

Abstract
After decades of investigation, the causes of essential hypertension remain obscure. The contribution of the nervous system has been excluded by some on the basis that baroreceptor mechanisms maintain blood pressure only over the short term. However, this point of view ignores one of the most powerful contributions of the brain in maintaining biological fitness—specifically, the ability to promote adaptation of behavioral and physiological responses to cope with new challenges and maintain this new capacity through processes involving neuroplasticity. We present a body of recent findings demonstrating that prior, short-term challenges can induce persistent changes in the central nervous system to result in an enhanced blood pressure response to hypertension-eliciting stimuli. This sensitized hypertensinogenic state is maintained in the absence of the inducing stimuli, and it is accompanied by sustained upregulation of components of the brain renin-angiotensin-aldosterone system and other molecular changes recognized to be associated with central nervous system neuroplasticity. Although the heritability of hypertension is high, it is becoming increasingly clear that factors beyond just genes contribute to the etiology of this disease. Life experiences and attendant changes in cellular and molecular components in the neural network controlling sympathetic tone can enhance the hypertensive response to recurrent, sustained, or new stressors. Although the epigenetic mechanisms that allow the brain to be reprogrammed in the face of challenges to cardiovascular homeostasis can be adaptive, this capacity can also be maladaptive under conditions present in different evolutionary eras or ontogenetic periods.
Keywords: renin-angiotensin-aldosterone systems, neural networks controlling blood pressure, slow pressor angiotensin-elicited hypertension, induction-delay-expression paradigm, salt sensitivity, aldosterone, cross-sensitization
“Physiologists were slow in analyzing the influence of the central nervous system on cardiovascular performance, despite the fact that blanching and blushing of the face and changed beating of the heart have, since the dawn of literature, been colourfully described as the mirror of man's joy and distress” [Folkow B and Neil E, p. 307, 1971 (60)].
understanding the mechanisms that determine arterial blood pressure (BP) over the course of a lifetime is of major interest to many regulatory and integrative physiologists. There are fundamental questions that provoke this attention. The first is what drives a progressive increase in BP from normotensive to pathological levels? Of individuals, aged 35 to 70, sampled in a cross-sectional study from communities in 17 high-, middle- and low-income countries, 40.8% had hypertension [HT; (32)]. Second, and most importantly, what is responsible for initially triggering this rise in pressure? Considerable effort has been expended attempting to identify a fundamental fault (e.g., genes, impaired renal function, salt intake, altered hemodynamics, altered membrane permeability, etc.) causing HT (155). However, in only 5 to 10% of hypertensives can the disorder be attributed to a specific complication (e.g., renal artery stenosis, renal failure, pheochromocytoma, hyperaldosteronism), and accordingly be classified as secondary HT. In the remaining cases in which HT has an undetermined etiology, this cardiovascular disease is referred to as primary, idiopathic, or essential hypertension (EH).
The proximal determinants of BP are cardiac output (CO) and total peripheral resistance (TPR; TPR × CO = BP), and the immediate factors affecting the function of the heart and vasculature have received extensive attention for their roles in the pathogenesis of HT (73, 94, 128, 138, 146, 174). As a result of such intense scrutiny, it is recognized that EH is not a unitary disorder, but one that should be parceled into disease subtypes or syndromes caused by multiple interacting factors (27).
The autonomic nervous system is widely acknowledged to be among the most important factors to play a key role in the pathology of EH (118). In both animal models of HT and human EH, there is evidence that autonomic disruption is associated with both the onset and maintenance of high BP (59, 70, 119, 123, 141, 144). Normotensive individuals with a family history of HT have increased circulating catecholamines and increased sympathetic nerve activity at rest and when challenged with physiological and psychological stressors (51, 124, 145, 202). On the basis of both the proportion of EH patients demonstrating elevated sympathetic tone and the number of cases in which sympatholytic drugs lower high BP, Essler et al. (49) have estimated that the syndrome of neurogenic EH accounts for no less than 50% of all cases of this disorder.
Several mechanisms are proposed to be root causes for initiating increased sympathetic drive and, in turn, EH. These include exogenous stressors; chemoarterial, cardiopulmonary, and arterial baroreflex dysfunction; various humoral factors such as angiotensin, leptin, and insulin (118); and impaired cerebral perfusion (29, 43). Associated with the latter possibility is the idea that central nervous system (CNS) receptors sensing low pressure (142) or hypoperfusion of the brain (29, 42) trigger enhanced sympathetic drive. Hypertension is also hypothesized to result from an upward resetting of a CNS embodied set-point that leads to increased sympathetic drive in response to what is detected as inappropriately low arterial BP (142). Although the ideas of a CNS controller and a variable set-point driving increased sympathetic tone and BP are provocative hypothetical constructs, what still needs to be understood is how such mechanisms are instantiated in the brain and how a set-point or gain is changed to act as an ultimate cause of EH.
Recent studies from our laboratory indicate that one of the fundamental properties of the CNS— the capacity of neuroplasticity to bring about adaptations in behavioral and physiological response systems as a consequence of prior experience—can operate in the expression of HT. These new findings implicate cellular and molecular changes in components of a neural network controlling BP that are altered to result in sensitization of the response to hypertensinogenic stimuli. This invited review describes this work and discusses the implications of neuroplasticity as a key factor in the etiology of EH.
We begin this review by providing background on 1) the function of the nervous system in the regulation of BP, 2) the characteristics of neuroplasticity and its involvement in reprogramming the expression of behavioral and physiological responses controlled by the CNS (i.e., response adaptation), and 3) the nature of the brain renin-angiotensin-aldosterone (RAAS) system.
Implication of the CNS in the Regulation of BP
Investigations of a role for the CNS in the regulation of BP began in earnest about a century-and-a-half ago. Research progressed over the next 100 years by employing transections of the spinal cord and brain and then with more focused lesions and electrical stimulation to identify regions that were referred to as cardiovascular centers (reviewed in Refs. 60 and 181). On the basis of work conducted in his laboratory in the late 1920s, Koch proposed that interruption of the peripheral nerves carrying afferent information from arterial baroreceptors caused acute hypertension because it removed a state of tonic inhibition of BP that was exerted by the CNS (reviewed by Ref. 102).
Beginning about 50 years ago, experimental findings began to support the idea that the CNS plays an important role in the pathogenesis of chronic HT (42). Early studies implicating CNS-driven neurogenic HT employed techniques such as chronic electrical stimulation of the hypothalamic defense area (61) and lesions of the solitary tract nuclei [NTS; (46)] to induce high BP. Studies showing that depletion of central catecholamines (55) or ablation of periventricular tissue surrounding the anteroventral third ventricle [AV3V; (14, 15)] block the induction of experimental HT lent support to the hypothesis that the integrity of specific neurochemical systems and brain areas is necessary for the expression of high BP.
The application of newly developed neuroanatomical and functional techniques facilitated the identification of key brain nuclei and their related interconnectivity to define a widely distributed neural network controlling BP (see Refs. 38 and 71 for reviews). The brain receives streams of BP-relevant information from both the external and internal environments. Sensory inputs from interoceptors located at various places in the body and brain serve as indices of blood volume, BP, and extracellular osmolarity. Both afferent nerves from the viscera and systemically generated humoral factors (e.g., hormones and osmolality) participate in the process of body-brain signaling of cardiovascular relevant information. Neural input from the viscera enters the CNS either through the dorsal roots of spinal nerves or by way of the IXth and Xth cranial nerves. Both sources of sensory information enter the brain proper by way of the NTS in the caudal medulla, and from there, this information flows into a neural network controlling BP.
Among the best characterized types of visceral input affecting BP are those that derive from systemic cardiovascular receptors located on both the arterial and venous sides of the circulation. High-pressure baroreceptors, located in the carotid sinus and aorta, generate input to the brain, reflecting arterial pressure on a moment-to-moment basis. Interruption of input from arterial baroreceptors removes inhibitory influences on the sympathetic nervous system and on the release of pressor hormones (e.g., vasopressin), producing acute HT. Low-pressure baroreceptors sensing distention of chambers of the heart, great veins, and components of the pulmonary circulation act to monitor blood volume and affect autonomic and endocrine control of BP through afferent nerves to the brain (64, 69).
Many humoral factors, such as extracellular osmolarity, hormones [e.g., ANG II and aldosterone (ALDO)], and cytokines, also impart information to the neural systems controlling BP. Hormones that cross the blood-brain barrier can exert their influence directly on components of this neural network, and substances that are excluded from the brain act through brain sensory circumventricular organs, which lack a blood-brain barrier (90). Two of these receptive structures, the subfornical organ (SFO) and the organum vasculosum (OVLT), are located in the forebrain along the lamina terminalis (LT), and the third, the area postrema (AP), is situated on the dorsal surface of the most caudal part of the medulla. Information from multiple sources is integrated by the network to produce a coordinated pattern of hormonal and autonomic responses that is optimal to correct or minimize the effects of disruptions in cardiovascular and body fluid homeostasis in a given context (e.g., environmental setting).
The neural network controlling BP is widely distributed throughout the neuraxis. Among the commonly recognized components of this control system are the NTS, rostral and caudal ventrolateral medulla (RVLM and CVLM), motor nucleus of the vagus (MNX), parabrachial nucleus, paraventricular hypothalamic nucleus (PVN), dorsomedial hypothalamic nucleus, OVLT, median preoptic nucleus (MnPO), SFO, and spinal cord intermediolateral cell column (IML; see Fig. 1 for the locations of some of these components). Each nucleus within the network projects to other brain regions, and there are many reciprocal connections allowing both feedforward and feedback loops for information processing among the components.
Fig. 1.

A schematic of selected components of the neural network controlling sympathetic tone that contributes to blood pressure regulation. Represented are key descending influences from forebrain structures located along the lamina terminalis [i.e., subfornical organ (SFO), median preoptic nucleus (MnPO), organum vasculosum (OVLT)] projecting to the hypothalamic paraventricular nucleus (PVN). The SFO and the OVLT have no blood-brain barrier and are primary sites receiving information about blood-borne factors and solutes in the extracellular fluid. The SFO is the primary forebrain target for ANG II and cells in the OVLT function as osmo- or Na+-receptors. The MnPO, which lies inside the blood-brain barrier, receives input from both the SFO and OVLT and probably functions to process information about the status of intracellular and extracellular fluid compartments and blood pressure. The SFO, MnPO, and OVLT all provide input to the PVN. In turn, the PVN integrates this information with input from other sources (not shown) to exert a major influence on preganglionic sympathetic neurons in the spinal cord [interomediolateral cell column (IML)] both directly and via the rostral ventrolateral medulla (RVLM). Mineralocorticoid receptors are distributed at many sites throughout the neuraxis. At the present time, it is not clear whether many of these receptors are accessed by circulating aldosterone or paracrine acting aldosterone synthesized locally (68). Represented are some additional areas implicated in cardiovascular control. These include the area postrema (AP), caudal ventrolateral medulla (CVLM), nucleus of the solitary tract (NTS) and parabrachial nucleus (PBN), which either directly or indirectly influence activity in the RVLM. AC, anterior commissure OC, optic chiasm.
Progress has been made in understanding the roles of, and interactions between, components of the neural network controlling BP. For example, medullary structures (i.e., NTS, AP, CVLM, RVLM, MNX) are importantly involved in the control of cardiac and vascular baroreceptor reflexes. The set-point for short-term BP maintenance by baroreceptor reflexes that is controlled by this hindbrain circuitry can be overridden under certain conditions, such as exercise or the presence of exogenous stressors, and reset to elevated levels by inputs that descend from structures higher in the neuraxis, such as the prefrontal cortex, amygdala, and hypothalamus (131, 170). Within the hypothalamus, the PVN is the primary component involved in controlling BP by exerting a major influence on sympathetic tone through projections to the RVLM and the IML. The PVN receives input not only from the hindbrain but also from descending projections from structures located along the LT (Fig. 1). It is by way of LT structures that a significant amount of the information derived from humoral factors (e.g., osmolality and hormones) is imparted to the CNS (15, 89, 91, 92, 169, 178).
For the most part, progress in establishing the components and connectivity of the neural network controlling BP has been comparable to the advances made in defining the neural substrates of other sensory and effector systems, such as those involved in somatic motor control, the primary senses, biological rhythms, pain, pleasure, reproduction, and learning and memory. However, compared with investigating the functional role of neural networks in other research areas, cardiovascular control has lagged behind in one particular realm. Specifically, surprisingly little research has focused on the effects of experience-induced changes in the neural network controlling BP and the potential impact of neuroplasticity on the long-term regulation of BP and EH.
Neuroplasticity and Mechanisms for Adaptation and Sustained Functional Changes in Behavioral and Physiological Systems
Sixty-six years ago, Donald Hebb (79) presented a seminal hypothesis relating memory to modifications in the CNS at the level of interactions between neurons. Hebb introduced the idea that a particular type of sustained change in neural function was required for the storage of information. Specifically, he proposed that establishing and maintaining memories involved the process of increasing the functional relationship between neurons, so that activation of a neuron axon terminal coinciding with increased activity in the cell that it innervates results in strengthening of the synaptic connection between the two cells. In other words, a change in synaptic strength between neurons provides the physical means to represent memories laid down as a result of experience. Within a few years Hebb's hypothesis received experimental support in anesthetized rabbits, in which brief, high-frequency stimulation of the perforant pathway into the hippocampal dentate gyrus produced sustained facilitation of extracellular electrical field potentials (10). This phenomenon, known as long-term potentiation (LTP), was demonstrated to last for months. Because the hippocampus had been implicated in processes related to learning and memory (e.g., Ref. 160), it was a simple logical step to couple the phenomenon of LTP with Hebb's hypothesized mechanism for information storage.
