

Abstract
Cancer cachexia is characterized by the progressive loss of skeletal muscle mass. While mouse skeletal muscle's response to an acute bout of stimulated low-frequency concentric muscle contractions is disrupted by cachexia, gaps remain in our understanding of cachexia's effects on eccentric contraction-induced muscle growth. The purpose of this study was to determine whether repeated bouts of stimulated high-frequency eccentric muscle contractions [high-frequency electrical muscle stimulation (HFES)] could stimulate myofiber growth during cancer cachexia progression, and whether this training disrupted muscle signaling associated with wasting. Male ApcMin/+ mice initiating cachexia (N = 9) performed seven bouts of HFES-induced eccentric contractions of the left tibialis anterior muscle over 2 wk. The right tibialis anterior served as the control, and mice were killed 48 h after the last stimulation. Age-matched C57BL/6 mice (N = 9) served as wild-type controls. ApcMin/+ mice lost body weight, muscle mass, and type IIA, IIX, and IIB myofiber cross-sectional area. HFES increased myofiber cross-sectional area of all fiber types, regardless of cachexia. Cachexia increased muscle noncontractile tissue, which was attenuated by HFES. Cachexia decreased the percentage of high succinate dehydrogenase activity myofibers, which was increased by HFES, regardless of cachexia. While cachexia activated AMP kinase, STAT3, and ERK1/2 signaling, HFES decreased AMP kinase phosphorylation, independent of the suppression of STAT3. These results demonstrate that cachectic skeletal muscle can initiate a growth response to repeated eccentric muscle contractions, despite the presence of a systemic cachectic environment.
Keywords: cancer cachexia, high-frequency electrical stimulation, muscle inflammation, myofiber growth, eccentric contractions
cancer cachexia is a wasting syndrome, characterized by the loss of skeletal muscle (53), and contributes to increased patient morbidity and mortality (55). Although many pharmacological therapeutics for treating cancer-induced muscle wasting have been proposed, and some tested in clinical trials, most have failed due to their singular specificity and adverse side effects (13). Exercise is a potential nonpharmacological treatment that improves indices of health related to muscle and systemic function in healthy individuals and also those with chronic disease (26, 63). While exercise has been widely discussed for therapeutic use in cancer patients, the mechanistic effect of exercise on wasting processes is currently being determined (18). Additionally, there are gaps in our understanding of how the cancer environment alters skeletal muscle's response to contraction, and the mechanistic basis of these effects are being examined in mouse models of cancer cachexia.
Treadmill exercise training in ApcMin/+ mice, a model of cancer cachexia, can prevent interleukin-6 (IL-6)-induced muscle mass loss (49, 59). Interestingly, these exercise-induced improvements in muscle mass and metabolic function do not require a reduction in muscle inflammatory signaling (49). Others have reported treadmill exercise, alone or in combination with nutritional provision, can reduce tumor growth and improve muscle mass in tumor-bearing rodents (14, 45, 50). Exercise feasibility is an issue with more severe cachexia, and cachectic mice are not capable of performing vigorous treadmill exercise training. While not entirely exercise, skeletal muscle's metabolic and growth response to contraction has been examined through the use of electrical muscle stimulation (4, 40, 48, 60). Low-frequency electrical muscle stimulation (LFES) examines acute and training adaptations to endurance-like muscle contractions (40, 44, 48). Cancer cachexia can disrupt acute LFES concentric contraction-induced metabolic signaling (48). In contrast, eccentric contractions induced by high-frequency electrical muscle stimulation (HFES) examine muscle signaling associated with hypertrophy (4, 12, 40, 60). Related to cancer cachexia, repeated bouts of eccentric contraction started at the time of C26 tumor implantation can prevent mouse extensor digitorum longus (EDL) muscle protein loss (2). The effect of cancer cachexia on eccentric contraction-induced growth signaling is not known. Additionally, it remains to be determined whether repeated eccentric contractions can induce myofiber hypertrophy after cachexia has been initiated and the cachectic environment is present.
It is well established that muscle signaling pathways related to inflammation, energy status, and proteolysis are disrupted with cachexia progression and have regulatory roles in the wasting process (16, 41). Many of these same pathways can be regulated by both acute and repeated bouts of exercise (15). For example, IL-6 and subsequent muscle signal transducer and activator of transcription 3 (STAT3) signaling through the gp130 receptor is elevated during the progression of cachexia (5, 9, 10, 56), and either STAT3 inhibition (9) or gp130 loss can attenuate muscle wasting in some mouse models of cachexia (47). Muscle IL-6 mRNA expression is transiently induced by acute exercise (28), and treadmill exercise disrupts IL-6-induced regulation of muscle mass loss in ApcMin/+ mice (49, 59). During the progression of cancer cachexia, muscle STAT3 signaling through the gp130 receptor can regulate the cellular energy-sensing enzyme 5′-adenosine monophosphate-activated protein kinase (AMPK), which is chronically activated in severely cachectic muscle (56). AMPK activation can regulate muscle protein turnover through mammalian (or the mechanistic) target of rapamycin (mTOR) inhibition (8), increased muscle-specific ubiquitin ligase expression (54), and autophagy processes (29). While AMPK activation can also promote mitochondrial biogenesis, chronic AMPK activation accompanies reduced mitochondrial biogenesis and content in severe cachexia (49, 56, 59). Additionally, improved metabolic function with treadmill exercise training during the initiation of cancer cachexia was accompanied by reduced AMPK activation (49). Related to muscle hypertrophy, AMPK activation can attenuate overload-induced muscle growth (39) and eccentric contraction-induced mTOR signaling (52). While multiple bouts of eccentric muscle contractions can increase muscle mass and protein content in tumor-bearing mice (2), whether repeated bouts of HFES can result in muscle hypertrophy in a microenvironment that possesses chronically elevated STAT3 and AMPK signaling is not known. Moreover, the effect of HFES-induced eccentric muscle contractions on myofiber hypertrophy in healthy or cachectic mice has yet to be established.
While initial evidence suggests repeated eccentric contractions can prevent EDL muscle protein loss in tumor-bearing mice (2), further work is needed to determine whether the development of cancer cachexia impacts muscle morphology and signaling related to myofiber growth normally induced by eccentric contractions. To our knowledge, whether cachectic muscle can initiate a growth response to repeated eccentric contractions in different fiber types after the initiation of cachexia is not known. The purpose of this study was to determine whether repeated bouts of stimulated high-frequency eccentric muscle contraction could stimulate myofiber growth during the progression of cachexia, and whether this training disrupted muscle signaling associated with the regulation of wasting processes. In addition, we determined the effect of eccentric contractions on morphological indices of muscle remodeling and oxidative metabolism during the progression of cachexia. It was hypothesized that eccentric muscle contractions would stimulate myofiber growth in all fiber types during the progression of cachexia. To test this hypothesis, ApcMin/+ mice that had initiated cachexia performed seven bouts of HFES, which elicit eccentric muscle contractions of the tibialis anterior (TA) muscle. Muscle morphology related to myofiber growth and muscle remodeling was examined in the TA muscle. In addition, muscle cachectic signaling pathways associated with wasting were examined. The results demonstrate that cachectic muscle can initiate myofiber growth in response to repeated bouts of eccentric muscle contraction, despite the presence of the cachectic environment.
