

Abstract
Impaired vasodilation in persistent pulmonary hypertension of the newborn (PPHN) is characterized by mitochondrial dysfunction. We investigated the hypothesis that a decreased endothelial nitric oxide synthase level leads to impaired mitochondrial biogenesis and function in a lamb model of PPHN induced by prenatal ductus arteriosus constriction. We ventilated PPHN lambs with 100% O2 alone or with inhaled nitric oxide (iNO). We treated pulmonary artery endothelial cells (PAECs) from normal and PPHN lambs with detaNONOate, an NO donor. We observed decreased mitochondrial (mt) DNA copy number, electron transport chain (ETC) complex subunit levels, and ATP levels in PAECs and lung tissue of PPHN fetal lambs at baseline compared with gestation matched controls. Phosphorylation of AMP-activated kinase (AMPK) and levels of peroxisome proliferator-activated receptor-γ coactivator 1-α (PGC-1α) and sirtuin-1, which facilitate mitochondrial biogenesis, were decreased in PPHN. Ventilation with 100% O2 was associated with larger decreases in ETC subunits in the lungs of PPHN lambs compared with unventilated PPHN lambs. iNO administration, which facilitated weaning of FiO2, partly restored mtDNA copy number, ETC subunit levels, and ATP levels. DetaNONOate increased eNOS phosphorylation and its interaction with heat shock protein 90 (HSP90); increased levels of superoxide dismutase 2 (SOD2) mRNA, protein, and activity; and decreased the mitochondrial superoxide levels in PPHN-PAECs. Knockdown of eNOS decreased ETC protein levels in control PAECs. We conclude that ventilation with 100% O2 amplifies oxidative stress and mitochondrial dysfunction in PPHN, which are partly improved by iNO and weaning of oxygen.
Keywords: biogenesis, SOD2, eNOS, mitochondria, oxidative stress
endothelial nitric oxide synthase (eNOS) plays a central role in postnatal pulmonary vasodilation, a physiologic event required for successful transition at birth (25). Expression of eNOS increases towards term gestation in fetal lungs to prime the neonatal lung for this adaptation (34). Exposure of the fetal lungs to higher oxygen tension at birth stimulates eNOS activity and increases nitric oxide (NO) release (18, 19, 22). The function of eNOS is regulated by interactions with the chaperone heat shock protein 90 (HSP90), phosphorylation at serine1179 and availability of arginine and tetrahydrobiopterin (BH4) and reactive oxygen species (ROS) (18, 33a, 36–37). NO has several downstream effects in the pulmonary circulation, including vasodilation, regulation of mitochondrial function, and angiogenesis (15, 27, 39).
Exposure of the fetal lung to higher oxygen levels at birth increases ATP production, which in turn increases eNOS activity (18, 19). Superoxide (O2−) anion, produced as a byproduct of mitochondrial oxidative phosphorylation, is scavenged by the mitochondrial superoxide dismutase (SOD2) (1, 2). Regulation of mitochondrial O2− levels by SOD2 is necessary for normal endothelial function and ATP synthesis (2). Development of adequate number and mass of mitochondria through biogenesis is required as fetal lungs prepare for postnatal transition and increase in ATP synthesis. NO is an important upstream regulator of mitochondrial biogenesis, which involves both nuclear and mitochondrial encoded genes (15, 27). Mitochondrial proteins produced by the nuclear genes induce mitochondrial DNA (mtDNA) transcription and replication and synthesis of electron transport chain (ETC) protein subunits (8, 26, 30–33). Peroxisome proliferator-activated receptor-γ coactivator 1-α (PGC-1α) is the master regulator of mitochondrial biogenesis and also integrates biogenesis with oxidative phosphorylation (3, 29). AMP-activated kinase (AMPK) and histone deacetylase, sirtulin-1 (SIRT1), together modulate PGC1α activity in a coordinated manner to maintain metabolic and redox homeostasis (6, 8, 26–27, 29).
Studies in the ductal constriction model of persistent pulmonary hypertension of the newborn (PPHN) have shown that eNOS expression is downregulated and levels of reactive oxygen species (ROS) are increased in the pulmonary arteries (28, 36). In addition, eNOS uncoupling from impaired HSP90 interactions with eNOS and decreased BH4 levels further decreases NO levels in PPHN (21, 39). In experimental and clinical studies, inhaled nitric oxide (iNO) induces pulmonary vasodilation, leading to sustained improvement in oxygenation in infants with PPHN (12, 40–41). However, mechanical ventilation and hyperoxia, even for a short period, increase ROS formation. ROS react with NO at a diffusion-limited rate, leading to rapid depletion of NO (5, 14).
We recently reported that mitochondrial oxidative stress is increased in pulmonary artery endothelial cells (PAECs) in PPHN (1). Exposure of PPHN infants with preexisting mitochondrial oxidative stress in their vessels to mechanical ventilation and hyperoxia could potentially exacerbate mitochondrial oxidative stress and vascular dysfunction (8, 23). The decrease in NO and increase in ROS can lead to loss of mitochondria via decrease in biogenesis and removal of dysfunctional mitochondria by mitophagy (27). PAECs in PPHN have significantly reduced ATP levels, suggesting impaired oxidative phosphorylation (2).
We, therefore, hypothesized that increased mitochondrial oxidative stress and decreased NO availability impair mitochondrial biogenesis in PPHN. Our objectives for this study are to 1) determine the potential mechanistic link between reduced NO levels and alterations in mitochondrial biogenesis in PPHN, 2) determine the impact of ventilation with 100% O2, typically used in PPHN infants, on mitochondrial biogenesis in PPHN, and 3) determine the effects of iNO therapy and subsequent weaning of FiO2, as typically used in the clinical management of PPHN, on mitochondrial biogenesis and redox balance in lambs with PPHN.
STUDY DESIGNS AND METHODS
Antibodies and chemicals.
Rabbit anti-SOD-2 (1:10,000) was obtained from Assay Designs-Enzo Life Science (Ann Arbor, MI). Mouse anti-β-actin (1:10,000), rabbit anti-eNOS (1:5,000), rabbit anti-eNOS serine1179 (1:1,000), rabbit anti-HSP90 (1:1,000), and eNOS specific siRNA were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse anti-mitochondrial ETC complex proteins were obtained from Mitoscience (Eugene, OR). The SOD2 activity detection kit was from Cayman Laboratory. ATP extraction reagent kit was obtained from Sigma-Aldrich (St. Louis, MO). Primers for sheep NADH dehydrogenase 1 (ND1), NADH dehydrogenase 2 (ND2), SOD2, and eNOS and detaNONOate were obtained from Life Sciences Technology, Invitrogen (Carlsbad, CA).
Creation of PPHN lamb model.