LTP is not a unique phenomenon associated only with the hippocampus, but it is inducible at many sites in the nervous system (117), including sympathetic ganglia (2, 33). By varying electrical stimulation parameters, it is possible to demonstrate different forms of neuroplasticity [e.g., low-frequency stimulation induces long-term depression (LTD) of activity] and to alter how long LTP is maintained after its induction [e.g., early-LTP vs. late-LTP; (20)]. Changes in synaptic efficiency in the short term can be mediated by the persistence of phosphorylation of intracellular second messengers regulated by protein kinases and phosphatases, whereas long-term changes involve the synthesis of new proteins.
Changes in synaptic efficiency, related to LTP and LTD, represent only one type of mechanism involved in neuroplasticity. There are other modifiable processes that are neuroplastic in nature and that have been implicated in memory formation and maintenance. These include the growth of existing dendrites and introduction of new dendritic spines, axonal sprouting, formation of new synapses, or even the genesis of new neurons (41, 152, 153). Neuroplasticity that depends upon controlling DNA expression is more complex and requires more time compared with neuroplasticity related to posttranscriptional or posttranslational regulation, but provides the possibility of greater permanence (189).
Various types of functional changes have been associated with alterations in synaptic efficiency. For example, in the case of habituation, the magnitude of an initial response to a neutral (innocuous) stimulus progressively declines with successive presentations. In contrast, stimuli associated with activation of systems that defend against insult or bodily injury often will produce progressive increases in responding over the course of repetitive presentations. Such an increase in strength or magnitude is referred to as response sensitization. Habituation and sensitization are considered to be simple forms of learning (28, 96, 147), and the neurobiological mechanisms that underlie these phenomena have been related to more complex forms of learning and memory associated with classical (Pavlovian) and instrumental (operant) conditioning (153). The nature and role of adaptive neural modifications resulting in changes of functional capacity are studied in relation to reflex motor responses (147), pain (148, 149), pleasure [i.e., motivation for and reinforcement of natural rewards, as well as drugs of abuse (86, 154, 190)], baroreflexes and chemoreflexes (99, 130), intermittent hypoxia (37), respiration (115), cough (12), stress (80), salt appetite (136), and exercise (129, 134). Progress in the investigation of the role of neuroplasticity in functional systems has been most rapid in cases in which the neural pathways and related neurochemical systems associated with those functions have been defined.
RAAS and the Coupling of the Systemic RAAS with the Brain RAAS
Renin, ANG II, and ALDO are recognized to be among the most prominent and best characterized hormones involved in cardiovascular control. The elucidation of a biochemical cascade for the formation of ANG II began with the discovery of the enzyme renin (177). Renin released from the kidney acts on a circulating glycoprotein, angiotensinogen, to generate ANG I that is then processed by angiotensin-converting enzyme (ACE, now referred to as ACE1) to form the primary effector peptide, ANG II. Circulating ANG II acts on two different G protein-coupled, seven-membrane-spanning receptors (R) [angiotensin type-1 (AT1R), and angiotensin type-2 (AT2R) receptors]. Since the discovery of renal renin, there has been steady progress in describing the biochemical characteristics, sites of synthesis and action, and functional roles of renin and ANG II. Such initial investigations of renin and ANG II defined what is now referred to as the circulating, or classic renin-angiotensin system (RAS).
Because of their actions as remarkable pressor agents, interest in circulating renin and ANG II was reinforced by the discoveries that renal HT could be modeled by constriction of the renal arteries (67), triggering the release of renin, and that ANG II was a major stimulus for the synthesis and release of the sodium-retaining mineralocorticoid, ALDO (39, 135). The combination of actions exerted by ANG II and ALDO led to a strong functional association of these hormones, so that they, along with renin, are now often referred to collectively as the RAAS (6).
In recent years, several new components of the circulating RAS have been identified. A homolog of ACE1 and ACE2 removes a COOH-terminal phenylalanine from ANG II to produce ANG1-7. This peptide interacts with another ANG receptor, the Mas receptor (Mas-R), through which ANG1-7 can sometimes produce responses that increase BP (e.g., 137, 156). However, there are relatively more reports that show ANG1-7 has actions that reduce BP so that, on balance, ACE2, ANG1-7, and the Mas-R are proposed to act as members of an anti-hypertensive or counter-regulatory arm of the RAS (6, 52, 157). It is also notable that ANG II itself may be associated with both the hypertensive and the antihypertensive arms of the RAS since ANG II acting on AT2R has effects opposing those produced by the interaction of ANG II with AT1R (6, 52).
Studies of the circulating RAS laid the groundwork for a major phase of progress of discovering broader mechanisms and functions of these systems. It became recognized that many or all of the components of the RAS were synthesized, retained, and acted in autocrine, intracrine and paracrine fashions within many tissues and organs. These sites include the brain, kidney, vascular wall, heart, adipose tissue, hemopoietic bone marrow, lungs, and the gastrointestinal tract (6, 53). The seminal finding that a renin-like material (iso-renin) is present in the brain spurred interest in tissue RAS (56, 63). Subsequent work by many contributors has led to a general consensus that renin, ANG II, and ANG-R are generated de novo within the mammalian CNS and constitute a brain RAS.
Prior to the first findings indicating the likely presence of a brain RAS, studies had shown that ANG II or renin administered directly to the CNS elicit both pressor responses (21, 161) and thirst (13, 48) in several species. Because the pressor and dipsogenic actions of circulating renin and ANG II were known (57, 177), the obvious questions that arose were does the circulating RAS exert influence on the brain, and if so, where and how? Because ANG II has a molecular mass of a little more than 1,000 Da, the circulating peptide is excluded from most of the brain and must act through one or more of the sensory circumventricular organs (90). Studies in the dog identified the AP as a central target in which circulating ANG II elicited cardiovascular actions (54), and in the rat, the SFO was implicated in mediating both pressor and drinking responses to blood-borne ANG II. Ablation of the SFO abolishes and significantly attenuates drinking and pressor responses, respectively, to systemically delivered ANG II (120, 167). However, when the SFO is ablated, drinking to intracebroventricularly injected ANG II remains (23, 110).
The SFO communicates with the rest of the brain, most notably the MnPO, PVN, and other forebrain nuclei, via a bundle of axons issuing from its ventral stalk (26, 111, 132). AV3V lesions, which interrupt most, if not all, of the efferent projections from the ventral stalk of the SFO, significantly attenuate the pressor responses to intravenous infusions of ANG II (22) and abolish the drinking response to systemically administered ANG II (24, 25). The same effects on BP and thirst responses to systemic ANG II occur after axotomy produced by a small knife cut beneath the SFO (108, 109). As with SFO lesions, drinking and BP responses to intracerebroventricular ANG II remain after the knife cuts under the SFO (108). These findings led us to hypothesize that ANG II acts on the SFO in the mode of a circulating hormone, where its action is coupled to efferents from the SFO that contain and use ANG II acting in the mode of a neurotransmitter (89, 93, 107, 108). Substantial evidence has developed to support the concept of humoral-neural coupling of the circulating RAS with the brain RAS through circumventricular organs (e.g., see Ref. 169 for review).
Circulating ANG II acts on the SFO to drive sympathetic activity through a descending multisynaptic pathway, including the MnPO, PVN, RVLM, and IML (Fig. 1). Information transmitted in this circuitry reflects the levels of circulating ANG II (50) and other humoral factors, including extracellular osmolality (178). Several neurotransmitters and neuromodulators, including ANG II (50, 87), glutamate (88), GABA (165), and reactive oxygen species (81), act at synapses in the chain of neurons comprising this descending pathway. Sustained changes in the actions, availability, and activity of such neuromodulators/neurotransmitters and their receptors, related intracellular messengers and transcription factors in the LT structures, PVN, RVLM, and IML may be involved in altering synaptic efficiency associated with sensitization of the hypertensive response.
Renin-Angiotensin-Aldosterone Systems Acting Centrally Produce Sensitization of the Hypertensive Response
To study the role of sensitization in the pathogenesis of high BP, we recently employed an experimental paradigm that is unique in HT research. In these studies, rats instrumented to record BP and heart rate (HR) were infused chronically with a low (non-pressor) dose of ANG II (10 ng/kg/min sc) during a 1-wk period to induce sensitization (this period is referred to as “induction”). A 1-wk rest period followed (i.e., a no-treatment period referred to as “delay”) to test for the persistence of the effects of “induction” and allow for the unequivocal metabolism of any exogenous ANG II. At the end of delay, the test phase of the experiment was initiated (referred to as “expression”). During expression, the rats were administered ANG II under conditions in which the dose (120 ng·kg−1·min−1 sc) does not immediately elicit HT but rather increases BP very gradually to reach hypertensive levels only after several days (see below for more details and discussion of mechanisms of the so-called slow-pressor model of ANG II-elicited HT). With this induction-delay-expression experimental paradigm, it was easy to demonstrate sensitization of the HT response, since animals that were exposed to a nonpressor dose of ANG II during induction showed a significantly enhanced pressor response during expression [(200); Fig. 2A]. Furthermore, we found that sensitization of the hypertensive response to ANG II directly involves the CNS. This was demonstrated first by showing that intracerebroventricular administration of the AT1R antagonist, irbesartan, along with subcutaneous ANG II during induction blocks sensitization of the ANG II-elicited hypertensive response during expression [(200); Fig. 2A], and second by showing that sensitization can be induced simply by administering a low dose of ANG II centrally (1 ng·kg−1·min−1 icv) during induction [(200); Fig. 2B].
Fig. 2.
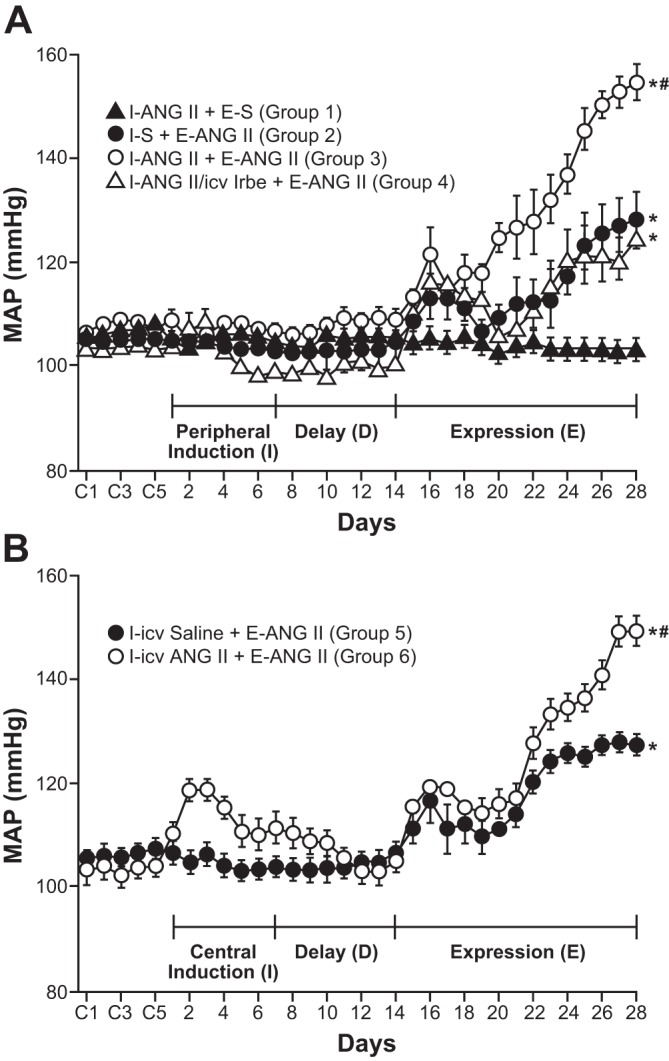
Pretreatment with either subcutaneous or intracerebroventricular ANG II during a 1-wk induction period (I) sensitized the hypertensive response elicited by a slow pressor infusion of ANG II (120 ng·kg−1·min−1 sc) administered during a 2-wk expression period (E). A: during the induction period, ANG II (10 ng·kg−1·min−1 sc; groups 1, 3, and 4) or saline (S; group 2) was administered. The induction period was followed by a 1-wk, no-treatment delay (D) before the start of the expression period (E). Animals in group 4 received irbesartin (Irbe), an angiotensin type 1 receptor antagonist (125 μg/day icv), along with the ANG II treatment during the induction period. Irbe blocked sensitization of the hypertensive response. B: sensitization of the ANG II-elicited hypertensive response during the expression phase (120 ng·kg−1·min−1 sc) was also produced by ANG II delivered intracerebroventricularly (1 ng·kg−1·min−1; group 6) during induction compared with rats in group 5 that received saline (S) intracerebroventricularly. *P < 0.05 vs. baseline or I-ANG II+E-S. #P < 0.05 vs. I-S+E-ANG II or I-ANG II/icv Irbe+E-ANG II. [With permission from Wolters Kluwer Health, Hypertension, Sensitization of slow pressor angtiontensin II (Ang II)-initiated hypertension: induction of sensitization by prior Ang II treatment, by Xue B, Zhang Z, Johnson RF, and Johnson AK, vol. 59, 2012 (200)].
ALDO Cross-Sensitizes the Hypertensive Response Elicited by the Slow-Pressor ANG II Model
Studies have shown that central pharmacological blockade or gene silencing using si-RNA of either mineralocorticoid receptors (MR) or AT1R in the brain prevents both ALDO- (194) and ANG II-induced HT (84, 194). These results indicate that ANG II and ALDO interact in the brain in a cooperative manner, so that functional integrity of both receptors is necessary for processing either ANG II- or ALDO-elicited HT. These findings prompted consideration of whether ALDO will cross-sensitize the slow-pressor ANG II-elicited hypertensive response. This was tested using the induction-delay-expression paradigm with ALDO given 750 ng/h sc during induction. After delay, the slow-pressor dose of ANG II (120 ng·kg−1·min−1) was used to test the hypertensive response during expression. As hypothesized, systemic ALDO sensitized ANG II-induced HT (Fig. 3). Evidence that the sensitizing actions of ALDO involve the CNS was demonstrated first by showing that ALDO-induced sensitization of slow-pressor ANG II-elicited HT was blocked by intracerebroventricular administration of the MR antagonist, RU28318 (1.1 μg/h), along with subcutaneous administration of ALDO during induction (Fig. 3); and second, by demonstrating that ALDO (10 ng/h) given intracerebroventricularly by itself during induction, sensitized the slow-pressor response to ANG II delivered during expression (201).