MATERIALS AND METHODS
Animals.
The ApcMin/+ mouse is a genetic model of colorectal cancer and cachexia (5, 36). These mice harbor a heterozygous mutation in the adenomatous polyposis coli (Apc) gene, which promotes the development of intestinal tumors, beginning as early as 4 wk of age (38). Mice develop an IL-6-dependent cancer cachexia phenotype between 3 and 6 mo of age (5, 36). Due to the slow onset and progression of body weight loss, this model is advantageous as treatments can be started after the initiation of cancer cachexia. Male ApcMin/+ mice on a C57BL/6 background were originally purchased from Jackson Laboratories (Bar Harbor, ME) and bred at the University of South Carolina's Animal Resource Facility. All mice used in the present study were obtained from the investigator's breeding colony within the Center for Colon Cancer Research Mouse Core. Mice were individually housed, kept on a 12:12-h light-dark cycle, and had access to standard rodent chow (no. 8604 Rodent Diet; Harlan Teklad, Madison, WI) and water ad libitum. Body weight measurements were taken weekly, and the percentage of body weight loss from peak body weight (∼10-14 wk of age) was calculated. Mice lacking the ApcMin/+ mutation (C57BL/6) served as controls for all experiments. The University of South Carolina's Institutional Animal Care and Use Committee approved all animal experimentation in this study.
HFES.
At 16 wk of age C57BL/6 and ApcMin/+ mice (N = 9/group) were subjected to multiple bouts of HFES over a period of 2 wk (Fig. 1A). HFES of the left hindlimb was performed as previously described, with slight modifications (4). During each stimulation procedure, mice were anesthetized via isoflurane (2% in O2 with 1.5% maintenance), the left leg/hip region was shaved, and two needle electrodes were placed on the left leg subcutaneously posterior to the femur to stimulate the sciatic nerve. Tetanic muscle contractions were generated using a Grass Stimulator (Grass Instruments, Quincy, MA) for 10 sets of 6 repetitions (100 Hz, 6–12 V, 1-ms duration, 3-s repetition duration). Ten seconds of rest were given between repetitions, and 50 s of rest were given between sets. The stimulation protocol recruits all motor units of the hindlimb. The maximal force production of the plantar flexors (gastrocnemius, soleus, and plantaris) are greater than the dorsiflexors (TA and EDL) (61, 62), which results in net plantar flexion of the ankle (4). Therefore, the dorsiflexors undergo lengthening eccentric muscle contractions against the shortening concentric plantar flexors. Each session lasted ∼22 min in duration. Following each stimulation procedure, mice were given an intraperitoneal injection of warm saline and returned to cages upon complete recovery. Mice performed a total of seven stimulation sessions, with each stimulation separated by 48 h. Mice were killed 48 h after the last stimulation procedure to minimize the effects of acute muscle contraction and were fasted 5 h before death but had free access to water ad libitum.
Fig. 1.
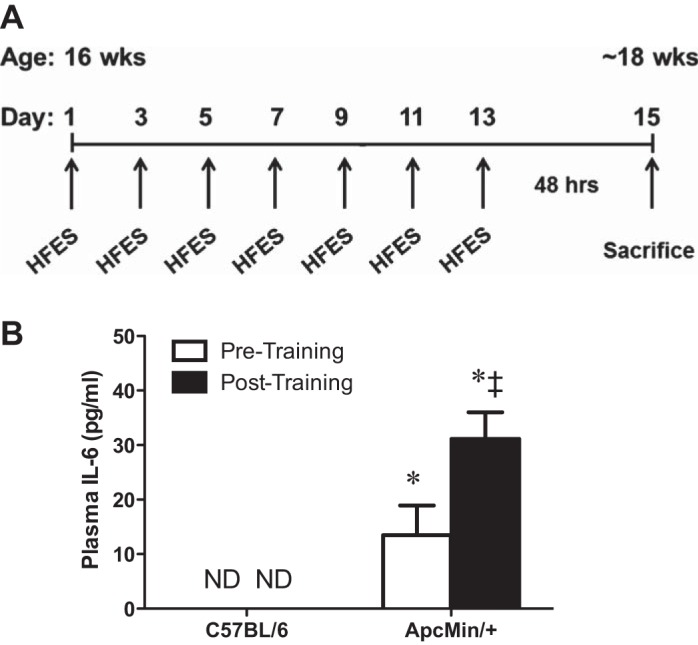
Effects of multiple bouts of high-frequency electrical stimulation (HFES) on circulating interleukin (IL)-6 in C57BL/6 and ApcMin/+ mice. A: experimental design. At 16 wk of age, male C57BL/6 and ApcMin/+ mice performed multiple bouts of HFES. Each stimulation procedure was separated by 48 h, and mice were killed 48 h after the last stimulation. Mice were fasted 5 h before death. B: plasma IL-6 levels pre- and posttraining. Values are means ± SE. Statistical significance was set at P < 0.05: difference between *C57BL/6 and ApcMin/+ mice, and ‡pre- and posttraining. ND, not detectable.
Tissue collection.
Mice were anesthetized with a subcutaneous injection of ketamine-xylazine-acepromazine cocktail (1.4 ml/kg body wt) at the time of death. Hindlimb muscles were rapidly excised, cleared of excess connective tissue, rinsed in PBS, weighed, and snap frozen in liquid nitrogen. The TA muscle was cut at the midbelly, placed in optimum cutting temperature compound, and frozen in liquid nitrogen. Blood was collected before muscle collection via retro-orbital eye bleed with heparinized capillary tubes, placed on ice, and centrifuged (10,000 g for 10 min at 4°C). The supernatant was removed and stored for plasma IL-6 analysis. Plasma and tissue samples were stored at −80°C until further analysis.
TA morphology.
Transverse muscle sections (∼10 μm) were cut from the midbelly of the TA on a cryostat at −20°C and stored at −80°C until further analysis. Hematoxylin and eosin (H&E) staining was performed to examine muscle morphology, as previously described (7, 34, 36). H&E-stained muscle sections were digitized and analyzed using ImageJ imaging software [National Institutes of Health (NIH), Bethesda, MD]. Deep and superficial regions of the TA muscle were included in all analysis performed (46). Therefore, at least 15 random non-overlapping digital images at ×25 magnification were taken from each H&E-stained muscle section per animal and were examined for centralized nuclei, as previously described (7). Centralized nuclei were defined as nuclei found equidistant from a well-defined sarcolemma and are expressed as the percentage of centralized nuclei per total number of myofibers (>900 myofibers were examined per animal). The noncontractile tissue area was examined on digital images (N = 15/animal) of H&E-stained muscle sections at ×40 magnification, as previously described (34, 36). The percentage of noncontractile tissue was determined by overlaying an 18 × 14 digital pixel grid and counting the number of pixels associated with the myofiber or extracellular matrix. Pixels that were clearly distinguishable and at least 75% in the extracellular matrix were counted. The total number of pixels associated with the extracellular matrix was divided by the total number of pixels to give the percentage noncontractile tissue. All images were coded, and analyses were performed by a researcher blinded to the treatment groups.