This study was approved by the Institutional Animal Care and Use Committees of the Medical College of Wisconsin and University of Buffalo and conformed to National Institutes of Health Guidelines for the Care and Use of Laboratory Animals. Pulmonary hypertension was induced by prenatal ductus arteriosus constriction as previously described (18–19). Time-dated pregnant ewes at 128 days of gestation (term ∼145 days) were anesthetized with inhaled isoflurane (2%). The fetal lamb was partially exteriorized by a midline laparotomy and hysterotomy, and an umbilical tape was tied around the ductus arteriosus. The fetal chest was then closed and the lamb was placed back in the uterus. Sham-operated and gestation-matched twin fetal lambs were used as controls. The ewe was then allowed to recover for 8 days. Previous studies have shown that these lambs consistently develop severe PPHN with this intervention (20, 23, 28).
Ventilation studies were done at the University of Buffalo. Lambs were ventilated for 24 h and killed. Right carotid and jugular venous lines were placed at the time of caesaran section at birth. Blood gases were measured every 10–15 min in the first hour and every hour for 24 h. Ventilator rates and pressures were adjusted to keep arterial partial pressure of CO2 (PaCO2) in the target range of 40–55 mmHg and pH >7.20 (Table 1). PPHN lambs received one dose of 3 ml/kg of Calfactant (Infasurf; ONY Laboratories, Amherst, NY, a gift from Dr. Edmund A. Egan), administered via endotracheal tube at birth to avoid the contribution of surfactant deficiency to hypoxemia, followed by mechanical ventilation and were assigned to two groups: 1) ventilation with 100% O2 for 24 h without weaning and without iNO (n = 7), or 2) ventilation with 20 parts per million (ppm) iNO administered via endotracheal tube, which was started between 6 and 12 h when the oxygenation index (OI = mean airway pressure × FiO2 × 100/PaO2 in mmHg) was >25 (n = 7). Inhaled NO was started at a mean of 7.6 ± 1.1 h and before 10 h of age in all seven lambs. The inspired O2 concentration was titrated to maintain PaO2 between 60 and 80 mmHg to simulate the clinical management of infants with PPHN.
Table 1.
Changes in the arterial blood gases and ventilator mean airway pressure and fractions of inspired oxygen concentrations in PPHN fetal lambs ventilated with oxygen alone or oxygen plus iNO
| Time, h |
|||||
|---|---|---|---|---|---|
| Variables/Group | 0 | 6 | 12 | 18 | 24 |
| MAP, cmH2O | |||||
| iNO | 13 (0.5) | 14 (0.5) | 11* (1.3) | 11* (1.1) | 11* (1.2) |
| Ventilation | 14 (1.5) | 15 (3.2) | 16 (2.0) | 15 (2.6) | 17 (1.5) |
| FiO2 | |||||
| iNO | 100 (0) | 90 (0) | 65* (0.28) | 59* (0.35) | 53* (0.38) |
| Ventilation | 100 (0) | 100 (0) | 100 (0) | 100 (0) | 100 (0) |
| PaO2 | |||||
| iNO | 23.7 (3.78) | 35.6 (25.1) | 104.6* (70.9) | 105.1* (39.2) | 78.9* (5.7) |
| Ventilation | 31.7 (5.3) | 33.2 (17.8) | 29.6 (18.5) | 27.2 (20.5) | 40.8 (3.74) |
| pH | |||||
| iNO | 7.43* (0.02) | 7.33 (0.05) | 7.36* (0.05) | 7.35 (0.01) | 7.36* (0.07) |
| Ventilation | 7.20 (0.01) | 7.31 (0.07) | 7.17 (0.06) | 7.22 (0.11) | 7.12 (0.06) |
| Pco2 | |||||
| iNO | 56 (5.40) | 46.1* (3.40) | 41.1* (0.35) | 41.8* (16.20) | 39.4* (0.92) |
| Ventilation | 45 (2.30) | 38 (0.56) | 56 (6.20) | 80 (49.70) | 69 (3.74) |
iNO, inhaled nitric oxide; AMP, mean arterial pressure; FiO2, fraction of inspired oxygen concentration; PPHN, persistent pulmonary hypertension of the newborn. Numbers in parentheses are standard deviations. *P < 0.05 from ventilated fetal lambs with PPHN with 100% O2 alone.
In the PPHN fetal lambs that responded well to iNO treatment, FiO2 was weaned by 10–15% based on PaO2 and oxygen saturation. When FiO2 was <60% and PaO2 remained >60, iNO was weaned by 5 ppm every 4 h until the dose was at 5 ppm and then by 1 ppm every 4 h. If the FiO2 requirement increased by >15% following a wean, the previous dose of iNO was resumed. All lambs were ventilated for 24 h and killed. Lung tissue samples were collected from the left apical lobes, washed in cold buffer, and snap frozen in liquid nitrogen.
Gestation-matched unventilated PPHN fetal lambs and normal fetal lambs were used as controls. Fetal lungs were harvested for the isolation of lung tissue that was snap frozen in liquid N2 for protein studies.
Cell culture.
Control and PPHN fetal lambs delivered at term gestation without ventilation at Medical College of Wisconsin were used for isolation of pulmonary arteries and PAECs, using techniques we described previously (1, 20). PAECs isolated from control (normal) and unventilated PPHN fetal lambs were cultured in DMEM with 20% FCS at 37°C in room air and 5% CO2. We used PAECs between passages 2 and 4 for our experiments. Endothelial cell identity was verified by staining for factor VIII antigen and acetylated LDL uptake (2).
NO donor studies.
PAECs isolated from normal fetal lambs and fetal lambs with PPHN were grown to 60% confluence in 60-mm well plates. Cells in selected plates were treated with 200 nM detaNONOate, which was added to the culture medium followed by incubation for 18 h at 37°C. We assessed the effects of detaNONOate on mitochondrial ETC subunit levels, levels of mitochondrial superoxide, SOD2 expression and activity, and cellular oxygen consumption as described below.
NO measurements.
The concentration of NO released by detaNONOate was assessed by measuring NO level in cultured medium of rapidly dividing PAECs following detaNONOate treatment using an NO analyzer (inNO-T-II; Innovative Instruments), connected to a computer. PAECs were grown in 100-mm tissue culture plates until 90% confluent. Cells were then transferred to the measuring platform in 21% O2 at room temperature (25°C) and allowed to equilibrate for 1 h before measurement. After sensor calibration, 200 nM detaNONOate were added to the medium, and NO level was measured for the next 18 h.
Western blot analysis of proteins.
PAECs were grown to 90% confluence and washed twice in ice-cold HBSS. The cells were lysed in modified RIPA buffer. Protein content of the lysates was determined by bicinchoninic acid (BCA) assay. Separated proteins were transferred to nitrocellulose membranes and were blotted with specific polyclonal antibodies for SOD2, eNOS, ETC I–V proteins, and β-actin, overnight at 4°C. The membranes were blotted with horseradish peroxidase (HRP)-conjugated anti-mouse IgG antibody (1:10,000; Bio-Rad) or HRP-conjugated anti-rabbit IgG antibody (1:10,000; Bio-Rad) and exposed to CL-XPosure film (Pierce) after treatment with SuperSignal West Pico (Pierce). The signals were analyzed with ImageJ and normalized to the expression of loading control, β-actin, for whole cell and tissue lysates.
