblasts by activating liver X receptors. Placenta 32: 183–191, 2011. [DOI] [PubMed] [Google Scholar]
- 7.Baptiste-Roberts K, Salafia CM, Nicholson WK, Duggan A, Wang NY, Brancati FL. Maternal risk factors for abnormal placental growth: the national collaborative perinatal project. BMC Pregnancy Childbirth 8: 44, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barros JS, Bairos VA, Baptista MG, Fagulha JO. Immunocytochemical localization of endothelin-1 in human placenta from normal and preeclamptic pregnancies. Hypertens Pregnancy 20: 125–137, 2001. [DOI] [PubMed] [Google Scholar]
- 9.Benirschke K. Remarkable placenta. Clin Anatomy 11: 194–205, 1998. [DOI] [PubMed] [Google Scholar]
- 10.Benkhoff S, Loot AE, Pierson I, Sturza A, Kohlstedt K, Fleming I, Shimokawa H, Grisk O, Brandes RP, Schroder K. Leptin potentiates endothelium-dependent relaxation by inducing endothelial expression of neuronal NO synthase. Arterioscler Thromb Vasc Biol 32: 1605–1612, 2012. [DOI] [PubMed] [Google Scholar]
- 11.Benyo DF, Miles TM, Conrad KP. Hypoxia stimulates cytokine production by villous explants from the human placenta. J Clin Endocrinol Metab 82: 1582–1588, 1997. [DOI] [PubMed] [Google Scholar]
- 12.Benyo DF, Smarason A, Redman CW, Sims C, Conrad KP. Expression of inflammatory cytokines in placentas from women with preeclampsia. J Clin Endocrinol Metab 86: 2505–2512, 2001. [DOI] [PubMed] [Google Scholar]
- 13.Bhattacharya S, Campbell DM, Liston WA, Bhattacharya S. Effect of Body Mass Index on pregnancy outcomes in nulliparous women delivering singleton babies. BMC Public Health 7: 168, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bodnar LM, Ness RB, Harger GF, Roberts JM. Inflammation and triglycerides partially mediate the effect of prepregnancy body mass index on the risk of preeclampsia. Am J Epidemiol 162: 1198–1206, 2005. [DOI] [PubMed] [Google Scholar]
- 15.Bodnar LM, Ness RB, Markovic N, Roberts JM. The risk of preeclampsia rises with increasing prepregnancy body mass index. Ann Epidemiol 15: 475–482, 2005. [DOI] [PubMed] [Google Scholar]
- 16.Bridges JP, Gilbert JS, Colson D, Gilbert SA, Dukes MP, Ryan MJ, Granger JP. Oxidative stress contributes to soluble fms-like tyrosine kinase-1 induced vascular dysfunction in pregnant rats. Am J Hypertens 22: 564–568, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brosens IA, Robertson WB, Dixon HG. The role of the spiral arteries in the pathogenesis of preeclampsia. Obstet Gynecol Ann 1: 177–191, 1972. [PubMed] [Google Scholar]
- 18.Bullo M, Garcia-Lorda P, Peinado-Onsurbe J, Hernandez M, Del Castillo D, Argiles JM, Salas-Salvado J. TNFalpha expression of subcutaneous adipose tissue in obese and morbid obese females: relationship to adipocyte LPL activity and leptin synthesis. Int J Obes Relat Metab Disord 26: 652–658, 2002. [DOI] [PubMed] [Google Scholar]
- 19.Bullo M, Garcia-Lorda P, Salas-Salvado J. Plasma soluble tumor necrosis factor alpha receptors and leptin levels in normal-weight and obese women: effect of adiposity and diabetes. Eur J Endocrinol 146: 325–331, 2002. [DOI] [PubMed] [Google Scholar]
- 20.Bytautiene E, Tamayo E, Kechichian T, Drever N, Gamble P, Hankins GD, Saade GR. Prepregnancy obesity and sFlt1-induced preeclampsia in mice: developmental programming model of metabolic syndrome. Am J Obstet Gynecol 204: 398 e391–e398, 2011. [DOI] [PubMed] [Google Scholar]
- 21.Cekmen MB, Erbagci AB, Balat A, Duman C, Maral H, Ergen K, Ozden M, Balat O, Kuskay S. Plasma lipid and lipoprotein concentrations in pregnancy induced hypertension. Clin Biochem 36: 575–578, 2003. [DOI] [PubMed] [Google Scholar]
- 22.Cervar M, Kainer F, Desoye G. Pre-eclampsia and gestational age differently alter binding of endothelin-1 to placental and trophoblast membrane preparations. Molec Cell Endocrinol 110: 65–71, 1995. [DOI] [PubMed] [Google Scholar]
- 23.Chacon MR, Miranda M, Jensen CH, Fernandez-Real JM, Vilarrasa N, Gutierrez C, Naf S, Gomez JM, Vendrell J. Human serum levels of fetal antigen 1 (FA1/Dlk1) increase with obesity, are negatively associated with insulin sensitivity and modulate inflammation in vitro. Int J Obes (Lond) 32: 1122–1129, 2008. [DOI] [PubMed] [Google Scholar]
- 24.Challier JC, Basu S, Bintein T, Minium J, Hotmire K, Catalano PM, Hauguel-de Mouzon S. Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta 29: 274–281, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chappell LC, Seed PT, Briley A, Kelly FJ, Hunt BJ, Charnock-Jones DS, Mallet AI, Poston L. A longitudinal study of biochemical variables in women at risk of preeclampsia. Am J Obstet Gynecol 187: 127–136, 2002. [DOI] [PubMed] [Google Scholar]
- 26.Chen G, Wilson R, Wang SH, Zheng HZ, Walker JJ, McKillop JH. Tumour necrosis factor-alpha (TNF-alpha) gene polymorphism and expression in pre-eclampsia. Clin Exp Immunol 104: 154–159, 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cho GJ, Roh GS, Kim HJ, Kim YS, Cho SH, Choi WJ, Paik WY, Kang SS, Choi WS. Differential expression of placenta growth factors and their receptors in the normal and pregnancy-induced hypertensive human placentas. J Korean Med Sci 18: 402–408, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi JW, Pai SH, Kim SK, Ito M, Park CS, Cha YN. Increases in nitric oxide concentrations correlate strongly with body fat in obese humans. Clin Chem 47: 1106–1109, 2001. [PubMed] [Google Scholar]
- 29.Cobellis L, Mastrogiacomo A, Federico E, Schettino MT, De Falco M, Manente L, Coppola G, Torella M, Colacurci N, De Luca A. Distribution of Notch protein members in normal and preeclampsia-complicated placentas. Cell Tissue Res 330: 527–534, 2007. [DOI] [PubMed] [Google Scholar]
- 30.Cornelius DC, Wallace K, Scott JD, Campbell N, Thomas A, Hogg JP, Moseley J, LaMarca B. [35-OR]: A role for TH17 cells and IL-17 in mediating the pathophysiology associated with preeclampsia. Pregnancy Hypertens 5: 17, 2015. [Google Scholar]