Immunohistochemistry for myosin heavy chain IIA, IIX, and IIB.
Immunohistochemistry for myosin heavy chain (MHC) type IIA, IIX, and IIB was performed as previously described (20). Transverse muscle sections of the TA were air dried for 10 min, fixed in cold acetone for 1 min, and washed in PBS for 5 min. Sections were quenched in 0.3% H2O2-methanol solution for 20 min and rinsed in PBS three times for 5 min. Sections were blocked in 10% normal goat serum (Vectastain ABC kit, Vector Laboratories, Burlingame, CA) in PBS for 1 h at room temperature and then incubated overnight at 4°C with primary antibodies [mouse IgG1 monoclonal anti-type IIA MHC (clone SC-71; 1:3), mouse IgM monoclonal anti-type IIB MHC (clone BF-F3, 1:1), or mouse IgM monoclonal MHC IIX (clone 6H1, 1:1)]. All MHC antibodies were obtained from the Developmental Studies Hybridoma Bank (University of Iowa, Ames, IA). The next morning, sections were washed three times in PBS for 5 min. Secondary antibodies (biotinylated anti-mouse IgG or IgM; Vector Laboratories) were incubated with the sections for 1 h at 37°C, and sections were washed again three times for 5 min in PBS. Avidin-biotin complex (ABC) system (Vector Laboratories) was used to detect the biotinylated secondary antibody by incubating ABC solution at room temperature for 30 min. Sections were washed three times for 5 min in PBS and visualized by incubating in diaminobenzidine solution for 6 min (Vectastain DAB kit, Vector Laboratories). The sections were rinsed with distilled H2O three times, dried, and mounted by coverslips with a mounting media. At least 10 random, non-overlapping digital images at ×40 magnification were taken, and myofiber cross-sectional area (CSA) was quantified in fibers stained positive for MHC type IIA, IIX, and IIB using imaging software (ImageJ; NIH). Images from deep and superficial regions of the TA muscle were included to exclude sampling bias due to regional fiber-type differences (46). Each fiber was traced with a handheld mouse, and the number of pixels traced was calibrated to a defined area in square micrometers. An average of 100 myofibers consisting of both deep and superficial muscle regions per animal were manually traced, which was determined to be an appropriate fiber number as there were no further changes in the standard deviation of myofiber areas observed. The analyses were performed by an investigator blinded to the treatment groups.
Succinate dehydrogenase activity.
Succinate dehydrogenase (SDH) enzyme activity was performed as previously described to determine muscle oxidative capacity (20). Briefly, frozen cross sections were air-dried for 10 min, followed by incubation in a solution containing 0.2 M phosphate buffer (pH 7.4), 0.1 M MgCl2, 2.4 mM nitroblue tetrazolium, and 0.2 M succinic acid for 45 min at 37°C. Sections were then washed in distilled H2O for 3 min, dehydrated in 50% ethanol for 2 min, and mounted for viewing with mounting media. Digital photographs were taken from each section at ×25 magnification, and fibers were manually traced with imaging software (ImageJ; NIH). Similar to MHC analysis, whole TA muscle cross sections were examined, since fibers with high oxidative capacity are more abundant in the deep region of the muscle compared with the superficial region (46). The images were converted to 8-bit gray scale (range of gray levels 0–255) images, and an integrated optical density was created by subtracting the background intensity from each myofiber. Thresholds corresponding to high SDH enzyme activity were set manually and uniformly across all images, and myofibers were classified as having high or low SDH enzyme activities. All muscle regions were included in analysis, and the percentage of high SDH enzyme activity myofibers is expressed as the percentage per total number of myofibers. The analyses were performed by an investigator blinded to the treatment groups.
Western blotting.
Western blot analysis was performed as previously described (24). Frozen TA muscle was homogenized in ice-cold Mueller buffer, and protein concentration was determined by the Bradford method. The nonstimulated TA muscle from the control leg of C57BL/6 and ApcMin/+ mice were run on the same gel to determine differences due to cancer cachexia. TA muscles from the nonstimulated leg and HFES leg were run on the same gel to determine the effect of HFES in ApcMin/+ mice. Crude muscle homogenates were fractionated on 6–15% SDS-polyacrylamide gels and transferred to polyvinylidene difluoride membranes overnight. Membranes were stained with Ponceau red to verify equal loading and transfer for each gel. Membranes were blocked at room temperature for 1–2 h in 5% Tris-buffered saline with 0.1% Tween-20 (TBST) milk. Primary antibodies for phospho-NF-κB (S468), total NF-κB, phospho-STAT3 (Y750), total STAT3, phospho-AMPK (T172), total AMPK, phospho-ERK1/2 (T202/Y204), total ERK1/2, and atrogin-1 (ECM Biosciences) were diluted 1:1,000 to 1:2,000 in 5% TBST-milk, followed by a 2-h incubation with membranes at room temperature. Anti-rabbit or anti-mouse IgG horseradish-peroxidase-conjugated secondary antibody was incubated with the membranes at 1:2,000 dilutions for 1 h in 5% TBST-milk at room temperature. Enhanced chemiluminescence (GE Healthcare Life Sciences, Piscataway, NJ) was used to visualize the antibody-antigen interactions. Images were digitally scanned and quantified by densitometry using imaging software (ImageJ; NIH). All antibodies were from Cell Signaling, unless otherwise stated.
Plasma IL-6 concentration.
Plasma IL-6 concentrations were determined as previously described (24). A commercially available IL-6 enzyme-linked immunosorbent assay kit was obtained from BD Biosciences (San Diego, CA), and the manufacturer's protocol was followed. Briefly, a Costar clear 96-well plate (Corning, NY) was coated with IL-6 capture antibody and allowed to incubate overnight. The next morning, the plate was blocked with assay diluent buffer and washed, and IL-6 standards and plasma samples were added in duplicate to the plate. The plate was again washed and streptavidin-horseradish peroxidase reagent was added to each well. After several washes, 3,3′,5,5′-tetramethylbenzidine substrate was added, and the reaction was developed for 20 min. The reaction was stopped with sulfuric acid, and absorbance was read in a Bio-Rad iMark plate reader (Hercules, CA) at 450 nm.
Statistical analysis.
Results are reported as the means ± SE. A repeated-measures two-way ANOVA was performed to determine differences between cachexia and HFES in C57BL/6 and ApcMin/+ mice using the control leg and stimulated leg from each mouse. Post hoc analyses were performed with Student-Newman-Keuls methods when appropriate. Unpaired Student's t-test was performed to determine differences in control leg TA protein expression between C57BL/6 and ApcMin/+ mice. Paired Student's t-test was performed to determine differences between the control leg and stimulated leg TA protein expression within ApcMin/+ mice. The accepted level of significance was set at P < 0.05 for all analysis. Statistical analysis was performed using SigmaStat version 3.5 (Systat Software, Richmond, CA).
RESULTS
Body weight, muscle mass, and plasma IL-6 levels in C57BL/6 and ApcMin/+ mice.