Peripheral lung tissue samples were flash frozen in liquid nitrogen, pulverized, and placed in modified RIPA buffer to obtain homogenates. An aliquot of protein (50 μg) was used for immunoblots for ETC complex I–V proteins, SOD2, eNOS, and β-actin as described above.
Quantification of mtDNA copy number.
We quantified mtDNA and nuclear DNA copy numbers using the iQ5 multicolor real-time PCR detection system (Bio-Rad). Genomic DNA was extracted from 100-μg tissue samples using Qiagen mini DNA extraction kit. 5 μg of the purified genomic DNA was used for the PCR as described previously (3). NADH dehydrogenases 1 and 2 (ND1 and ND2) genes were selected from the D-loop of mtDNA and PCR primers were designed using Primer3 with sequence as follows: ND1: forward 5′-GGAGGACTGAACCAAACCCA-3′ and reverse 5′-GCCTGAGGATAGGGGAAT-3′ and ND2: forward 5′-TACTTCAACCCATCGCCGAC-3′ and reverse 5′-ACGGCTAGGCTTGA-TATGGC-3′. The specificity of each primer was confirmed by performing a blast search to ensure that the primer sequences are unique for the template sequence. The PCR cycle was started at 95°C for 3 min followed by 40 cycles of 95°C for 10 s and then 58°C for 1 min. Melting temperatures were monitored for each pair of primers, and single-peak melting temperature was observed for all of the primer pairs. The mtDNA copy number was normalized to 18S and β-actin copy numbers. The number of the threshold cycle (Ct) for each target DNA was corrected against the corresponding Ct of 18S and β-actin to obtain the Ct, and then 2−ΔΔCt was calculated against the corresponding control for estimation of DNA abundance.
Quantification of PGC1α, SOD2, and eNOS mRNA.
RNA was extracted via the TRIzol (Sigma) method from lung tissue and the contaminated DNA was removed by the TURBO DNA-free Kit (Ambion) in a 37°C water bath for 60 min. cDNA was synthesized from the extracted RNA using the iScript cDNA synthesis kit (Bio-Rad). The PCR primers were designed using Primer3 as previously described and sequences are as follows: PGC1α (forward 5′-CAGACCTGACACAACGC-3′ and reverse 5′CTTGAAAAATT GCTTGCGT-3′), SOD2 (forward 5′-ATTGCTGGAAGCCCATCAAAC-3′ and reverse 5′-AGCAGGGGGATAAGACCT-3′), and eNOS (forward 5′-CCTCACCGCTACAAC-3′ and reverse 5′-GCACAGCCAGGTTGATCT-3′). Real-time RT-PCR was performed using the iQ5 multicolor real-time PCR detection system (Bio-Rad) as described above.
ATP quantification.
Lung tissue samples were homogenized in ATP-releasing agent using ATP extraction kit by Sigma-Aldrich. Lysates were analyzed for ATP concentration using firefly luciferase bioluminescence method. ATP levels in each sample were expressed in nanomoles per milligram of protein.
Measurement of SOD2 activity.
Lung tissue samples were homogenized in HEPES buffer and SOD2 specific activity was quantified using colorimetric method as described previously (1, 2). SOD2 specific activity was obtained by treatment of cell lysates with potassium cyanide (KCN) to inhibit cytosolic and extracellular SOD isoforms. One unit of SOD activity was defined as the amount of enzyme needed to exhibit 50% dismutation of the O2−, and SOD2 activity was corrected against protein content measured using BCA method.
Coimmunoprecipitation studies.
eNOS-HSP90 interaction and eNOS serine1179 phosphorylated fraction of total eNOS protein were assessed in lung tissue using immunoprecipitation as previously described (2). eNOS was immunoprecipitated from lung tissue. The immunoprecipitate was blotted for eNOS, HSP90, and serine1179 phosphorylated eNOS.
eNOS knockdown.
We used small-interfering RNA (siRNA; Santa Cruz Biotechnology) to knock down eNOS expression in PAECs. Control PAECs were seeded in antibiotic-free medium supplemented with 20% FCS. Ten microliters of eNOS siRNA duplex were diluted in 1 ml of Opti-MEM1 and 10 μl of Lipofectamine RNAiMax (Invitrogen). After 15 min at room temperature, cells suspended in 4 ml of antibiotic free culture medium were added to the diluted siRNA mixture and incubated at 37°C in a CO2 incubator. After 24 h of incubation at 37°C in a CO2 incubator, the medium was replaced with fresh 1× normal regular growth medium and incubated for another 24 h before the cells were used for experiments. PAECs transfected with nonsilencing RNA (sc-37007) were used as controls.
Assessment of mitochondrial O2− levels.
We used mitochondrial targeted hydro-ethidine epifluorescence (MitoSOX; Molecular Probes, Sigma) to quantify O2− levels in the mitochondria. Control PAECs were grown to 50–60% confluence in eight-well chamber slides. Control cells in selected wells were transduced with siRNA targeted to eNOS. After 24 h of siRNA transduction, growth medium was replaced with 1× normal regular growth medium. Control PAECs transduced with eNOS siRNA and PPHN-PAECs were incubated with or without 200 nM detaNONOate for 18 h. After treatments, cells were incubated with 10 μM Mito-HE for 30 min to detect mitochondrial O2− levels as described previously (2). Fluorescence signals were quantified using fluorescence detection microscopy and analyzed with MetaView software.
Assessment of mitochondrial function.
The effects of NO-induced changes in mitochondrial bioenergetics were assessed using Seahorse Bioscience XF24 technology (Seahorse Bioscience, Billerica, MA). Before the studies, empirical optimization of cell density, doses of oligomycin, FCCP, and antimycin for PAECs were determined. PAECs, 3 × 104 in count, from control and PPHN were grown in 24-well plates. Specified wells containing control and PPHN cells were treated with 200 nM detaNONOate for 18 h in normal culture medium. Just before the experiment, culture medium was replaced with Seahorse media and left to equilibrate at 37°C for 1–2 h. Baseline O2 consumption rate (OCR) was then established and measured. Oligomycin was then injected to measure OCR due to proton leak. Maximum OCR was measured after injection of FCCP. Nonmitochondrial O2 consumption was measured after antimycin was injected. Cells were washed in HBSS and protein was quantified using BCA method. Results were normalized to protein content.
Statistical analyses.
Data are shown as means ± SE. Student's t-test was used for normally distributed data, and Mann-Whitney U-test was used for data that did not pass the normality test for comparison between two groups. One-way ANOVA with Student-Newman-Keuls post hoc test was used when more than two groups were compared. Data were analyzed with Graph Pad Prism (Graph Pad Software, La Jolla, CA). A P < 0.05 was considered significant.