- 31.Craici IM, Wagner SJ, Bailey KR, Fitz-Gibbon PD, Wood-Wentz CM, Turner ST, Hayman SR, White WM, Brost BC, Rose CH, Grande JP, Garovic VD. Podocyturia predates proteinuria and clinical features of preeclampsia: longitudinal prospective study. Hypertension 61: 1289–1296, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cursio R, Miele C, Filippa N, Van Obberghen E, Gugenheim J. Liver HIF-1 alpha induction precedes apoptosis following normothermic ischemia-reperfusion in rats. Transplant Proc 40: 2042–2045, 2008. [DOI] [PubMed] [Google Scholar]
- 33.D'Ippolito S, Tersigni C, Scambia G, Di Simone N. Adipokines, an adipose tissue and placental product with biological functions during pregnancy. Biofactors 38: 14–23, 2012. [DOI] [PubMed] [Google Scholar]
- 34.da Silva AA, Kuo JJ, Tallam LS, Hall JE. Role of endothelin-1 in blood pressure regulation in a rat model of visceral obesity and hypertension. Hypertension 43: 383–387, 2004. [DOI] [PubMed] [Google Scholar]
- 35.Darmochwal-Kolarz D, Kludka-Sternik M, Tabarkiewicz J, Kolarz B, Rolinski J, Leszczynska-Gorzelak B, Oleszczuk J. The predominance of Th17 lymphocytes and decreased number and function of Treg cells in preeclampsia. J Reprod Immunol 93: 75–81, 2012. [DOI] [PubMed] [Google Scholar]
- 36.Davis JR, Giardina JB, Green GM, Alexander BT, Granger JP, Khalil RA. Reduced endothelial NO-cGMP vascular relaxation pathway during TNF-alpha-induced hypertension in pregnant rats. Am J Physiol Regul Integr Comp Physiol 282: R390–R399, 2002. [DOI] [PubMed] [Google Scholar]
- 37.de Grauw TJ, Myers RE, Scott WJ. Fetal growth retardation in rats from different levels of hypoxia. Biol Neonate 49: 85–89, 1986. [DOI] [PubMed] [Google Scholar]
- 38.Dey M, Arora D, Narayan N, Kumar R. Serum cholesterol and ceruloplasmin levels in second trimester can predict development of pre-eclampsia. North Am J Medical Sci 5: 41–46, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Di Benedetto A, D'Anna R, Cannata ML, Giordano D, Interdonato ML, Corrado F. Effects of prepregnancy body mass index and weight gain during pregnancy on perinatal outcome in glucose-tolerant women. Diabetes Metab 38: 63–67, 2012. [DOI] [PubMed] [Google Scholar]
- 40.Dietz PM, Callaghan WM, Sharma AJ. High pregnancy weight gain and risk of excessive fetal growth. Am J Obstet Gynecol 201: 51 e51–56, 2009. [DOI] [PubMed] [Google Scholar]
- 41.Donato AJ, Henson GD, Morgan RG, Enz RA, Walker AE, Lesniewski LA. TNF-alpha impairs endothelial function in adipose tissue resistance arteries of mice with diet-induced obesity. Am J Physiol Heart Circ Physiol 303: H672–H679, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dubova EA, Pavlov KA, Borovkova EI, Bayramova MA, Makarov IO, Shchegolev AI. Vascular endothelial growth factor and its receptors in the placenta of pregnant women with obesity. Bull Exp Biol Med 151: 253–258, 2011. [DOI] [PubMed] [Google Scholar]
- 43.El-Chaar D, Finkelstein SA, Tu X, Fell DB, Gaudet L, Sylvain J, Tawagi G, Wen SW, Walker M. The impact of increasing obesity class on obstetrical outcomes. J Obstet Gynaecol Can 35: 224–233, 2013. [DOI] [PubMed] [Google Scholar]
- 44.Endresen MJ, Tosti E, Heimli H, Lorentzen B, Henriksen T. Effects of free fatty acids found increased in women who develop pre-eclampsia on the ability of endothelial cells to produce prostacyclin, cGMP and inhibit platelet aggregation. Scand J Clin Lab Invest 54: 549–557, 1994. [DOI] [PubMed] [Google Scholar]
- 45.Fabbrini E, deHaseth D, Deivanayagam S, Mohammed BS, Vitola BE, Klein S. Alterations in fatty acid kinetics in obese adolescents with increased intrahepatic triglyceride content. Obesity (Silver Spring) 17: 25–29, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferraro ZM, Barrowman N, Prud'homme D, Walker M, Wen SW, Rodger M, Adamo KB. Excessive gestational weight gain predicts large for gestational age neonates independent of maternal body mass index. J Matern Fetal Neonatal Med 25: 538–542, 2012. [DOI] [PubMed] [Google Scholar]
- 47.Fiore G, Capasso A. Effects of vitamin E and C on placental oxidative stress: an in vitro evidence for the potential therapeutic or prophylactic treatment of preeclampsia. Med Chem 4: 526–530, 2008. [DOI] [PubMed] [Google Scholar]
- 48.Flegal KM, Carroll MD, Kit BK, Ogden CL. Prevalence of obesity and trends in the distribution of body mass index among US adults, 1999–2010. JAMA 307: 491–497, 2012. [DOI] [PubMed] [Google Scholar]
- 49.Flegal KM, Carroll MD, Ogden CL, Curtin LR. Prevalence and trends in obesity among US adults, 1999–2008. JAMA 303: 235–241, 2010. [DOI] [PubMed] [Google Scholar]
- 50.Founds SA, Powers RW, Patrick TE, Ren D, Harger GF, Markovic N, Roberts JM. A comparison of circulating TNF-alpha in obese and lean women with and without preeclampsia. Hypertension Pregnancy 27: 39–48, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frederick IO, Williams MA, Sales AE, Martin DP, Killien M. Pre-pregnancy body mass index, gestational weight gain, and other maternal characteristics in relation to infant birth weight. Matern Child Health J 12: 557–567, 2008. [DOI] [PubMed] [Google Scholar]
- 52.Frias AE, Morgan TK, Evans AE, Rasanen J, Oh KY, Thornburg KL, Grove KL. Maternal high-fat diet disturbs uteroplacental hemodynamics and increases the frequency of stillbirth in a nonhuman primate model of excess nutrition. Endocrinology 152: 2456–2464, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frohlich JD, Huppertz B, Abuja PM, Konig J, Desoye G. Oxygen modulates the response of first-trimester trophoblasts to hyperglycemia. Am J Pathol 180: 153–164, 2012. [DOI] [PubMed] [Google Scholar]
- 54.Genbacev O, Zhou Y, Ludlow JW, Fisher SJ. Regulation of human placental development by oxygen tension. Science 277: 1669–1672, 1997. [DOI] [PubMed] [Google Scholar]
- 55.George EM, Colson D, Dixon J, Palei AC, Granger JP. Heme Oxygenase-1 Attenuates Hypoxia-Induced sFlt-1 and Oxidative Stress in Placental Villi through Its Metabolic Products CO and Bilirubin. Int J hypertens 2012: 486053, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ghosh SK, Raheja S, Tuli A, Raghunandan C, Agarwal S. Serum placental growth factor as a predictor of early onset preeclampsia in overweight/obese pregnant women. J Am Soc Hypertens 7: 137–148, 2013. [DOI] [PubMed] [Google Scholar]