Male C57BL/6 and ApcMin/+ mice performed seven bouts of HFES over 2 wk (Fig. 1A). There were no differences in peak body weight between C57BL/6 and ApcMin/+ before training (Table 1). ApcMin/+ mice had initiated cachexia at the beginning of HFES and continued to lose body weight during the training period (Table 1). ApcMin/+ mice had smaller TA and gastrocnemius muscle mass (−26% and −30%, respectively) at death compared with C57BL/6 mice. HFES increased the eccentrically contracted TA muscle mass, regardless of genotype, but there was no effect on the concentrically contracted gastrocnemius muscle mass (Table 1). Plasma IL-6 was elevated before training and was further increased posttraining in ApcMin/+ mice (Fig. 1B). Plasma IL-6 was below the level of detection pre- and posttraining in C57BL/6 mice. There were no differences in tibia length, a measure of body size, between C57BL/6 and ApcMin/+ mice.
Table 1.
Effect of multiple bouts of HFES on body weight and muscle mass in C57BL/6 and ApcMin/+ mice
| C57BL/6 | ApcMin/+ | |
|---|---|---|
| Body weight, g | ||
| Peak | 26.0 ± 0.3 | 24.8 ± 0.4 |
| Pretraining | 25.8 ± 0.8 | 23.0 ± 0.6 a |
| Posttraining | 25.8 ± 0.7 | 22.1 ± 0.6 a,b,c |
| Body weight change, %change from peak | ||
| Pretraining | −1 ± 1 | −7 ± 2 b |
| Posttraining | −1 ± 1 | −11 ± 2 b |
| Tibialis anterior, mg | ||
| Control | 44 ± 2 | 33 ± 3 d |
| HFES | 47 ± 1 e | 34 ± 2 de |
| Gastrocnemius, mg | ||
| Control | 120 ± 5 | 84 ± 8 d |
| HFES | 119 ± 4 | 83 ± 9 d |
| Tibia length, mm | 17.1 ± 0.1 | 17.0 ± 0.1 |
| n | 9 | 9 |
Values are means ± SE; n, no. of mice. HFES, high-frequency electrical stimulation. Statistical significance was set at P < 0.05: significantly different from
peak body weight,
C57BL/6, and
pretraining body weight; main effect of
ApcMin/+ and
HFES.
Effect of cachexia and HFES on TA MHC type IIA, IIX, and IIB myofiber CSA in C57BL/6 and ApcMin/+ mice.
The CSA and size distribution of MHC type IIA, IIX, and IIB myofibers were examined in the TA muscles of C57BL/6 and ApcMin/+ mice. Cachexia decreased the mean CSA of all fiber types compared with C57BL/6 mice (Fig. 2). HFES increased the mean CSA of all fiber types, regardless of genotype (Fig. 2). When myofiber size distribution is examined, which can demonstrate remodeling related to small and large myofibers, HFES increased the percentage of large-diameter myofibers and decreased the percentage of small-diameter myofibers in all fiber types, regardless of genotype (data not shown). These data demonstrate that all fiber types can hypertrophy after HFES, regardless of cachexia severity.
Fig. 2.
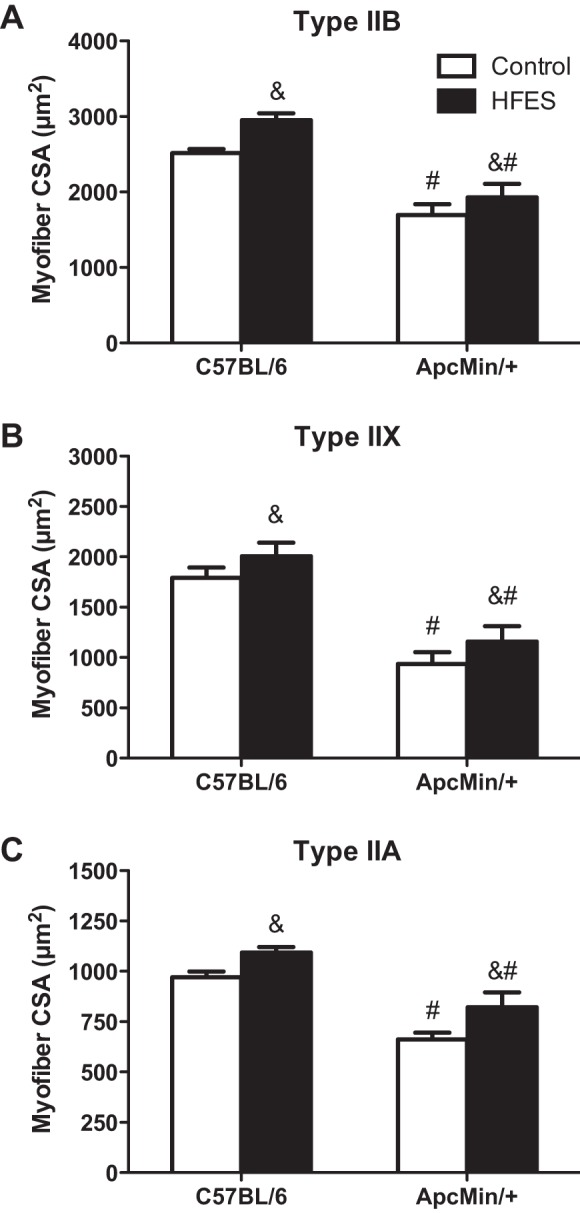
Effects of HFES on tibialis anterior (TA) myosin heavy chain type IIB (A), IIX (B), and IIA (C) myofiber cross-sectional area (CSA) in C57BL/6 and ApcMin/+ mice. All analysis was performed in the TA muscle. CSA values are means ± SE. Statistical significance was set at P < 0.05: main effect of &HFES and #ApcMin/+ mice.
Effect of cachexia and HFES on TA muscle extracellular matrix remodeling, regeneration, and myofiber oxidative capacity in C57BL/6 and ApcMin/+ mice.
TA muscle extracellular matrix remodeling and regeneration were examined in C57BL/6 and ApcMin/+ mice. While there was an increase in the percentage of noncontractile tissue in ApcMin/+ mice compared with C57BL/6 mice (Fig. 3, A and B), HFES attenuated the increase in noncontractile tissue in ApcMin/+ mice (Fig. 3, A and B). The percentage of myofibers containing centralized nuclei was not different between ApcMin/+ and C57BL/6 mice (Fig. 3C). HFES did not affect the percentage of myofibers containing centralized nuclei in C57BL/6 and ApcMin/+ mice (Fig. 3C). SDH activity, an index of myofiber oxidative metabolism (20, 56), was examined in the TA muscle of C57BL/6 and ApcMin/+ mice. There was a decrease in the percentage of high SDH activity myofibers in ApcMin/+ mice compared with C57BL/6 mice (Fig. 3D). HFES increased the percentage of high SDH activity myofibers, regardless of genotype (Fig. 3D). Cachectic muscle had a lower percentage of high SDH activity myofibers and an increased extracellular matrix area, but there was not evidence for myofiber regeneration, as indicated by centrally located nuclei. Coinciding with the induction of myofiber hypertrophy, HFES suppressed extracellular matrix remodeling and induced SDH activity in cachectic muscle without inducing myofiber regeneration.