RESULTS
Mitochondrial biogenesis is impaired in the lungs of PPHN fetal lambs.
Copy numbers for ND1 and ND2 DNA, components of mitochondrial ETC complex-I, were decreased at baseline in the lungs of unventilated PPHN fetal lambs compared with gestation-matched normal fetal lamb controls (Fig. 1A). This observation demonstrates that mtDNA copy number was decreased in PPHN. Mitochondrial ETC protein subunit proteins were also decreased in PPHN, accompanied by reduced ATP levels in PPHN fetal lambs when compared with controls (Fig. 1, B and C). These findings are consistent with our previous observation that AMPK function and ATP levels are decreased in PPHN-PAECs (2, 38).
Fig. 1.

Mitochondrial biogenesis is impaired in the lungs of fetal lambs with persistent pulmonary hypertension (PPHN). Copy number for NADH dehydrogenases 1 and 2 (ND1 and ND2)and components of the mitochondrial DNA (mtDNA) (A) and mitochondrial electron transport chain (ETC) complexes I–V (B) are significantly decreased in lungs of PPHN fetal lambs compared with gestation matched normotensive fetal lambs (control). This is accompanied by a decrease in ATP levels (C). Ventilation with 100% O2 caused a larger decrease in ETC complexes I–V, with no significant additional change in ND1 and ND2 DNA copy numbers in the lungs of PPHN fetal lambs. Addition of inhaled nitric oxide (iNO) in ventilated PPHN fetal lambs significantly increased ND1 and ND2 DNA copy numbers and ETC complex protein levels compared with unventilated PPHN lambs and PPHN lambs ventilated with 100% O2 alone (n = 6, P < 0.05). #P < 0.05, from normal fetal lambs. ΩP < 0.05, from unventilated PPHN fetal lambs. *P < 0.05, from PPHN lambs ventilated with 100% O2 alone.
Effects of mechanical ventilation and hyperoxia with or without iNO on systemic oxygenation in PPHN fetal lambs.
Seven lambs with PPHN were ventilated without iNO and the FiO2 was kept at 1.0 for the entire duration of 24-h ventilation. Seven other lambs with PPHN were ventilated with addition of iNO after 6 h of age and the FiO2 was weaned, based on arterial blood gases to maintain arterial Po2 in the target range. PPHN lambs had severe hypoxemia with OI of 37 ± 7 at birth. There was no difference in the baseline OI between the two groups at birth. Administration of iNO resulted in a significant reduction in the mean airway pressure (MAP) and FiO2, based on improved oxygenation, compared with PPHN lambs ventilated with oxygen alone (Table 1). The arterial Pco2 was significantly lower and pH was higher in PPHN lambs that received iNO compared with ventilated PPHN lambs with O2 alone (Table 1).
Ventilation with 100% O2 exacerbates the mitochondrial dysfunction in PPHN.
Compared with unventilated PPHN fetal lambs, ventilation with 100% O2 for 24 h induced a further decrease in ETC subunits (Fig. 1B); however, we observed no significant change in ATP levels from unventilated PPHN lambs (Fig. 1C). This suggests that an extra-mitochondrial source of ATP, such as aerobic glycolysis, is induced to maintain the ATP levels in these lambs.
iNO with weaning FiO2 restores mitochondrial function in PPHN.
iNO treatment for 12–18 h in ventilated PPHN fetal lambs resulted in a decrease in mean FiO2 to <0.7 and mean OI to <30 within 2–6 h of iNO initiation (Table 1). This strategy was associated with increased mitochondrial DNA copy number and ETC subunits (Fig. 1, A and B). Since increased mitochondrial number and protein content do not imply functional integrity of new mitochondria, we measured ATP levels in these samples. Compared with ventilation with 100% O2 alone, iNO + lower FiO2 use was also associated with higher ATP levels (Fig. 1C). These data suggest improved mitochondrial function in the group assigned to iNO use and FiO2 wean in PPHN.
Ventilation with 100% oxygen impairs and iNO restores PGC1α function in PPHN. The decreased mitochondrial copy number and ETC were associated with decreases in levels of PGC1α, a transcription cofactor that regulates mitochondrial biogenesis and phospho-AMPKα and Sirt1, which regulate the function of PGC-1α, in PPHN. The protein level of AMPKα did not change with PPHN in the absence of ventilation (Fig. 2A). Ventilation with 100% O2 led to decrease in AMPKα and further decreases in p-AMPKα, PGC1α, and Sirt-1 protein levels compared with unventilated PPHN samples (Fig. 2, B and C). These data suggest that ventilation with 100% O2 induces downregulation of transcriptional activators that regulate mitochondrial biogenesis.
Fig. 2.

iNO restores mitochondrial biogenesis and oxidative phosphorylation by increasing the expression levels of peroxisome proliferator-activated receptor-γ coactivator 1-α (PGC-1α) in ventilated PPHN fetal lambs. A: in lung lysates of PPHN fetal lambs without ventilation, expression of SIRT 1, PGC1α as well as p-AMPKα were decreased. B: ventilation with 100% O2 alone led to a further decrease in p-AMPKα and SIRT1 protein levels and PGC1α protein and mRNA levels and a decrease in AMPKα in PPHN lambs. Inhaled NO significantly increased the levels of p-AMPKα, AMPKα, SIRT1, as well as PGC1α in ventilated PPHN fetal lambs compared with PPHN lambs with or without ventilation. (n = 6 P < 0.05). #P < 0.05, from normal fetal lambs. ΩP < 0.05, from unventilated PPHN lamb. *P < 0.05, from PPHN fetal lambs ventilated with 100% oxygen alone.
iNO treatment for 12–18 h in ventilated PPHN fetal lambs resulted in an increase in PGC1α, as well as p-AMPKα, AMPKα, and Sirt1 expression compared with ventilation with 100% O2 alone and to unventilated PPHN fetal lambs (Fig. 2, B and C).
NO-induced mitochondrial biogenesis improves cellular respiration in PPHN-PAECs.
We recently reported that PPHN-PAECs have lower ATP/higher ADP levels, suggesting uncoupling of respiration and ATP synthesis (2). We tested the impact of NO-induced mitochondrial biogenesis on cellular respiration by measuring oxygen consumption and utilization for ATP synthesis in PPHN-PAECs. Previous studies reported that 1 μM detaNONOate releases 4 nM NO in the medium with a half-life of 22 h (4). Our data show that addition of 200 nM detaNONOate releases ∼3 nM of NO over 18 h consistently into the culture medium, which will then be available to the PAECs (Fig. 3A). At baseline, basal oxygen consumption rate and extra-mitochondrial O2 consumption rate were higher in PPHN-PAECs compared with normotensive PAECs (control). In contrast, ATP-linked O2 consumption rate was lower in PPHN-PAECs. In normotensive PAECs, treatment with detaNONOate for 18 h decreased the mitochondrial biogenesis and ATP synthesis (data not shown). In contrast, treatment with detaNONOate induced an increase in ATP-linked O2 consumption, which was associated with a decrease in OCR, extra-mitochondrial O2 consumption and proton leak in PPHN-PAECs (Fig. 3B). These data suggest that PPHN is associated with decreased mitochondrial biogenesis, uncoupling of respiration, and decreased ATP synthesis. These changes were reversed by treatment with an NO donor.