- 57.Giardina JB, Green GM, Cockrell KL, Granger JP, Khalil RA. TNF-alpha enhances contraction and inhibits endothelial NO-cGMP relaxation in systemic vessels of pregnant rats. Am J Physiol Regul Integr Comp Physiol 283: R130–R143, 2002. [DOI] [PubMed] [Google Scholar]
- 58.Gilbert JS, Babcock SA, Granger JP. Hypertension produced by reduced uterine perfusion in pregnant rats is associated with increased soluble fms-like tyrosine kinase-1 expression. Hypertension 50: 1142–1147, 2007. [DOI] [PubMed] [Google Scholar]
- 59.Gilbert JS, Verzwyvelt J, Colson D, Arany M, Karumanchi SA, Granger JP. Recombinant vascular endothelial growth factor 121 infusion lowers blood pressure and improves renal function in rats with placentalischemia-induced hypertension. Hypertension 55: 380–385, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gillis EE, Williams JM, Garrett MR, Mooney JN, Sasser JM. The Dahl salt sensitive rat is a spontaneous model of superimposed preeclampsia. Am J Physiol Regul Integr Comp Physiol 309: R62–R70, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gonzalez-Perez RR, Xu Y, Guo S, Watters A, Zhou W, Leibovich SJ. Leptin upregulates VEGF in breast cancer via canonic and non-canonical signalling pathways and NFkappaB/HIF-1alpha activation. Cell Signal 22: 1350–1362, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gu P, Xu A. Interplay between adipose tissue and blood vessels in obesity and vascular dysfunction. Rev Endocr Metab Disord 14: 49–58, 2013. [DOI] [PubMed] [Google Scholar]
- 63.Gupte M, Thatcher SE, Boustany-Kari CM, Shoemaker R, Yiannikouris F, Zhang X, Karounos M, Cassis LA. Angiotensin converting enzyme 2 contributes to sex differences in the development of obesity hypertension in C57BL/6 mice. Arterioscler Thromb Vasc Biol 32: 1392–1399, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hayes EK, Lechowicz A, Petrik JJ, Storozhuk Y, Paez-Parent S, Dai Q, Samjoo IA, Mansell M, Gruslin A, Holloway AC, Raha S. Adverse fetal and neonatal outcomes associated with a life-long high fat diet: role of altered development of the placental vasculature. PLos One 7: e33370, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hendler I, Blackwell SC, Mehta SH, Whitty JE, Russell E, Sorokin Y, Cotton DB. The levels of leptin, adiponectin, and resistin in normal weight, overweight, and obese pregnant women with and without preeclampsia. Am J Obstet Gynecol 193: 979–983, 2005. [DOI] [PubMed] [Google Scholar]
- 66.Herse F, Fain JN, Janke J, Engeli S, Kuhn C, Frey N, Weich HA, Bergmann A, Kappert K, Karumanchi SA, Luft FC, Muller DN, Staff AC, Dechend R. Adipose tissue-derived soluble fms-like tyrosine kinase 1 is an obesity-relevant endogenous paracrine adipokine. Hypertension 58: 37–42, 2011. [DOI] [PubMed] [Google Scholar]
- 67.Higgins L, Mills TA, Greenwood SL, Cowley EJ, Sibley CP, Jones RL. Maternal obesity and its effect on placental cell turnover. J Matern Fetal Neonatal Med 26: 783–788, 2013. [DOI] [PubMed] [Google Scholar]
- 68.Hofmann AP, Gerber SA, Croy BA. Uterine natural killer cells pace early development of mouse decidua basalis. Mol Human Reproduct 20: 66–76, 2014. [DOI] [PubMed] [Google Scholar]
- 69.Holdsworth-Carson SJ, Lim R, Mitton A, Whitehead C, Rice GE, Permezel M, Lappas M. Peroxisome proliferator-activated receptors are altered in pathologies of the human placenta: gestational diabetes mellitus, intrauterine growth restriction and preeclampsia. Placenta 31: 222–229, 2010. [DOI] [PubMed] [Google Scholar]
- 70.Hubel CA, McLaughlin MK, Evans RW, Hauth BA, Sims CJ, Roberts JM. Fasting serum triglycerides, free fatty acids, and malondialdehyde are increased in preeclampsia, are positively correlated, and decrease within 48 hours post partum. Am J Obstet Gynecol 174: 975–982, 1996. [DOI] [PubMed] [Google Scholar]
- 71.Hung TH, Burton GJ. Hypoxia and reoxygenation: a possible mechanism for placental oxidative stress in preeclampsia. Taiwan J Obstet Gynecol 45: 189–200, 2006. [DOI] [PubMed] [Google Scholar]
- 72.Hung TH, Charnock-Jones DS, Skepper JN, Burton GJ. Secretion of tumor necrosis factor-alpha from human placental tissues induced by hypoxia-reoxygenation causes endothelial cell activation in vitro: a potential mediator of the inflammatory response in preeclampsia. Am J Pathol 164: 1049–1061, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hung TH, Chen SF, Liou JD, Hsu JJ, Li MJ, Yeh YL, Hsieh TT. Bax, Bak and mitochondrial oxidants are involved in hypoxia-reoxygenation-induced apoptosis in human placenta. Placenta 29: 565–583, 2008. [DOI] [PubMed] [Google Scholar]
- 74.Hung TH, Skepper JN, Burton GJ. In vitro ischemia-reperfusion injury in term human placenta as a model for oxidative stress in pathological pregnancies. Am J Pathol 159: 1031–1043, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hung TH, Skepper JN, Charnock-Jones DS, Burton GJ. Hypoxia-reoxygenation: a potent inducer of apoptotic changes in the human placenta and possible etiological factor in preeclampsia. Circ Res 90: 1274–1281, 2002. [DOI] [PubMed] [Google Scholar]
- 76.Hunkapiller NM, Gasperowicz M, Kapidzic M, Plaks V, Maltepe E, Kitajewski J, Cross JC, Fisher SJ. A role for Notch signaling in trophoblast endovascular invasion and in the pathogenesis of pre-eclampsia. Development 138: 2987–2998, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huppertz B, Weiss G, Moser G. Trophoblast invasion and oxygenation of the placenta: measurements versus presumptions. J Reprod Immunol 101–102: 74–79, 2014. [DOI] [PubMed] [Google Scholar]
- 78.Jebelovszki E, Kiraly C, Erdei N, Feher A, Pasztor ET, Rutkai I, Forster T, Edes I, Koller A, Bagi Z. High-fat diet-induced obesity leads to increased NO sensitivity of rat coronary arterioles: role of soluble guanylate cyclase activation. Am J Physiol Heart Circ Physiol 294: H2558–H2564, 2008. [DOI] [PubMed] [Google Scholar]