Fig. 3.

Effects of HFES on TA muscle extracellular matrix remodeling, regeneration, and myofiber oxidative capacity in C57BL/6 and ApcMin/+ mice. A: representative images of hematoxylin- and eosin-stained TA muscle cross sections. Scale bar, 50 μm. The percentage of noncontractile tissue (B), centralized nuclei (C), and high succinate dehydrogenase (SDH) enzyme activity myofibers (D) in C57BL/6 and ApcMin/+ mice is shown. All analysis was performed in the TA muscle. Values are means ± SE. Statistical significance was set at P < 0.05: †difference between all groups; ‡difference between all groups; &main effect of HFES; and #main effect of ApcMin/+ mice.
Effect of cachexia and HFES on TA muscle cachectic signaling in ApcMin/+ mice.
Cachectic signaling related to muscle wasting was examined in the control TA muscle of C57BL/6 and ApcMin/+ mice. Cachexia increased control muscle STAT3 (Y705) phosphorylation ninefold, AMPK (T172) phosphorylation sixfold, ERK1/2 (T202/Y204) phosphorylation eightfold, and atrogin-1 protein expression ninefold compared with C57BL/6 mice (Fig. 4A). The phosphorylation of NF-κB p65 (S468) was highly variable in ApcMin/+ mice and did not reach statistical significance compared with C57BL/6 mice (Fig. 4A). The effect of HFES on cachectic muscle signaling was examined in ApcMin/+ mice. While NF-κB p65 (S468) phosphorylation was highly variable in the control muscle of ApcMin/+ mice, HFES decreased NF-κB p65 (S468) phosphorylation in ApcMin/+ mice (Fig. 4B). Furthermore, AMPK (T172) phosphorylation was decreased by HFES in ApcMin/+ mice (Fig. 4B). STAT3 (Y705) phosphorylation, ERK1/2 (T202/Y204) phosphorylation, and atrogin-1 protein expression were not altered by HFES in ApcMin/+ mice (Fig. 4B). In summary, while cachexia activated several signaling pathways associated with skeletal muscle wasting, repeated eccentric muscle contractions induced by HFES attenuated muscle signaling related to inflammation and energy status in cachectic muscle.
Fig. 4.

Effects of HFES on TA muscle cachectic signaling in ApcMin/+ mice. A: representative Western blot (right) and quantification of the ratio (left) of phosphorylated (p) and total forms of NF-κB, signal transducer and activator of transcription 3 (STAT3), 5′-adenosine monophosphate-activated protein kinase (AMPK), and extracellular signal-regulated kinase-1/2 (ERK1/2), and total atrogin-1 protein expression in the nonstimulated control leg of C57BL/6 and ApcMin/+ mice. Samples from the nonstimulated control leg of C57BL/6 and ApcMin/+ mice were run on the same gel. Data are normalized to the nonstimulated control leg of C57BL/6 mice. Values are means ± SE. Statistical significance was set at P < 0.05. *difference between C57BL/6 and ApcMin/+ mice. B: representative Western blot (right) and quantification of the ratio (left) of phosphorylated and total forms of NF-κB, STAT3, AMPK and ERK1/2, and total atrogin-1 protein expression in the nonstimulated control leg and HFES leg of ApcMin/+ mice. Samples from the nonstimulated control leg and HFES leg of ApcMin/+ mice were run on the same gel. Data are normalized to the nonstimulated control leg of ApcMin/+ mice. All analysis was performed in the TA muscle. Values are means ± SE. Statistical significance was set at P < 0.05: *difference between nonstimulated control leg and HFES leg. IOD, integrated optical density.
DISCUSSION
Physical activity and exercise interventions have clear therapeutic implications for treating and preventing muscle wasting associated with cancer cachexia. Whole body treadmill exercise can prevent muscle mass loss in tumor-bearing mice (14, 45, 50) and also rescue suppressed muscle oxidative metabolism at the initiation of cachexia (49, 59). However, the inability of severely cachectic mice to perform voluntary exercise has limited our understanding of how cachexia alters the muscle response to exercise. Stimulated low-frequency concentric muscle contractions have demonstrated that cachectic muscle's metabolic response is disrupted compared with healthy muscle (48); however, the growth response to stimulated high-frequency eccentric muscle contractions is not well understood. While repeated bouts of eccentric muscle contraction performed during the duration of tumor development in mice can prevent muscle protein loss (2), the capacity of cancer cachexia to disrupt the muscle's growth response to eccentric contractions has not been determined. We report the novel findings that repeated eccentric contractions after the initiation of cachexia can induce myofiber growth in the cachectic TA muscle. This growth occurred in the presence of the cancer environment that induced severe wasting in the contralateral noncontracted muscle. Collectively, these results add to the field by demonstrating that cachectic muscle has the capacity to initiate myofiber growth in response to repeated bouts of eccentric muscle contraction, despite the presence of the cachectic environment.
HFES of the sciatic nerve produces eccentric contractions of the rodent TA and EDL muscles and has been used to prevent muscle protein loss in tumor-bearing mice (2). However, cachectic skeletal muscle's capacity for myofiber growth induced by HFES after the initiation of cachexia is not currently understood. The ApcMin/+ mice in our present study demonstrated characteristics consistent with the initiation of cachexia at the start of HFES, and cachexia symptoms progressed during the 2-wk treatment period. While body weight was reduced and plasma IL-6 levels were elevated before the first bout of HFES, ApcMin/+ mice continued to lose body weight, and circulating IL-6 levels continued to increase during the treatment period. There was also significant muscle wasting as myofiber CSA was decreased in all fiber types of ApcMin/+ mice. Although fiber type has been reported to affect wasting susceptibility (32), we did not find preferential wasting due to fiber type, which is consistent with our laboratory's previous studies in ApcMin/+ mice (6). While functional overload-induced muscle growth involves the hypertrophy of all fiber types (17), specific fiber-type hypertrophy after HFES has not been reported in either healthy or cachectic muscle. We report that HFES induced myofiber growth in all fiber types examined in mice, regardless of cachexia. These results demonstrate that repeated bouts of HFES can induce a muscle growth response in the presence of the tumor-induced cachectic environment and appreciably extend previous observations related to overload-induced muscle growth in tumor-bearing rats (42, 43). While there is a growing body of research demonstrating that overload can induce growth in tumor-bearing rodents, there is currently an extremely limited understanding of the cachectic environment-induced signaling mechanisms that muscle contraction could counteract. Whether HFES could override cachexia-induced suppression of anabolic signaling or suppress catabolic signaling in muscle, despite the presence of the cachectic systemic environment, remains to be investigated.