Fig. 3.

DetaNONOate increased the coupling of respiration and oxidative phosphorylation in PPHN-pulmonary artery endothelial cells (PAECs). In PPHN-PAECs, the basal O2 consumption rate (OCR), proton leak, as well as extra-mitochondrial oxygen consumption rate were higher compared with normotensive PAECs (controls), but the ATP-linked O2 consumption was lower in PPHN-PAECs. Treatment with 200 nM detaNONOate, an NO donor, for 18 h released ∼3 nM NO (Fig. 2A) and decreased OCR, proton leak, and extra-mitochondrial O2 consumption rates in PPHN-PAECs, while specifically increasing the ATP-linked OCR in PPHN-PAECs (n = 6, P < 0.05). #P < 0.05, from normal PAECs. *P < 0.05, from untreated PPHN-PAECs.
iNO and DetaNONOate improve redox balance in PPHN-PAECs.
Previous studies have shown that PGC-1α overexpression upregulates mitochondrial biogenesis and SOD2 function in cultured neurons. In this study, we observed that iNO increased SOD2 mRNA and protein and SOD2-specific activity in the lungs of PPHN fetal lambs compared with unventilated PPHN fetal lambs (Fig. 4, A–C). In contrast, ventilation with 100% O2 alone had no significant impact on SOD2 expression and activity when compared with nonventilated PPHN fetal lambs. NO-induced increase in oxidative capacity and ATP synthesis could potentially lead to increased mitochondrial O2− levels as a by-product of oxidative phosphorylation. We therefore, investigated the effects of NO on mitochondrial O2− levels. Treatment for 18 h with detaNONOate decreased MitoSOX fluorescence intensity in PPHN-PAECs when compared with untreated PPHN-PAECs (Fig. 5).
Fig. 4.

iNO improves mitochondrial Redox balance in PPHN. SOD2 protein and mRNA levels were unchanged in the lungs of PPHN fetal lambs, whereas SOD2 activity was significantly decreased in PPHN compared with the age-matched normal fetal lambs. Ventilation with 100% O2 alone further decreased SOD2 mRNA, protein, and activity levels in PPHN fetal lambs. Treatment with iNO increased SOD2 mRNA, protein, and activity levels compared with PPHN fetal lambs ventilated with 100% O2 alone or unventilated PPHN lambs (n = 6, P < 0.05). #P < 0.05, from control lambs. *P < 0.05, from PPHN fetal lambs ventilated with 100% O2 alone.
Fig. 5.
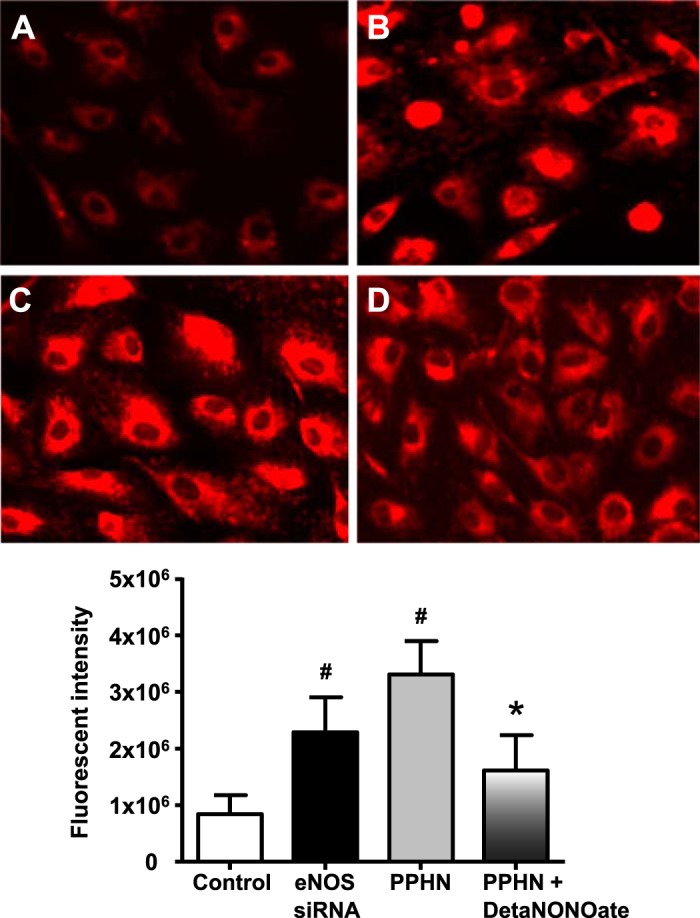
DetaNONOate decreased mitochondrial oxidative stress in PPHN-PAECs. Representative micrograph showing MitoSOX fluorescence in control PAECs (A), eNOS knockdown of control PAECs (B), PPHN-PAECs (C), and detaNONOate-treated PPHN-PAECs (D). MitoSOX fluorescence intensity was increased in eNOS knockdown control PAECs and in PPHN-PAECs compared with controls. DetaNONOate significantly decreased MitoSOX fluorescence in PPHN-PAECs compared with untreated PPHN-PAECs (n = 4, P < 0.05). #P < 0.05, from control PAECs. *P < 0.05, from eNOS knockdown and from untreated PPHN-PAECs.
Role of eNOS and NO in mitochondrial biogenesis and redox regulation.
Knockdown of eNOS in PAECs from control fetal lambs decreased eNOS protein level by ∼80% and markedly reduced ETC subunits and SOD2 expression compared with nonsilencing RNA transfected cells (Fig. 6, A and B). Knockdown of eNOS in control cells also increased MitoSOX fluorescence intensity compared with controls (Fig. 5B). This indicates the critical role of eNOS in the regulation of mitochondrial redox balance and function (Fig. 5). Upregulation of SOD2 function may help attenuate the mitochondrial O2− from NO-induced increase in oxidative phosphorylation.
Fig. 6.

Endothelial nitric oxide synthase (eNOS) and NO regulate mitochondrial biogenesis and Redox balance in the PAECs: siRNA-mediated eNOS knockdown decreased SOD2 protein level (A) and electron transport chain (ETC) complexes I–V (B) in control PAECs compared with nonsilencing (NS) siRNA (n = 12, P < 0.05). *P < 0.05 from nonsilencing siRNA (controls).
iNO restores eNOS function in PPHN fetal lambs.