- 79.Kakogawa J, Sumimoto K, Kawamura T, Minoura S, Kanayama N. Noninvasive monitoring of placental oxygenation by near-infrared spectroscopy. Am J Perinatol 27: 463–468, 2010. [DOI] [PubMed] [Google Scholar]
- 80.Kakogawa J, Sumimoto K, Kawamura T, Minoura S, Kanayama N. Transabdominal measurement of placental oxygenation by near-infrared spectroscopy. Am J Perinatol 27: 25–29, 2010. [DOI] [PubMed] [Google Scholar]
- 81.Kawamura T, Kakogawa J, Takeuchi Y, Takani S, Kimura S, Nishiguchi T, Sugimura M, Sumimoto K, Kanayama N. Measurement of placental oxygenation by transabdominal near-infrared spectroscopy. Am J Perinatol 24: 161–166, 2007. [DOI] [PubMed] [Google Scholar]
- 82.Khamaisi M, Skarzinski G, Mekler J, Zreik F, Damouni R, Ariel I, Bursztyn M. Hyperinsulinemia increases placenta endothelin-converting enzyme-1 expression in trophoblasts. Am J Hypertens 25: 109–114, 2012. [DOI] [PubMed] [Google Scholar]
- 83.Kiprono LV, Wallace K, Moseley J, Martin J Jr, Lamarca B. Progesterone blunts vascular endothelial cell secretion of endothelin-1 in response to placental ischemia. Am J Obstet Gynecol 209: 44 e41–46, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kobayasi R, Akamine EH, Davel AP, Rodrigues MA, Carvalho CR, Rossoni LV. Oxidative stress and inflammatory mediators contribute to endothelial dysfunction in high-fat diet-induced obesity in mice. J Hypertens 28: 2111–2119, 2010. [DOI] [PubMed] [Google Scholar]
- 85.Kruger H, Arias-Stella J. The placenta and the newborn infant at high altitudes. Am J Obstet Gynecol 106: 586–591, 1970. [DOI] [PubMed] [Google Scholar]
- 86.Kueht ML, McFarlin BK, Lee RE. Severely obese have greater LPS-stimulated TNF-alpha production than normal weight African-American women. Obesity (Silver Spring) 17: 447–451, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kuo JJ, Jones OB, Hall JE. Inhibition of NO synthesis enhances chronic cardiovascular and renal actions of leptin. Hypertension 37: 670–676, 2001. [DOI] [PubMed] [Google Scholar]
- 88.LaMarca B, Parrish M, Ray LF, Murphy SR, Roberts L, Glover P, Wallukat G, Wenzel K, Cockrell K, Martin JN Jr, Ryan MJ, Dechend R. Hypertension in response to autoantibodies to the angiotensin II type I receptor (AT1-AA) in pregnant rats: role of endothelin-1. Hypertension 54: 905–909, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.LaMarca B, Speed J, Fournier L, Babcock SA, Berry H, Cockrell K, Granger JP. Hypertension in response to chronic reductions in uterine perfusion in pregnant rats: effect of tumor necrosis factor-alpha blockade. Hypertension 52: 1161–1167, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.LaMarca B, Wallace K, Herse F, Wallukat G, Martin JN Jr, Weimer A, Dechend R. Hypertension in response to placental ischemia during pregnancy: role of B lymphocytes. Hypertension 57: 865–871, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.LaMarca B, Wallukat G, Llinas M, Herse F, Dechend R, Granger JP. Autoantibodies to the angiotensin type I receptor in response to placental ischemia and tumor necrosis factor alpha in pregnant rats. Hypertension 52: 1168–1172, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.LaMarca BB, Bennett WA, Alexander BT, Cockrell K, Granger JP. Hypertension produced by reductions in uterine perfusion in the pregnant rat: role of tumor necrosis factor-alpha. Hypertension 46: 1022–1025, 2005. [DOI] [PubMed] [Google Scholar]
- 93.LaMarca BB, Cockrell K, Sullivan E, Bennett W, Granger JP. Role of endothelin in mediating tumor necrosis factor-induced hypertension in pregnant rats. Hypertension 46: 82–86, 2005. [DOI] [PubMed] [Google Scholar]
- 94.Langford A, Joshu C, Chang JJ, Myles T, Leet T. Does gestational weight gain affect the risk of adverse maternal and infant outcomes in overweight women? Matern Child Health J 15: 860–865, 2011. [DOI] [PubMed] [Google Scholar]
- 95.Lee H, Park H, Kim YJ, Kim HJ, Ahn YM, Park B, Park JH, Lee BE. Expression of lectin-like oxidized low-density lipoprotein receptor-1 (LOX-1) in human preeclamptic placenta: possible implications in the process of trophoblast apoptosis. Placenta 26: 226–233, 2005. [DOI] [PubMed] [Google Scholar]
- 96.Liang C, DeCourcy K, Prater MR. High-saturated-fat diet induces gestational diabetes and placental vasculopathy in C57BL/6 mice. Metabolism 59: 943–950, 2010. [DOI] [PubMed] [Google Scholar]
- 97.Liang CY, Wang LJ, Chen CP, Chen LF, Chen YH, Chen H. GCM1 regulation of the expression of syncytin 2 and its cognate receptor MFSD2A in human placenta. Biol Reprod 83: 387–395, 2010. [DOI] [PubMed] [Google Scholar]
- 98.Liu H, Wu Y, Qiao F, Gong X. Effect of leptin on cytotrophoblast proliferation and invasion. J Huazhong Univ Sci Technolog Med Sci 29: 631–636, 2009. [DOI] [PubMed] [Google Scholar]
- 99.Lobato NS, Neves KB, Filgueira FP, Fortes ZB, Carvalho MH, Webb RC, Oliveira AM, Tostes RC. The adipokine chemerin augments vascular reactivity to contractile stimuli via activation of the MEK-ERK1/2 pathway. Life Sci 91: 600–606, 2012. [DOI] [PubMed] [Google Scholar]
- 100.Lorentzen B, Drevon CA, Endresen MJ, Henriksen T. Fatty acid pattern of esterified and free fatty acids in sera of women with normal and pre-eclamptic pregnancy. Br J Obstet Gynaecol 102: 530–537, 1995. [DOI] [PubMed] [Google Scholar]
- 101.Macdonald-Wallis C, Tilling K, Fraser A, Nelson SM, Lawlor DA. Gestational weight gain as a risk factor for hypertensive disorders of pregnancy. Am J Obstet Gynecol 209: 327.e1–327.e17, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Maebayashi Asanuma A, Yamamoto T, Azuma H, Kato E, Yamamoto N, Murase T, Chishima F, Suzuki M. Expression of placenta growth factor, soluble fms-like tyrosine kinase-1, metal-responsive transcription factor-1, heme oxygenase 1 and hypoxia inducible factor-1alpha mRNAs in pre-eclampsia placenta and the effect of pre-eclampsia sera on their expression of choriocarcinoma cells. J Obstet Gynaecol Res 40: 2095–2103, 2014. [DOI] [PubMed] [Google Scholar]