The muscle's microenvironment includes the local levels of secreted cytokines and growth factors, which regulate the activity of many cell types found in muscle (11). This regulatory mix can alter the muscle's extracellular matrix and affect the capacity for muscle regeneration and growth (33, 51). There are currently gaps in our understanding of how cancer cachexia affects the muscle microenvironment, and whether cancer-induced alterations to extracellular matrix remodeling could impair growth-related processes. Increased muscle regeneration has been reported in human pancreatic cancer and C26 tumor-bearing mice (1, 3, 23). We report the novel finding that cachexia progression increased TA muscle noncontractile tissue, and this induction of noncontractile tissue was not associated with histological evidence of muscle degeneration and regeneration. This is in contrast to reported findings in the cachectic oxidative soleus muscle, which demonstrated muscle regeneration without extracellular matrix expansion (36). These differences between the glycolytic TA muscle and oxidative soleus muscle may represent the differential effect of muscle phenotype on the cachectic response. Interestingly, we demonstrate in the present study that the dysregulation of the extracellular matrix did not disrupt HFES-induced myofiber growth, and HFES resulted in the attenuation of noncontractile tissue expansion in cachectic skeletal muscle. Further research is needed to determine the implications of disrupted extracellular remodeling during the progression of cancer cachexia and the role of muscle contraction on extracellular matrix regulation in cachectic skeletal muscle. Unaccustomed eccentric muscle contractions promote greater muscle damage compared with concentric muscle contractions, and muscle injury and inflammatory cell infiltration have been reported in rat TA muscle following an acute bout of HFES (35). Interestingly, we did not find evidence of overt muscle damage 48 h following the last stimulation procedure in either wild-type or ApcMin/+ mice. It is reasonable that early eccentric contraction-induced muscle damage was resolved before the end of the training study, or the initial contraction stimulus elicited protection from subsequent eccentric contraction-induced damage. Additional studies are needed to clearly establish how cachexia severity influences the susceptibility to eccentric contraction-induced muscle damage. Nonetheless, progressive eccentric exercise training can increase strength and mobility in cancer survivors (30, 31), and these same benefits can be achieved in prostate cancer survivors on androgen deprivation therapy (19). While these studies provide initial evidence for the use of eccentric muscle contractions in cancer patients, the therapeutic potential of resistance exercise to improve muscle mass and function in cachectic cancer patients remains to be determined.
AMPK is a potent regulator of skeletal muscle metabolism (27), which can be activated by cellular energy status, calcium levels, and IL-6/STAT3 signaling (21). Related to muscle wasting, AMPK has a role in the regulation of mitochondrial biogenesis, autophagy, and protein turnover (37). While several mouse models of cancer cachexia demonstrate chronically elevated AMPK activity in skeletal muscle (47, 48, 57, 58), the direct signals activating AMPK during the progression of cachexia have not been established. In the present study, cachectic ApcMin/+ mice had elevated AMPK signaling in the nonstimulated control muscle, which is consistent with previous studies (48, 57, 58). The physiological consequences of this activation are also not fully understood. In healthy muscle, exercise can stimulate AMPK activity and subsequent induction of autophagy and mitochondrial biogenesis (22, 25). Interestingly, mitochondrial biogenesis was not associated with the induction of AMPK in cachectic muscle, where muscle oxidative metabolism was suppressed (56, 57). Consistent with prior studies, we report that the induction of muscle AMPK phosphorylation was associated with chronically elevated STAT3. The direct relationship between AMPK activation and STAT3 signaling in cachectic muscle has not been firmly established. Our laboratory has previously found that IL-6-induced STAT3 and AMPK activity coincide with mTOR suppression in vivo and in vitro (58). However, AMPK inhibition, but not STAT3 inhibition, can rescue IL-6-induced suppression of mTOR signaling in myotubes (58). Additionally, treadmill exercise at the initiation of cachexia can attenuate AMPK phosphorylation, independent of reductions in elevated muscle STAT3 signaling (49). We have extended these observations by demonstrating that repeated HFES suppressed chronic activation of AMPK, although STAT3 signaling remained significantly increased in severely cachectic skeletal muscle. While we provide initial evidence that eccentric contractions attenuated AMPK signaling in cachectic muscle, mechanistic studies are needed to determine whether these changes were associated with altered regulation of mitochondrial biogenesis, autophagy, and protein turnover. Additionally, further work is needed to determine the relationship between AMPK and STAT3 signaling for the suppression of anabolic signaling and muscle oxidative metabolism in cachectic skeletal muscle.
In summary, we demonstrate that cachectic skeletal muscle retains the anabolic plasticity to initiate growth in response to eccentric muscle contractions. We report that repeated eccentric contractions after the initiation of cachexia can induce myofiber growth, regardless of fiber type in cachectic muscle. Coinciding with myofiber growth were reduced extracellular matrix remodeling and the suppression of chronically activated AMPK signaling. These data suggest that cachectic skeletal muscle can initiate growth in response to repeated bouts of eccentric muscle contractions, despite the presence of a systemic cachectic environment. Further research is needed to determine whether the initial improvements in myofiber growth can be sustained over time with the progression of cancer cachexia. Additionally, whether exercise training enhances skeletal muscle's response to an anabolic stimulus, such as feeding, will improve our efforts for treating the cachectic cancer patient.
GRANTS
This work was supported by National Institutes of Health Grants R01 CA-121249A501 and P20 RR-017698 (J. A. Carson), SPARC Graduate Research Grant from the Office of the Vice President for Research at the University of South Carolina (J. P. Hardee), and an ACSM Foundation Research Grant from the American College of Sports Medicine Foundation (J. P. Hardee).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.P.H., S.S., and J.A.C. conception and design of research; J.P.H., J.E.M., S.G., S.S., K.L.H., M.J.P., and D.K.F. performed experiments; J.P.H., J.E.M., and J.A.C. analyzed data; J.P.H. and J.A.C. interpreted results of experiments; J.P.H. and J.A.C. prepared figures; J.P.H., J.E.M., S.G., S.S., K.L.H., M.J.P., D.K.F., and J.A.C. drafted manuscript; J.P.H., J.E.M., S.G., S.S., K.L.H., M.J.P., D.K.F., and J.A.C. edited and revised manuscript; J.P.H., J.E.M., S.G., S.S., K.L.H., M.J.P., D.K.F., and J.A.C. approved final version of manuscript.
ACKNOWLEDGMENTS
The type IIA and IIB MHC monoclonal antibodies developed by S. Schiaffino, and the type IIX MHC monoclonal antibodies developed by C. Lucas were obtained from the Developmental Studies Hybridoma Bank, created by the National Institute of Child Health and Human Development of the National Institutes of Health and maintained at The University of Iowa, Department of Biology, Iowa City, IA 52242.