Fig. 3.
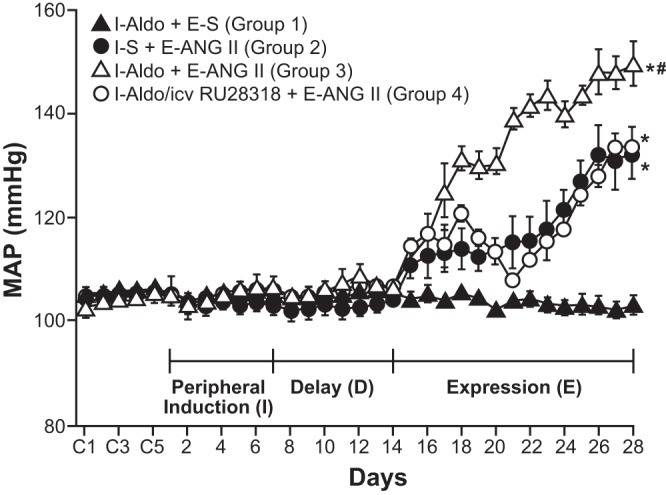
Pretreatment with aldosterone (Aldo) during a 1-wk induction period (I) cross-sensitized the hypertensive response elicited by a slow pressor infusion of ANG II (120 ng·kg−1·min−1 sc) administered during a 2-wk expression period (E). The induction period was followed by a 1-wk, no-treatment delay period (D) before the start of the expression period (E). During the induction period Aldo (750 ng/h sc; groups 1, 3, and 4) or saline (S; group 2) was administered. Animals in group 4 received RU283, a mineralocorticoid receptor antagonist, (1.1 μg/n icv) along with the subcutaneous Aldo treatment during the induction period. RU28318 blocked cross-sensitization of the hypertensive response. *P < 0.05 vs. baseline or I-Aldo+E-S. #P < 0.05 vs. I-S+E-ANG II, or I-Aldo/icv RU28318+E-ANG II. [With permission from Wolters Kluwer Health, Hypertension, Sensitization of slow pressor angtiontensin II (Ang II)-initiated hypertension: induction of sensitization by prior Ang II treatment, by Xue B, Zhang Z, Roncari CF, and Johnson AK, vol. 60, 2012 (201)].
MR for ALDO are widespread in the CNS (5), including the SFO (1). King et al. (98), found that ANG II binding was increased in the septal-AV3V region in brains from rats pretreated systemically with a mineralocorticoid agonist, DOCA. Furthermore, using an in vivo electrophysiological approach, Thorton and Nicolaïdais (176) found that MnPO and medial septal neurons showed enhanced firing to iontophoretically applied ANG II when their rats were pretreated with systemic DOCA. More recently, in vitro imaging studies of dissociated SFO neurons found that overnight conditioning with ALDO enhanced the increase in [Ca2+]i in response to ANG II (201). Taken together, current findings are consistent with the conclusion that one-way ALDO and ANG interact cooperatively and synergistically in the CNS to produce sensitization is through actions on the same neurons.
On the basis of binding studies conducted in rat kidney, two types of receptors were described for corticosteroids (see Ref. 62 for review). The classification of Type I sites, or MR, was assigned to those that bound ALDO with high affinity, and Type II sites to those that bound the mineralocorticoid with lower affinity. The Type II binding sites in renal tissue were found to be the same as those that bind the glucocorticoid (GC), dexamethasone, with a high affinity in a number of other tissues and are referred to as glucocorticoid-R (GR).
Cortisol and corticosterone, the primary GC in humans and in mice and rats, respectively, circulate at 100- to 1,000-fold the concentration of ALDO. Consequently, under physiological conditions, the majority of MR receptors throughout the body are occupied by GC. The opportunity for interaction of ALDO with MR is afforded by the presence of the enzyme 11β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) in cells, which converts corticosterone and cortisol to an inactive metabolite, thereby allowing binding of the mineralocorticoid to MR. As with MR, 11β-HSD2 is expressed throughout the brain (151, 191). The concentration of 11β-HSD2 is highest in the fetus, progressively decreasing in neonates and into adulthood (191). In summarizing several studies of MR and 11β-HSD2 localization at the level of brain structures (i.e., nuclei), Gomez-Sanchez and Gomez-Sanchez (68) report that both are present in adults in the PVN, ventromedial hypothalamic nucleus, the amygdala, locus coeruleus, medial vestibular nucleus, and subcommissural organ. There is extensive discussion as to which brain regions contain individual cells housing both MR and 11β-HSD2. On the basis of a histochemical survey of the brain, Geerling and Loewy (65) indicate that the only region where they found both MR and 11β-HSD2 to be coexpressed in the same cells was in a portion of the NTS.
When contemplating the availability and central actions of ALDO, one alternative to consider is that ALDO might be formed and act in the brain in a paracrine fashion. In such a case, mineralocorticoids could be generated locally at concentrations sufficient to displace bound GC from MR. All of the constituents necessary for the synthesis of ALDO from cholesterol are present in human and rat brains (reviewed in Ref. 68).
Several Types of Treatments Cross-Sensitize the Hypertensive Response
To begin to determine whether other types of challenges are capable of inducing sensitization of HT, the ANG II slow-pressor response was tested following two distinct conditions: 1) activation of the circulating RAAS following sodium deprivation and 2) suppression of the circulating RAAS following high dietary sodium (199). Four groups of animals were studied. During 2 wk of induction, two groups were maintained on a standard diet, a third was placed on a sodium-deficient diet, and a fourth group was maintained on a diet containing 2% NaCl. After induction, all groups received normal diet, and three of the groups were given ANG II (120 ng/kg/min sc) during 2 wk of expression, while one of the two groups that were given normal chow during induction was given vehicle subcutaneously during this period. Under both conditions, in which dietary sodium was manipulated (i.e., both high- and low-salt intake), the animals responded to ANG II with significantly enhanced HT during expression (Fig. 4). Together, these findings raise the interesting question of whether the dietary manipulations have some common aspect (e.g., both being general stressors) that induces sensitization.
Fig. 4.

Pretreatment of rats with either a sodium-deficient diet (LS) or a 2% high-salt diet (HS) during a 2-wk induction period (I) cross-sensitized the hypertensive response elicited by a slow pressor infusion of ANG II (120 ng·kg−1·min−1 sc) administered during a 2-wk expression period (E) compared with animals that were maintained on normal rat chow (NS) during I. (Note that all animals were on normal rat chow during E). *P < 0.05 vs. baseline or I-NS+E-Saline. #P < 0.05 vs. I-NS+E-ANG II.
Ongoing studies are testing the possibility that a wide range of challenges administered during induction can sensitize the hypertensive response. We find that intracerebroventricular administration of either leptin (193), or a proinflammatory cytokine, tumor necrosis factor-α (196), or 3 wk of feeding a high-fat diet (197) sensitizes the response to ANG II-elicited HT. All of these sensitizing challenges are proposed to be prohypertensinogenic (see e.g., 47, 183).
One of the obvious questions that was apparent to us following our initial findings of sensitization and cross-sensitization was how long can the sensitized state be sustained once animals have undergone induction? We recently began to test these limits (195) under conditions that would provide useful information about the efficacy of treatments administered during the perinatal period. It has long been recognized that challenges delivered during critical periods can have long-term consequences on physiology and behavior (106, 159). Treatments given during the perinatal period are especially effective in producing enduring effects on biological systems, including the cardiovascular system. For example, the stress of short periods of separation of nursing rat pups from their mother produces sustained changes in the autonomic control of the heart (179) and adult BP (180) and enhances the hypertensive response to ANG II in adults (113, 114). In this context, we recently tested whether induction of a sensitized hypertensive response can be elicited in offspring by producing hypertension in the mother during pregnancy. In these studies, aortic BP and HR were measured in mothers and their offspring by telemetry. Dams were infused with ANG II subcutaneously throughout pregnancy, and shortly after the birth of their pups, peptide delivery to the mother was terminated. The offspring were weaned at 21 days of age and given a 7-wk delay before they were studied during expression using the slow-pressor model of ANG II-elicited HT. By the end of expression (ANG II 120 ng·kg−1·min−1 sc for 2 wk), the male offspring of hypertensive dams showed a significantly enhanced hypertensive response (Δ41.6 ± 2.6 mmHg) compared with the male offspring of dams that were infused with saline during pregnancy (Δ17.1 ± 1.3 mmHg). These results indicate that a sensitized hypertensive response can be induced beginning in the prenatal period and that the underlying changes that mediate the sensitized response are sustained for a remarkably long period of time. In addition, these results indicate that perinatal programming of a predisposition toward hypertension appears to be very similar to cross sensitization of the hypertensive response seen in adults. It seems reasonable to speculate that the sensitized state producing a predisposition to HT can last a lifetime, particularly when it is induced during a critical development period.
The finding that the interval between induction and expression of the HT response can be quite long raises the interesting prospect that prophylactic interventions delivered during induction and/or delay might prevent or attenuate high BP in a manner similar to what has been shown in genetically programmed models of HT [reviewed in (103, 204)]. For example, Harrap et al. (77) found that administering the converting enzyme inhibitor, perindopril, for several weeks to young, but not more mature rats, produced sustained reductions in the BP of adult spontaneously hypertensive rats, even after the antihypertensive treatment was stopped. In adult males and ovariectomized females, we have recently found that coadministration of estrogen along with a sensitizing dose of ANG II blocks the enhanced HT response elicited during expression (198). Recognizing the possibility of using such prophylactic interventions indicates that it will be important to explore the full range of types and times of treatments that can be administered during the induction and delay to block the consequences of sensitization of the HT response.
Induction of Salt-Sensitive Hypertension
Dietary salt (i.e., sodium chloride) has been argued by many to be an important contributing factor to the etiology and progression of HT (e.g., 40, 116). The likelihood of BP increasing in response to salt loading varies among humans and members of nonhuman species. Rigorously defined protocols have been used to characterize salt sensitivity (184), and it is estimated that in the United States about 51% of hypertensive patients and about 26% of normotensives are salt sensitive (95).
There is growing evidence indicating that regions of the brain controlling sympathetic tone play an important role in salt sensitivity (16, 85, 143). In recent experiments, we examined whether ANG II or ALDO treatments given during induction would produce salt sensitivity (35). These studies employed a method to test for salt sensitivity that used 2% NaCl as the sole drinking fluid (58, 112). During induction, rats instrumented chronically to record BP and HR were given 1-wk treatments for induction with either vehicle or nonpressor infusions of either ANG II (10 ng·kg−1·min−1 sc) or ALDO (750 ng/h). After 1 wk of delay, the rats were given 2% NaCl as their sole drinking fluid without additional ANG II or ALDO during expression. All animals had a significant rise in BP during high salt intake. However, the increase in BP was significantly greater in those that had been treated with ANG II or ALDO during induction (Fig. 5, A and B). When 2% saline was replaced with water, BP fell to preexposure levels. A further study showed that either ANG II (1 ng·kg−1·min−1) or ALDO (10 ng/h) administered intracerebroventricularly during induction also produces salt sensitivity (Fig. 5, C and D).
Fig. 5.

Either angiotensin (ANG II) or aldosterone (Aldo) administered either subcutaneously or intracerebroventricularly during a 1-wk induction period (Induction) cross-sensitized the hypertensive response to 2% hypertonic saline administered as the sole drinking fluid during a 1-wk expression period following a 1-wk delay period. The blood pressure of rats that received either ANG II (10 ng·kg−1·min−1), Aldo (750 ng/h sc) or vehicle (control) during Induction (A) are shown, as well as the difference scores from baseline (B). C: blood pressure of rats that received ANG II (1 ng·kg−1·min−1 icv), Aldo (10 ng/h icv), or vehicle (control, icv) during Induction are shown. D: difference scores from baseline are shown. *P < 0.05 vs. controls; n = 9 or 10/each group. [Adapted from Ref. 35].
Brain RAAS in the LT and PVN and the Sensitization/Cross-Sensitization of ANG II-Induced HT
As a means to assess the effects of sensitization induced by low doses of systemic ANG II or ALDO on the neural networks controlling sympathetic tone and BP, quantitative PCR (q-PCR) was used to study changes in mRNA expression within two key brain regions in different phases of the induction-delay-expression paradigm (200, 201). In these studies the PVN and LT structures were dissected from brains at the end of delay and at the end of expression to determine changes in mRNA in animals sensitized with subcutaneous ANG II or cross-sensitized with subcutaneous ALDO. Sensitizing doses of either ANG II or ALDO produced significant increases in the expression of mRNA of many components of the RAAS in the LT at both the end of delay and after expression, but in the PVN only at the end of expression.
The patterns of increased mRNA expression differed between the sensitizing treatments. Considering first the hypertensive arm of the RAAS, at the end of delay, 1) AT1R, MR, and ACE 1 were all increased after either ANG II or ALDO, 2) angiotensinogen was increased only after ALDO, and 3) ALDO synthase was increased only after ANG II. The increase in AT1R after either of the sensitizing treatments is consistent with several other in vivo and in vitro studies showing upregulation, or increased binding of ANG II immediately after treatments that elevate circulating ANG II (7, 30, 31, 133, 140, 158, 186) or administration of a mineralocorticoid agonist (98, 163, 164, 187). Although not totally unique, it is important to recognize that the capacity of ANG II and ALDO to upregulate a primary receptor is an important property of the brain RAAS. It is usually the case that agonists downregulate their receptors. The functional significance of agonist-induced receptor upregulation in the RAAS is that it constitutes a feed-forward mechanism that can participate in response amplification (sensitization) to drive a heightened hypertensive response.