- 103.Magann EF, Doherty DA, Sandlin AT, Chauhan SP, Morrison JC. The effects of an increasing gradient of maternal obesity on pregnancy outcomes. Aust N Z J Obstet Gynaecol 53: 250–257, 2013. [DOI] [PubMed] [Google Scholar]
- 104.Mao L, Zhou Q, Zhou S, Wilbur RR, Li X. Roles of apolipoprotein E (ApoE) and inducible nitric oxide synthase (iNOS) in inflammation and apoptosis in preeclampsia pathogenesis and progression. PLos One 8: e58168, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Margerison Zilko CE, Rehkopf D, Abrams B. Association of maternal gestational weight gain with short- and long-term maternal and child health outcomes. Am J Obstet Gynecol 202: 574 e571–e578, 2010. [DOI] [PubMed] [Google Scholar]
- 106.Mariman EC, Wang P. Adipocyte extracellular matrix composition, dynamics and role in obesity. Cell Mol Life Sci 67: 1277–1292, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Martinez Abundis E, Gonzalez Ortiz M, Quinones Galvan A, Ferrannini E. Hyperinsulinemia in glucose-tolerant women with preeclampsia. A controlled study. Am J Hypertens 9: 610–614, 1996. [DOI] [PubMed] [Google Scholar]
- 108.Masuyama H, Nobumoto E, Segawa T, Hiramatsu Y. Severe superimposed preeclampsia with obesity, diabetes and a mild imbalance of angiogenic factors. Acta Med Okayama 66: 171–175, 2012. [DOI] [PubMed] [Google Scholar]
- 109.Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, Epstein FH, Sukhatme VP, Karumanchi SA. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest 111: 649–658, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mbah AK, Kornosky JL, Kristensen S, August EM, Alio AP, Marty PJ, Belogolovkin V, Bruder K, Salihu HM. Super-obesity and risk for early and late pre-eclampsia. BJOG 117: 997–1004, 2010. [DOI] [PubMed] [Google Scholar]
- 111.McCarthy FP, Drewlo S, English FA, Kingdom J, Johns EJ, Kenny LC, Walsh SK. Evidence implicating peroxisome proliferator-activated receptor-gamma in the pathogenesis of preeclampsia. Hypertension 58: 882–887, 2011. [DOI] [PubMed] [Google Scholar]
- 112.McDonald SD, Han Z, Mulla S, Beyene J, Knowledge Synthesis Group. Overweight and obesity in mothers and risk of preterm birth and low birth weight infants: systematic review and meta-analyses. BMJ 341: c3428, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.McGuane JT, Danielson LA, Debrah JE, Rubin JP, Novak J, Conrad KP. Angiogenic growth factors are new and essential players in the sustained relaxin vasodilatory pathway in rodents and humans. Hypertension 57: 1151–1160, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mendieta Zeron H, Garcia Solorio VJ, Nava Diaz PM, Garduno Alanis A, Santillan Benitez JG, Dominguez Garcia V, Escobar Briones C, Denova Gutierrez E. Hyperleptinemia as a prognostic factor for preeclampsia: a cohort study. Acta Medica 55: 165–171, 2012. [DOI] [PubMed] [Google Scholar]
- 115.Mise H, Sagawa N, Matsumoto T, Yura S, Nanno H, Itoh H, Mori T, Masuzaki H, Hosoda K, Ogawa Y, Nakao K. Augmented placental production of leptin in preeclampsia: possible involvement of placental hypoxia. J Clin Endocrinol Metab 83: 3225–3229, 1998. [DOI] [PubMed] [Google Scholar]
- 116.Moll SJ, Jones CJ, Crocker IP, Baker PN, Heazell AE. Epidermal growth factor rescues trophoblast apoptosis induced by reactive oxygen species. Apoptosis 12: 1611–1622, 2007. [DOI] [PubMed] [Google Scholar]
- 117.Mundy AL, Haas E, Bhattacharya I, Widmer CC, Kretz M, Baumann K, Barton M. Endothelin stimulates vascular hydroxyl radical formation: effect of obesity. Am J Physiol Regul Integr Comp Physiol 293: R2218–R2224, 2007. [DOI] [PubMed] [Google Scholar]
- 118.Murata M, Fukushima K, Takao T, Seki H, Takeda S, Wake N. Oxidative stress produced by xanthine oxidase induces apoptosis in human extravillous trophoblast cells. J Reproduct Dev 59: 7–13, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Murphy JG, Herrington JN, Granger JP, Khalil RA. Enhanced [Ca2+]i in renal arterial smooth muscle cells of pregnant rats with reduced uterine perfusion pressure. Am J Physiol Heart Circ Physiol 284: H393–H403, 2003. [DOI] [PubMed] [Google Scholar]
- 120.Murphy SR, LaMarca B, Cockrell K, Arany M, Granger JP. l-arginine supplementation abolishes the blood pressure and endothelin response to chronic increases in plasma sFlt-1 in pregnant rats. Am J Physiol Regul Integr Comp Physiol 302: R259–R263, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Murphy SR, LaMarca BB, Cockrell K, Granger JP. Role of endothelin in mediating soluble fms-like tyrosine kinase 1-induced hypertension in pregnant rats. Hypertension 55: 394–398, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Murphy SR, LaMarca BB, Parrish M, Cockrell K, Granger JP. Control of soluble fms-like tyrosine-1 (sFlt-1) production response to placental ischemia/hypoxia: role of tumor necrosis factor-α. Am J Physiol Regul Integr Comp Physiol 304: R130–R135, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nova A, Sibai BM, Barton JR, Mercer BM, Mitchell MD. Maternal plasma level of endothelin is increased in preeclampsia. Am J Obstet Gynecol 165: 724–727, 1991. [DOI] [PubMed] [Google Scholar]
- 124.Novotny SR, Wallace K, Heath J, Moseley J, Dhillon P, Weimer A, Wallukat G, Herse F, Wenzel K, Martin JN Jr, Dechend R, Lamarca B. Activating autoantibodies to the angiotensin II type I receptor play an important role in mediating hypertension in response to adoptive transfer of CD4+ T lymphocytes from placental ischemic rats. Am J Physiol Regul Integr Comp Physiol 302: R1197–R1201, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Omole Ohonsi AaA AO. Pre-eclampsia-a study of risk factors. AJOL 53: 99–102, 2008. [Google Scholar]
- 126.Palei A, Spradley FT, Granger JP. Chronic hyperleptinemia results in the development of hypertension in pregnant rats. Am J Physiol Regul Integr Comp Physiol 308: R855–R861, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Palei AC, Spradley FT, Granger JP. Euglycemic hyperinsulinemia increases blood pressure in pregnant rats independent of placental antiangiogenic and inflammatory factors. Am J Hypertens 26: 1445–1451, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Pardo F, Silva L, Saez T, Salsoso R, Gutierrez J, Sanhueza C, Leiva A, Sobrevia L. Human supraphysiological gestational weight gain and fetoplacental vascular dysfunction. Int J Obes (Lond) 39: 1264–1273, 2015. [DOI] [PubMed] [Google Scholar]