REFERENCES
- 1.Acharyya S, Butchbach ME, Sahenk Z, Wang H, Saji M, Carathers M, Ringel MD, Skipworth RJ, Fearon KC, Hollingsworth MA, Muscarella P, Burghes AH, Rafael-Fortney JA, Guttridge DC. Dystrophin glycoprotein complex dysfunction: a regulatory link between muscular dystrophy and cancer cachexia. Cancer Cell 8: 421–432, 2005. [DOI] [PubMed] [Google Scholar]
- 2.al-Majid S, McCarthy DO. Resistance exercise training attenuates wasting of the extensor digitorum longus muscle in mice bearing the colon-26 adenocarcinoma. Biol Res Nurs 2: 155–166, 2001. [DOI] [PubMed] [Google Scholar]
- 3.Aulino P, Berardi E, Cardillo VM, Rizzuto E, Perniconi B, Ramina C, Padula F, Spugnini EP, Baldi A, Faiola F, Adamo S, Coletti D. Molecular, cellular and physiological characterization of the cancer cachexia-inducing C26 colon carcinoma in mouse. BMC Cancer 10: 363, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baar K, Esser K. Phosphorylation of p70(S6k) correlates with increased skeletal muscle mass following resistance exercise. Am J Physiol Cell Physiol 276: C120–C127, 1999. [DOI] [PubMed] [Google Scholar]
- 5.Baltgalvis KA, Berger FG, Pena MM, Davis JM, Muga SJ, Carson JA. Interleukin-6 and cachexia in ApcMin/+ mice. Am J Physiol Regul Integr Comp Physiol 294: R393–R401, 2008. [DOI] [PubMed] [Google Scholar]
- 6.Baltgalvis KA, Berger FG, Pena MM, Mark Davis J, White JP, Carson JA. Activity level, apoptosis, and development of cachexia in ApcMin/+ mice. J Appl Physiol (1985) 109: 1155–1161, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bandstra ER, Thompson RW, Nelson GA, Willey JS, Judex S, Cairns MA, Benton ER, Vazquez ME, Carson JA, Bateman TA. Musculoskeletal changes in mice from 20–50 cGy of simulated galactic cosmic rays. Radiat Res 172: 21–29, 2009. [DOI] [PubMed] [Google Scholar]
- 8.Bolster DR, Crozier SJ, Kimball SR, Jefferson LS. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J Biol Chem 277: 23977–23980, 2002. [DOI] [PubMed] [Google Scholar]
- 9.Bonetto A, Aydogdu T, Jin X, Zhang Z, Zhan R, Puzis L, Koniaris LG, Zimmers TA. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. Am J Physiol Endocrinol Metab 303: E410–E421, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bonetto A, Aydogdu T, Kunzevitzky N, Guttridge DC, Khuri S, Koniaris LG, Zimmers TA. STAT3 activation in skeletal muscle links muscle wasting and the acute phase response in cancer cachexia. PLoS One 6: e22538, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Charge SB, Rudnicki MA. Cellular and molecular regulation of muscle regeneration. Physiol Rev 84: 209–238, 2004. [DOI] [PubMed] [Google Scholar]
- 12.Chen YW, Nader GA, Baar KR, Fedele MJ, Hoffman EP, Esser KA. Response of rat muscle to acute resistance exercise defined by transcriptional and translational profiling. J Physiol 545: 27–41, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cohen S, Nathan JA, Goldberg AL. Muscle wasting in disease: molecular mechanisms and promising therapies. Nat Rev Drug Discov 14: 58–74, 2015. [DOI] [PubMed] [Google Scholar]
- 14.Deuster PA, Morrison SD, Ahrens RA. Endurance exercise modifies cachexia of tumor growth in rats. Med Sci Sports Exerc 17: 385–392, 1985. [PubMed] [Google Scholar]
- 15.Egan B, Zierath JR. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab 17: 162–184, 2013. [DOI] [PubMed] [Google Scholar]
- 16.Fearon KC, Glass DJ, Guttridge DC. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab 16: 153–166, 2012. [DOI] [PubMed] [Google Scholar]
- 17.Goodman CA, Kotecki JA, Jacobs BL, Hornberger TA. Muscle fiber type-dependent differences in the regulation of protein synthesis. PLoS One 7: e37890, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grande AJ, Silva V, Riera R, Medeiros A, Vitoriano SG, Peccin MS, Maddocks M. Exercise for cancer cachexia in adults. Cochrane Database Syst Rev 11: CD010804, 2014. [DOI] [PubMed] [Google Scholar]
- 19.Hansen PA, Dechet CB, Porucznik CA, LaStayo PC. Comparing eccentric resistance exercise in prostate cancer survivors on and off hormone therapy: a pilot study. PM R 1: 1019–1024, 2009. [DOI] [PubMed] [Google Scholar]
- 20.Hardee JP, Puppa MJ, Fix DK, Gao S, Hetzler KL, Bateman TA, Carson JA. The effect of radiation dose on mouse skeletal muscle remodeling. Radiol Oncol 48: 247–256, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13: 251–262, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, An Z, Loh J, Fisher J, Sun Q, Korsmeyer S, Packer M, May HI, Hill JA, Virgin HW, Gilpin C, Xiao G, Bassel-Duby R, Scherer PE, Levine B. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 481: 511–515, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He WA, Berardi E, Cardillo VM, Acharyya S, Aulino P, Thomas-Ahner J, Wang J, Bloomston M, Muscarella P, Nau P, Shah N, Butchbach ME, Ladner K, Adamo S, Rudnicki MA, Keller C, Coletti D, Montanaro F, Guttridge DC. NF-kappaB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J Clin Invest 123: 4821–4835, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hetzler KL, Hardee JP, Puppa MJ, Narsale AA, Sato S, Davis JM, Carson JA. Sex differences in the relationship of IL-6 signaling to cancer cachexia progression. Biochim Biophys Acta 1852: 816–825, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hood DA. Invited Review: contractile activity-induced mitochondrial biogenesis in skeletal muscle. J Appl Physiol (1985) 90: 1137–1157, 2001. [DOI] [PubMed] [Google Scholar]
- 26.Hurley BF, Hanson ED, Sheaff AK. Strength training as a countermeasure to aging muscle and chronic disease. Sports Med 41: 289–306, 2011. [DOI] [PubMed] [Google Scholar]
- 27.Jager S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A 104: 12017–12022, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keller P, Keller C, Carey AL, Jauffred S, Fischer CP, Steensberg A, Pedersen BK. Interleukin-6 production by contracting human skeletal muscle: autocrine regulation by IL-6. Biochem Biophys Res Commun 310: 550–554, 2003. [DOI] [PubMed] [Google Scholar]
- 29.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13: 132–141, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lastayo PC, Larsen S, Smith S, Dibble L, Marcus R. The feasibility and efficacy of eccentric exercise with older cancer survivors: a preliminary study. J Geriatr Phys Ther 33: 135–140, 2010. [PMC free article] [PubMed] [Google Scholar]