Decreased NO bioavailability is a key factor in the development of PPHN (40). Previous studies have shown that impaired eNOS interactions with HSP90 increased O2− formation instead of NO (16, 20). Consistent with these reports, we observed a decrease in eNOS mRNA, protein, HSP90/eNOS ratio, and serine1179 phosphorylated eNOS in the unventilated PPHN lung samples (Fig. 7, A–D). Ventilation of PPHN lambs with 100% O2 led to a twofold increase in eNOS mRNA and no increases in eNOS protein levels, HSP90/eNOS ratio, and serine1179 phosphorylation compared with unventilated PPHN fetal lambs (Fig. 7, A–D). Use of iNO with weaning of FiO2 was associated with an eightfold increase in eNOS mRNA, threefold increase in protein levels, and increases in eNOS-HSP90 association and serine1179 phosphorylation, compared with ventilation with 100% O2 alone (Fig. 7, A–D). These data demonstrate that administration of iNO and weaning of FiO2 is associated with conditions that favor a coupled function of eNOS in the lung.
Fig. 7.

iNO increased endothelial nitric oxide synthase (eNOS) expression in PPHN fetal lambs. eNOS mRNA (A) and protein levels (B) were decreased in PPHN lungs. Ventilation of PPHN fetal lambs with 100% oxygen increased eNOS mRNA, but not protein levels. iNO given with the weaning of FiO2 increased both eNOS mRNA and protein levels (A and B) (n = 4, P < 0.05). Heat shock protein 90 (HSP90)-eNOS association (C) and serine1179 phosphorylation of eNOS (D) were decreased in the lungs of PPHN lambs compared with gestation-matched control lambs and did not change further during ventilation with 100% O2. Addition of iNO was associated with increases in HSP90/eNOS association (C) and levels of Phos-serine1177 eNOS in PPHN lambs (D) (n = 4, P < 0.05). #P < 0.05, from control. ΩP < 0.05, from unventilated PPHN lungs. *P < 0.05, from ventilation with 100% oxygen.
DISCUSSION
Our studies demonstrate that mitochondrial biogenesis and ATP synthesis are impaired in PPHN. The decrease in ATP level is associated with decreased eNOS activity and impaired pulmonary vasodilation, which was previously reported in PPHN (2, 35). This finding may be central to the metabolic alterations in PPHN and may have important therapeutic implications. Ventilation with high FiO2 exacerbated the mitochondrial metabolic abnormalities in PPHN. We observed that iNO treatment, which allowed decreased FiO2 use, restored mitochondrial biogenesis and redox balance in PPHN. These data suggest that the mitochondrial redox state is a critical part of the complex adaptive responses during postnatal transition.
PPHN represents a failure of normal cardiopulmonary adaption at birth (10, 28). The ductal constriction model of PPHN simulates a known mechanism for PPHN and also reproduces the hemodynamic and structural alterations observed in neonates with PPHN (23). PPHN is frequently associated with parenchymal lung diseases such as meconium aspiration, sepsis, and respiratory distress syndrome, causing ventilation-perfusion (V/Q) mismatch and severe hypoxemia (9, 10). Use of high inspired oxygen concentrations in the affected infants may worsen the pulmonary vascular dysfunction (23). Although O2 is known to stimulate eNOS activity and NO production (18), mechanical ventilation and hyperoxia increase ROS formation in a synergistic and cumulative manner. We observed that decreases in mitochondrial ETC subunit proteins in PPHN were associated with an increase in both mitochondrial and extra-mitochondrial oxygen consumption in PPHN. NO regulates mitochondrial oxygen consumption via reversible inhibition of cytochrome c oxidase (complex IV) enzyme (27). Our studies suggest that decreased NO levels led to a loss of this inhibitory effect of NO on cellular oxygen consumption in PPHN. Furthermore, in PPHN, the ETC subunit proteins may be dysfunctional, which may lead to increased electron leak during electron transport in oxidative phosphorylation. This can lead to an increase in superoxide anion formation inside mitochondria in PPHN-PAECs. The observed decrease in oxidative phosphorylation, coupled with increases in mitochondrial O2− levels in PPHN (2), suggests a potential increase in oxidative stress during exposure to hyperoxia in PPHN. Since most infants with PPHN are not diagnosed prenatally, initiation of iNO therapy is often delayed. Our data demonstrate that ventilation with 100% O2 worsens the mitochondrial function in PPHN. This may lead to poor response to iNO and worsening of the vascular dysfunction. These observations suggest a potential mechanism for a lack of response to iNO in some PPHN infants.
Inhaled NO use led to improved oxygenation and reduced inspired oxygen concentrations and restored the mitochondrial biogenesis and Redox balance in ventilated PPHN lambs. The relative contribution of iNO and decreased exposure to O2 in restoring mitochondrial biogenesis is not clear from our study. Previous studies have shown that NO can decrease DNA damage in yeast caused by ROS (30a). Randomized controlled trials have shown that earlier initiation of iNO therapy decreases extracorporeal membrane oxygenation rates in infants with PPHN (12). Our data on decreased ETC protein subunits and SOD2 expression and function following eNOS knock down support the role of eNOS in the regulation of mitochondrial biogenesis and Redox homeostasis. Our findings are also consistent with previous observations in eNOS−/− mice, which demonstrated that endogenous NO regulates mitochondrial biogenesis in vivo (11).
We observed that iNO increased mitochondrial oxidative capacity and ATP synthesis in PPHN. In addition to increasing eNOS activity, ATP plays important roles in facilitating transport of substances across cell membranes. We reported previously that inducible hsp70 chaperones SOD2 to the mitochondria after synthesis in the cytosol via ATP-dependent mechanisms (2). ATP increases SOD2 interactions with HSP70 and facilitates SOD2 import to the mitochondria, where it is activated and scavenges O2−. Inhaled NO increased SOD2 at both transcriptional and posttranslational levels. An increase in mitochondrial biogenesis from NO exposure can increase ATP synthesis by oxidative phosphorylation. In the absence of iNO, it was difficult to wean the FiO2 from 1.0 while maintaining PaO2 in the target range in our PPHN lambs (Table 1). iNO use allowed us to wean the FiO2 and mean airway pressure, which also potentially contributed to favorable effects on mitochondrial redox state and biogenesis.