Importantly, at the end of delay, there were no significant changes in mRNA expression of RAAS components in the PVN. This suggests that, at least with the parameters used to this point in our induction-delay-expression studies, LT structures, but not the PVN, are involved in the initiation of the process of sensitization. These findings do not rule out the likely possibility that the PVN and other downstream nuclei (e.g., RVLM, IML, sympathetic ganglion) may be recruited with stronger stimuli or a longer induction.
The significant increases in mRNA of several RAAS components in both the LT and PVN at the end of expression might suggest that there is a role for downstream elements during the elicitation of HT (200, 201). However, these changes in mRNA must be interpreted with caution because of the differences in BP between the sensitized vs. nonsensitized groups that occur over the course of expression, which could be a confounding factor.
Implications of RAAS-Driven Neuroplasticity for the Sensitization of Hypertension
The systemic administration of high doses of ANG II produces an abrupt increase in BP. As reviewed by Simon et al. (166), acute HT induced by high doses of ANG II is initially accompanied by salt and water retention and ALDO secretion (3, 18, 75), but that the infusion of low, initially nonpressor doses of ANG II (i.e., the slow-pressor model) gradually elicits HT by means other than mechanisms related to expansion of extracellular fluid. ANG II delivered at moderately low doses that do not produce an immediate rise in BP will gradually elicit HT if the administration is maintained.
The slow-pressor model of ANG II-elicited HT was first demonstrated by Dickinson and colleagues (44, 45) in the rabbit, and then subsequently reproduced in dog (127), rat (17), sheep (82), and human (3). The ability of repeated or sustained administration of low doses of ANG II to progressively shift the dose-response curve upward to produce a greater BP response was originally defined as “angiotensin auto-potentiation” (11, 66). The capacity of ANG II to auto-potentiate or produce response sensitization is recognized for several of its actions, including contraction of vascular smooth muscle (66), thirst (133), and salt appetite (19). Neuroplasticity and the upregulation of components of the hypertensive arm of the RAAS by ANG II and ALDO are another plausible example of auto-potentiation.
CNS-mediated sensitization may be an important mechanism accounting for the rapid increase in BP observed when reclipping a renal artery one or two days after it has been unclipped in rats with established two-kidney, one-clip renal HT [2K-1C; (168, 175)]. Enhanced responsiveness to administration of exogenous renin or ANG II is also seen in unclipped 2K-1C hypertensive rats shortly after the renal artery clip is removed or the clipped kidney is ablated (4, 168, 175). ten Berg and de Jong (175) implicated the nervous system in their renal artery clipping-unclipping study by demonstrating that after destroying the spinal cord there was no enhanced renin-induced pressor response in acutely unclipped 2K-1C rats. Taken together, the clipping-unclipping studies seem to indicate that a cardiovascular control system behaved as if it had a memory for some factor induced by renal clipping. Because the 2K-1C rat model of renal HT elevates circulating ANG II chronically (121), it is plausible that ANG II acted on and through a reprogrammed CNS to trigger a sensitized HT response when the renal artery was reclipped.
The induction-delay-expression paradigm permits a means to temporally dissociate the cellular and molecular changes involved in the process of sensitization from those responsible for the actual expression of HT. Commonly used procedures to induce experimental HT conflate multiple underlying processes involved in the induction of sensitization with those involved in the expression of the hypertensive response. Conventional methods used to elicit HT typically begin with a fixed procedure (e.g., renal artery constriction or baroreceptor denervation) or treatment (e.g., administration of a fixed dose of a pressor agent or feeding a dietary component such as high fat or salt) and then allow the consequence of the intervention to run its course until frank HT is present. As such, the progression of events before the appearance of HT is largely out of the control of the investigator. The conceptually and methodologically simple induction-delay-expression paradigm provides remarkable leverage to probe the mechanisms mediating induction vs. expression over the course of development of HT. The mechanisms involved in increasing the probability for expression of an enhanced hypertensive response as a result of CNS plasticity and those underlying the actual expression of the disorder are likely to be different with each having different time courses and unique underlying molecular and cellular processes. In addition, the induction-delay-expression paradigm affords the opportunity to investigate the mechanisms that maintain the sensitized state across delay. Understanding these mechanisms may lead to the discovery of interventions to prevent the exaggerated expression of high BP that occurs on subsequent encounters with stimuli that elicit frank HT.
The capacity of ANG II for auto-potentiation and the discovery that very low doses of ANG II can act through the CNS to progressively sensitize its own pressor response potentially explains an apparent paradox observed in many individuals with EH. Specifically, the BP-lowering efficacy of antagonists of the RAS is not correlated with patients' pretreatment levels of circulating renin (125). In addition, plasma renin activity does not have to be significantly elevated to maintain high BP in patients with so-called medium renin EH (reviewed in Ref. 105). It seems reasonable to speculate that such patients may have a CNS that was reprogrammed by prior exposure to ANG II—or some other hypertensinogenic stimulus—so that modest levels of circulating ANG II and ALDO are sufficient to elicit a sensitized hypertensive response through their actions on the brain.
Molecular Candidates Commonly Implicated in Sensitization Are Prime Candidates for Roles in the Sensitization of the Hypertensive Response
As previously noted, both sensitization and its underlying neuroplasticity have been investigated extensively over the last 40-plus years. In looking broadly across this massive body of research, there are four features common to nearly every model in which neuroplasticity is implicated in the modification of functional responses.
First, the controlling neural network seems to have at least one or two “signature neuromodulators” with unique roles in the neuroplasticity of that functional system. For example, substance P and calcitonin gene-related peptide play important roles in information signaling and in sensitization in the neural network subserving pain (148, 149, 162). Similarly, dopamine is associated with neuroplasticity of networks involved in behavioral motivation and reward (188). From what was described earlier, it seems that ANG II and ALDO are likely to fulfill the role of signature neuromodulators in the neural network controlling BP and are likely to play a role in the neuroplasticity affecting BP.
Second, in virtually every case that they have been studied, glutamate and glutamate receptors [particularly, N-methyl-d-aspartate (NMDA-R) and (aminomethyl) phosphonic acid (AMPA-R)] are linked to neuroplasticity (34, 153). Glutamate, acting on AMPA-R and working in concert with signature neuromodulators, is critical for producing a rapid depolarization of postsynaptic cells. This initial change in membrane potential is key to initiating the removal of Mg2+ that blocks a nonselective cation channel associated with the NMDA-R. This open channel results in large increases in [Ca2+]i, promoting further depolarization and signaling for phosphorylation of proteins that induce and maintain neuroplasticity (185).
Third, one or more growth factors are implicated in playing multiple roles in neuroplasticity (e.g., altering membrane potentials, increasing transcription, promoting cell viability, and morphological changes). Probably, the most notable growth factor is brain-derived neurotrophic factor (BDNF), which acts through tropomyosin-related kinase B-R (TrkB-R). BDNF has been associated with almost every aspect of neural and functional plasticity (203).
There are multiple signaling cascades activated by BDNF acting through the TrkB-R, which include the RAS-mitogen activated protein kinase (MAPK), the phosphatidylinositol 3 -kinase (PI3K)-Akt and the PLCγ-Ca2+ pathways (97). The MAPK signaling pathway plays a role in synaptic plasticity and memory (172). Signaling through the Ras-MAPK along with other pathways can activate transcription factors, such as cAMP response element-binding protein (CREB), which binds to cAMP response elements (CRE) in DNA to effect transcription of message for several molecules, including renin, BDNF, some immediate early genes and some glutamate-R subunits.
Fourth, long-term neuroplastic changes require alterations in gene expression and the synthesis of new proteins. Transcription factors, such as c-Fos, FOSB, ΔFOSB, zif268, and arc, are important for the synthesis of new receptors, ion channels, structural proteins, and components of enzymes that sustain enhanced excitability (e.g., increased postsynaptic AMPA-R) and structural changes (e.g., increased size of dendritic spines). For example, cellular processes controlling the synthesis and the number of AMPA-R in postsynaptic membranes are important for the long-term maintenance of plasticity (153). Inducible transcription factors vary in the latency for onset of their expression and persistence after stimuli trigger a cascade of neuroplasticity-related cellular events (126).
In neurons, different members of the c-Fos family of immediate early gene (IEG) transcription factors are activated by acute stimuli. The products of many IEG are induced rapidly and are degraded within a few hours. However, ΔFOSB is one IEG that appears in neurons only after a delay, but it accumulates with repeated or sustained challenges and then declines slowly with a long persistence after the end of stimulation (126). The sustained activity of ΔFOSB as a transcription factor has been proposed to be a mechanism for neuroplasticity and long-term storage of information (126).
Recent studies on the role of FosB/ΔFOSB in chronic intermittent hypoxia and BP are particularly relevant to neuroplasticity and the sensitization of a form of HT that is dependent on the integrity of the brain RAS (101). Cunningham et al. (37, 100) have shown that chronic intermittent hypoxia (8 h/day for 7 days) increases both BP and ΔFOSB in the SFO, MnPO, OVLT, NTS, RVLM, and parvocellular portions of the PVN. Administration of a dominant-negative mRNA construct into the MnPO to inhibit the transcriptional actions of ΔFOSB significantly reduced the increase in BP produced by chronic intermittent hypoxia (37).
We have investigated whether there are changes in the expression or activation of key molecular indicators of neuroplasticity in LT structures (35). Specifically, we looked for changes in 1) two growth factors, BDNF and vascular endothelial growth factor (VEGF); 2) the TrkB receptor; 3) one of the classes of MAPK, p38 mitogen-activated protein kinase (p38 MAPK); and 4) the transcription factor CREB at the end of delay following induction with either subcutaneous ANG II or ALDO. The result of the sensitizing treatments was an upregulation of mRNA and protein for BDNF (Fig. 6, A and B), but not for VEGF or TrkB. There were no changes in the total amounts of protein for either p38 MAPK or CREB, but there were shifts from the unphosphorylated to the phosphorylated state for both, which is consistent with greater functional activation of these signaling molecules. In addition, following induction with either subcutaneous ALDO or subcutaneous ANG II, we have recently found increased phosphorylated protein (Fig. 6, C and D) and message for the GluN1 subunit of the N-methyl-d-aspartate (NMDA) receptor after delay (Zhang Z, Xue B, and Johnson AK, unpublished findings).
Fig. 6.

Protein expression of brain-derived neurotrophic factor (BDNF) and of phosphorylated N-methyl-d-aspartate-receptor (NMDA-R) subunit GluN1 in the collective structures of the lamina terminalis (LT) at the end of delay after induction with either subcutaneous ANG II or subcutaneous aldosterone (Aldo). A: Western blots of BDNF expression in LT. B: mean data showing that the LT of Aldo- and ANG II-infused rats had higher expression of total BDNF, which included both the precursor pro-BDNF, corresponding to the 27-kDa band and the total BDNF corresponding to the 14-kDa band. (*P < 0.05 vs. controls; n = 3/group). C, top: Western blots indicating increased phosphorylated GluN1 subunit of the NMDA-R after Control (vehicle, sc) or ANG II (10 ng·kg−1·min−1 sc) administered during induction followed by delay. C, bottom: relative amount of phosphorylated GluN1 subunit of the NMDA-R in the LT of sensitized rats compared with Control at the end of delay following induction. D, top: Western blots indicating increased phosphorylated GluN1 subunit of the NMDA-R after Control (vehicle, sc) or Aldo (750 ng/h sc) administered during induction followed by delay. D, bottom: Relative amount of phosphorylated GluN1 subunit of the NMDA-R in the LT of sensitized rats compared with Control at the end of delay following induction. [Parts A and B adapted from Ref. 35; Parts C and D, from Zhang Z, Xue B, and Johnson AK, unpublished data.]
Figure 7 summarizes some of the signaling mechanisms that are likely to be involved in the LT neuroplasticity underlying sensitization of HT. On the basis of ongoing studies, we believe that sensitization of the hypertensinogenic response follows several of the common “rules” of neuroplasticity that operate in other behavioral and physiological systems. Furthermore, it is reasonable to hypothesize that changes in one or more of the LT structures is responsible for increasing the magnitude of hypertensive responses following experience with hypertensinogenic stimuli. In future work, it will be important to determine in which of the LT structures the changes in message and protein occur, whether similar molecular changes are seen in more caudal components of the BP control network, the persistence of such changes, and the sequence of changes over the course of induction-delay-expression. In addition, it will be necessary to pursue other mechanisms implicated in the sustained storage of information, such as those associated with epigenetic processes.
Fig. 7.
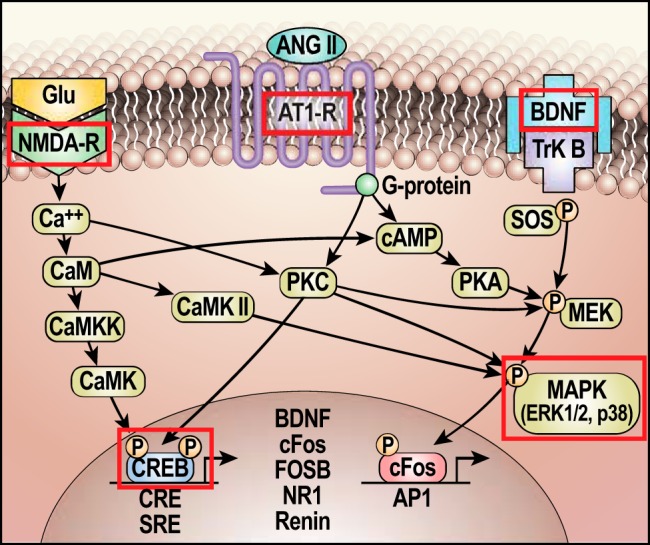
Schematic of hypothesized molecular mechanisms involved in neuroplasticity underlying the sensitization of hypertension by prior experience. Outlined in red are components found to have sustained changes at the end of the delay period following the induction of sensitization of the hypertensive response. Glu, glutamate; CaM, calmodulin; CaMK II, Ca2+/calmodulin-dependent kinase II; CaMKK, calmodulin-dependent protein kinase kinase; CaMK, Ca2+/calmodulin-dependent protein kinase; AT1R, angiotensin type 1 receptor; CRE, cAMP response element; CREB, CRE-binding protein; cAMP, cyclic adenosine monophosphate; BDNF, brain-derived neurotrophic factor; ERK1/2, extracellular signal regulating kinase; TrkB, tropomyosin-related kinase B; SOS, son of sevenless (guanine nucleotide exchange factor); SRE, serum response element; MEK, MAP kinase kinase (a.k.a. MAPKK); MAPK, mitogen-activated protein kinase; c-Fos, FOSB, FBJ murine osteosarcoma viral oncogene homolog B; AP1, activator protein 1; PKA, protein kinase A; NMDA-R, a glutamate ionotropic receptor; ANG II, angiotensin II; NR1, NMDA-R subunit.