- 130.Parrish MR, Murphy SR, Rutland S, Wallace K, Wenzel K, Wallukat G, Keiser S, Ray LF, Dechend R, Martin JN, Granger JP, LaMarca B. The effect of immune factors, tumor necrosis factor-alpha, and agonistic autoantibodies to the angiotensin II type I receptor on soluble fms-like tyrosine-1 and soluble endoglin production in response to hypertension during pregnancy. Am J Hypertens 23: 911–916, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pavan L, Hermouet A, Tsatsaris V, Therond P, Sawamura T, Evain-Brion D, Fournier T. Lipids from oxidized low-density lipoprotein modulate human trophoblast invasion: involvement of nuclear liver X receptors. Endocrinology 145: 4583–4591, 2004. [DOI] [PubMed] [Google Scholar]
- 132.Peltier MR, Gurzenda EM, Murthy A, Chawala K, Lerner V, Kharode I, Arita Y, Rhodes A, Maari N, Moawad A, Hanna N. Can oxygen tension contribute to an abnormal placental cytokine milieu? Am J Reprod Immunol 66: 279–285, 2011. [DOI] [PubMed] [Google Scholar]
- 133.Peralta C, Jimenez-Castro MB, Gracia-Sancho J. Hepatic ischemia and reperfusion injury: effects on the liver sinusoidal milieu. J Hepatol 59: 1094–1106, 2013. [DOI] [PubMed] [Google Scholar]
- 134.Perez-Sepulveda A, Torres MJ, Khoury M, Illanes SE. Innate immune system and preeclampsia. Front Immunol 5: 244, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Perlow JH, Morgan MA, Montgomery D, Towers CV, Porto M. Perinatal outcome in pregnancy complicated by massive obesity. Am J Obstet Gynecol 167: 958–962, 1992. [DOI] [PubMed] [Google Scholar]
- 136.Podjarny E, Bernheim J, Katz B, Green J, Mekler J, Bursztyn M. Chronic exogenous hyperinsulinemia in pregnancy: a rat model of pregnancy-induced hypertension. J Am Soc Nephrol 9: 9–13, 1998. [DOI] [PubMed] [Google Scholar]
- 137.Qiu C, Baylis C. Endothelin and angiotensin mediate most glomerular responses to nitric oxide inhibition. Kidney Int 55: 2390–2396, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Radke AL, Reynolds LE, Melo RC, Dvorak AM, Weller PF, Spencer LA. Mature human eosinophils express functional Notch ligands mediating eosinophil autocrine regulation. Blood 113: 3092–3101, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Radulescu L, Munteanu O, Popa F, Cirstoiu M. The implications and consequences of maternal obesity on fetal intrauterine growth restriction. J Med Life 6: 292–298, 2013. [PMC free article] [PubMed] [Google Scholar]
- 140.Rajakumar A, Doty K, Daftary A, Harger G, Conrad KP. Impaired oxygen-dependent reduction of HIF-1alpha and -2alpha proteins in pre-eclamptic placentae. Placenta 24: 199–208, 2003. [DOI] [PubMed] [Google Scholar]
- 141.Rajasingam D, Seed PT, Briley AL, Shennan AH, Poston L. A prospective study of pregnancy outcome and biomarkers of oxidative stress in nulliparous obese women. Am J Obstet Gynecol 200: 395 e391–e399, 2009. [DOI] [PubMed] [Google Scholar]
- 142.Ramsay JE, Ferrell WR, Crawford L, Wallace AM, Greer IA, Sattar N. Maternal obesity is associated with dysregulation of metabolic, vascular, and inflammatory pathways. J Clin Endocrinol Metab 87: 4231–4237, 2002. [DOI] [PubMed] [Google Scholar]
- 143.Rana S, Karumanchi SA, Lindheimer MD. Angiogenic factors in diagnosis, management, and research in preeclampsia. Hypertension 63: 198–202, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Raymond D, Peterson E. A critical review of early-onset and late-onset preeclampsia. Obstet Gynecol Surv 66: 497–506, 2011. [DOI] [PubMed] [Google Scholar]
- 145.Redman CW, Sargent IL. Immunology of pre-eclampsia. Am J Reprod Immunol 63: 534–543, 2010. [DOI] [PubMed] [Google Scholar]
- 146.Roberts JM, Bodnar LM, Patrick TE, Powers RW. The role of obesity in preeclampsia. Pregnancy Hypertens 1: 6–16, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Roberts KA, Riley SC, Reynolds RM, Barr S, Evans M, Statham A, Hor K, Jabbour HN, Norman JE, Denison FC. Placental structure and inflammation in pregnancies associated with obesity. Placenta 32: 247–254, 2011. [DOI] [PubMed] [Google Scholar]
- 148.Roberts L, LaMarca BB, Fournier L, Bain J, Cockrell K, Granger JP. Enhanced endothelin synthesis by endothelial cells exposed to sera from pregnant rats with decreased uterine perfusion. Hypertension 47: 615–618, 2006. [DOI] [PubMed] [Google Scholar]
- 149.Robson A, Harris LK, Innes BA, Lash GE, Aljunaidy MM, Aplin JD, Baker PN, Robson SC, Bulmer JN. Uterine natural killer cells initiate spiral artery remodeling in human pregnancy. FASEB J 26: 4876–4885, 2012. [DOI] [PubMed] [Google Scholar]
- 150.Rode L, Nilas L, Wojdemann K, Tabor A. Obesity-related complications in Danish single cephalic term pregnancies. Obstet Gynecol 105: 537–542, 2005. [DOI] [PubMed] [Google Scholar]
- 151.Roldan M, Macias-Gonzalez M, Garcia R, Tinahones FJ, Martin M. Obesity short-circuits stemness gene network in human adipose multipotent stem cells. FASEB J 25: 4111–4126, 2011. [DOI] [PubMed] [Google Scholar]
- 152.Russell Z, Salihu HM, Lynch O, Alio AP, Belogolovkin V. The association of prepregnancy body mass index with pregnancy outcomes in triplet gestations. Am J Perinatol 27: 41–46, 2010. [DOI] [PubMed] [Google Scholar]
- 153.Rust OA, Bofill JA, Zappe DH, Hall JE, Burnett JC Jr, Martin JN Jr. The origin of endothelin-1 in patients with severe preeclampsia. Obstet Gynecol 89: 754–757, 1997. [DOI] [PubMed] [Google Scholar]
- 154.Saben J, Lindsey F, Zhong Y, Thakali K, Badger TM, Andres A, Gomez-Acevedo H, Shankar K. Maternal obesity is associated with a lipotoxic placental environment. Placenta 35: 171–177, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sahin Z, Acar N, Ozbey O, Ustunel I, Demir R. Distribution of Notch family proteins in intrauterine growth restriction and hypertension complicated human term placentas. Acta Histochem 113: 270–276, 2011. [DOI] [PubMed] [Google Scholar]