PGC1α appears to play a critical role in iNO-induced mitochondrial biogenesis and redox balance. We observed decreases in the levels of AMPKα, Sirt-1, pAMPKα, and eNOS proteins when PPHN fetal lambs were exposed to mechanical ventilation with hyperoxia. Expression of PGC1α and mitochondrial number and function were also decreased during ventilation with 100% O2. Increasing NO levels either by treatment with detaNONOate in cultured PAECs or iNO therapy in lambs increased PGC1α levels and mitochondrial biogenesis. We speculate that increased ROS formation in hyperoxia contributes to depletion of NO, thereby attenuating PGC1α.
iNO treatment restored eNOS expression and activity in PPHN. Previous studies have shown that eNOS expression and activity and eNOS-HSP90 interaction are decreased in PPHN. (16, 21). We observed that iNO treatment in ventilated PPHN fetal lambs increased eNOS mRNA and protein levels, serine1179 phosphorylation and eNOS interactions with HSP90. To verify that these effects are due to NO, we investigated the effect of the NO donor detaNONOate on endothelial cells from PPHN lambs. These studies demonstrated that NO donor alone can restore eNOS expression and function in PAECs, consistent with the in vivo effects observed in ventilated lambs. Our data suggest that NO has an independent effect in improving eNOS function and mitochondrial biogenesis.
There are some limitations to this study. We cannot determine whether ventilation with iNO, weaning to lower inspired oxygen concentrations, or reduced volume and barotrauma from weaning MAP or a combination of the three factors resulted in the observed beneficial effects on mitochondrial biogenesis and eNOS activity. Our attempts to ventilate PPHN lambs with lower levels of inspired oxygen without iNO led to poor survival due to severe hypoxemia (data not shown). We have previously shown that ventilation with iNO and 100% oxygen increases eNOS expression by twofold in PPHN lambs (21). In the current study, we demonstrate that iNO and weaning inspired oxygen increase eNOS expression fourfold. We speculate that iNO and weaning inspired oxygen may have an additive effect on eNOS expression. Another limitation to our study is that we did not measure pulmonary blood flow and pulmonary arterial pressures in our ventilated lambs. Therefore, we are unable to verify whether increased mitochondrial biogenesis in iNO-treated lambs is associated with improved pulmonary vasodilation. A thoracotomy and probe placement for monitoring pulmonary hemodynamics significantly increased morbidity leading to increased mortality in PPHN lambs after 6–12 h of ventilation.
In conclusion, PPHN is associated with impaired mitochondrial biogenesis and function and decreased ATP synthesis in pulmonary arteries in our fetal lamb model. Ventilation with 100% O2 may worsen this vascular dysfunction in PPHN. Administration of iNO allowed for a rapid wean of FiO2 and MAP. This strategy restored mitochondrial biogenesis and improved redox balance. Further studies are needed to determine whether early initiation of iNO therapy in patients with PPHN restores vascular function and improves vasodilation and metabolic balance in the affected infants.
GRANTS
This work was supported by National Institutes of Health Grants R03-HD-065841 and R01-HL-057268 (to G. G. Konduri) and 5R01-HD-072929 (to S Lakshminrusimha), Muma Endowed Chair in Neonatology (to G. G. Konduri), and an investigator initiated grant from Ikaria, LLC (to S. Lakshminrusimha).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
A.J.A. and G.G.K. conception and design of research; A.J.A., A.E., M.A., T.M., and S.L. performed experiments; A.J.A. and T.M. analyzed data; A.J.A., A.E., and R.-J.T. interpreted results of experiments; A.J.A. and M.A. prepared figures; A.J.A. drafted manuscript; A.J.A., R.-J.T., S.L., and G.G.K. edited and revised manuscript; A.J.A., S.L., and G.G.K. approved final version of manuscript.
REFERENCES
- 1.Afolayan AJ, Eis A, Teng RJ, Bakhutashvili I, Kaul S, Davis JM, Konduri GG. Decreases in manganese superoxide dismutase expression and activity contribute to oxidative stress in persistent pulmonary hypertension of the newborn. Am J Physiol Lung Cell Mol Physiol 303: L870–L879, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Afolayan AJ, Teng RJ, Eis A, Rana U, Broniowska KA, Corbett JA, Pritchard K, Konduri GG. Inducible HSP70 regulates superoxide dismutase-2 and mitochondrial oxidative stress in the endothelial cells from developing lungs. Am J Physiol Lung Cell Mol Physiol 306: L351–L360, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Austin S, St-Pierre J. PGC1a and mitochondrial metabolism–emerging concepts and relevance in ageing and neurodegenerative disorders. J Cell Sci 125: 4963–4971, 2012. [DOI] [PubMed] [Google Scholar]
- 4.Bebok Z, Varga K, Hicks JK, Venglarik CJ, Kovacs T, Chen L, Hardiman KM, Collawn JF, Sorscher EJ, Matalon S. Reactive oxygen nitrogen species decrease cystic fibrosis transmembrane conductance regulator expression and camp-mediated Cl− secretion in airway epithelia. J Biol Chem 277: 43041–43049, 2002. [DOI] [PubMed] [Google Scholar]
- 5.Beckman JS, Koppenol WH. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and the ugly. Am J Physiol Cell Physiol 271: C1424–C1437, 1996. [DOI] [PubMed] [Google Scholar]
- 6.Bergeron R, Ren JM, Cadman KS, Moore IK, Perret P, Pypaert M, Young LH, Semenkovich CF, Shulman GI. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am J Physiol Endocrinol Metab 281: E1340–E1346, 2001. [DOI] [PubMed] [Google Scholar]
- 7.Canto C, Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol 20: 98–105, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dromparis P, Michelakis ED. Mitochondria in vascular health and disease. Annu Rev Physiol 75: 95–126, 2013. [DOI] [PubMed] [Google Scholar]
- 9.Fanaroff AA. Meconium aspiration syndrome: historical aspects. J Perinatol 28: S3–S7, 2008. [DOI] [PubMed] [Google Scholar]
- 10.Fox WW, Gewitz MH, Dinwiddie R, Drummond WH, Peckham GJ. Pulmonary hypertension in perinatal aspiration syndromes. Pediatrics 59: 205–211, 1977. [PubMed] [Google Scholar]
- 11.Gesing A, Bartke A, Wang F, Karbownik-Lewinska M, Masternak MM. Key regulators of mitochondrial biogenesis are increased in kidneys of growth hormone receptor knockout (GHRKO) mice. Cell Biochem Funct 29: 459–467, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.González A, Fabres J, D′Apremont I, Urcelay G, Avaca M, Gandolfi C, Kattan J. Randomized controlled trial of early compared with delayed use of inhaled nitric oxide in newborns with a moderate respiratory failure and pulmonary hypertension. J Perinatol 30: 420–424, 2010. [DOI] [PubMed] [Google Scholar]