The “Missing Heritability” of HT, Epigenetics and the Adaptive Role of Neuroplasticity
With the availability of methods to screen for genetic variants in genome-wide association studies, came the belief that genetic differences accounting for phenotypic variation associated with many complex diseases would be found. HT clusters in families and has a high heritability. Depending on the study, it is estimated that inheritance accounts for 25 to 65% of the variance in BP (27, 78, 192). However, as with many other complex disorders, genome-wide association studies of HT find that genetic variants account for only a small portion of phenotypic variation and disease risk. Summing the incremental increases to BP contributed by each of the identified so-called “pressor genes” fails to provide a full accounting for the hypertensive phenotype (36). Apparent discrepancies between genotypes and disorder phenotypes have been referred to as “the missing heritability of complex diseases” (122). Several strategies have been proposed attempting to bring heritability and gene variation studies into register with one another (104, 205). One hypothesis to account for the missing heritability component of hypertension is that epigenetic mechanisms cause the mismatch (36).
Epigenetics, literally meaning “above and beyond” the gene, was originally proposed to account, in a very broad, theoretical sense, for how the identical genetic information contained in all somatic cells comes to be differentially expressed to generate a broad array of cellular and physiological phenotypes (182). Functionally, epigenetic processes include DNA methylation and histone modification (both referred to as “marking”), and actions of noncoding RNAs, but do not involve altering the sequence of DNA bases. Such mechanisms modifying gene expression allow cells or cell lines to become phenotypically distinct and maintain that distinction.
Since the term was first introduced, the definition of epigenetics has evolved to have varied meanings for different fields within biology (9, 74). From one perspective, epigenetics is the study of mechanisms that produce heritable cellular and physiological traits present in daughter cells that are not due to changes in the sequence of DNA bases (150). From this point of view, epigenetic investigation does not involve neurons because, for the most part, nerve cells in adult mammals do not divide. However, there are other points of view that extend the concept of epigenetics to consider all types of chromatin modification affecting a phenotype regardless of whether cell replication is involved. The NIH “Roadmap Epigenomics Project” takes this broader view by stating, “For purposes of this program, epigenetics refers to both heritable changes in gene activity and expression (in the progeny of cells or of individuals) and also stable, long-term alterations in the transcriptional potential of a cell that are not necessarily heritable.” (http://www.roadmapepigenomics.org/overview). Marking of the components of chromatin can take place in nondividing cells like neurons to affect the proteins they express and, in turn, determine neuronal structure and function. Such biochemical modifications can be sustained for a lifetime and, as a consequence, epigenetics is recognized to be important in the neurosciences. Epigenetic mechanisms in the nervous system are investigated in the areas of behavioral development, learning, cognition, drug addiction, and psychopathology (173). Work in this area collectively is represented by the newly emerging field of neuroepigenetics (171).
In a recent report of a National Heart, Lung and Blood Institute working group on epigenetics and HT, the authors addressed the issue of the missing heritability of HT, focusing on the importance of epigenetic mechanisms in the pathogenesis of EH (36). The report included several hypotheses to account for the missing heritability and made many important recommendations, including the need to pursue various approaches and entertain new hypotheses. However, the report did not discuss the role of the nervous system in EH and the potential of neuroepigenetics for understanding some forms of high BP. Recognizing the capacity that neuroplasticity and molecular memory have in the sensitization of the hypertensive response, it is reasonable to assume that neuroepigenetics will become increasingly important for gaining deeper insight into the etiology of EH. It seems reasonable to hypothesize that a portion of the missing heritability of HT not accounted for by “hypertensive genes” per se, may ultimately prove to be attributable to the heritability of genes determining neuroplasticity where chromatin has been marked by environmental experience to modify activity in the neural network controlling BP.
Summary and Perspectives
The primary purpose for developing the current review was to summarize recent findings indicating that prior experience can enhance the hypertensive response to subsequent challenges and to recognize that this form of sensitization is accompanied by sustained changes in the CNS. Converging results of several studies indicate that this sensitization involves lasting changes in the brain RAAS, as well as increased expression or activation of several molecular markers associated with CNS neuroplasticity. Thus far, the molecular changes modified in association with the induction of sensitization are found in structures of the LT that are part of the CNS neural network controlling sympathetic tone and BP. The discoveries of sensitization and memory-related processes that affect the long-term regulation of BP have important implications for understanding, preventing and treating EH.
Earlier assessments of the potential control mechanisms involved in the long-term regulation of BP questioned the importance of the nervous system in such a role. This was mainly because of a limited perspective of what the nervous system has the capacity to do. Much of the past consideration of the neural contribution to the regulation of BP was restricted to assessing the ability of arterial baroreceptor reflexes to correct perturbations in BP. For example, using an engineering systems approach led to the conclusion that baroreceptor mechanisms alone are only moderately effective over the short term (i.e., having only a modest gain) in maintaining or restoring BP and ineffective over the long term because they adapt (72). However, dismissing a role for the nervous system in cardiovascular control on just this basis does not take into account one of the most powerful roles of the CNS—the capacity for adaptation and altering the magnitude of behavioral and physiological responses as a result of prior experience. In systems analysis terms, the demonstration that the hypertensive response can be sensitized provides an instantiation of an adaptive controller for the regulation of BP that operates in the CNS. That is, it is a controller that learns as a result of prior experience (83, 102). Recognition that the brain can act as an adaptive controller to increase the gain of the hypertensive response as a result of neuroplastic changes must engender new views on the regulation of BP and etiology of EH.
The capacity of behavioral and physiological systems to adapt as a result of experience and deliver stronger responses to subsequent challenges has the potential to increase biological fitness. Inherent in this ability are mechanisms related to information acquisition and storage. These are recognized properties of the immune system (memory B and T cells) and the central nervous system, which are responsible for mediating experience-initiated changes in behavioral and physiological responses. Phenotypic changes in neurons can be induced by environmental experiences that entail interactions among the genome, epigenome, and nervous system and include neuronal activity-dependent mechanisms (i.e., activity-dependent neuroplasticity). Epigenetic changes in the nervous system are most prevalent during development, but also occur throughout life (189). Such lifelong changes can provide adaptive plasticity and have the capability of enhancing fitness of an individual in the face of fluctuating environmental conditions (8).
Although adaptive mechanisms can have beneficial effects, sustained or repeated activation of responses under inappropriate conditions can lead to pathology. This can be seen both in neurally based systems such as those associated with pain (e.g., hyperalgesia and allodynia) or drug abuse (e.g., drug craving) and in disordered immune responses (e.g., autoimmune disease and allergy). The same may be true for the capacity to sensitize the hypertensive response. The sensitization of pressor mechanisms may have played an important adaptive role at one point in mankind's evolutionary history or during an earlier time in the life of an individual, but a sensitized response can become inappropriate and pathogenic when evoked under different circumstances [i.e., in different evolutionary eras or under different ecological conditions (139), or at later times in life (76)].
HT research is in a new era. The recognition that factors beyond the genotype can interact with genes to contribute to the pathogenesis of EH in most cases (36) brings into focus the need to understand how prior environmental experiences and conditions can induce sustained changes in CNS pathways controlling sympathetic activity and a sensitized hypertensive response. To fully comprehend the etiology of EH, it will be necessary to recognize the need for a paradigm shift that involves a new perspective on the role of the CNS and, hence, new experimental approaches. Attention needs to be focused on the full capacity of the nervous system in determining the long-term regulation of BP. The induction-delay-expression experimental model has great potential for playing an important role in such anticipated and necessary explorations.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: A.K.J., Z.Z., and B.X. conception and design of research; A.K.J., Z.Z., S.C.C., and B.X. analyzed data; A.K.J., Z.Z., S.C.C., S.W.H., R.L.T., and B.X. interpreted results of experiments; A.K.J., Z.Z., S.C.C., and B.X. prepared figures; A.K.J. drafted manuscript; A.K.J., T.G.B., S.W.H., R.L.T., and B.X. edited and revised manuscript; A.K.J., Z.Z., S.C.C., T.G.B., S.W.H., R.L.T., and B.X. approved final version of manuscript; Z.Z., S.C.C., T.G.B., and B.X. performed experiments.
ACKNOWLEDGMENTS
The authors and research from their laboratories presented in this review were supported in part by National Institutes of Health Grants HL-14388, HL-098207, and MH-08241. The authors thank Marilyn Dennis for her assistance with preparation of the manuscript.
REFERENCES
- 1.Ahima R, Krozowski Z, Harlan R. Type I corticosteroid receptor-like immunoreactivity in the rat CNS: distribution and regulation by corticosteroids. J Comp Neurol 313: 522–538, 1991. [DOI] [PubMed] [Google Scholar]
- 2.Alkadhi KA, Alzoubi KH, Aleisa AM. Plasticity of synaptic transmission in autonomic ganglia. Prog Neurobiol 75: 83–108, 2005. [DOI] [PubMed] [Google Scholar]
- 3.Ames RP, Borkowski AJ, Sicinski AM, Laragh JH. Prolonged infusions of angiotensin II and norepinephrine and blood pressure, electrolyte balance, and aldosterone and cortisol secretion in normal man and in cirrhosis with ascites. J Clin Invest 44: 1171–1186, 1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aoki K, Masson GM. Pressor responsiveness to renin and angiotensin in renal hypertensive rats. Nephron 6: 484–497, 1969. [DOI] [PubMed] [Google Scholar]
- 5.Arriza JL, Simerly RB, Swanson LW, Evans RM. The neuronal mineralocorticoid receptor as a mediator of glucocorticoid response. Neuron 1: 887–900, 1988. [DOI] [PubMed] [Google Scholar]
- 6.Bader M. Tissue renin-angiotensin-aldosterone systems: targets for pharmacological therapy. Annu Rev Pharmacol Toxicol 50: 439–465, 2010. [DOI] [PubMed] [Google Scholar]
- 7.Barth SW, Gerstberger R. Differential regulation of angiotensinogen and AT1A receptor mRNA within the rat subfornical organ during dehydration. Brain Res Mol Brain Res 64: 151–164, 1999. [DOI] [PubMed] [Google Scholar]
- 8.Beldade P, Mateus AR, Keller RA. Evolution and molecular mechanisms of adaptive developmental plasticity. Mol Ecol 20: 1347–1363, 2011. [DOI] [PubMed] [Google Scholar]
- 9.Bird A. Perceptions of epigenetics. Nature 447: 396–398, 2007. [DOI] [PubMed] [Google Scholar]
- 10.Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol 232: 331–356, 1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bohr DF. Angiotensin in vascular smooth muscle. In: Angiotensin: Handbook of Experimental Pharmacology, edited by Page IH, and Bumpus FM. New York: Springer-Verlag, 1974, p. 424. [Google Scholar]
- 12.Bonham AC, Sekizawa S, Chen CY, Joad JP. Plasticity of brainstem mechanisms of cough. Respir Physiol Neurobiol 152: 312–319, 2006. [DOI] [PubMed] [Google Scholar]