- 156.Schildroth J, Rettig-Zimmermann J, Kalk P, Steege A, Fahling M, Sendeski M, Paliege A, Lai EY, Bachmann S, Persson PB, Hocher B, Patzak A. Endothelin type A and B receptors in the control of afferent and efferent arterioles in mice. Nephrol Dial Transplant 26: 779–789, 2011. [DOI] [PubMed] [Google Scholar]
- 157.Schonkeren D, van der Hoorn ML, Khedoe P, Swings G, van Beelen E, Claas F, van Kooten C, de Heer E, Scherjon S. Differential distribution and phenotype of decidual macrophages in preeclamptic versus control pregnancies. Am J Pathol 178: 709–717, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Shah D, Frazer M, Badr KF. Circulating endothelin-1 is not increased in severe preeclampsia. J Maternal Fetal Neonatal Med 1: 177–180, 1992. [Google Scholar]
- 159.Shah TJ, Leik CE, Walsh SW. Neutrophil infiltration and systemic vascular inflammation in obese women. Reprod Sci 17: 116–124, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Shek EW, Brands MW, Hall JE. Chronic leptin infusion increases arterial pressure. Hypertension 31: 409–414, 1998. [DOI] [PubMed] [Google Scholar]
- 161.Shen H, Liu H, Chen H, Guo Y, Zhang M, Xu X, Xiang W. Analysis of placental growth factor in placentas of normal pregnant women and women with hypertensive disorders of pregnancy. J Huazhong Univ Sci Technolog Med Sci 26: 116–119, 2006. [DOI] [PubMed] [Google Scholar]
- 162.Shi Z, Brooks VL. Leptin differentially increases sympathetic nerve activity and its baroreflex regulation in female rats: role of estrogen. J Physiol 593: 1633–1647, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Shigematsu K, Nakatani A, Kawai K, Moriuchi R, Katamine S, Miyamoto T, Niwa M. Two subtypes of endothelin receptors and endothelin peptides are expressed in differential cell types of the rat placenta: in vitro receptor autoradiographic and in situ hybridization studies. Endocrinology 137: 738–748, 1996. [DOI] [PubMed] [Google Scholar]
- 164.Sibley CP, Pardi G, Cetin I, Todros T, Piccoli E, Kaufmann P, Huppertz B, Bulfamante G, Cribiu FM, Ayuk P, Glazier J, Radaelli T. Pathogenesis of intrauterine growth restriction (IUGR)-conclusions derived from a European Union Biomed 2 Concerted Action project “Importance of Oxygen Supply in Intrauterine Growth Restricted Pregnancies”-a workshop report. Placenta 23, Suppl A: S75–S79, 2002. [DOI] [PubMed] [Google Scholar]
- 165.Siegmund B, Sennello JA, Jones-Carson J, Gamboni-Robertson F, Lehr HA, Batra A, Fedke I, Zeitz M, Fantuzzi G. Leptin receptor expression on T lymphocytes modulates chronic intestinal inflammation in mice. Gut 53: 965–972, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Sisino G, Bouckenooghe T, Aurientis S, Fontaine P, Storme L, Vambergue A. Diabetes during pregnancy influences Hofbauer cells, a subtype of placental macrophages, to acquire a pro-inflammatory phenotype. Biochim Biophys Acta 1832: 1959–1968, 2013. [DOI] [PubMed] [Google Scholar]
- 167.Sivakumar K, Bari MF, Adaikalakoteswari A, Guller S, Weickert MO, Randeva HS, Grammatopoulos DK, Bastie CC, Vatish M. Elevated fetal adipsin/acylation-stimulating protein (ASP) in obese pregnancy: novel placental secretion via Hofbauer cells. J Clin Endocrinol Metab 98: 4113–4122, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Skarzinski G, Khamaisi M, Bursztyn M, Mekler J, Lan D, Evdokimov P, Ariel I. Intrauterine growth restriction and shallower implantation site in rats with maternal hyperinsulinemia are associated with altered NOS expression. Placenta 30: 898–906, 2009. [DOI] [PubMed] [Google Scholar]
- 169.Solomon CG, Carroll JS, Okamura K, Graves SW, Seely EW. Higher cholesterol and insulin levels in pregnancy are associated with increased risk for pregnancy-induced hypertension. Am J Hypertens 12: 276–282, 1999. [DOI] [PubMed] [Google Scholar]
- 170.Soma H, Yoshida K, Mukaida T, Tabuchi Y. Morphologic changes in the hypertensive placenta. Contrib Gynecol Obstet 9: 58–75, 1982. [PubMed] [Google Scholar]
- 171.Spradley FT, Palei AC, Granger JP. Obese melanocortin-4 receptor-deficient rats exhibit augmented angiogenic balance and vasorelaxation during pregnancy. Physiol Reports 1: e00081, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Stepan H, Philipp A, Roth I, Kralisch S, Jank A, Schaarschmidt W, Lossner U, Kratzsch J, Bluher M, Stumvoll M, Fasshauer M. Serum levels of the adipokine chemerin are increased in preeclampsia during and 6 months after pregnancy. Regul Pept 168: 69–72, 2011. [DOI] [PubMed] [Google Scholar]
- 173.Stewart FM, Freeman DJ, Ramsay JE, Greer IA, Caslake M, Ferrell WR. Longitudinal assessment of maternal endothelial function and markers of inflammation and placental function throughout pregnancy in lean and obese mothers. J Clin Endocrinol Metab 92: 969–975, 2007. [DOI] [PubMed] [Google Scholar]
- 174.Straughen JK, Misra DP, Kumar P, Misra VK. The influence of overweight and obesity on maternal soluble fms-like tyrosine kinase 1 and its relationship with leptin during pregnancy. Reprod Sci 20: 269–275, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Tache V, Ciric A, Moretto Zita M, Li Y, Peng J, Maltepe E, Milstone DS, Parast M. Hypoxia and trophoblast differentiation: a key role for PPARgamma. Stem Cells Dev 22: 2815–2824, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Tal R, Shaish A, Barshack I, Polak-Charcon S, Afek A, Volkov A, Feldman B, Avivi C, Harats D. Effects of hypoxia-inducible factor-1alpha overexpression in pregnant mice: possible implications for preeclampsia and intrauterine growth restriction. Am J Pathol 177: 2950–2962, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Tam Tam KB, George E, Cockrell K, Arany M, Speed J, Martin JN Jr, Lamarca B, Granger JP. Endothelin type A receptor antagonist attenuates placental ischemia-induced hypertension and uterine vascular resistance. Am J Obstet Gynecol 204: 330 e331–e334, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Tam Tam KB, Lamarca B, Arany M, Cockrell K, Fournier L, Murphy S, Martin JN Jr, Granger JP. Role of reactive oxygen species during hypertension in response to chronic antiangiogenic factor (sFlt-1) excess in pregnant rats. Am J Hypertens 24: 110–113, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Tanoue Y, Herijgers P, Meuris B, Verbeken E, Leunens V, Lox M, Flameng W. Ischemic preconditioning reduces unloaded myocardial oxygen consumption in an in-vivo sheep model. Cardiovasc Res 55: 633–641, 2002. [DOI] [PubMed] [Google Scholar]