- 13.Handschin C, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev 27: 728–735, 2006. [DOI] [PubMed] [Google Scholar]
- 14.Hirata Y, Satonaka H. Hypertension and oxidative stress. JAMA 4: 540–545, 2001. [Google Scholar]
- 15.Kelly DP, Scarpulla R. Since NO is the upstream regulator of mitochondrial biogenesis, C. transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev 18: 357–368, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Konduri GG, Ou JS, Shi Y, Pritchard KA Jr. Decreased association of HSP90 impairs endothelial nitric oxide synthase in fetal lambs with persistent pulmonary hypertension. Am J Physiol Heart Circ Physiol 285: H204–H211, 2003. [DOI] [PubMed] [Google Scholar]
- 17.Konduri GG, Sokol GM, Van Meurs KP, Singer J, Ambalavanan N, Lee T, Solimano A. Impact of early surfactant and inhaled nitric oxide therapies on outcomes in term/late preterm neonates with moderate hypoxic respiratory failure. J Perinatol 33: 944–949, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Konduri GG, Mital S. Adenosine and ATP cause nitric oxide dependent pulmonary vasodilation in fetal lambs. Biol Neonate 78: 220–229, 2000. [DOI] [PubMed] [Google Scholar]
- 19.Konduri GG, Mattei J. Role of oxidative phosphorylation and ATP release in mediating birth related pulmonary vasodilation in fetal lambs. Am J Physiol Heart Circ Physiol 283: H1600–H1608, 2002. [DOI] [PubMed] [Google Scholar]
- 20.Konduri GG, Solimano A, Sokol GM, Singer J, Ehrenkranz RA, Singhal N, Wright LL, Van Meurs K, Stork E, Kirpalani H, Peliowski A; Neonatal Inhaled Nitric Oxide Study Group. A randomized trial of early versus standard inhaled nitric oxide therapy in term and near-term newborn infant with hypoxic respiratory failure. Pediatrics 113: 559–564, 2004. [DOI] [PubMed] [Google Scholar]
- 21.Konduri GG, Bakhutashvili I, Eis A, Pritchard K Jr. Oxidant stress from uncoupled nitric oxide synthase impairs vasodilation in fetal lambs with persistent pulmonary hypertension. Am J Physiol Heart Circ Physiol 292: H1812–H1820, 2007. [DOI] [PubMed] [Google Scholar]
- 22.Lakshminrusimha S, Swartz D, Gugino SF, Ma CX, Wynn KA, Ryan RM, Russell JA, Steinhorn RH. Oxygen concentration and pulmonary hemodynamics in newborn lambs with pulmonary hypertension. Pediatr Res 66: 539–544, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lakshminrusimha S, Russell JA, Steinhorn RH, Ryan RM, Gugino SF, Morin FC, Swartz DD, Kumar VH. Pulmonary arterial contractility in neonatal lambs increases with 100% oxygen resuscitation. Pediatr Res 59: 137–141, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lévy M, Maurey C, Dinh-Xuan AT, Vouhé P, Israël-Biet D. Developmental expression of vasoactive, and growth factors in human lung. role in pulmonary vascular resistance adaptation at birth. Pediatr Res 57: 21R–25R, 2005. [DOI] [PubMed] [Google Scholar]
- 26.Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1alpha. J Biol Chem 280: 16456–16460, 2005. [DOI] [PubMed] [Google Scholar]
- 27.Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valerio A, Francolini M, Moncada S, Carruba MO. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science 299: 896–899, 2003. [DOI] [PubMed] [Google Scholar]
- 28.Papamatheakis DG, Chundu M, Blood AB, Wilson SM. Prenatal programming of pulmonary hypertension induced by chronic hypoxia or ductal ligation in sheep. Pulmon Circ 3: 757–780, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Puigserver P. Tissue-specific regulation of metabolic pathways through the transcriptional coactivator PGC1-alpha. Int J Obes (Lond) 29, Suppl 1: S5–9, 2005. [DOI] [PubMed] [Google Scholar]
- 30.Reznick RM, Shulman GI. The role of AMP-activated protein kinase in mitochondrial biogenesis. J Physiol 574: 33–39, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30a.Rowe LA, Degtyareva RN, Doetsch PW. DNA damage-induced reactive oxygen species (ROS) stress response in Saccharomyces cerevisiae. Free Radic Biol Med 45: 1167–1177, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scarpulla RC. Transcriptional activators and coactivators in the nuclear control of mitochondrial function in mammalian cells. Gene 286: 81–89, 2002. [DOI] [PubMed] [Google Scholar]
- 32.Scarpulla RC. Nuclear activators and coactivators in mammalian mitochondrial biogenesis. Biochim Biophys Acta 1576: 1–14, 2002. [DOI] [PubMed] [Google Scholar]
- 33.Schreiber SN, Emter R, Hock MB, Knutti D, Cardenas J, Podvinec M, Oakeley EJ, Kralli A. The estrogen-related receptor alpha (ERR alpha) functions in PPAR gamma co-activator 1alpha (PGC-1alpha)-induced mitochondrial biogenesis. Proc Natl Acad Sci USA 101: 6472- 6477, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33a.Sessa WC. Regulation of endothelial derived nitric oxide in health and disease. Mem Inst Oswaldo Cruz 100, Suppl 1: 15–18, 2005. [DOI] [PubMed] [Google Scholar]
- 34.Shaul PW, Afshar S, Gibson LL, Sherman TS, Kerecman JD, Grubb PH, Yoder BA, McCurnin DC. Devlopmental changes in nitric oxide synthase isoform expression, and nitric oxide production in fetal baboon lung. Am J Physiol Lung Cell Mol Physiol 283: L1192–L1199, 2002. [DOI] [PubMed] [Google Scholar]
- 35.Smeitink J, van den Heuvel L, DiMauro S. The genetics and pathology of oxidative phosphorylation. Nat Rev Genet 2: 342–352, 2001. [DOI] [PubMed] [Google Scholar]
- 36.Steinhorn RH, Wedgwood S, Russell JA, Gugino SF, Lakshminrusimha S, Black S. Increased production of reactive oxygen species in a lamb model of persistent pulmonary hypertension (Abstract). Pediatr Res 49: 353A, 2001. [Google Scholar]
- 37.Takahashi S, Mendelsohn ME. Synergistic activation of endothelial nitric-oxide synthase (eNOS) by HSP90 and Akt. J Biol Chem 278: 30821–30827, 2003. [DOI] [PubMed] [Google Scholar]
- 38.Teng RJ, Du J, Afolayan AJ, Eis A, Shi Y, Konduri GG. AMP kinase activation improves angiogenesis in pulmonary artery endothelial cells with in utero pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 304: L29–L42, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Teng RJ, Du J, Xu H, Bakhutashvili I, Eis A, Shi Y, Pritchard KA Jr, Konduri GG. Sepiapterin improves angiogenesis of pulmonary artery endothelial cells with in utero pulmonary hypertension by recoupling endothelial nitric oxide synthase. Am J Physiol Lung Cell Mol Physiol 301: L334–L345, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.The Franco-Belgium Collaborative NO Trial Group. Early compared with delayed inhaled nitric oxide in moderately hypoxemic neonates with respiratory failure: a randomized controlled trial. Lancet 354: 1066–1071, 1999. [PubMed] [Google Scholar]
- 41.UK Collaborative ECMO Trial Group. UK collaborative randomized trial of neonatal extracorporeal membrane oxygenation. Lancet 348: 75–82, 1996. [PubMed] [Google Scholar]