- 13.Booth DA. Mechanism of action of norepinephrine in eliciting an eating response on injection into the rat hypothalamus. J Pharmacol Exp Ther 160: 336–348, 1968. [PubMed] [Google Scholar]
- 14.Brody MJ, Fink GD, Buggy J, Haywood JR, Gordon FJ, Johnson AK. The role of the anteroventral third ventricle (AV3V) region in experimental hyeprtension. Circ Res 43: I2–I13, 1978. [DOI] [PubMed] [Google Scholar]
- 15.Brody MJ, Johnson AK. Role of the anteroventral third ventricle region in fluid and electrolyte balance, arterial pressure regulation, and hypertension. In: Frontiers in Neuroendocrinology, edited by Martini L, Ganong WF. New York: Raven Press, 1980. [Google Scholar]
- 16.Brooks VL, Scrogin KE, McKeogh DF. The interaction of angiotensin II and osmolality in the generation of sympathetic tone during changes in dietary salt intake. an hypothesis. Ann NY Acad Sci 940: 380–394, 2001. [DOI] [PubMed] [Google Scholar]
- 17.Brown AJ, Casals-Stenzel J, Gofford S, Lever AF, Morton JJ. Comparison of fast and slow pressor effects of angiotensin II in the conscious rat. Am J Physiol Heart Circ Physiol 241: H381–H388, 1981. [DOI] [PubMed] [Google Scholar]
- 18.Brown JJ, Casals-Stenzel J, Cumming AM, Davies DL, Fraser R, Lever AF, Morton JJ, Semple PF, Tree M, Robertson JI. Angiotensin II, aldosterone and arterial pressure: a quantitative approach. Arthur C Corcoran Memorial Lecture. Hypertension 1: 159–179, 1979. [DOI] [PubMed] [Google Scholar]
- 19.Bryant RW, Epstein AN, Fitzsimons JT, Fluharty SJ. Arousal of a specific and persistent sodium appetite in the rat with continuous intracerebroventricular infusion of angiotensin II. J Physiol 301: 365–382, 1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bryne JH. Learning and memory: basic mechanisms. In: Fundamental Neuroscience, edited by Squire LR, Berg D, Bloom FE, du Lac S, Ghosh A, Spitzer N. Boston, MA: Academic Press/Elsevier, 2008, p. 1133–1152. [Google Scholar]
- 21.Buckley JP, Bickerton RK, Halliday RP, Kato H. Central effects of peptides on the cardiovascular system. Ann NY Acad Sci 104: 299–311, 1963. [DOI] [PubMed] [Google Scholar]
- 22.Buggy J, Fink GD, Johnson AK, Brody MJ. Prevention of the development of renal hypertension by anteroventral third ventricular tissue lesions. Circ Res 40: I110–I117, 1977. [PubMed] [Google Scholar]
- 23.Buggy J, Fisher AE, Hoffman WE, Johnson AK, Phillips MI. Ventricular obstruction: effect on drinking induced by intracranial injection of angiotensin. Science 190: 72–74, 1975. [DOI] [PubMed] [Google Scholar]
- 24.Buggy J, Johnson AK. Anteroventral third ventricle periventricular ablation: Temporary adipsia and persisting thirst deficits. Neurosci Lett 5: 177–182, 1977. [DOI] [PubMed] [Google Scholar]
- 25.Buggy J, Johnson AK. Preoptic-hypothalamic periventricular lesions: Thirst deficits and hypernatremia. Am J Physiol Regul Integr Comp Physiol 233: R44–R52, 1977. [DOI] [PubMed] [Google Scholar]
- 26.Carithers J, Bealer SL, Brody MJ, Johnson AK. Fine structural evidence of degeneration in supraoptic nucleus and subfornical organ of rats with lesions in the anteroventral third ventricle. Brain Res 201: 1–12, 1980. [DOI] [PubMed] [Google Scholar]
- 27.Carretero OA, Oparil S. Essential hypertension. Part I: definition and etiology. Circulation 101: 329–335, 2000. [DOI] [PubMed] [Google Scholar]
- 28.Castellucci V, Pinsker H, Kupfermann I, Kandel ER. Neuronal mechanisms of habituation and dishabituation of the gill-withdrawal reflex in aplysia. Science 167: 1745–1748, 1970. [DOI] [PubMed] [Google Scholar]
- 29.Cates MJ, Dickinson CJ, Hart EC, Paton JF. Neurogenic hypertension and elevated vertebrobasilar arterial resistance: is there a causative link? Curr Hypertens Rep 14: 261–269, 2012. [DOI] [PubMed] [Google Scholar]
- 30.Charron G, Laforest S, Gagnon C, Drolet G, Mouginot D. Acute sodium deficit triggers plasticity of the brain angiotensin type 1 receptors. FASEB J 16: 610–612, 2002. [DOI] [PubMed] [Google Scholar]
- 31.Chen Y, da Rocha MJ, Morris M. Osmotic regulation of angiotensin AT1 receptor subtypes in mouse brain. Brain Res 965: 35–44, 2003. [DOI] [PubMed] [Google Scholar]
- 32.Chow CK, Teo KK, Rangarajan S, Islam S, Gupta R, Avezum A, Bahonar A, Chifamba J, Dagenais G, Diaz R, Kazmi K, Lanas F, Wei L, Lopez-Jaramillo P, Fanghong L, Ismail NH, Puoane T, Rosengren A, Szuba A, Temizhan A, Wielgosz A, Yusuf R, Yusufali A, McKee M, Liu L, Mony P, Yusuf S. Prevalence, awareness, treatment, and control of hypertension in rural and urban communities in high-, middle-, and low-income countries. JAMA 310: 959–968, 2013. [DOI] [PubMed] [Google Scholar]
- 33.Cifuentes F, Arias ER, Morales MA. Long-term potentiation in mammalian autonomic ganglia: an inclusive proposal of a calcium-dependent, trans-synaptic process. Brain Res Bull 97: 32–38, 2013. [DOI] [PubMed] [Google Scholar]
- 34.Citri A, Malenka RC. Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology 33: 18–41, 2008. [DOI] [PubMed] [Google Scholar]
- 35.Clayton SC, Zhang Z, Beltz T, Xue B, Johnson AK. CNS neuroplasticity and salt-sensitive hypertension induced by prior treatment with subpressor doses of ANG II or aldosterone. Am J Physiol Regul Integr Comp Physiol 306: R908–R917, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cowley AW Jr Nadeau JH, Baccarelli A, Berecek K, Fornage M, Gibbons GH, Harrison DG, Liang M, Nathanielsz PW, O'Connor DT, Ordovas J, Peng W, Soares MB, Szyf M, Tolunay HE, Wood KC, Zhao K, Galis ZS. Report of the National Heart, Lung, and Blood Institute Working Group on epigenetics and hypertension. Hypertension 59: 899–905, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cunningham JT, Knight WD, Mifflin SW, Nestler EJ. An essential role for ΔFosB in the median preoptic nucleus in the sustained hypertensive effects of chronic intermittent hypoxia. Hypertension 60: 179–187, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dampney RA. Functional organization of central pathways regulating the cardiovascular system. Physiol Rev 74: 323–364, 1994. [DOI] [PubMed] [Google Scholar]
- 39.Davis JO, Ayers CR, Carpenter CC. Renal origin of an aldosterone-stimulating hormone in dogs with thoracic caval constriction and in sodium-depleted dogs. J Clin Invest 40: 1466–1474, 1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Wardener H, MacGregor GA. Sodium intake and mortality. Lancet 351: 1508; author reply 1509–1510, 1998. [DOI] [PubMed] [Google Scholar]
- 41.Deng W, Aimone JB, Gage FH. New neurons and new memories: how does adult hippocampal neurogenesis affect learning and memory? Nat Rev Neurosci 11: 339–350, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dickinson CJ. Neurogenic Hypertension. Blackwell: Oxford, 1965. [Google Scholar]
- 43.Dickinson CJ, Thomason AD. Vertebral and internal carotid arteries in relation to hypertension and cerebrovascular disease. Lancet 2: 46–48, 1959. [DOI] [PubMed] [Google Scholar]
- 44.Dickinson CJ, Yu R. The progressive pressor response to angiotensin in the rabbit. J Physiol 190: 91–99, 1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dickinson DM, Lawrence JR, Adelaide MB. A slowly developing pressor response to small concentrations of angiotensin: its bearing on the pathogenesis of chronic renal hyeprtension. Lancet 281: 1354–1356, 1963. [DOI] [PubMed] [Google Scholar]
- 46.Doba N, Reis DJ. Acute fulminating neurogenic hypertension produced by brainstem lesions in the rat. Circ Res 32: 584–593, 1973. [DOI] [PubMed] [Google Scholar]
- 47.Dubinion JH, da Silva AA, Hall JE. Enhanced blood pressure and appetite responses to chronic central melanocortin-3/4 receptor blockade in dietary-induced obesity. J Hypertens 28: 1466–1470, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Epstein AN, Fitzsimons JT, Rolls BJ. Drinking induced by injection of angiotensin into the brain of the rat. J Physiol 210: 457–474, 1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Esler M, Lambert E, Schlaich M. Chronic activation of the sympathetic nervous system is the dominant contributor to systemic hypertension. J Appl Physiol (1985) 109: 1996–1998, discussion 2016, 2010. [DOI] [PubMed] [Google Scholar]
- 50.Ferguson AV. Angiotensinergic regulation of autonomic and neuroendocrine outputs: critical roles for the subfornical organ and paraventricular nucleus. Neuroendocrinology 89: 370–376, 2009. [DOI] [PubMed] [Google Scholar]
- 51.Ferrara LA, Moscato TS, Pisanti N, Marotta T, Krogh V, Capone D, Mancini M. Is the sympathetic nervous system altered in children with familial history of arterial hypertension? Cardiology 75: 200–205, 1988. [DOI] [PubMed] [Google Scholar]
- 52.Ferrario CM. New physiological concepts of the renin-angiotensin system from the investigation of precursors and products of angiotensin I metabolism. Hypertension 55: 445–452, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ferrario CM, Ahmad S, Nagata S, Simington SW, Varagic J, Kon N, Dell'italia LJ. An evolving story of angiotensin-II-forming pathways in rodents and humans. Clin Sci (Lond) 126: 461–469, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ferrario CM, Gildenberg PL, McCubbin JW. Cardiovascular effects of angiotensin mediated by the central nervous system. Circ Res 30: 257–262, 1972. [DOI] [PubMed] [Google Scholar]
- 55.Finch L, Haeusler G, Thoenen H. Failure to induce experimental hypertension in rats after intraventricular injection of 6-hydroxydopamine. Br J Pharmacol 44: 356P–357P, 1972. [PMC free article] [PubMed] [Google Scholar]
- 56.Fischer-Ferraro C, Nahmod VE, Goldstein DJ, Finkielman S. Angiotensin and renin in rat and dog brain. J Exp Med 133: 353–361, 1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fitzsimons JT. The kidney as a thirst receptor. J Physiol 191: 128P–129P, 1967. [PubMed] [Google Scholar]
- 58.Florin M, Lo M, Liu KL, Sassard J. Salt sensitivity in genetically hypertensive rats of the Lyon strain. Kidney Int 59: 1865–1872, 2001. [DOI] [PubMed] [Google Scholar]
- 59.Folkow B. Physiological aspects of primary hypertension. Physiol Rev 62: 347–504, 1982. [DOI] [PubMed] [Google Scholar]
- 60.Folkow B, Neil E. Circulation. New York: Oxford, 1971. [Google Scholar]
- 61.Folkow B, Rubinstein EH. Cardiovascular effects of acute and chronic stimulation of the hypothalamic defense area in the rat. Acta Physiol Scand 68: 48–57, 1966. [Google Scholar]
- 62.Funder JW, Sheppard K. Adrenocortical steroids and the brain. Annu Rev Physiol 49: 397–411, 1987. [DOI] [PubMed] [Google Scholar]
- 63.Ganten D, Minnich JL, Granger P, Hayduk K, Brecht HM, Barbeau A, Boucher R, Genest J. Angiotensin-forming enzyme in brain tissue. Science 173: 64–65, 1971. [DOI] [PubMed] [Google Scholar]
- 64.Gauer OH, Henry JP, Behn C. The regulation of extracellular fluid volume. Annu Rev Physiol 32: 547–595, 1970. [DOI] [PubMed] [Google Scholar]
- 65.Geerling JC, Loewy AD. Aldosterone in the brain. Am J Physiol Renal Physiol 297: F559–F576, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Godfraind T. Angiotensin auto-potentiation. Br J Pharmacol 40: 542P–543P, 1970. [PMC free article] [PubMed] [Google Scholar]
- 67.Goldblatt H, Lynch J, Hanzal RF, Summerville WW. Studies on experimental hypertension: I. The production of persisten elevation of systolic blood pressure by means of renal ischemia. J Exp Med 59: 347–379, 1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gomez-Sanchez EP, Gomez-Sanchez CE. Central regulation of blood pressure by the mineralocorticoid receptor. Mol Cell Endocrinol 350: 289–298, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Grassi G, Gavazzi C, Cesura AM, Picotti GB, Mancia G. Changes in plasma catecholamines in response to reflex modulation of sympathetic vasoconstrictor tone by cardiopulmonary receptors. Clin Sci (Lond) 68: 503–510, 1985. [DOI] [PubMed] [Google Scholar]
- 70.Grassi G, Mark A, Esler M. The sympathetic nervous system alterations in human hypertension. Circ Res 116: 976–990, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Guyenet PG. The sympathetic control of blood pressure. Nat Rev Neurosci 7: 335–346, 2006. [DOI] [PubMed] [Google Scholar]
- 72.Guyton AC. Circulatory Physiology III: Arterial Pressure and Hypertension. Philadelphia, PA: W.B. Saunders, 1980. [Google Scholar]
- 73.Guyton AC. Long-term arterial pressure control: an analysis from animal experiments and computer and graphic models. Am J Physiol Regul Integr Comp Physiol 259: R865–R877, 1990. [DOI] [PubMed] [Google Scholar]
- 74.Haig D. Commentary: The epidemiology of epigenetics. Int J Epidemiol 41: 13–16, 2012. [DOI] [PubMed] [Google Scholar]
- 75.Hall JE. Control of sodium excretion by angiotensin II: intrarenal mechanisms and blood pressure regulation. Am J Physiol Regul Integr Comp Physiol 250: R960–R972, 1986. [DOI] [PubMed] [Google Scholar]