- 31.LaStayo PC, Marcus RL, Dibble LE, Smith SB, Beck SL. Eccentric exercise versus usual-care with older cancer survivors: the impact on muscle and mobility–an exploratory pilot study. BMC Geriatr 11: 5, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li P, Waters RE, Redfern SI, Zhang M, Mao L, Annex BH, Yan Z. Oxidative phenotype protects myofibers from pathological insults induced by chronic heart failure in mice. Am J Pathol 170: 599–608, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Y, Foster W, Deasy BM, Chan Y, Prisk V, Tang Y, Cummins J, Huard J. Transforming growth factor-beta1 induces the differentiation of myogenic cells into fibrotic cells in injured skeletal muscle: a key event in muscle fibrogenesis. Am J Pathol 164: 1007–1019, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McClung JM, Mehl KA, Thompson RW, Lowe LL, Carson JA. Nandrolone decanoate modulates cell cycle regulation in functionally overloaded rat soleus muscle. Am J Physiol Regul Integr Comp Physiol 288: R1543–R1552, 2005. [DOI] [PubMed] [Google Scholar]
- 35.McLoughlin TJ, Mylona E, Hornberger TA, Esser KA, Pizza FX. Inflammatory cells in rat skeletal muscle are elevated after electrically stimulated contractions. J Appl Physiol (1985) 94: 876–882, 2003. [DOI] [PubMed] [Google Scholar]
- 36.Mehl KA, Davis JM, Berger FG, Carson JA. Myofiber degeneration/regeneration is induced in the cachectic ApcMin/+ mouse. J Appl Physiol (1985) 99: 2379–2387, 2005. [DOI] [PubMed] [Google Scholar]
- 37.Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol 13: 1016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 247: 322–324, 1990. [DOI] [PubMed] [Google Scholar]
- 39.Mounier R, Lantier L, Leclerc J, Sotiropoulos A, Pende M, Daegelen D, Sakamoto K, Foretz M, Viollet B. Important role for AMPKalpha1 in limiting skeletal muscle cell hypertrophy. FASEB J 23: 2264–2273, 2009. [DOI] [PubMed] [Google Scholar]
- 40.Nader GA, Esser KA. Intracellular signaling specificity in skeletal muscle in response to different modes of exercise. J Appl Physiol (1985) 90: 1936–1942, 2001. [DOI] [PubMed] [Google Scholar]
- 41.Narsale AA, Carson JA. Role of interleukin-6 in cachexia: therapeutic implications. Curr Opin Support Palliat Care 8: 321–327, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Norton JA, Lowry SF, Brennan MF. Effect of work-induced hypertrophy on skeletal muscle of tumor- and nontumor-bearing rats. J Appl Physiol Respir Environ Exercise Physiol 46: 654–657, 1979. [DOI] [PubMed] [Google Scholar]
- 43.Otis JS, Lees SJ, Williams JH. Functional overload attenuates plantaris atrophy in tumor-bearing rats. BMC Cancer 7: 146, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Patel TJ, Cuizon D, Mathieu-Costello O, Friden J, Lieber RL. Increased oxidative capacity does not protect skeletal muscle fibers from eccentric contraction-induced injury. Am J Physiol Regul Integr Comp Physiol 274: R1300–R1308, 1998. [DOI] [PubMed] [Google Scholar]
- 45.Penna F, Busquets S, Pin F, Toledo M, Baccino FM, Lopez-Soriano FJ, Costelli P, Argiles JM. Combined approach to counteract experimental cancer cachexia: eicosapentaenoic acid and training exercise. J Cachexia Sarcopenia Muscle 2: 95–104, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pullen AH. The distribution and relative sizes of three histochemical fibre types in the rat tibialis anterior muscle. J Anat 123: 1–19, 1977. [PMC free article] [PubMed] [Google Scholar]
- 47.Puppa MJ, Gao S, Narsale AA, Carson JA. Skeletal muscle glycoprotein 130's role in Lewis lung carcinoma-induced cachexia. FASEB J 28: 998–1009, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Puppa MJ, Murphy EA, Fayad R, Hand GA, Carson JA. Cachectic skeletal muscle response to a novel bout of low-frequency stimulation. J Appl Physiol (1985) 116: 1078–1087, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Puppa MJ, White JP, Velazquez KT, Baltgalvis KA, Sato S, Baynes JW, Carson JA. The effect of exercise on IL-6-induced cachexia in the Apc (Min/+) mouse. J Cachexia Sarcopenia Muscle 3: 117–137, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Salomao EM, Toneto AT, Silva GO, Gomes-Marcondes MC. Physical exercise and a leucine-rich diet modulate the muscle protein metabolism in Walker tumor-bearing rats. Nutr Cancer 62: 1095–1104, 2010. [DOI] [PubMed] [Google Scholar]
- 51.Sato K, Li Y, Foster W, Fukushima K, Badlani N, Adachi N, Usas A, Fu FH, Huard J. Improvement of muscle healing through enhancement of muscle regeneration and prevention of fibrosis. Muscle Nerve 28: 365–372, 2003. [DOI] [PubMed] [Google Scholar]
- 52.Thomson DM, Fick CA, Gordon SE. AMPK activation attenuates S6K1, 4E-BP1, and eEF2 signaling responses to high-frequency electrically stimulated skeletal muscle contractions. J Appl Physiol (1985) 104: 625–632, 2008. [DOI] [PubMed] [Google Scholar]
- 53.Tisdale MJ. Mechanisms of cancer cachexia. Physiol Rev 89: 381–410, 2009. [DOI] [PubMed] [Google Scholar]
- 54.Tong JF, Yan X, Zhu MJ, Du M. AMP-activated protein kinase enhances the expression of muscle-specific ubiquitin ligases despite its activation of IGF-1/Akt signaling in C2C12 myotubes. J Cell Biochem 108: 458–468, 2009. [DOI] [PubMed] [Google Scholar]
- 55.Veasey Rodrigues H, Baracos VE, Wheler JJ, Parsons HA, Hong DS, Naing A, Fu S, Falchoock G, Tsimberidou AM, Piha-Paul S, Chisholm G, Kurzrock R. Body composition and survival in the early clinical trials setting. Eur J Cancer 49: 3068–3075, 2013. [DOI] [PubMed] [Google Scholar]
- 56.White JP, Baltgalvis KA, Puppa MJ, Sato S, Baynes JW, Carson JA. Muscle oxidative capacity during IL-6-dependent cancer cachexia. Am J Physiol Regul Integr Comp Physiol 300: R201–R211, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.White JP, Baynes JW, Welle SL, Kostek MC, Matesic LE, Sato S, Carson JA. The regulation of skeletal muscle protein turnover during the progression of cancer cachexia in the Apc(Min/+) mouse. PLoS One 6: e24650, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.White JP, Puppa MJ, Gao S, Sato S, Welle SL, Carson JA. Muscle mTORC1 suppression by IL-6 during cancer cachexia: a role for AMPK. Am J Physiol Endocrinol Metab 304: E1042–E1052, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.White JP, Puppa MJ, Sato S, Gao S, Price RL, Baynes JW, Kostek MC, Matesic LE, Carson JA. IL-6 regulation on skeletal muscle mitochondrial remodeling during cancer cachexia in the ApcMin/+ mouse. Skelet Muscle 2: 14, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Witkowski S, Lovering RM, Spangenburg EE. High-frequency electrically stimulated skeletal muscle contractions increase p70s6k phosphorylation independent of known IGF-I sensitive signaling pathways. FEBS Lett 584: 2891–2895, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wong TS, Booth FW. Protein metabolism in rat gastrocnemius muscle after stimulated chronic concentric exercise. J Appl Physiol (1985) 69: 1709–1717, 1990. [DOI] [PubMed] [Google Scholar]
- 62.Wong TS, Booth FW. Skeletal muscle enlargement with weight-lifting exercise by rats. J Appl Physiol (1985) 65: 950–954, 1988. [DOI] [PubMed] [Google Scholar]
- 63.Zinna EM, Yarasheski KE. Exercise treatment to counteract protein wasting of chronic diseases. Curr Opin Clin Nutr Metab Care 6: 87–93, 2003. [DOI] [PubMed] [Google Scholar]