- 180.Tavana TZ, J; Madadi G. The relationship between maternal serum highly sensitive C-reactive protein, leptin and hypertensive disorders of pregnancy. Internet J Endocrinol 6: 1, 2011. [Google Scholar]
- 181.Tracy TA, Miller GL. Obstetric problems of the massively obese. Obstet Gynecol 33: 204–208, 1969. [PubMed] [Google Scholar]
- 182.Valsamakis G, Kumar S, Creatsas G, Mastorakos G. The effects of adipose tissue and adipocytokines in human pregnancy. Ann NY Acad Sci 1205: 76–81, 2010. [DOI] [PubMed] [Google Scholar]
- 183.van Drongelen J, van Koppen A, Pertijs J, Gooi JH, Parry LJ, Sweep FC, Lotgering FK, Smits P, Spaanderman ME. Impaired vascular responses to relaxin in diet-induced overweight female rats. J Appl Physiol 112: 962–969, 2012. [DOI] [PubMed] [Google Scholar]
- 184.Verlohren S, Geusens N, Morton J, Verhaegen I, Hering L, Herse F, Dudenhausen JW, Muller DN, Luft FC, Cartwright JE, Davidge ST, Pijnenborg R, Dechend R. Inhibition of trophoblast-induced spiral artery remodeling reduces placental perfusion in rat pregnancy. Hypertension 56: 304–310, 2010. [DOI] [PubMed] [Google Scholar]
- 185.von Versen-Hoeynck FM, Powers RW. Maternal-fetal metabolism in normal pregnancy and preeclampsia. Front Biosci 12: 2457–2470, 2007. [DOI] [PubMed] [Google Scholar]
- 186.Vrijkotte TG, Krukziener N, Hutten BA, Vollebregt KC, van Eijsden M, Twickler MB. Maternal lipid profile during early pregnancy and pregnancy complications and outcomes: the ABCD study. J Clin Endocrinol Metab 97: 3917–3925, 2012. [DOI] [PubMed] [Google Scholar]
- 187.Wallace JM, Horgan GW, Bhattacharya S. Placental weight and efficiency in relation to maternal body mass index and the risk of pregnancy complications in women delivering singleton babies. Placenta 33: 611–618, 2012. [DOI] [PubMed] [Google Scholar]
- 188.Wallace K, Richards S, Dhillon P, Weimer A, Edholm ES, Bengten E, Wilson M, Martin JN Jr, LaMarca B. CD4+ T-helper cells stimulated in response to placental ischemia mediate hypertension during pregnancy. Hypertension 57: 949–955, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Wallis AB, Saftlas AF, Hsia J, Atrash HK. Secular trends in the rates of preeclampsia, eclampsia, and gestational hypertension, United States, 1987–2004. Am J Hypertens 21: 521–526, 2008. [DOI] [PubMed] [Google Scholar]
- 190.Wang T, Zhou R, Gao L, Wang Y, Song C, Gong Y, Jia J, Xiong W, Dai L, Zhang L, Hu H. Elevation of urinary adipsin in preeclampsia: correlation with urine protein concentration and the potential use for a rapid diagnostic test. Hypertension 64: 846–851, 2014. [DOI] [PubMed] [Google Scholar]
- 191.Wang Y, Walsh SW. TNF alpha concentrations and mRNA expression are increased in preeclamptic placentas. J Reprod Immunol 32: 157–169, 1996. [DOI] [PubMed] [Google Scholar]
- 192.Warrington JP, George EM, Palei AC, Spradley FT, Granger JP. Recent advances in the understanding of the pathophysiology of preeclampsia. Hypertension 62: 666–673, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Weed S, Bastek JA, Anton L, Elovitz MA, Parry S, Srinivas SK. Examining the correlation between placental and serum placenta growth factor in preeclampsia. Am J Obstet Gynecol 207: 140.e1–140.e6, 2012. [DOI] [PubMed] [Google Scholar]
- 194.Weil BR, Westby CM, Van Guilder GP, Greiner JJ, Stauffer BL, DeSouza CA. Enhanced endothelin-1 system activity with overweight and obesity. Am J Physiol Heart Circ Physiol 301: H689–H695, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Weisbrod RM, Griswold MC, Du Y, Bolotina VM, Cohen RA. Reduced responsiveness of hypercholesterolemic rabbit aortic smooth muscle cells to nitric oxide. Arterioscler Thromb Vasc Biol 17: 394–402, 1997. [DOI] [PubMed] [Google Scholar]
- 196.Weiss HR, Grayson J, Liu X, Barsoum S, Shah H, Chi OZ. Cerebral ischemia and reperfusion increases the heterogeneity of local oxygen supply/consumption balance. Stroke 44: 2553–2558, 2013. [DOI] [PubMed] [Google Scholar]
- 197.Wermter AK, Scherag A, Meyre D, Reichwald K, Durand E, Nguyen TT, Koberwitz K, Lichtner P, Meitinger T, Schafer H, Hinney A, Froguel P, Hebebrand J, Bronner G. Preferential reciprocal transfer of paternal/maternal DLK1 alleles to obese children: first evidence of polar overdominance in humans. Eur J Hum Genet 16: 1126–1134, 2008. [DOI] [PubMed] [Google Scholar]
- 198.Whitley GS, Dash PR, Ayling LJ, Prefumo F, Thilaganathan B, Cartwright JE. Increased apoptosis in first trimester extravillous trophoblasts from pregnancies at higher risk of developing preeclampsia. Am J Pathol 170: 1903–1909, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Wyrwoll CS, Mark PJ, Waddell BJ. Directional secretion and transport of leptin and expression of leptin receptor isoforms in human placental BeWo cells. Mol Cell Endocrinol 241: 73–79, 2005. [DOI] [PubMed] [Google Scholar]
- 200.Yu X, Yang Z, Yu M. Correlation of tumor necrosis factor alpha and interleukin 6 with hypertensive renal damage. Ren Fail 32: 475–479, 2010. [DOI] [PubMed] [Google Scholar]
- 201.Yu Z, Han S, Zhu J, Sun X, Ji C, Guo X. Pre-pregnancy body mass index in relation to infant birth weight and offspring overweight/obesity: a systematic review and meta-analysis. PLos One 8: e61627, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Zeck W, Widberg C, Maylin E, Desoye G, Lang U, McIntyre D, Prins J, Russell A. Regulation of placental growth hormone secretion in a human trophoblast model–the effects of hormones and adipokines. Pediatr Res 63: 353–357, 2008. [DOI] [PubMed] [Google Scholar]