- 76.Hanson MA, Gluckman PD. Early developmental conditioning of later health and disease: physiology or pathophysiology? Physiol Rev 94: 1027–1076, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Harrap SB, Van der Merwe WM, Griffin SA, Macpherson F, Lever AF. Brief angiotensin converting enzyme inhibitor treatment in young spontaneously hypertensive rats reduces blood pressure long-term. Hypertension 16: 603–614, 1990. [DOI] [PubMed] [Google Scholar]
- 78.Havlik RJ, Garrison RJ, Feinleib M, Kannel WB, Castelli WP, McNamara PM. Blood pressure aggregation in families. Am J Epidemiol 110: 304–312, 1979. [DOI] [PubMed] [Google Scholar]
- 79.Hebb D. The Organization of Behavior. New York: Wiley & Sons, 1949. [Google Scholar]
- 80.Herman JP, Flak J, Jankord R. Chronic stress plasticity in the hypothalamic paraventricular nucleus. Prog Brain Res 170: 353–364, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hirooka Y, Kishi T, Sakai K, Takeshita A, Sunagawa K. Imbalance of central nitric oxide and reactive oxygen species in the regulation of sympathetic activity and neural mechanisms of hypertension. Am J Physiol Regul Integr Comp Physiol 300: R818–R826, 2011. [DOI] [PubMed] [Google Scholar]
- 82.Hood SG, Cochrane T, McKinley MJ, May CN. Investigation of the mechanisms by which chronic infusion of an acutely subpressor dose of angiotensin II induces hypertension. Am J Physiol Regul Integr Comp Physiol 292: R1893–R1899, 2007. [DOI] [PubMed] [Google Scholar]
- 83.Houk JC. Control strategies in physiological systems. FASEB J 2: 97–107, 1988. [DOI] [PubMed] [Google Scholar]
- 84.Huang BS, Ahmadi S, Ahmad M, White RA, Leenen FH. Central neuronal activation and pressor responses induced by circulating ANG II: role of the brain aldosterone-“ouabain” pathway. Am J Physiol Heart Circ Physiol 299: H422–H430, 2010. [DOI] [PubMed] [Google Scholar]
- 85.Huang BS, Amin MS, Leenen FH. The central role of the brain in salt-sensitive hypertension. Curr Opin Cardiol 21: 295–304, 2006. [DOI] [PubMed] [Google Scholar]
- 86.Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci 29: 565–598, 2006. [DOI] [PubMed] [Google Scholar]
- 87.Ito S, Komatsu K, Tsukamoto K, Kanmatsuse K, Sved AF. Ventrolateral medulla AT1 receptors support blood pressure in hypertensive rats. Hypertension 40: 552–559, 2002. [DOI] [PubMed] [Google Scholar]
- 88.Ito S, Komatsu K, Tsukamoto K, Sved AF. Tonic excitatory input to the rostral ventrolateral medulla in Dahl salt-sensitive rats. Hypertension 37: 687–691, 2001. [PubMed] [Google Scholar]
- 89.Johnson AK. The periventricular anteroventral third ventricle (AV3V): its relationship with the subfornical organ and neural systems involved in maintaining body fluid homeostasis. Brain Res Bull 15: 595–601, 1985. [DOI] [PubMed] [Google Scholar]
- 90.Johnson AK, Gross PM. Sensory circumventricular organs and brain homeostatic pathways. FASEB J 7: 678–686, 1993. [DOI] [PubMed] [Google Scholar]
- 91.Johnson AK, Thunhorst RL. The neuroendocrinology of thirst and salt appetite: visceral sensory signals and mechanisms of central integration. Front Neuroendocrinol 18: 292–353, 1997. [DOI] [PubMed] [Google Scholar]
- 92.Johnson AK, Thunhorst RL. The neuroendocrinology, neurochemistry and molecular biology of thirst and salt appetite. In: Handbook of Neurochemistry and Molecular Neurobiology: Behavioral Neurochemistry, Neuroendocrinology and Molecular Neurobiology, edited by Lajtha A, Blaustein JD. New York: Springer, 2007. [Google Scholar]
- 93.Johnson AK, Wilkin LD. The lamina terminalis. In: Circumventricular Organs and Body Fluids, edited by Gross P. Boca Raton: CRC Press, 1987. [Google Scholar]
- 94.Julius S. Tachycardia in hypertension: a saga of progress despite prejudice, confusion, and inertia. Prog Cardiovasc Dis 52: 26–30, 2009. [DOI] [PubMed] [Google Scholar]
- 95.Kanbay M, Chen Y, Solak Y, Sanders PW. Mechanisms and consequences of salt sensitivity and dietary salt intake. Curr Opin Nephrol Hypertens 20: 37–43, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kandel ER, Dudai Y, Mayford MR. The molecular and systems biology of memory. Cell 157: 163–186, 2014. [DOI] [PubMed] [Google Scholar]
- 97.Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol 10: 381–391, 2000. [DOI] [PubMed] [Google Scholar]
- 98.King SJ, Harding JW, Moe KE. Elevated salt appetite and brain binding of angiotensin II in mineralocorticoid-treated rats. Brain Res 448: 140–149, 1988. [DOI] [PubMed] [Google Scholar]
- 99.Kline DD. Plasticity in glutamatergic NTS neurotransmission. Respir Physiol Neurobiol 164: 105–111, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Knight WD, Little JT, Carreno FR, Toney GM, Mifflin SW, Cunningham JT. Chronic intermittent hypoxia increases blood pressure and expression of FosB/ΔFosB in central autonomic regions. Am J Physiol Regul Integr Comp Physiol 301: R131–R139, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Knight WD, Saxena A, Shell B, Nedungadi TP, Mifflin SW, Cunningham JT. Central losartan attenuates increases in arterial pressure and expression of FosB/ΔFosB along the autonomic axis associated with chronic intermittent hypoxia. Am J Physiol Regul Integr Comp Physiol 305: R1051–R1058, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Korner P. Essential Hypertension and Its Causes: Neural and Non-Neural Mechanisms. New York: Oxford University Press, 2007. [Google Scholar]
- 103.Kuneš J, Kadlecová M, Vaněčková I, Zicha J. Critical developmental periods in the pathogenesis of hypertension. Physiol Res 61 Suppl 1: S9–S17, 2012. [DOI] [PubMed] [Google Scholar]
- 104.Lander ES. Initial impact of the sequencing of the human genome. Nature 470: 187–197, 2011. [DOI] [PubMed] [Google Scholar]
- 105.Laragh JH, Sealey JE. The plasma renin test reveals the contribution of body sodium-volume content (V) and renin-angiotensin (R) vasoconstriction to long-term blood pressure. Am J Hypertens 24: 1164–1180, 2011. [DOI] [PubMed] [Google Scholar]
- 106.Levine S, Mullins RF Jr. Hormonal influences on brain organization in infant rats. Science 152: 1585–1592, 1966. [DOI] [PubMed] [Google Scholar]
- 107.Lind RW, Johnson AK. Central and peripheral mechanisms mediating angiotensin-induced thirst. In: The Renin Angiotensin System in the Brain, edited by Ganten D, Printz M, Phillips MI, Scholkens BA. New York: Springer-Verlag, 1982, p. 353–364. [Google Scholar]
- 108.Lind RW, Johnson AK. Subfornical organ-median preoptic connections and drinking and pressor responses to angiotensin II. J Neurosci 2: 1043–1051, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lind RW, Ohman LE, Lansing MB, Johnson AK. Transection of subfornical organ neural connections diminishes the pressor response to intravenously infused angiotensin II. Brain Res 275: 361–364, 1983. [DOI] [PubMed] [Google Scholar]
- 110.Lind RW, Thunhorst RL, Johnson AK. The subfornical organ and the integration of multiple factors in thirst. Physiol Behav 32: 69–74, 1984. [DOI] [PubMed] [Google Scholar]
- 111.Lind RW, Van Hoesen GW, Johnson AK. An HRP study of the connections of the subfornical organ of the rat. J Comp Neurol 210: 265–277, 1982. [DOI] [PubMed] [Google Scholar]
- 112.Lo M, Liu KL, Clemitson JR, Sassard J, Samani NJ. Chromosome 1 blood pressure QTL region influences renal function curve and salt sensitivity in SHR. Physiol Genomics 8: 15–21, 2002. [DOI] [PubMed] [Google Scholar]
- 113.Loria AS, D'Angelo G, Pollock DM, Pollock JS. Early life stress downregulates endothelin receptor expression and enhances acute stress-mediated blood pressure responses in adult rats. Am J Physiol Regul Integr Comp Physiol 299: R185–R191, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Loria AS, Ho DH, Pollock JS. A mechanistic look at the effects of adversity early in life on cardiovascular disease risk during adulthood. Acta Physiol (Oxf) 210: 277–287, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lovett-Barr MR, Satriotomo I, Muir GD, Wilkerson JE, Hoffman MS, Vinit S, Mitchell GS. Repetitive intermittent hypoxia induces respiratory and somatic motor recovery after chronic cervical spinal injury. J Neurosci 32: 3591–3600, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.MacGregor GA, de Wardener HE. Salt, Diet, and Health. Cambridge: Cambridge University Press, 1998. [Google Scholar]
- 117.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron 44: 5–21, 2004. [DOI] [PubMed] [Google Scholar]
- 118.Mancia G, Grassi G. The autonomic nervous system and hypertension. Circ Res 114: 1804–1814, 2014. [DOI] [PubMed] [Google Scholar]
- 119.Mancia G, Grassi G, Giannattasio C, Seravalle G. Sympathetic activation in the pathogenesis of hypertension and progression of organ damage. Hypertension 34: 724–728, 1999. [DOI] [PubMed] [Google Scholar]
- 120.Mangiapane ML, Simpson JB. Subfornical organ: forebrain site of pressor and dipsogenic action of angiotensin II. Am J Physiol Regul Integr Comp Physiol 239: R382–R389, 1980. [DOI] [PubMed] [Google Scholar]
- 121.Mann JF, Johnson AK, Ganten D. Plasma angiotensin II: dipsogenic levels and angiotensin-generating capacity of renin. Am J Physiol Regul Integr Comp Physiol 238: R372–R377, 1980. [DOI] [PubMed] [Google Scholar]
- 122.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TF, McCarroll SA, Visscher PM. Finding the missing heritability of complex diseases. Nature 461: 747–753, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mark AL. The sympathetic nervous system in hypertension: a potential long-term regulator of arterial pressure. J Hypertens Suppl 14: S159–S165, 1996. [PubMed] [Google Scholar]
- 124.Masuo K, Mikami H, Ogihara T, Tuck ML. Sympathetic nerve hyperactivity precedes hyperinsulinemia and blood pressure elevation in a young, nonobese Japanese population. Am J Hypertens 10: 77–83, 1997. [DOI] [PubMed] [Google Scholar]
- 125.McAreavey D, Robertson JI. Angiotensin-converting enzyme inhibitors and moderate hypertension. Drugs 40: 326–345, 1990. [DOI] [PubMed] [Google Scholar]
- 126.McClung CA, Ulery PG, Perrotti LI, Zachariou V, Berton O, Nestler EJ. ΔFosB: a molecular switch for long-term adaptation in the brain. Brain Res Mol Brain Res 132: 146–154, 2004. [DOI] [PubMed] [Google Scholar]
- 127.McCubbin JW, DeMoura RS, Page IH, Olmsted F. Arterial hypertension elicited by subpressor amounts of angiotensin. Science 149: 1394–1395, 1965. [DOI] [PubMed] [Google Scholar]
- 128.Mendelsohn ME. In hypertension, the kidney is not always the heart of the matter. J Clin Invest 115: 840–844, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Michelini LC, Stern JE. Exercise-induced neuronal plasticity in central autonomic networks: role in cardiovascular control. Exp Physiol 94: 947–960, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mifflin SW. Short-term potentiation of carotid sinus nerve inputs to neurons in the nucleus of the solitary tract. Respir Physiol 110: 229–236, 1997. [DOI] [PubMed] [Google Scholar]
- 131.Miki K, Yoshimoto M, Tanimizu M. Acute shifts of baroreflex control of renal sympathetic nerve activity induced by treadmill exercise in rats. J Physiol 548: 313–322, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Miselis RR, Shapiro RE, Hand PJ. Subfornical organ efferents to neural systems for control of body water. Science 205: 1022–1025, 1979. [DOI] [PubMed] [Google Scholar]
- 133.Moellenhoff E, Blume A, Culman J, Chatterjee B, Herdegen T, Lebrun CJ, Unger T. Effect of repetitive icv injections of ANG II on c-Fos and AT1-receptor expression in the rat brain. Am J Physiol Regul Integr Comp Physiol 280: R1095–R1104, 2001. [DOI] [PubMed] [Google Scholar]
- 134.Mueller PJ. Exercise training and sympathetic nervous system activity: evidence for physical activity dependent neural plasticity. Clin Exp Pharmacol Physiol 34: 377–384, 2007. [DOI] [PubMed] [Google Scholar]
- 135.Mulrow PJ, Ganong WF. Stimulation of aldosterone secretion by angiotensisn. II. A preliminary report. Yale J Biol Med 33: 386–395, 1961. [PMC free article] [PubMed] [Google Scholar]
- 136.Na ES, Morris MJ, Johnson RF, Beltz TG, Johnson AK. The neural substrates of enhanced salt appetite after repeated sodium depletions. Brain Res 1171: 104–110, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nakagaki T, Hirooka Y, Ito K, Kishi T, Hoka S, Sunagawa K. Role of angiotensin-(1–7) in rostral ventrolateral medulla in blood pressure regulation via sympathetic nerve activity in Wistar-Kyoto and spontaneous hypertensive rats. Clin Exp Hypertens 33: 223–230, 2011. [DOI] [PubMed] [Google Scholar]
- 138.Navar LG. Counterpoint: Activation of the intrarenal renin-angiotensin system is the dominant contributor to systemic hypertension. J Appl Physiol 109: 1998–2000, discussion 2015, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Nesse RM, Ganten D, Gregory TR, Omenn GS. Evolutionary molecular medicine. J Mol Med (Berl) 90: 509–522, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Nunes FC, Braga VA. Chronic angiotensin II infusion modulates angiotensin II type I receptor expression in the subfornical organ and the rostral ventrolateral medulla in hypertensive rats. J Renin Angiotensin Aldosterone Syst 12: 440–445, 2011. [DOI] [PubMed] [Google Scholar]
- 141.Oparil S. The sympathetic nervous system in clinical and experimental hypertension. Kidney Int 30: 437–452, 1986. [DOI] [PubMed] [Google Scholar]
- 142.Osborn JW. Hypothesis: set-points and long-term control of arterial pressure. A theoretical argument for a long-term arterial pressure control system in the brain rather than the kidney. Clin Exp Pharmacol Physiol 32: 384–393, 2005. [DOI] [PubMed] [Google Scholar]
- 143.Osborn JW, Fink GD, Sved AF, Toney GM, Raizada MK. Circulating angiotensin II and dietary salt: converging signals for neurogenic hypertension. Curr Hypertens Rep 9: 228–235, 2007. [DOI] [PubMed] [Google Scholar]
- 144.Palatini P, Julius S. The role of cardiac autonomic function in hypertension and cardiovascular disease. Curr Hypertens Rep 11: 199–205, 2009. [DOI] [PubMed] [Google Scholar]
- 145.Perini C, Muller FB, Rauchfleisch U, Battegay R, Buhler FR. Effects of psychological and physical covariates on plasma catecholamines in borderline hypertensives and offspring of hypertensive parents. Clin Exp Hypertens A 12: 137–150, 1990. [DOI] [PubMed] [Google Scholar]
- 146.Pettersen KH, Bugenhagen SM, Nauman J, Beard DA, Omholt SW. Arterial stiffening provides sufficient explanation for primary hypertension. PLoS Comput Biol 10: e1003634, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Pinsker HM, Hening WA, Carew TJ, Kandel ER. Long-term sensitization of a defensive withdrawal reflex in aplysia. Science 182: 1039–1042, 1973. [DOI] [PubMed] [Google Scholar]
- 148.Ren K, Dubner R. Central nervous system plasticity and persistent pain. J Orofac Pain 13: 155–163; discussion 164–171, 1999. [PubMed] [Google Scholar]
- 149.Ren K, Dubner R. Pain facilitation and activity-dependent plasticity in pain modulatory circuitry: role of BDNF-TrkB signaling and NMDA receptors. Mol Neurobiol 35: 224–235, 2007. [DOI] [PubMed] [Google Scholar]
- 150.Riggs AD, Martienssen RA, Russo VEA. Introduction. In: Epigenetic Mechanisms of Gene Regulation, edited by Russo VEA, Martienssen RA, Riggs AD. Plainview, NY: Cold Spring Harbor Laboratory Press, 1996. [Google Scholar]
- 151.Roland BL, Li KX, Funder JW. Hybridization histochemical localization of 11 β-hydroxysteroid dehydrogenase type 2 in rat brain. Endocrinology 136: 4697–4700, 1995. [DOI] [PubMed] [Google Scholar]
- 152.Rosenzweig MR, Bennett EL. Psychobiology of plasticity: effects of training and experience on brain and behavior. Behav Brain Res 78: 57–65, 1996. [DOI] [PubMed] [Google Scholar]
- 153.Rudy JW. The Neurobiology of Learning and Memory. New York: Sinaur, 2014. [Google Scholar]
- 154.Russo SJ, Mazei-Robison MS, Ables JL, Nestler EJ. Neurotrophic factors and structural plasticity in addiction. Neuropharmacology 56 Suppl 1: 73–82, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Sambhi MP. Fundamental Fault in Hypertension. Hingham, MA: M. Nijhoff Kluwer, 1984. [Google Scholar]
- 156.Santos RA, Campagnole-Santos MJ. Central and peripheral actions of angiotensin-(1–7). Braz J Med Biol Res 27: 1033–1047, 1994. [PubMed] [Google Scholar]
- 157.Santos RA, Ferreira AJ. Angiotensin-(1–7) and the renin-angiotensin system. Curr Opin Nephrol Hypertens 16: 122–128, 2007. [DOI] [PubMed] [Google Scholar]
- 158.Sanvitto GL, Johren O, Hauser W, Saavedra JM. Water deprivation upregulates ANG II AT1 binding and mRNA in rat subfornical organ and anterior pituitary. Am J Physiol Endocrinol Metab 273: E156–E163, 1997. [DOI] [PubMed] [Google Scholar]
- 159.Scott JP. Critical periods in behavioral development. Science 138: 949–958, 1962. [DOI] [PubMed] [Google Scholar]
- 160.Scoville WB, Milner B. Loss of recent memory after bilateral hippocampal lesions. 1957. J Neuropsychiatry Clin Neurosci 12: 103–113, 2000. [DOI] [PubMed] [Google Scholar]
- 161.Severs WB, Daniels AE, Buckley JP. On the central hypertensive effect of angiotensin II. Int J Neuropharmacol 6: 199–205, 1967. [DOI] [PubMed] [Google Scholar]
- 162.Seybold VS. The role of peptides in central sensitization. Hand Exp Pharmacol 194: 451–491, 2009. [DOI] [PubMed] [Google Scholar]
- 163.Shelat SG, Flanagan-Cato LM, Fluharty SJ. Glucocorticoid and mineralocorticoid regulation of angiotensin II type 1 receptor binding and inositol triphosphate formation in WB cells. J Endocrinol 162: 381–391, 1999. [DOI] [PubMed] [Google Scholar]
- 164.Shelat SG, King JL, Flanagan-Cato LM, Fluharty SJ. Mineralocorticoids and glucocorticoids cooperatively increase salt intake and angiotensin II receptor binding in rat brain. Neuroendocrinology 69: 339–351, 1999. [DOI] [PubMed] [Google Scholar]
- 165.Shinohara K, Hirooka Y, Kishi T, Sunagawa K. Reduction of nitric oxide-mediated gamma-amino butyric acid release in rostral ventrolateral medulla is involved in superoxide-induced sympathoexcitation of hypertensive rats. Circ J 76: 2814–2821, 2012. [DOI] [PubMed] [Google Scholar]
- 166.Simon G, Abraham G, Cserep G. Pressor and subpressor angiotensin II administration. Two experimental models of hypertension. Am J Hypertens 8: 645–650, 1995. [DOI] [PubMed] [Google Scholar]
- 167.Simpson JB, Epstein AN, Camardo JS Jr. Localization of receptors for the dipsogenic action of angiotensin II in the subfornical organ of rat. J Comp Physiol Psychol 92: 581–601, 1978. [DOI] [PubMed] [Google Scholar]
- 168.Skulan TW, Brousseau AC, Leonard KA. Accelerated induction to two-kidney hypertension in rats and renin-angiotensin sensitivity. Circ Res 35: 734–741, 1974. [DOI] [PubMed] [Google Scholar]
- 169.Smith PM, Ferguson AV. Circulating signals as critical regulators of autonomic state—central roles for the subfornical organ. Am J Physiol Regul Integr Comp Physiol 299: R405–R415, 2010. [DOI] [PubMed] [Google Scholar]
- 170.Spyer KM. Central nervous integration of cardiovascular control. J Exp Biol 100: 109–128, 1982. [DOI] [PubMed] [Google Scholar]
- 171.Sweatt JD. The emerging field of neuroepigenetics. Neuron 80: 624–632, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Sweatt JD. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol 14: 311–317, 2004. [DOI] [PubMed] [Google Scholar]
- 173.Sweatt JD, Meaney MJ, Nestler EJ, Schahram A. Epigentic Regulation in the Nervous System: Basic Mechanisms and Clinical Impact. San Diego, CA: Elsevier, 2013. [Google Scholar]
- 174.Tarazi RC. The role of the heart in hypertension. In: Fundamental Fault in Hypertension, edited by Sambhi MP. Boston, MA: Martinus Nijhoff Publishers, 1984. [Google Scholar]
- 175.ten Berg R, de Jong W. Mechanism of enhanced blood pressure rise after reclipping following removal of a renal artery clip in rats. Hypertension 2: 4–13, 1980. [DOI] [PubMed] [Google Scholar]
- 176.Thornton SN, Nicolaidis S. Long-term mineralocorticoid-induced changes in rat neuron properties plus interaction of aldosterone and ANG II. Am J Physiol Regul Integr Comp Physiol 266: R564–R571, 1994. [DOI] [PubMed] [Google Scholar]
- 177.Tigerstedt R, Bergmann PG. Niere und Kreislauf. Skandinav Arch Physiol 8: 223, 1898. [Google Scholar]
- 178.Toney GM, Chen QH, Cato MJ, Stocker SD. Central osmotic regulation of sympathetic nerve activity. Acta Physiol Scand 177: 43–55, 2003. [DOI] [PubMed] [Google Scholar]
- 179.Tucker DC, Bhatnagar RK, Johnson AK. Genetic and environmental influences on developing autonomic control of heart rate. Am J Physiol Regul Integr Comp Physiol 246: R578–R586, 1984. [DOI] [PubMed] [Google Scholar]
- 180.Tucker DC, Johnson AK. Influence of neonatal handling on blood pressure, locomotor activity, and preweanling heart rate in spontaneously hypertensive and Wistar-Kyoto rats. Dev Psychobiol 17: 587–600, 1984. [DOI] [PubMed] [Google Scholar]
- 181.Uvnas B. Central cardiovascular control. In: Handbook of Physiology, Section I: Neurophysiology, Central Cardiovascular Control. Baltimore: Williams and Wilkins, 1960, p. 1131–1162. [Google Scholar]
- 182.Waddington CH. The epigenotype. 1942. Int J Epidemiol 41: 10–13, 2012. [DOI] [PubMed] [Google Scholar]
- 183.Wei SG, Zhang ZH, Beltz TG, Yu Y, Johnson AK, Felder RB. Subfornical organ mediates sympathetic and hemodynamic responses to blood-borne proinflammatory cytokines. Hypertension 62: 118–125, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Weinberger MH, Miller JZ, Luft FC, Grim CE, Fineberg NS. Definitions and characteristics of sodium sensitivity and blood pressure resistance. Hypertension 8: II127–II134, 1986. [DOI] [PubMed] [Google Scholar]
- 185.Wiegert JS, Bading H. Activity-dependent calcium signaling and ERK-MAP kinases in neurons: a link to structural plasticity of the nucleus and gene transcription regulation. Cell Calcium 49: 296–305, 2011. [DOI] [PubMed] [Google Scholar]
- 186.Wilson KM, Sumners C, Fregly MJ. Effects of increased circulating angiotensin II (AII) on fluid exchange and binding of AII in the brain. Brain Res Bull 20: 493–501, 1988. [DOI] [PubMed] [Google Scholar]
- 187.Wilson KM, Sumners C, Hathaway S, Fregly MJ. Mineralocorticoids modulate central angiotensin II receptors in rats. Brain Res 382: 87–96, 1986. [DOI] [PubMed] [Google Scholar]
- 188.Wise RA. Forebrain substrates of reward and motivation. J Comp Neurol 493: 115–121, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Wolf C, Linden DE. Biological pathways to adaptability–interactions between genome, epigenome, nervous system and environment for adaptive behavior. Genes Brain Behav 11: 3–28, 2012. [DOI] [PubMed] [Google Scholar]
- 190.Wolf ME. The role of excitatory amino acids in behavioral sensitization to psychomotor stimulants. Prog Neurobiol 54: 679–720, 1998. [DOI] [PubMed] [Google Scholar]
- 191.Wyrwoll CS, Holmes MC, Seckl JR. 11β-hydroxysteroid dehydrogenases and the brain: from zero to hero, a decade of progress. Front Neuroendocrinol 32: 265–286, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Xu X, Su S, Treiber FA, Vlietinck R, Fagard R, Derom C, Gielen M, Loos RJ, Snieder H, Wang X. Specific genetic influences on nighttime blood pressure. Am J Hypertens 28: 440–443, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Xue B, Beltz TG, Guo F, Johnson AK. Central leptin sensitization of angiotensin II-induced hypertension. FASEB J 28: 858.853, 2014. [Google Scholar]
- 194.Xue B, Beltz TG, Yu Y, Guo F, Gomez-Sanchez CE, Hay M, Johnson AK. Central interactions of aldosterone and angiotensin II in aldosterone- and angiotensin II-induced hypertension. Am J Physiol Heart Circ Physiol 300: H555–H564, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Xue B, Guo F, Beltz TG, Thunhorst RL, Hay M, Johnson AK. Sensitization of angiotensin (ANG) II-elicited hypertension in offspring of hypertensive pregnant rats: evidence of sex differences. FASEB J 29: 813.811, 2015. [Google Scholar]
- 196.Xue B, Guo F, Yu Y, Beltz TG, Felder RB, Johnson AK. Central tumor necrosis factor-α of angiotensin II-elicited hypertension. Hypertension 64: AMP02, 2014. [Google Scholar]
- 197.Xue B, Yang Y, Guo F, Beltz TG, Felder RB, Johnson AK. Central inflammation induced by high-fat diet sensitizes angiotensin II hypertension. FASEB J 29: 986.983, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Xue B, Zhang Z, Beltz TG, Guo F, Hay M, Johnson AK. Estrogen regulation of the brain renin-angiotensin system in protection against angiotensin II-induced sensitization of hypertension. Am J Physiol Heart Circ Physiol 307: H191–H198, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Xue B, Zhang Z, Guo F, Johnson AK. Either sodium deprivation or sodium excess sensitizes angiotensin (ANG) II-induced hyeprtension. FASEB J 27: 695.612, 2013. [Google Scholar]
- 200.Xue B, Zhang Z, Johnson RF, Johnson AK. Sensitization of slow pressor angiotensin II (Ang II)-initiated hypertension: induction of sensitization by prior Ang II treatment. Hypertension 59: 459–466, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Xue B, Zhang Z, Roncari CF, Guo F, Johnson AK. Aldosterone acting through the central nervous system sensitizes angiotensin II-induced hypertension. Hypertension 60: 1023–1030, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Yamada Y, Miyajima E, Tochikubo O, Matsukawa T, Shionoiri H, Ishii M, Kaneko Y. Impaired baroreflex changes in muscle sympathetic nerve activity in adolescents who have a family history of essential hypertension. J Hypertens Suppl 6: S525–S528, 1988. [DOI] [PubMed] [Google Scholar]
- 203.Yirmiya R, Goshen I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav Immun 25: 181–213, 2011. [DOI] [PubMed] [Google Scholar]
- 204.Zicha J, Kuneš J. Ontogenetic aspects of hypertension development: analysis in the rat. Physiol Rev 79: 1227–1282, 1999. [DOI] [PubMed] [Google Scholar]
- 205.Zuk O, Hechter E, Sunyaev SR, Lander ES. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc Natl Acad Sci USA 109: 1193–1198, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]